
Method Article
Капли на основе штриховое кодирование одну ячейку Transcriptomics взрослых тканей млекопитающих
* Эти авторы внесли равный вклад
В этой статье
Резюме
Этот протокол описывает общие процессы и контроль качества проверяет необходимые для подготовки здоровых взрослых клеток млекопитающих одной капли основе, высокая пропускная способность одной ячейки РНК-Seq препаратов. Также предоставляются последовательности параметров, чтения выравнивание и вниз по течению одноклеточных bioinformatic анализа.
Аннотация
Анализ экспрессии генов одну ячейку на тысячи отдельных ячеек в ткани или микроокружения является ценным инструментом для выявления клеточный состав, дискриминации функциональных состояний и молекулярные пути, лежащие в основе наблюдаемых ткани функции и поведения животных. Однако изоляции нетронутыми, здоровые клетки одного из взрослых тканей млекопитающих для последующего течению одноклеточного молекулярного анализа может быть сложным. Этот протокол описывает общие процессы и контроль качества проверяет, что необходимо для получения высокого качества взрослые одноклеточного препараты от нервной системы или кожи, включен последующих беспристрастной одноклеточного РНК последовательности и анализа. Также предоставляются рекомендации по течению bioinformatic анализа.
Введение
С развитием выдвижений в удобной Биоинформатика инструменты в течение последнего десятилетия3и высокую пропускную способность одной ячейки технологии1,2 возникло новое поле с высоким разрешением ген выражение анализа – Одноместный клеточной РНК последовательности (scRNA-Seq). Изучение экспрессии генов в одной ячейке впервые была разработана для выявления неоднородности в пределах определенных клеточных популяций, такие как в стволовых клеток или раковые клетки, или для выявления редких популяции клеток4,5, которые были недостижимой с помощью традиционных массовых РНК последовательности методов. Инструменты bioinformatic позволили идентификации новых подгрупп населения (сера)2, Визуализация ордена клетки вдоль psuedotime пространства (монокль)6, определение активных сигналов сетей внутри или между населением ( ЖИВОПИСНЫЕ)7, предсказание Ассамблеи сингл-клеток в искусственной 3D пространстве (сера и более)8. Эти новые и захватывающие анализы доступны для научного сообщества scRNA-Seq быстро становится новый стандартный подход для анализа выражения гена.
Несмотря на огромный потенциал scRNA-Seq технические skillsets, требуется производить чистую набора данных и точно интерпретировать результаты может быть сложным для новичков. Здесь основные, но всеобъемлющего протокола, начиная от изоляции единичных клеток от всей первичной тканей для визуализации и представления данных для публикации представлен (рис. 1). Во-первых изоляции здоровых единичных клеток может считаться сложным, как различные ткани различаются по степени их чувствительности к ферментативного пищеварения и последующие механические диссоциации. Этот протокол обеспечивает руководство в эти меры изоляции и идентифицирует важные качества контрольно-пропускных пунктах на протяжении всего процесса. Во-вторых понимание совместимость и требования между технологией одну ячейку и секвенирование нового поколения может быть запутанным. Этот протокол обеспечивает руководящие принципы для реализации платформы удобной, на основе капелька штриховое кодирование одноклеточных и выполнения последовательности. Наконец компьютерное программирование является важной предпосылкой для анализа транскриптомики Одноячеистые наборы данных. Этот протокол предоставляет ресурсы для начала работы с языком программирования R и предоставляет руководство по реализации двух популярных scRNA-Seq-R пакетов. Вместе этот протокол может направлять новичков в анализ scRNA-Seq для получения четких, интерпретируемых результатов. Этот протокол может корректироваться в большинстве тканей в мыши и главное, может быть изменен для использования с другими организмами, включая человеческие ткани. Коррективы в зависимости от ткани и пользователю будет необходимо.
Есть несколько соображений, чтобы иметь в виду, в то время как после этого протокола; включая, 1) после всех руководящих принципов контроля качества в шаги 1 и 2 настоящего Протокола рекомендуется обеспечить жизнеспособные одноклеточного подвеска всех клеток в образце интерес обеспечивая точное общее число клеток (кратко в Рисунок 2 ). После того, как это достигается, и если все оптимизированные условия соблюдаются, контроль качества шаги могут быть удалены (чтобы сэкономить время - сохранение качества РНК и сокращение ячеек потери). Подтверждение успешной изоляции одной высокой жизнеспособности клеток из ткани интерес очень рекомендую перед любой вниз по течению обработки. 2) так как некоторые типы клеток более чувствительны, чем другие, чтобы подчеркнуть, чрезмерная разобщенность методы могут непреднамеренно смещения населения, поэтому смешанным течению анализа. Нежный диссоциации без ненужных сотовой стрижка и пищеварение имеет решающее значение для достижения высоких урожаев сотовой и точное представление о составе ткани. Сдвига происходят во время действия Тритурация, СУИМ и ресуспендирования. 3) как с любой работой РНК, лучше представить, как мало дополнительных РНКазы в выборку можно во время подготовки. Это будет способствовать поддержанию высокого качества РНК. Используйте решения Ингибитор рибонуклеазы с промывки чистые инструменты и оборудования, которое не является РНКазы бесплатно, но избежать DEPC-лечение продуктами. 4) выполняют подготовку как можно быстрее. Это поможет сохранить высокое качество РНК и уменьшить смерти клетки. В зависимости от длины рассечение тканей и животных номер рассмотреть возможность начала несколько Анатомирование/препараты в то же время. 5) Подготовьте клетки на льду, когда можно поддерживать высокое качество РНК, сократить гибель клеток и медленно ячейки сигнализации и транскрипционный анализ активности. Хотя, ледяной обработка идеально подходит для большинства типов клеток, некоторые типы клеток (например, нейтрофилов) работают лучше при обработке при комнатной температуре. 6) Избегайте кальция, магния, ЭДТА и DEPC-лечение продукты во время подготовки ячейки.
протокол
Все протоколы, описанные здесь, в соответствии с и утверждена Комитетом уход животных в Университете Калгари.
1. разъединять ткани (1 день)
- Усыпить мышей с передозировкой Пентобарбитал натрия (и.п., 50 мг/кг) или в соответствующих случаях согласно протоколу животных этики. Затем удалить нежелательные волосы от спины и ног мыши и этанол стерилизуйте региона рассечение.
- Вскрыть ткани или микроокружения интерес. Для этого протокола мы используем кожи и нервных тканей для демонстрации обобщения капли на основе штриховое кодирование одну ячейку transcriptomics после взрослых ткани диссоциации.
- Для седалищного нерва используйте подробный протокол на Страттон et al. 9. Вкратце, вырезать кожу от задних региона обратно/ноги мыши. Сделайте надрез по длине бедра с лезвием стерильным скальпель. Использование тонкой щипцами и ножницы для выявления и удаления седалищного нерва.
- Для задней кожи используйте подробный протокол, найдены в Biernaskie и др. 10. Вкратце, вскрыть задней спинной кожи, делая надрезы с плечом к плечу, крестца и на спине с помощью тонкой щипцами и ножницы. Вырежьте кожу тонкими ломтиками (толщиной 0,5 см) с помощью лезвия стерильным скальпель.
- Вымойте ткань 2 раза с ледяной HBSS и удалить нежелательные соединительной ткани, жировые отложения или мусор под микроскопом рассечения.
- Для только дермы кожи плавают ломтики в dispase (5 мг/мл, 5 ед/мл) в HBSS за 30-40 мин при 37 ° C. Хирургическим отделяет эпидермис от дермы. Дальнейшее отделить использованием трипсина если интерес или отказаться от эпидермиса.
- Фарш образца на куски 1-2 мм с помощью лезвия стерильным скальпель и положить в свежей талой 2 мг/мл холодной коллагеназы IV фермента (2 мг/мл, 125 CDU/мг, в СМИ F12).
- Для нервных используйте ~ 500 мкл в 2 x седалищного нервов. Для кожи используйте ~ 8 мл на 1 x мышь назад кожи.
Примечание: Ткани необходимо полностью погружать в коллагеназы IV решения. Важно, что любые ферменты пищеварения обрабатываются, хранятся и надлежащим образом подготовлены. Если ферменты остаются при комнатной температуре для длительных периодов времени, одноклеточных изоляции потребует чрезмерных механических Тритурация и уменьшить жизнеспособность клеток. Коллагеназы-IV может также состоять в СМИ культуры клеток, где жизнеспособность клеток является наиболее оптимальным. Однако это может изменить активность фермента или transcriptional подписи так должны быть оптимизированы пользователь.
- Для нервных используйте ~ 500 мкл в 2 x седалищного нервов. Для кожи используйте ~ 8 мл на 1 x мышь назад кожи.
- Инкубируйте образец в фермента в ванне с 37 ° C за 30 минут с нежным пожимая каждые 10 мин. Шейкер, размещенных при 37 ° C является также соответствующие альтернативы.
- Нарезанных с P1000 дозаторов 20 - 30 раз в 30 мин после фермент дополнение.
- Повторите Тритурация каждые 30 мин до тех пор, пока решение появляется облачно и куски ткани главным образом отделить.
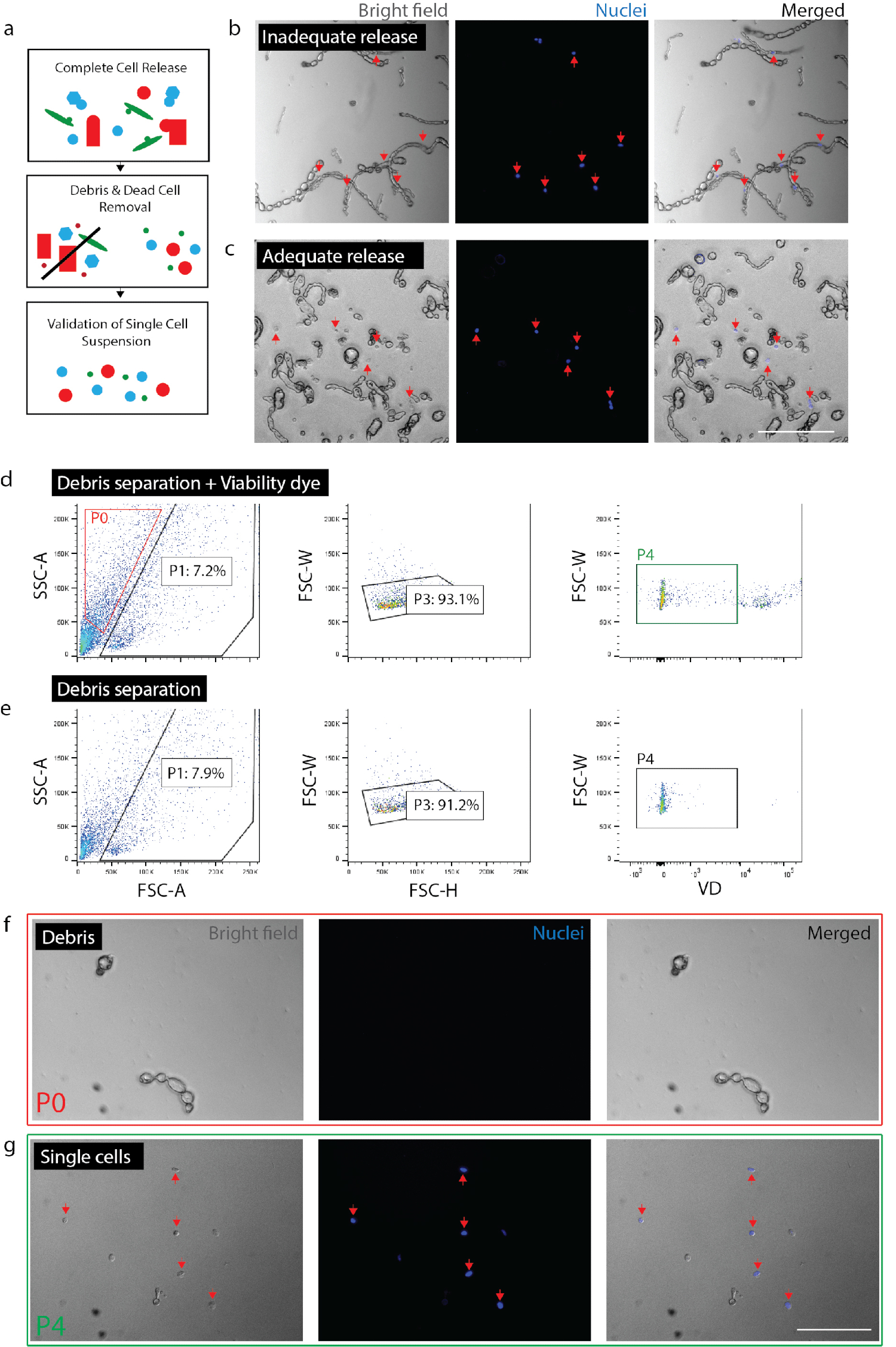
Примечание: Обеспечить полное освобождение клеток (рис. 2b, 2 c). Для подтверждения полного освобождения, плиты клетки с Nuc голубой (2 капли на 1 мл) и после 20 минут, проверьте под микроскопом, чтобы обеспечить все ядра связаны с одной клетки, а не мусор. Важно, чтобы проверить степень клеток выпуска в рамках данного эксперимента для каждого типа ткани или условие. В фиброзных тканей (например, хронической травмы) или ранен взрослых ткани релиз клеток будет резко отличаться от острой травмы или эмбриональных тканей. Это особенно важно, потому что некоторые типы клеток менее вероятно освободить от ткани, чем другие, исключая таким образом преференциально те клетки от течению анализа.- Для нервных, отделить ткани на 0,5-1,5 часов. Для кожи, отделить ткани для всего 2 часов (в последний час инкубации, добавить DNase (1 мг/мл) в образце кожи).
- Фильтр дважды с 40 мкм фильтром. Промойте фильтр с ледяной 1% BSA/HBSS.
- Центрифуга на 260 x g 8 мин. Затем удалите супернатант.
- Ресуспензируйте Пелле клеток в HBSS, содержащей 1% BSA, используя кончик широкий родила и место на льду. Объем ресуспендирования основывается на ткани тома (800 мг живого веса для кожи = 800 мкл тома; 10 мг мокрой вес для нервов = 100 мкл объем).
- При необходимости начать с низким ресуспендирования тома и затем отрегулировать при необходимости на основе расхода (события в секунду) в сортировщике СУИМ. Наиболее эффективно упорядочить плотности (максимизировать количество клеток, собранные во время сокращению времени) для коллекций – 3000-7000 события в секунду.
- При использовании жизнеспособности краситель, вывезти subaliquot для неокрашенных элемента управления. Затем добавить краситель жизнеспособность 1:15,000 (складе: 20 000 Нм/мкл) образца (1.3 Нм/мкл конечная концентрация) с использованием широкий родила подсказка для уменьшения стрижки.
Примечание: Очень важно для проверки степени гибели клеток в пределах данного эксперимента для каждого типа ткани или условие. Некоторые типы клеток в образце скорее умереть, чем другие, поэтому преференциально исключаются из вниз по течению анализа.- Проинкубируйте образцы с жизнеспособность краситель для 5-10 мин на льду в темноте. Затем добавить 4 мл ледяной 1% BSA/HBSS образца. Центрифуга на 260 x g на 8 минут, чтобы удалить избыток жизнеспособности красителя. Лечить subaliquot с не жизнеспособности красителя неокрашенных управления таким же образом.
2. изоляция жизнеспособных и здоровые клетки (1 день)
- Убедитесь, что средство СУИМ соответствует соответствующие активированных флуоресцированием клеток (FACS) параметры сортировки.
- Подготовка машины СУИМ заранее, чтобы обеспечить, что оно готово после завершения окончательного центрифуги на шаге 1 и убедитесь, что отсек коллекции хранится холодная с использованием блоков льда.
- Используйте следующие параметры: скорость потока: 1.0 (соответствует примерно 10 мкл/мин); Фильтр: 1.5 НД; Размер сопла: 100 мкм; Форвард разброс: 80-180 V (изменение при необходимости для того, чтобы отличить размер событий); Сторона разброс: 150-220 V (изменение при необходимости для того, чтобы отличить гранулярности/форма событий); Лазер: 100-400 V (изменение при необходимости для того, чтобы отличать жизнеспособности краситель положительных против негативные события и проверить это против управления краска не жизнеспособности); Ворота: Изменить по мере необходимости, чтобы убедиться, что собраны все клетки. Смотрите Рисунок 2d-2 g.
Примечание: Параметры СУИМ сильно зависит от типов клеток и сортировщика занятых и поэтому нужно быть оптимизированы пользователь.
- Подготовка 15 мл узкой нижней трубы с 8 мл ледяной 1% BSA/HBSS для образца коллекций. Статический внутри трубки и поверхностное натяжение может повлиять на эффективность улавливания. Инвертировать трубы до коллекций для обеспечения интерфейса между поверхностью жидкости и внутри трубки влажной.
Примечание: При работе с очень низкой сотовый номера, приспособиться к небольшая коллекция судна при необходимости. - После того, как собраны все клетки, центрифуга образца на 260 x g 8 мин.
Примечание: Перед центрифугированием, добавить 1% BSA/HBSS добиваться и мыть ячейки вниз от боковой поверхности и инвертировать/микс трубки сразу после СУИМ. - Ресуспензируйте Пелле клеток в 1% BSA/HBSS и держать на льду. Максимальный объем в образец, который совместим с шаг 3 обработки составляет 33,8 мкл, поэтому убедитесь, что объем разрежения/ресуспендирования окончательный ячейки подходит для получения идеального ячейки номер в 33,8 мкл. Другие варианты СМИ разрежения для этого шага (и все предыдущие разведений в 1% BSA/HBSS) включают DMEM и до 40% сыворотки, но избежать кальция, магния или ЭДТА, содержащих реагентов.
- Оставьте клетки на льду за минимальное количество времени. Идеально сотрудник должен подготовить все оборудование и реагенты для следующий шаг (шаг 3) во время окончательные шаги Шаг 2.
- Мобильный Подготовка критических проверки
- Подтверждают оценки числа клеток, полученные от СУИМ. В зависимости от типа ткани и диссоциации длины, мусора и клетки могут быть очень похожи по размеру и форме. Таким образом если не используется флуоресцентные репортер, СУИМ нельзя исключать все мусора. Рекомендуется, что счетчик окончательный клеток после того, как осуществляется сбор СУИМ понять какой процент событий (в зависимости от СУИМ), на самом деле клетки для данного препарата (рис. 2г). Выполните число ячеек с помощью Горяева или счетчик автоматизированных клеток (повторять дважды) и рассчитать процент жизнеспособных клеток, представленного всего событий, собранных согласно СУИМ машины.
- Проверка подготовка клетки. Проверить, что без крупных частиц (> 100 мкм) присутствуют, как они могут засорить оборудование в нижнем течении шагов. Неадекватные удаления мусора может угрожать опасность засорения одноклеточных microfluidic чип. Пластина оставшиеся ячейки с Nuc синий (как выше) чтобы убедиться, что нет большой мусора фрагменты присутствуют. Это также позволит для подтверждения, что клетки являются Сингулярные (т.е., не склеивая) дает уверенность что течению одноклеточного генетический анализ представляет отдельные клетки, вместо того, чтобы несколько ячеек.
- Решение относительно числа клеток в последовательности: существует широкий спектр взрослых клеток, тканей, полученных чисел на сэмпл, которые могут быть загружены в систему с до 8 образцов, которые могут выполняться в одно время. Авторы везде загружаются из 500 – 50 000 ячеек на сэмпл и получил хорошее качество scRNA-Seq наборов данных. Более дискуссия относительно наиболее подходящего номера ячейки для загрузки можно найти в разделе обсуждения. Окончательный вывод чисел последовательности ячеек в значительной степени зависит от качества сингл клетки изолированы. Загрузка 10 000 взрослых клеток, тканей, полученных может возвращать везде от 1000 до 4000 виртуализированного клетки (10-40% возврат). Если вы заинтересованы в последовательности номера высокого ячейки (~ 10 000 ячеек, максимальное количество, рекомендованных для этой системы), затем Загрузка 25 000-100 000 ячеек будет необходимо.
3. драгоценный камень поколения (гель шарик в эмульсии) и штриховое кодирование (1 день)
Примечание: Шаги 3-6 настоящего Протокола предназначены для использования в сочетании с наиболее распространенными на основе микрокапель одноклеточных платформы, производимый 10 X Genomics. Подробные руководящие принципы для шаги 3 и 4 изложены в производителя протокола (см. Протокол 3' хром одной клетки)11,12 и должны применяться в сочетании с настоящим Протоколом. Для достижения наилучших результатов необходимо выполнить шаг 3 сразу же после диссоциации (шаг 1) и клеток изоляции (шаг 2) шаги на день 1 настоящего Протокола.
- Подготовьте чип согласно производителя протокол 11,12. Эта платформа на основе микрокапель одноклеточных использует технологию что образцы ~ 750 000 штрихкоды отдельно проиндексировать транскриптом каждой ячейки. Это достигается путем разделения клетки на шарик геля в эмульсии (ГСМОС), где сгенерированный cDNA разделяют общие штрих-кодов. Во время генерации GEM доставляются клетки так, что большинство (90-99%) созданный камней содержат не клетки, а остальные, по большей части, содержат одну ячейку.
- Поместите фишку в держатель чип.
- Подготовка клетки хозяина микс на льду.
- Добавьте глицерин 50% неиспользуемых скважин и мкл 90 клетки хозяина смеси хорошо 1, 90 мкл гель бисера хорошо 2 и 270 мкл секционирования масло хорошо 3.
- Обложка чип с уплотнением.
- Загрузить чип и запустить в контроллере с одной ячейкой.
- Извлечь лоток, поместите фишку в лоток, убрать лоток и нажмите Play. Шарик в камень ячейку 3' гель включает грунты, содержащий последовательность частичного Illumina R1 (читать 1 Последовательность праймера), 16 нуклеотидов (nt) 10 x Barcode, 10 nt уникальной молекулярной идентификатор (UMI) и последовательность праймера поли dT. Во время выполнения гель бусинки в контроллере выпущен и смешивают с ячейки lysate и Мастер микс.
- Сбор 100 мкл пример и место в ПЦР-пробирку.
- Место ПЦР трубы в заранее установленные ПЦР машины и запустить ПЦР согласно комплект. После инкубации, будет включать камни полнометражных, перепутываются cDNA от Поли adenylated мРНК.
- После запуска место в-20 ° C ночь до 1 недели до предшествующего следующему шагу.
4. Очистка, усилители, строительство библиотеки и библиотеки количественной оценки (день 2 года)
Примечание: Подробные руководящие принципы для 4 шаги изложены в производителя протокол 11,12 и должны соблюдаться в связи с настоящим Протоколом.
- Используйте силана магнитные шарики для удаления оставшихся биохимические реагенты/грунты из реакционной смеси GEM.
- Усилить полнометражных, перепутываются cDNA для создания достаточной массы на строительство библиотеки.
- Оценки доходности ДНК. До начала строительства библиотеки оцените доходность ДНК образца. Это будет определить, сколько циклов для использования в шаг вниз по течению ПЦР (пример индекс PCR во время строительства библиотеки). В зависимости от содержания РНК данного образца, который может изменяться в зависимости от активации государств (например, управления vs ранения, и т.д.), тип клеток и клеток урожайности, рекомендованные цикла число может измениться.
- Для секвенирования ~ 3000 тканей, полученных клетки (никакого отношения к активации государств), авторы обнаружили, что 14 циклов (примеры: ~ 10-100 нг ДНК) является стандартным.
- Используйте Bioanalyzer для анализа ДНК. Обратитесь к пользователю руководство13.
- Фрагмент образца и выберите размер ДНК. До начала строительства библиотеки используйте ферментативные фрагментации и размер выделения протоколы для получения соответствующих cDNA ампликон размера.
- Подготовка образца для строительства библиотеки. В то время как R1 (читать 1 Последовательность праймера) добавляется к молекулам во время инкубации GEM; P5, P7 (образец индекса), и R2 (читать 2 Последовательность праймера) добавляются во время строительства библиотеки.
- Оценки доходности ДНК. Большинство объектов последовательности требуют представления окончательного библиотек, которые включают ДНК урожайность и качество информации. Таким образом запустите bioanalyzer после завершения всего протокола и перед транспортировкой для объекта последовательности.
- Хранить образцы на-80 ° C до 2 месяцев.
- До последовательности, количественно образцов, используя набор для количественного определения ДНК. Это может быть сделано на объекте последовательности.
5. Библиотека виртуализации (день 3 года)
Примечание: Платформа штриховое кодирование транскриптом одной ячейки, используемые в настоящем Протоколе генерирует Illumina совместимый в паре конец библиотеки начиная и заканчивая P5 и P7 последовательности. Хотя минимальная глубина необходимо разрешить тип ячейки личность может быть как мало, как 10 000 – 50 000 читает/ячейки15,16, ~ 100000 чтения/ячейку рекомендуется как оптимальный компромисс покрытия расходов для взрослых в естественных условиях клетки (имея в виду некоторые ячейки типы или минимально активированные клетки государства достигнет насыщенности на 30000-50000 чтение/ячейку).
- Транспортные кДНК библиотек на сухой лед на объект последовательности, оснащены соответствующей Illumina секвенсор.
- Предоставьте следующую информацию в объекте последовательности:
- Предоставить образец детали: образец индекса идентификаторы, соответствующие каждой библиотеки; видов; геномные базы данных для основной сборки (то есть, GRCm38 для мыши); electropherogram, показаны размеры фрагмента из bioanalyzer (между 200 и 9000 bp); cDNA концентрации (нг/мкл) и общая библиотека концентрации (Общая урожайность диапазон от 200-1400 нг); объем (мкл) образца.
- Предоставить последовательность запросов: количественно образцов, используя набор ДНК количественной оценки; Тип адаптера/индекса (TruSeq ДНК); Вид плиты (Eppendorf twin.tec, широкая юбка - рекомендуется для ДНК); секвенирование библиотеки технологии типа (10 x, полной последовательности инструкций и рекомендаций цикла)17.
- Запуск мелкой последовательности (опционально): исследования, анализ несколько биологических образцов выиграют от объединения образцов (агрегирование) для создания одного гена штрих матрица, содержащая данные из всех образцов. Свести к минимуму последствия партии между образцами при объединении, должны быть стандартизированы чтения глубина между различными библиотеками. Для того, чтобы сделать это, необходимо точное приближение одноклеточного чисел. Секвенсор MiSeq позволит мелкой последовательности и является экономически эффективных, практических способом для получения точных клеток оценок.
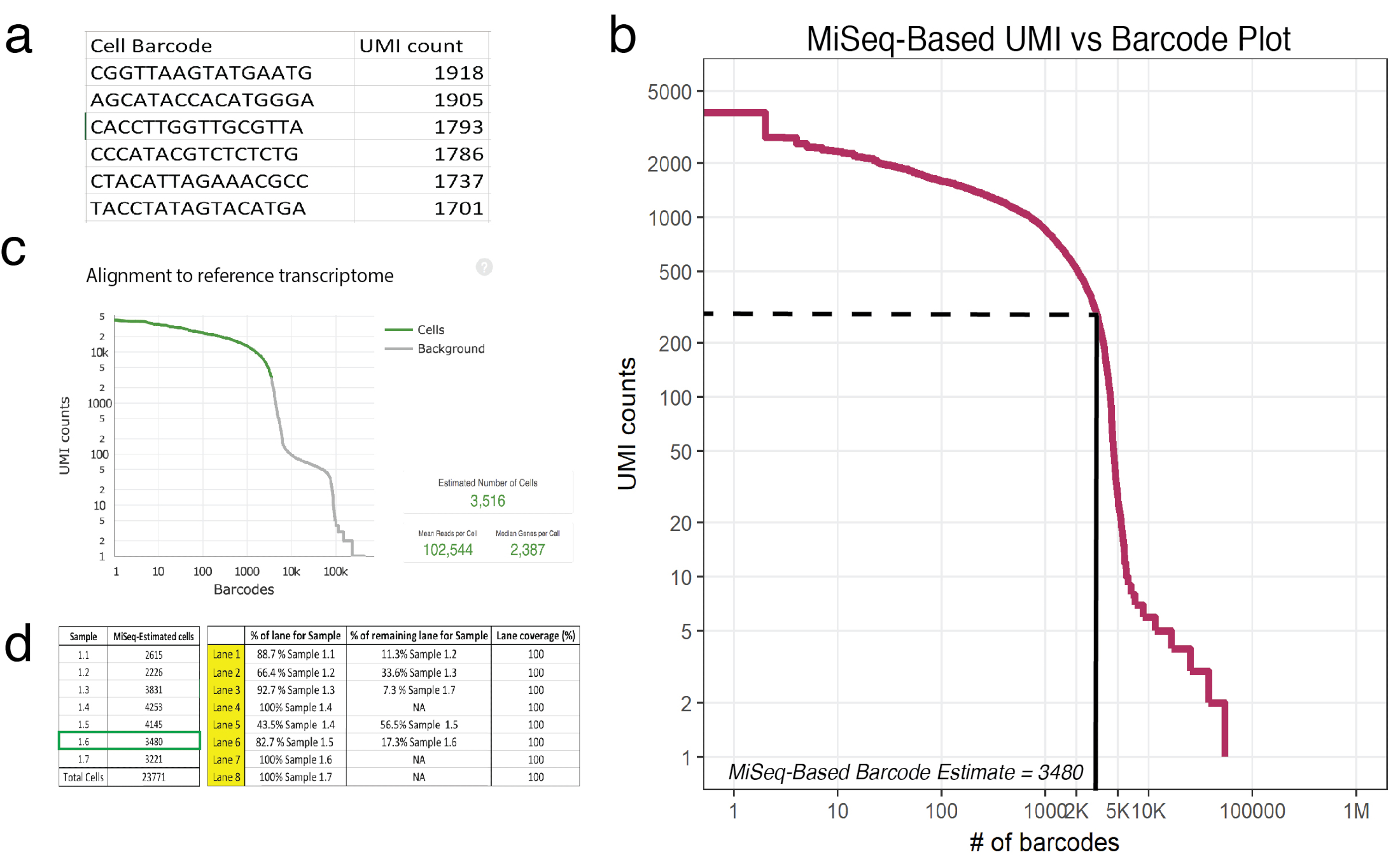
Примечание: Один проход с помощью программы sequencer MiSeq SR50 обеспечивает достаточный охват точно оценить приблизительно 20 000 ячеек. Этот запуск будет приблизительное количество UMI возместить за каждый уникальный штрих-код. На рисунке 3aпоказан заголовок пример (образец 1.6) вывода (.csv), штрих-код в листинге и его соответствующий UMI подсчитывает как определяется уверенно сопоставленных читает.- Проконсультироваться с bioinformatician познакомиться с R, язык программирования. Обратитесь к DataCamp учебники для больше информации18.
- Оценить необработанные данные, полученные от программы sequencer, используя предоставленный сценарий R как шаблон19. Необработанные данные относится к количеству UMIs сопоставляется каждый уникальный мобильный штрих-код. Сценарий читает что подсчитывает в CSV-файл, где первый столбец списка штрихкодов и второй столбец соответствующего UMI. Этот сценарий будет предоставлять участок (рис. 3b) а также предполагаемое количество перепутываются клеток в каждом образце. Настройка сценария убедитесь, что введенное количество UMI для данного образца в одной третьей точке первого резкое падение. В рисунке 3bэтот локоть падает около 225 UMIs соответствующий 3,480 перепутываются клеток.
- Сопоставимые на полную глубину виртуализации с помощью HiSeq (где 3,516 клетки были успешно виртуализации, рис. 3 c), мелкие последовательности оценки предсказали 3,480 клетки.
- Использования клеток приближений восстановления (от шага 5.3) или использования восстановления диаграммы по направлению производителя протокол20 планировать распределение Лейн для глубокого последовательности. Каждый образец должны получать сопоставимые покрытие, так что если мелкой секвенирование показывает, что существуют различные числа клеток в каждом образце (что бывает часто) затем Лейн распределения должен рассчитываться соответственно. Одна ячейка потока HiSeq (которая состоит из 8 дорожек) может последовательность читает пользовательские парных конца до 2,4 млрд. Пример потока клеток set-up представлен в 3d рисунок.
6. обработка чтения файлов
Примечание: Секвенирования одну ячейку 3' библиотеки, используя этот протокол создает необработанные данные в формате двоичного базового вызова (BCL). Клетки Ranger пакета используется для создания текстовых файлов FASTQ из BCL файлов, выполнять геномных и транскриптомики рядов, Джин графов, демультиплексирование и совокупность образцов. В этом разделе представлены основные шаги, которые позволяют пользователям загружать сырые BCL данные из объекта последовательности и создавать отфильтрованные ген штрих матрицы готовы к течению биоинформатики.
- Используйте централизованный сервер для запуска программы. BCL файлов FASTQ и большинство течению биоинформатики обработки требует значительной вычислительной мощности.
- Скачайте все raw чтения файлов на сервер (или FASTQ файлов, если они доступны).
- Проконсультироваться с администратором сервера, чтобы настроить учетную запись на централизованном сервере или кластере, а также познакомиться с Unix21.
- Используйте команду выборки для сервера операционной системы для загрузки всех файлов из объекта последовательности сервера.
- Большинство объектов последовательности предоставляют команду для загрузки файлов из безопасного пути, который может быть запущен из командной строки (см. пример ниже).
- Заменить «< имя_пользователя >» и «< пароль >» заполнители в командной строке с учетные данные.
Wget - O - «https://your_sequencing_facilitys_server.com/path_to_raw_read_files/--без печенье--без чек сертификат--после данных ' j_username = имя пользователя & j_password = пароль ' | Wget--без печенье--без чек сертификат--после данных ' j_username = имя пользователя & j_password = пароль ' - ди -
- Если только абсолютный путь к файлам предоставляется (т.е. https://your_sequencing_facilitys_server.com/path_to_raw_read_files/), вставьте этот путь в команда fetch.
- Распакуйте файлы: Если скачали конце файлов с расширением «.gz», он был сжат с помощью команды «gzip». Чтобы распаковать, запустить распакуйте команды в командной строке (см. пример ниже).
gunzip raw_read_files.gz - Скачайте последнюю версию клеток Ranger на сервер как автономные .tar22.
-
Критические: До загрузки убедитесь, что система Linux удовлетворяет минимальные требования23. Обеспечить как минимум 8-ядерный процессор с ОЗУ 64 ГБ и 1 ТБ свободного дискового пространства.
Примечание: Ranger клеток обеспечивает готовую человека и грызунов ссылку transcriptomes. Они могут быть изменены с помощью команды cellranger mkref для обнаружения генов как GFP24.
-
Критические: До загрузки убедитесь, что система Linux удовлетворяет минимальные требования23. Обеспечить как минимум 8-ядерный процессор с ОЗУ 64 ГБ и 1 ТБ свободного дискового пространства.
- Создавать файлы FASTQ из секвенсор в базовый вызов файлов (BCL) с помощью команды mkfastq cellranger.
Примечание: Программа выравнивания сырой считывает ссылку генома (из файлов FASTQ) и генерировать ген ячейки матрицы для течению анализа. Он использует STAR каппу, выполняющий сплайсинга сознающий выравнивание читает генома ссылку. Только уверенно сопоставленных читает (т.е., читает совместим с одного гена аннотации) используются для подсчета UMI.- Например используйте команду mkfastq cellranger:
cellranger mkfastq --id = sample_name \
--Запуск = / путь/к/образца \
--csv=csv_file_containing_lane_sample_index.csv
- Например используйте команду mkfastq cellranger:
- Запустите cellranger фото на FASTQ файлы, созданные с помощью mkfastq для создания графов одноклеточных гена.
- Например используйте команду cellranger Фото:
cellranger игр --id = sample_name \
--транскриптом = refdata-cellranger мм10-1.2.0 \
--fastqs = / абсолютной/путь/к/fastq/файлы \
--Пример = same_sample_name_supplied_to_cellranger_mkfastq \
--localcores = 30
- Например используйте команду cellranger Фото:
- Мульти Библиотека агрегации (опционально): объединить образцы, бассейн cellranger количество выходов с помощью cellranger АГР. Это приводит в матрице одного гена-штрих-код, содержащий данные объединены из нескольких библиотек. Пример cellranger АГР команду:
Cellranger АГР--id = sample_name \
--csv = csv_with_libraryID_ & _path_to_molecule_h5.csv \
--нормализовать = сопоставлены
Примечание: Библиотеки может быть статистически вычислена с помощью трех режимов нормализации (сопоставленных, сырые, нет). Сопоставлены рекомендуется, так как он подсэмплы выше глубины библиотеки до тех пор, пока все библиотеки имеют равные последовательности глубина25. - Для непосредственной визуализации/анализ данных импортируйте в выходной файл .cloupe (созданная с помощью cellranger игр или cellranger АГР) в 10 x лупа мобильный браузер26.
7. Расширенный анализ scRNA-Seq наборов данных
Примечание: Полное scRNA-Seq инструменты базы данных можно найти в scRNA инструменты3,27. Ниже рамки для неконтролируемого клеток pseudotemporal и кластеризации с использованием Сёра2 заказ с использованием монокль6. Хотя большая часть этой работы может быть сделано на локальном компьютере, в следующих шагах предполагается, что вычисление будет завершен с помощью институциональных сервера.
- Скачайте последнюю версию Miniconda на учетной записи сервера с использованием платформы Linux28.
- Установите последнюю версию с помощью Конда29Р.
- Печать данных, используя предоставленный сценарий Сёра R как шаблон30.
Примечание: Сера является на основе R инструментарий, позволяющий проверок контроля качества, кластеризации, дифференциальный анализ выражения гена, маркер идентификации генов, снижение размерности и визуализации данных scRNA-Seq. Всестороннее описание Сёра кодирования и руководства можно найти на веб-сайте Satija лаборатории31. - Печать данных, используя предоставленный сценарий монокль R как шаблон32.
Примечание: Монокль является другой на основе R инструментарий, который позволяет визуализации изменения выражения над pseudotime и определяет, лежащие в основе решения судьбы клетки генов. Всестороннее описание монокль кодирования и руководства можно найти на веб-сайте монокль33. - R-пакеты, такие как kBET могут быть использованы для тестирования и исправления пакета эффекты в результате объединения наборов данных34.
8. NCBI GEO и SRA представлений
Примечание: Так как легкий доступ к файлам сырой последовательности обеспечить воспроизводимость и реанализа, поступающей представлений онлайн публично доступные репозитории рекомендуется или требуется до представления рукописи. Национальный центр биотехнологической информации (NCBI) Омнибус выражение гена (GEO) и Архив чтения последовательности (SRA) являются публично доступных данных хранилищ для высокопроизводительного секвенирования данных35,36.
- Зарегистрироваться для NCBI GEO отправитель счета37.
- Представление полная ГЭП, который включает в себя три компоненты, скомпилированные в каталог/папку (под названием как GEO отправитель имя пользователя): 1) запись метаданных (одну таблицу за представление проекта); 2) файлов необработанных данных; 3) обрабатываются файлы данных.
- Скачайте и заполните таблицу метаданных38. Следующие представления общественности GEO может использоваться как руководство (GSE100320)39. Место таблицы в каталоге.
- Место Raw данных файлы, созданные из сценария cellranger игр для всех библиотек в каталог.
- Место обработки данных файлы (отфильтрованные barcodes.tsv, genes.tsv и matrix.mtx) генерируется из сценария cellranger игр для всех библиотек в каталог.
- Используйте учетные данные для сервера FTP GEO заявителя для передачи каталог, содержащий все три компонента. Для пользователей Linux/Unix: ncftp, lftp, ftp, sftp и ncftpput могут быть использованы.
- Уведомлять GEO для всех переводов38.
Результаты
Репертуар открытым исходным кодом упаковок, предназначенных для анализа наборов данных scRNA-Seq резко увеличилось40 с большинством этих пакетов использования языков на основе R3. Здесь представлены представителя результаты, с использованием двух из этих пакетов: оценки без присмотра группировки одной ячейки, основанные на выражении гена и заказ единичных клеток по траектории с целью разрешить мобильный неоднородность и разобрать биологического процессы.
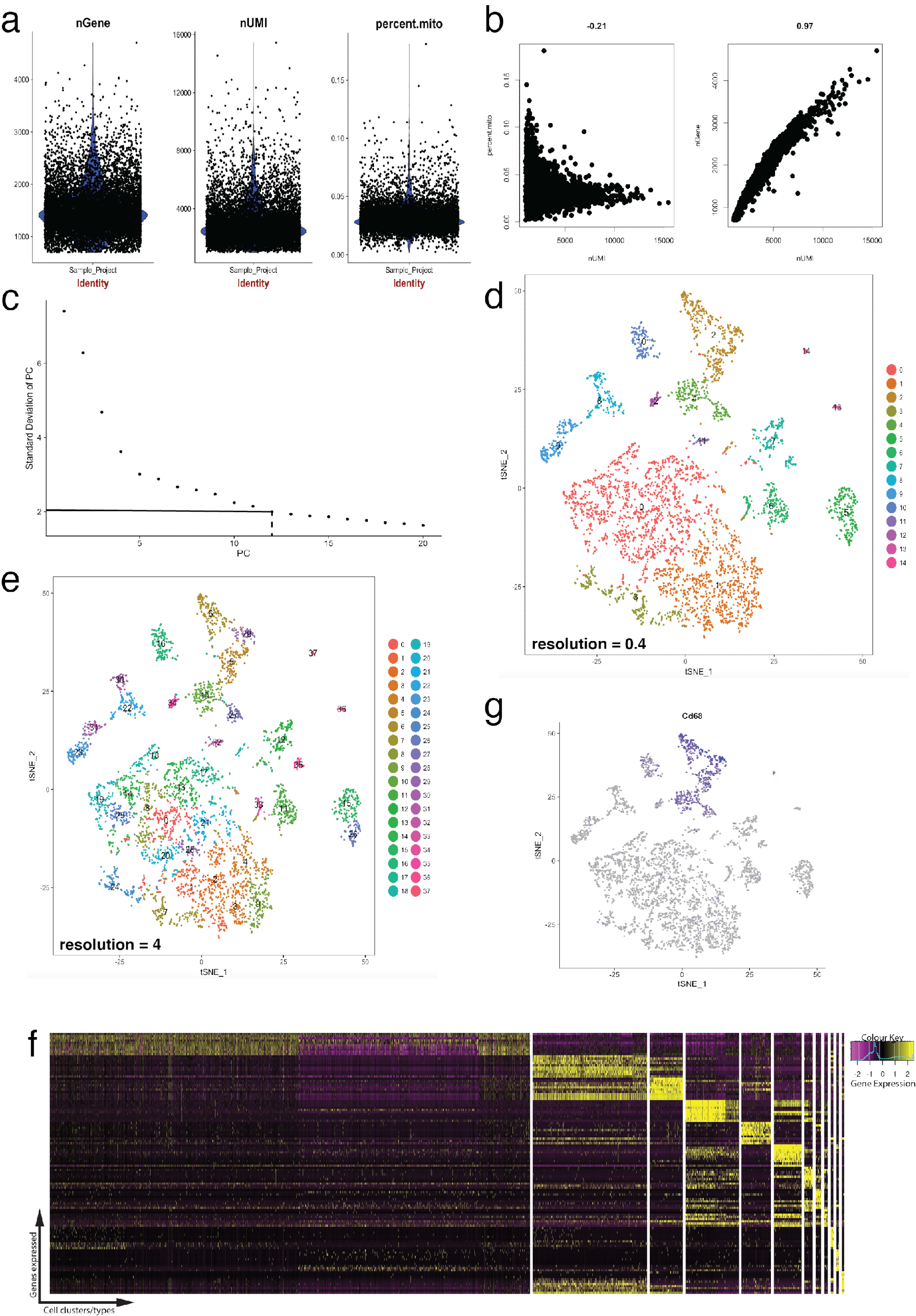
Рисунок 4 иллюстрирует использование Сёра для предварительной обработки, проверки качества и анализ течению биоинформатики. Во-первых фильтрации и удаления девиантного клеток от анализа имеет важное значение для проверки качества. Это было сделано с помощью скрипки (рис. 4a) и точечные участки (Рисунок 4b) визуализировать процент митохондриальных генов, количество генов (nGene) и количество UMI (Нуми) для идентификации ячейки Дуплеты и промахи. Любую ячейку с четкой останец количество генов, UMI или процент митохондриальных генов был удален с помощью Seurat FilterCells функции. Так как Сёра использует основной компонент (PC) анализа оценки кластеры клеток, определения статистически ПК включить является важным шагом. Отвод участков (рис. 4 c) были использованы для выбора ПК, в которых компьютеры за пределами плато стандартное отклонение ПК оси были исключены. Резолюции кластеризации также было манипулировать, продемонстрировав что количество кластеров может быть изменен, начиная от 0.4 (низкое разрешение, ведущих к меньше ячейка кластеров, Рисунок 4 d) до 4 (высокое разрешение приводит к выше ячейки кластеров, рисунке 4e ). С низким разрешением вполне вероятно, что каждый кластер представляет тип определенной ячейки, в то время как с высоким разрешением, это может также представлять подтипы или переходного состояния популяции клеток. В этом случае параметры кластера с низким разрешением были использованы для дальнейшего анализа выражения карты (с помощью функции DoHeatmap Seurat) для выявления наиболее выраженной генов в данной группе (Рисунок 4f). В данном случае наиболее сильно выраженной гены были определены путем оценки дифференциальной выражения в данной группе против всех других кластеров комбинированные, демонстрируя, что каждый кластер представлял однозначно определенных генов. Кроме того отдельные кандидат гены могут быть визуализированы на tSNE участках, с помощью функции FeaturePlot Seurat (Рисунок 4 g). Это позволило расшифровка существуют ли кластеры, которые представлены макрофагов. С помощью FeaturePlot, мы обнаружили, что оба кластер 2 и 4 выражая Cd68 - Пан макрофагального маркер.
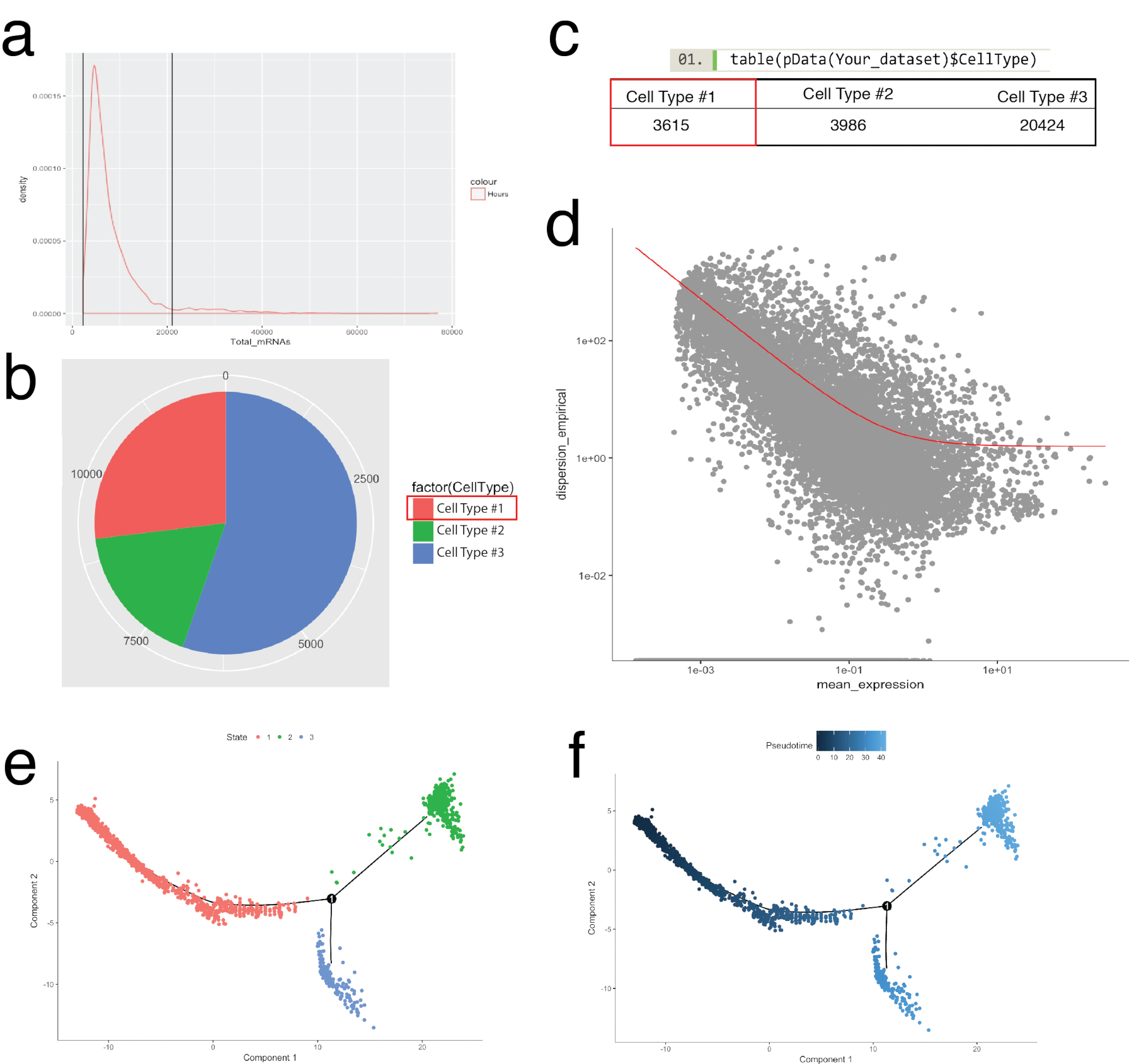
Монокль пакет был использован для соответствующей ячейки кластеры, выявленные в Сёра и для построения клеток траекторий, или pseudotemporal заказ, резюмировать биологических процессов (рис. 5). Pseudotemporal заказа может использоваться для образцов где одноклеточных выражение профили, как ожидается, следовать курсом биологического времени. Клетки можно заказать вдоль pseudotemporal континуум решить промежуточных состояний, точек бифуркации, два альтернативных клеток судеб и идентификации генов подписей лежащие в основе приобретение каждого судьбы. Во-первых, аналогично фильтрации Seurat, низкое качество клетки были удалены таким образом, чтобы распределение мРНК для всех ячеек было нормальное и упал между верхней и нижней границы, как это определено в Рисунок 5aжурнала. Затем с помощью функции newCellTypeHierarchy монокль в отдельные клетки были классифицированы и рассчитывает с помощью известных линии маркерных генов (Рисунок 5b, 5 c). Например клетки, выражая PDGF рецепторов альфа или фибробластов конкретных белок 1 были переданы ячейки тип #1 для создания критерия для определения фибробластов. Далее эта группа населения (ячейки типа #1) оценивали расшифровать фибробластов траекторий. Для этого был использован монокль в дифференциальной GeneTest функция, которая сравнению клетки, представляющие экстремальных государства в рамках населения и нашел дифференциального генов для заказа оставшиеся ячейки в численности населения (рис. 5 d). Путем применения методов коллектор обучения (тип уменьшения нелинейных размерность) для всех ячеек, был назначен координату вдоль pseudotemporal пути. Эта траектория был затем визуализирована состояния клеток (рис. 5e) и pseudotime (рис. 5f).

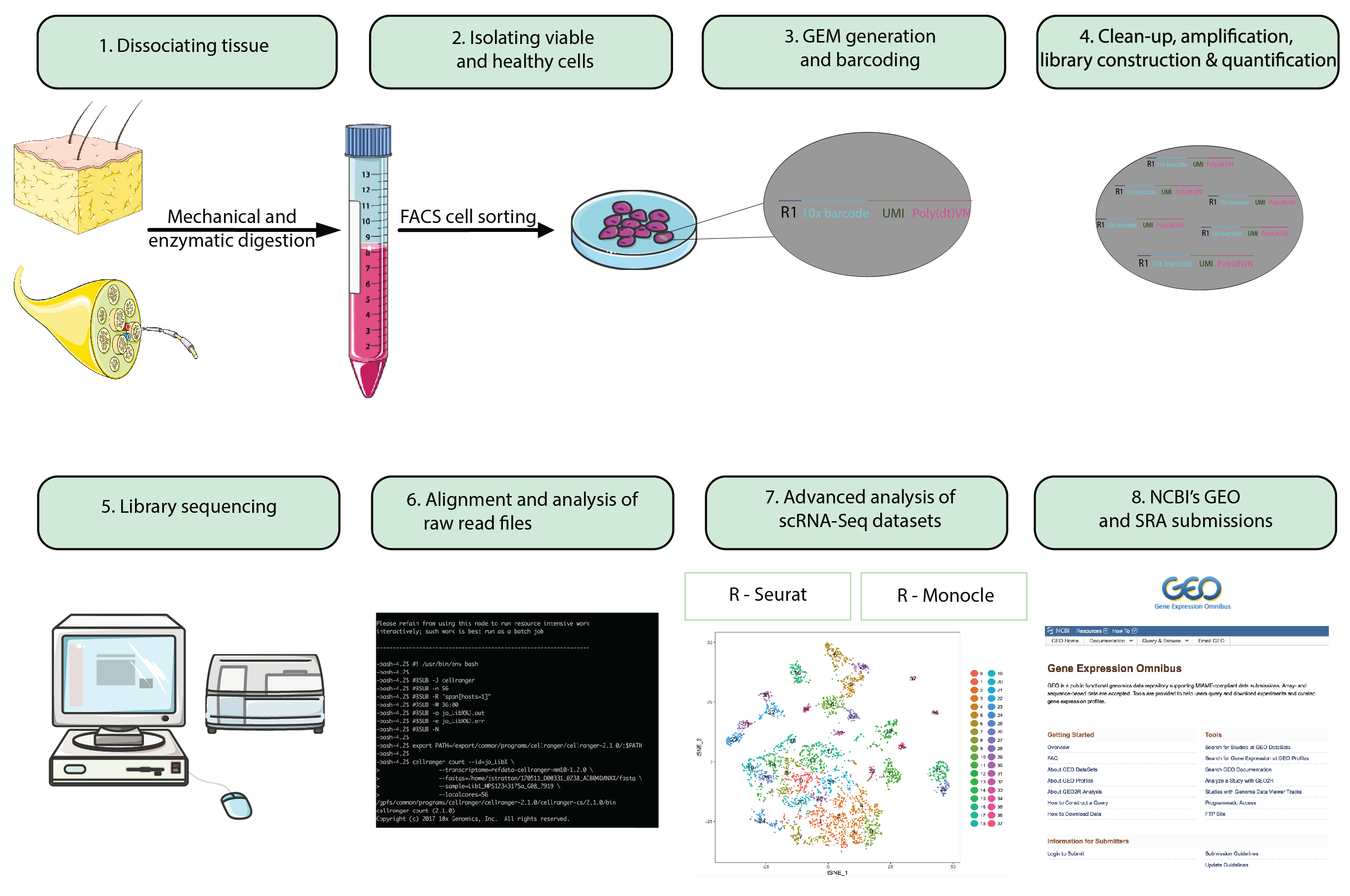
Рис 1: диаграмма. Шагах от всей животных подготовки к анализу одной ячейки РНК-Seq наборов данных для представления окончательного наборов данных в хранилище общедоступных. Гель бусинки в эмульсии (ГСМОС) относятся к бусины с перепутываются олигонуклеотиды, инкапсулирующие тысячи одиночных клеток. Пожалуйста, нажмите здесь, чтобы посмотреть большую версию этой фигуры.
{kind=link}

Рисунок 2: создание жизнеспособных одноклеточного подвеска из нервных тканей. (a) мультфильм Обзор проверок контроля качества. (b) клеток и мусора с клетки по-прежнему включены в мусора (красные стрелки). (c) клетки, освобождены от мусора (красные стрелки). (d) изоляция клетки, СУИМ. P0: мусора фракция; P1: клетки как фракция; P3: исключение триоли; P4: жизнеспособности краситель (Sytox оранжевый) отрицательные дроби. (e) не жизнеспособности управления красителя. (f) изображение фракции P0, представляющий изолированных мусора. (g) изображение фракции P4, представляющие изолированных жизнеспособных клеток (красные стрелки). (b) (c) (f) и (g) ядерных краситель, добавил 20 минут до изображений. Масштаб баров: 80 мкм. пожалуйста, нажмите здесь, чтобы посмотреть большую версию этой фигуры.
{kind=link}

Рисунок 3: мелкие последовательности предсказывает количество восстановленных клеток в 10 X обработанные образцы. () пример (образец 1.6) MiSeq созданные csv список ячейки штрих-кода и его соответствующий UMI подсчитывает как определяется уверенно сопоставленных читает. (b) ранга участок штрих-кодов для образца 1.6 показывает одно значительное падение в UMI count как функция ячейки штрих-кода. Пунктирная и твердой линии представляют отсечки между клетками и фона определяется визуального осмотра. (c) клетки штрих отмечено, используя ячейки Рейнджер конвейер пост HiSeq показывает, что мелкие последовательности точно аппроксимировать количество клеток для образца 1.6. (d) пример потока клеточной структуры, основанные на мелкие последовательности полученных оценок клетки. Для образца 1.6, поскольку мелкие последовательности предсказал 3480 клетки, 1.17 переулки были назначены для обеспечения > 100000 чтения за охват клетки последовательности в HiSeq. Примечание: Все полосы необходимо добавить до 100%. Пожалуйста, нажмите здесь, чтобы посмотреть большую версию этой фигуры.
{kind=link}

Рисунок 4: контроль качества и биоинформатики одноклеточных РНК-Seq набора данных с помощью пакета Сёра Р. (a) участков контроля качества метрик, которые включают в себя количество генов, молекулярные уникальных идентификаторов (UMIs) и процент стенограммы, сопоставление митохондриального генома. (b) образец гена участков детектирования клетки с девиантным уровнями митохондриальной стенограммы и UMIs. (c) образец локоть участок используется для специального определения статистически шт. Пунктирная и точка пунктирной линии представляют отсечки, где четкое «колено» становится очевидным в графе. PC размеры до этого локоть включены в анализ вниз по течению. (d, e) Граф на основе клеток кластеры визуализирована на двух различных резолюций в низкоразмерных пространства с помощью tSNE участок. (f) топ маркерных генов (желтая) для каждого кластера на heatmap выражения, с помощью функции DoHeatmap Seurat. (g) визуализации маркер выражения гена, например, Cd68 представляющих макрофагов (фиолетовый) с помощью функции FeaturePlot Seurat. Это предполагает, что этот кластер 2 и 4 (Группа d) этого набора данных представляет макрофагов. Пожалуйста, нажмите здесь, чтобы посмотреть большую версию этой фигуры.
{kind=link}

Рисунок 5: сотовый категоризации и заказ по peudotemporal траектории, используя Инструментарий монокль. a проверять распределение мРНК (выведен из графов UMI) для всех ячеек в образце. Только клетки с мРНК между 0 - ~ 20000 были использованы для анализа ниже по течению. (b, c) Назначение и подсчета клеток типы, основанные на известных lineage маркеров ячейки. Например, клетки, выражая PDGF рецепторов альфа или фибробластов конкретных белок 1 были назначены в ячейке тип #1 представляющих Пан фибробластов с помощью функции newCellTypeHierarchy в монокль. Количество различных типов клеток могут быть визуализированы как на круговой диаграмме (b) и как таблицы (c). (d) с помощью ячейки типа #1 (фибробласты) как, например, используемые для сортировки клеток могут быть визуализированы с помощью точечной, демонстрирующий гена дисперсии против означает выражение генов. Красная кривая показывает отсечки для генов используется для заказа рассчитанные среднеквадратическое отклонение модели с помощью функции estimateDispersions в монокль. Гены, которые отвечают этой отсечки были использованы для вниз по течению pseudotime заказа. (e, f) Визуализация траекторий клеток в цветные ячейки «государства» (e) и назначен монокль «Pseudotime» (f) сокращение двумерном пространстве. Пожалуйста, нажмите здесь, чтобы посмотреть большую версию этой фигуры.
{kind=link}
Обсуждение
Этот протокол демонстрирует соответствующую подготовку одиночных клеток может раскрыть транскрипционный анализ гетерогенности тысячи одиночных клеток и дискриминацию функциональных состояний или уникальных сотовых тождества в ткани. Протокол не требует репортер флуоресцентные белки или трансгенных инструментов и может быть применен к изоляции одиночных клеток из различных тканей, представляющих интерес, в том числе от людей; Имея в виду каждая ткань уникальна, и этот протокол потребует определенной перестройки/модификации.
Разнообразных и весьма динамичный транскрипционный анализ программ внутри клетки подчеркивали значение одной ячейки геномики. Помимо изоляции высокого качества РНК, критических образца Подготовка шаг, необходимый для высокого качества наборов данных является обеспечение того, что клетки полностью освобождаются от ткани и клетки являются здоровыми и нетронутыми. Это довольно прямо вперед для сбора клеток, которые легко выпущена, например циркулирующих клеток или тканей, где слабо сохраняются клетки, такие как лимфоидной ткани. Но это может быть сложным для других взрослых тканях, благодаря высокоразвитой сотовой архитектуры, охватывающих большие расстояния, окружающих внеклеточного матрикса и часто жесткой цитоскелета белков, участвующих в поддержании клеточной структуры. Даже с соответствующим диссоциации методов для полного отпуска клеток есть потенциал, что строгие и часто продолжительной обработки требуется изменит качество и ячейки целостность мРНК. Кроме того высокие температуры, используемые для фермента помощь диссоциации также влияют на transcriptional подписей29,30. Целью протокола является в настоящее время контроль качества проверяет, используя ткани, такие как Миелинизированные нервные взрослых и внеклеточной матрицы богатых взрослых кожи, чтобы продемонстрировать, как оптимизация может помочь преодолеть эти препятствия.
Главным соображением при разработке любого эксперимента scRNA-Seq является выбор последовательности глубины. Последовательность может быть весьма мультиплексированием и читать глубина может варьироваться от очень низкий, с помощью Drop-Seq2 до 5 миллионов читает/ячейку14 полнометражных методом РНК-Seq как смарт-последующие Большинство scRNA-Seq эксперименты можно обнаружить умеренно высокий выражение стенограммы с последовательности как низко как 10 000 читает/клетки, которые обычно достаточно для ячейки типа классификации41,42. Глубина мелкой последовательности имеет значение сэкономить на стоимости последовательности при попытке обнаружить редкие клеточных популяций через сложные ткани, где тысячи клеток может потребоваться приписать уверенно редких населения. Но глубине последовательности не адекватные, когда необходима подробная информация о экспрессии генов и процессы, связанные с тонкими transcriptional подписей. В настоящее время предполагается, что большая часть генов в клетке обнаружены с 500.000 читает/клетки, но это может варьироваться в зависимости от протокола и ткани типа43,-44. Хотя полнометражного Стенограмма последовательности обходит необходимость сборки и может таким образом, обнаружить роман или редкие сращивания варианты, секвенирование расходы часто ограничивают масштабирования таких подходов для изучения тысяч клеток, включающий систему сложных тканей. В отличие от 3' тегами одноклеточных библиотек такие, как те, указанных в настоящем Протоколе обычно ниже сложности и требуют меньшую последовательности. Важно отметить, что на одном из пяти поддерживаемых синтезаторов можно упорядочивать библиотеки, созданные с помощью описанных протокол: 1) NovaSeq, 2) HiSeq 3000/4000, 3) HiSeq 2500 быстрый запуск и высокий выход, 4) NextSeq 500/550 и 5) MiSeq.
Альтернативный подход к одной ячейке РНК-Seq, что снижает потребность в деликатных тканей и клеток, обработки еще не поддерживает некоторые из преимуществ одной ячейки РНК-Seq, является анализ РНК от одного ядра45. Этот подход позволяет более быстрой обработки, сокращения масштабов деградации РНК и более экстремальные меры для обеспечения надлежащего релиз ядра и таким образом вероятно позволяет более уверенно захвата транскрипционный анализ профилей, представляющий все ячейки в пределах данной ткани. Это конечно, только бы часть транскрипционный анализ деятельности в рамках данной ячейки, таким образом, в зависимости от того, каковы цели экспериментальных интерес этот подход может быть или не быть соответствующие.
Кроме полной характеристики сотовых самобытности в рамках данной ткани один из самых ценных анализов для scRNA-Seq наборов данных является оценка промежуточных транскрипционный анализ государств через «определенных» клеточных популяций. Эти промежуточные государства могут распространять идеи в линии связи между клетки внутри выявленных групп населения, которые не удалось с традиционными навалом, РНК-Seq подходов. Несколько scRNA-Seq bioinformatic инструменты были разработаны для выяснения этого. Такие инструменты могут оценить процессы, занимающиеся, например, раковые клетки, переходит к онкогенных/метастатического состояние, стволовые клетки, созревания в разнообразных терминала судьбы или иммунных клеток, курсируя между активным и покоя государствами. Транскриптом тонкие различия в клетках может также свидетельствовать о lineage предубеждения, что недавно разработанных bioinformatic инструменты, такие как FateID, может вывести47. Поскольку различия между переход клеток может быть трудным для выяснения учетом транскрипционный анализ различий может быть тонким, глубже последовательности может быть необходимым46. К счастью освещение неглубоко виртуализированного библиотеки может быть увеличена, если заинтересованы в зондирующего дальнейшего набора данных, повторно запустив библиотеке на другую ячейку потока.
Взятые вместе, этот протокол обеспечивает легко адаптировать рабочий процесс, который позволяет пользователям транскрипционно профиль сотни тысяч сингл клеток в пределах одного эксперимента. Конечное качество набора scRNA-Seq опирается на оптимизированные ячейки изоляции, проточной цитометрии, кДНК библиотеки поколения и интерпретации сырья ген штрих матриц. С этой целью этот протокол обеспечивает всесторонний обзор всех ключевых шагов, которые могут быть легко изменены для включения исследования тканей различных типов.
Раскрытие информации
Нет раскрытия информации
Благодарности
Мы признаем вспомогательного персонала на объекте UCDNA услуг, а также животных уход сотрудников Фонда в Университете Калгари. Мы благодарим Matt Workentine за его поддержку биоинформатики и Йенс Durruthy его технической поддержки. Эта работа финансируется грантом КНИИЗ (р.м. и J.B.), КНИИЗ новую награду следователь J.B., и Альберта детей здоровья исследовательский институт стипендий (J.S.).
Материалы
| Name | Company | Catalog Number | Comments |
| Products | |||
| RNAse out | Biosciences | 786-70 | |
| Pentobarbital sodium | Euthanyl | 50mg/kg | |
| HBSS | Gibco | 14175-095 | |
| Dispase 5U/ml | StemCell Technologies | 7913 | 5 mg/ml |
| Collagenase-4 125 CDU/mg | Sigma-Aldrich | C5138 | 2 mg/ml |
| DNAse | Sigma-Aldrich | DN25 | 10mg/ml |
| BSA | Sigma-Aldrich | A7906 | |
| 15 ml Narrow bottom tube VWR® High-Performance Centrifuge Tubes | VWR | 89039-666 | |
| Sytox Orange Viability Dye | Molecular Probes | 11320972 | 1.3 nM/µl |
| Nuc Blue Live ReadyProbes | Invitrogen | R37605 | |
| Agilent 2100 Bioanalyzer High senitivity DNA Reagents | Agilent | 5067-4626 | |
| Kapa DNA Quantification Kit | Kapa Biosystems | KK4844 | |
| Chromium Single Cell 3' reagents | 10x Genomics | ||
| Equipment | |||
| BD FACSAria III | BD Biosciences | ||
| Agilent 2100 Bioanalyzer Platform | Agilent | ||
| Illumina® HiSeq 4000 | Illumina | ||
| Illumina® MiSeq SR50 | Illumina | ||
| 10X Controller + accessories | 10x Genomics | ||
| Software | |||
| The Cell Ranger | 10x GENOMICS | support.10xgenomics.com/single-cell-gene-expression/software/overview/welcome | |
| Loupe Cell Browser | 10x GENOMICS | support.10xgenomics.com/single-cell-gene-expression/software/downloads/latest | |
| R | https://anaconda.org/r/r |
Ссылки
- Shalek, A. K., et al. Single-cell RNA-seq reveals dynamic paracrine control for cellular variation. Nature. 510, 363-369 (2014).
- Macosko, E. Z., et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell. 161, 1202-1214 (2015).
- Zappia, L., Phipson, B., Oshlack, A. Exploring the single-cell RNA-seq analysis landscape with the scRNA-tools database. bioRxiv:206573. , (2018).
- Dulken, B. W., Leeman, D. S., Boutet, S. C., Hebestreit, K., Brunet, A. Single cell transcriptomic analysis defines heterogeneity and transcriptional dynamics in the adult neural stem cell lineage. Cell Reports. 18 (3), 777-790 (2017).
- Llorens-Bobadilla, E., et al. Single-Cell Transcriptomics Reveals a Population of Dormant Neural Stem Cells that Become Activated upon Brain Injury. Cell Stem Cell. 17 (3), 329-340 (2015).
- Trapnell, C., et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nature Biotechnology. 32, 381-386 (2014).
- Aibar, S., et al. SCENIC: single-cell regulatory network inference and clustering. Nature Methods. 14, 1083-1086 (2017).
- Mayer, C., et al. Developmental diversification of cortical inhibitory interneurons. Nature. 555 (7697), 457-462 (2018).
- Stratton, J. A., et al. Purification and Characterization of Schwann Cells from Adult Human Skin and Nerve. eNeuro. 4 (3), (2017).
- Biernaskie, J. A., McKenzie, I. A., Toma, J. G., Miller, F. D. Isolation of skin-derived precursors (SKPs) and differentiation and enrichment of their Schwann cell progeny. Nature Protocols. 1 (6), 2803-2812 (2007).
- 10X Genomics. User Guides. , Available from: https://www.10xgenomics.com/resources/user-guides/ (2018).
- 10X Genomics. Chromium Single Cell 3' Training Module. , Available from: http://go.10xgenomics.com/training-modules/single-cell-gene-expression (2018).
- Agilent. , Available from: https://www.agilent.com/en-us/library/usermanuals?N=135 (2018).
- Kolodziejczyk, A. A. Single Cell RNA-Sequencing of Pluripotent States Unlocks Modular Transcriptional Variation. Cell Stem Cell. 17, 471-485 (2015).
- Jaitin, D. A., et al. Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types. Science. 343, 776-779 (2014).
- Pollen, A. A., et al. Low-coverage single-cell mRNA sequencing reveals cellular heterogeneity and activated signaling pathways in developing cerebral cortex. Nature Biotechnology. 32, 1053-1058 (2014).
- 10X Genomics. Sequencing Requirements for Single Cell 3'. , Available from: https://support.10xgenomics.com/single-cell-gene-expression/sequencing/doc/specifications-sequencing-requirements-for-single-cell-3 (2018).
- Datacamp. Introduction to R. , Available from: https://www.datacamp.com/courses/free-introduction-to-r (2018).
- Droplet-based, high-throughput single cell transcriptional analysis of adult mouse tissue using 10X Genomics#39; Chromium Single Cell 3' (v2) system: From tissue preparation to bioinformatic analysis. , Available from: https://figshare.com/s/97b83e649e5eefd01357 (2018).
- 10X Genomics. User Guides. , Available from: https://www.10xgenomics.com/resources/user-guides/ (2018).
- UNIX Tutorial for Beginners. , Available from: http://www.ee.surrey.ac.uk/Teaching/Unix/ (2018).
- 10X Genomics. Creating a Reference Package with cellranger mkref. , Available from: https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/advanced/references (2018).
- 10X Genomics. System Requirements. , Available from: https://support.10xgenomics.com/single-cell-gene-expression/software/overview/system-requirements (2018).
- 10X Genomics. Software Downloads. , Available from: https://support.10xgenomics.com/single-cell-gene-expression/software/downloads/latest (2018).
- 10X Genomics. Aggregating Multiple Libraries with cellranger aggr. , Available from: https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/using/aggregate#depth_normalization (2018).
- 10X Genomics. Loupe Cell Browser Gene Expression Tutorial. , Available from: https://support.10xgenomics.com/single-cell-gene-expression/software/visualization/latest/tutorial (2018).
- scRNA-tools. A table of tools for the analysis of single-cell RNA-seq data. , Available from: https://www.scrna-tools.org/ (2018).
- Conda. Downloading conda. , Available from: https://conda.io/docs/user-guide/install/download.html (2018).
- Anaconda. r / packages / r 3.5.1. , Available from: https://anaconda.org/r/r (2018).
- Droplet-based, high-throughput single cell transcriptional analysis of adult mouse tissue using 10X Genomics' Chromium Single Cell 3' (v2) system: From tissue preparation to bioinformatic analysis. , Available from: https://figshare.com/s/97b83e649e5eefd01357 (2018).
- Satija Lab. Seurat - Guided Clustering Tutorial. , https://satijalab.org/seurat/pbmc3k_tutorial.html (2018).
- Droplet-based, high-throughput single cell transcriptional analysis of adult mouse tissue using 10X Genomics' Chromium Single Cell 3' (v2) system: From tissue preparation to bioinformatic analysis. , Available from: https://figshare.com/s/97b83e649e5eefd01357 (2018).
- Monocle. , Available from: http://cole-trapnell-lab.github.io/monocle-release/docs/#constructing-single-cell-trajectories (2018).
- Github. An R package to test for batch effects in high-dimensional single-cell RNA sequencing data. , (2018).
- Edgar, R. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Research. 30, 207-210 (2002).
- Leinonen, R., Sugawara, H., Shumway, M. The sequence read archive. Nucleic Acids Research. 39, D19-D21 (2011).
- NIH. GenBank Submission Portal Wizards. , Available from: https://www.ncbi.nlm.nih.gov/account/register/?back_url=/geo/submitter/ (2018).
- NIH. Submitting data. , Available from: https://submit.ncbi.nlm.nih.gov/geo/submission/ (2018).
- Shah, P. T., et al. Single-Cell Transcriptomics and Fate Mapping of Ependymal Cells Reveals an Absence of Neural Stem Cell Function. Cell. 173, 1045-1057 (2018).
- Anon, Method of the Year 2013. Nature Methods. 11, 1(2013).
- Adam, M., Potter, A. S., Potter, S. S. Psychrophilic proteases dramatically reduce single-cell RNA-seq artifacts: a molecular atlas of kidney development. Development. 144, 3625-3632 (2017).
- Wu, Y. E., Pan, L., Zuo, Y., Li, X., Hong, W. Detecting activated cell populations using single-cell RNA-seq. Neuron. 96, 313-329 (2017).
- Zeigenhain, C., et al. Comparative Analysis of Single-Cell RNA Sequencing Methods. Molecular Cell. 65 (4), 631-643 (2017).
- Wu, A. R., et al. Quantitative assessment of single-cell RNA-sequencing methods. Nature Methods. 11 (1), 41-46 (2014).
- Habib, N., et al. Div-Seq: Single-nucleus RNA-Seq reveals dynamics of rare adult newborn neurons. Science. 353 (6302), 925-928 (2016).
- Janes, K. A. Single-cell states versus single-cell atlases - two classes of heterogeneity that differ in meaning and method. Current Opinions in Biotechnology. 39, 120-125 (2016).
- Herman, J. S., Sagar,, Grün, D. FateID infers cell fate bias in multipotent progenitors from single-cell RNA-seq data. Nature Methods. 15 (5), 379-386 (2018).
Перепечатки и разрешения
Запросить разрешение на использование текста или рисунков этого JoVE статьи
Запросить разрешениеThis article has been published
Video Coming Soon
Авторские права © 2025 MyJoVE Corporation. Все права защищены