
Method Article
드롭릿 바코드 기반 단일 셀 Transcriptomics 성인 포유류 직물의
* 이 저자들은 동등하게 기여했습니다
요약
이 프로토콜에서는 일반적인 프로세스를 설명 합니다 및 품질 관리 검사 드롭릿 기반, 높은 처리량 단일 셀 RNA-Seq 준비에 대 한 건강 한 성인 포유류 단일 세포를 준비에 필요한. 시퀀싱 매개 변수, 읽기 정렬 및 다운스트림 단일 셀 bioinformatic 분석 또한 제공 됩니다.
초록
조직 또는 microenvironment 내에서 개별 셀의 수천에 걸쳐 단일 세포 유전자 발현의 분석은 식별 셀 구성, 기능 및의 밑에 관찰 된 조직 하는 분자 경로 차별 하기 위한 유용한 도구 함수 및 동물성 행동입니다. 그러나, 후속 다운스트림 단일 세포 분자 분석에 대 한 성인 포유류 조직에서 그대로, 건강 한 세포의 고립 전하실 수 있습니다. 이 프로토콜에서는 일반 프로세스 및 품질 관리 신 시스템에서 높은-품질 성인 단일 셀 준비 하거나 피부를 위해 필요한 설정 이후 편견된 단일 셀 RNA 시퀀싱 및 분석 검사. 다운스트림 bioinformatic 분석에 대 한 가이드도 제공 됩니다.
서문
높은 처리량 단일 셀 기술1,2 와 지난 10 년간3사용자 친화적인 생물 정보학 도구 발전의 개발, 고해상도 유전자 표정 분석의 새로운 필드가 나왔다- 단일 셀 RNA 시퀀싱 (scRNA-Seq) 단일 셀 유전자 발현의 연구 처음 줄기 세포 또는 암 세포, 정의 된 세포 인구 내에서 식별 하거나 식별 어려운 사용 하는 셀의4,5, 드문 인구 개발 되었다 전통적인 대량 RNA 시퀀싱 기법입니다. 소설 부분 모집단 (Seurat)2, psuedotime 공간 (Monocle)6, 인구 (또는 내 활성 신호 네트워크의 정의 따라 셀 순서의 시각화의 식별을 가능 하 게 Bioinformatic 도구 아름 다운)7, 단일-셀 (Seurat, 그리고 더 많은) 인공 3D 공간에서의 조립의 예측8. 이러한 새롭고 흥미로운 분석 과학 지역 사회에 사용할 수 있는, scRNA-Seq 빠른-되고있다 유전자 표정 분석에 대 한 새로운 표준 접근을.
ScRNA-Seq의 광대 한 잠재력에도 불구 하 고 깨끗 한 데이터 집합을 생성 하 고 결과 정확 하 게 해석 하는 데 필요한 기술 skillsets 이민자를 전하실 수 있습니다. 여기, 기본적인, 하지만 포괄적인 프로토콜 (그림 1) 제시 시각화를 전체 기본 조직에서 단일 세포의 분리 및 게시에 대 한 데이터의 프레 젠 테이 션에서 시작 합니다. 첫째, 건강 한 세포의 격리 수 있습니다 간주 도전, 효소 소화와 후속 기계적 분리에 감도의 그들의 정도에 따라 다릅니다 다른 조직. 이 프로토콜 이러한 격리 단계에 제공 하 고 과정 전반에 걸쳐 중요 한 품질 관리 검사점을 식별 합니다. 둘째, 호환성 및 단일 셀 기술 및 차세대 시퀀싱 사이 요구 사항 이해 복잡할 수 있다. 이 프로토콜 구현 하는 사용자 친화적인, 드롭릿 기반 단일 셀 barcoding 플랫폼 및 시퀀싱을 수행 하는 지침을 제공 합니다. 마지막으로, 컴퓨터 프로그래밍은 단일 셀 transcriptomic 데이터 집합을 분석 하기 위한 중요 한 전제 조건입니다. 이 프로토콜 R 프로그래밍 언어에서 시작 하기 위한 리소스를 제공 하 고 구현 하는 두 개의 인기 있는 scRNA Seq 관련 R 패키지 지침을 제공 합니다. 함께,이 프로토콜 명확 하 고, 해석할 결과 얻기 위해 scRNA-Seq 분석 수행에 신규 이민자 가이드 수 있습니다. 이 프로토콜, 마우스에서 대부분 조직에 조정 될 수 있다 그리고 중요 한 것은 인간 직물을 포함 한 다른 유기 체와 함께 사용 하기 위해 수정할 수 있습니다. 조직 및 사용자에 따라 조정 해야 합니다.
다음이 프로토콜; 하는 동안 마음에 계속 몇 가지 고려 사항이 포함 하 여, 정확한 요약 셀 숫자 카운트 ( 그림 2에에서 요약 하면서 관심의 샘플 내에서 모든 셀의 가능한 단일 세포 현 탁 액을 되도록 권장 1) 다음 단계 1에서 모든 품질 관리 지침과이 프로토콜의 2 ). 일단이 이루어집니다, 그리고 품질 관리 단계를 삭제할 수 있습니다 모든 최적화 된 조건을 따른다면 (에 시간-절약 RNA 품질을 유지 하 고 감소 셀 손실). 높은 생존의 조직에서 단일 세포의 성공적인 격리 확인는 매우 추천 전에 다운스트림 처리 합니다. 2) 일부 세포 유형 스트레스를 다른 사람 보다 더 민감한 때문에, 과도 한 분리 기술 인구, 따라서 다운스트림 분석을 혼동 편견 실수로 수 있습니다. 불필요 한 세포 전단 및 소화 하지 않고 부드러운 분리는 높은 세포 수율과 조직 구성의 정확한 표현을 달성 하기 위한 중요 합니다. 전단 세력 분쇄, FACS, 물의 resuspension 단계 동안 발생합니다. 3)로 모든 RNA 일 그것은 최고의 작은 추가 RNase 가능한 샘플으로 준비 하는 동안 소개 하. 이 높은-품질 RNA를 유지 하는 데 도움이 됩니다. 청소 도구 및 모든 장비는 하지 rinsing ribonuclease 억제제 솔루션 사용 RNase 무료 DEPC 처리 제품을 피 하지만. 4) 가능한 한 빨리 준비를 수행 합니다. 이 높은-품질 RNA를 유지 하 고 세포 죽음을 줄이기 위해 도움이 됩니다. 조직 절 개 길이 동물 숫자에 따라 동시에 여러 해/준비를 시작 하는 것이 좋습니다. 5)을 유지 하는 고품질 RNA, 세포 죽음, 천천히 가능한 세포 신호 그리고 transcriptional 활동 때 얼음에 셀을 준비 합니다. 이기는 하지만, 얼음 처리는 대부분 세포 유형, 실 온에서 처리 될 때 일부 세포 유형 (예를들면, 중 성구) 더 나은 수행 합니다. 6) 피하 칼슘, 마그네슘, EDTA, DEPC 처리 제품 셀 준비 중.
프로토콜
여기에 설명 된 모든 프로토콜에 있으며 캘거리 대학 동물 관리 위원회에 의해 승인.
1. 출신과 조직 (주 1)
- 마우스 또는 동물 윤리 프로토콜에 따라 적절 한 나트륨 pentobarbital (i.p., 50 mg/kg)의 과다와 안락사 다음 다시 마우스의 다리에서 원치 않는 머리카락을 제거 하 고 에탄올 소독 해 부의 지역.
- 조직 또는 관심의 microenvironment 해 부. 이 프로토콜에 대 한 사용 하 여 피부와 신경 조직 드롭릿 바코드 기반 단일 셀 transcriptomics 성인 조직 분리 다음의 generalizability를 보여 줍니다.
- 좌 골 신경에 대 한 stratton 은 그 외 에 상세한 프로토콜 사용 하 여 9. 잠시, 마우스의 뒤로/다리의 뒷 영역에서 피부를 잘라. 살 균 메스 블레이드와 허벅지의 길이 따라 절 개를 확인 합니다. 좋은 집게와가 위를 사용 하 여 노출 하 고 좌 골 신경 제거.
- 다시 피부에 대 한 Biernaskie 외 에 상세한 프로토콜 사용 하 여 10. 짧게, 해 부 등 뒤 피부 절 개 하 여 어깨에서 어깨, 엉덩이 걸쳐 위아래로 미세 집게와가 위를 사용 하 여 다시. 살 균 메스 블레이드를 사용 하 여 얇게 (0.5 c m 두께)로 피부를 잘라.
- 2 회와 차가운 HBSS, 조직 세척 하 고 원치 않는 결합 조직, 지방 예금 또는 해 현미경 파편 제거.
- 피부 진 피만,에 대 한 37 ° c.에 30-40 분 HBSS에서 dispase (5 mg/mL, 5 U/mL)에서 슬라이스를 플 로트 수술은 진 피에서 표 피를 분리 합니다. 표 피를 삭제 하거나 추가 관심의 경우 트립 신을 사용 하 여 분리 합니다.
- 살 균 메스를 사용 하 여 1-2 m m 조각으로 샘플을 말하다와 갓 해 동 2 mg/mL 차가운 콜라-4 효소 (2 mg/mL, 125 CDU/mg, F12 미디어에서)에 넣어.
- 신경에 대 한 2 x 좌 골 신경 당 ~ 500 µ L을 사용 합니다. 피부에 대 한 1 x 마우스 다시 피부 당 ~ 8 mL을 사용 합니다.
참고: 조직 해야 합니다 수 완전히 침수 콜라 IV 솔루션. 그것은 중요 한 그 어떤 소화 효소 처리, 저장, 그리고 적절 하 게 준비입니다. 효소는 시간의 오랜 기간 동안 실 온에서 남아 있다, 단일 셀 절연 과도 한 기계적 분쇄를 필요로 하 고 세포 생존 능력을 감소 것입니다. 콜라-4 세포 생존 능력은 가장 최적의 셀 문화 미디어에서 만들 수 있습니다. 그러나,이 효소의 활동을 변경할 수 또는 transcriptional 서명 그래서 사용자가 최적화 되어야 합니다.
- 신경에 대 한 2 x 좌 골 신경 당 ~ 500 µ L을 사용 합니다. 피부에 대 한 1 x 마우스 다시 피부 당 ~ 8 mL을 사용 합니다.
- 부드러운 떨고 모든 10 분으로 30 분 동안 37 ° C 목욕에 효소에 샘플을 품 어. 37 ° C에 통 적절 한 대안 이기도 합니다.
- 20-30 시간 30 분 후 효소 추가에서 P1000 pipettor와 triturate.
- 분쇄 30 분 마다를 반복 하 여 솔루션 흐린 나타나고 조직의 덩어리는 크게 해리.
참고: 셀 (그림 2b, 2c)의 전체 버전을 확인 합니다. 완전 한 릴리스를 확인 하려면 셀 Nuc 블루 (2 방울 1 mL 당) 접시 20 분 후 모든 핵 파편 보다는 단일 셀에 연관 되도록 현미경 검사. 그것은 각 조직 유형 또는 상태에 대 한 주어진된 실험 내에서 셀 분리의 정도 확인 하는 중요 한입니다. 거리 조직 (즉, 만성 부상) 또는 손상 되지 않은 성인 조직, 세포의 릴리스는 급성 부상 또는 배아 조직에서 극적으로 달라 집니다. 특정 세포 유형, 다른 사람 보다 조직에서 출시 하는 따라서 우선적으로 다운스트림 분석에서 셀 제외 하기 때문에 이것이 특히 중요 합니다.- 신경에 대 한 분열 조직 0.5-1.5 시간 총. 총 2 시간에 대 한 조직, 피부에 대 한 해리 (외피의 마지막 시간에서 추가 DNase (1 mg/mL) 피부 샘플을).
- 두 번 40 µ m 필터와 필터. 린스 차가운 1% 필터 BSA/HBSS.
- 8 분 260 x g에서 원심. 상쾌한 제거.
- HBSS 1 %BSA 넓은 구멍 팁을 사용 하 여 포함 된 셀 펠 릿을 resuspend 하 고 얼음에 놓습니다. 물의 resuspension 볼륨 조직 볼륨 기반 (피부에 대 한 800 밀리 그램 젖은 무게 800 µ L = 볼륨, 신경에 대 한 무게를 젖은 10 mg = 100 µ L 볼륨).
- 필요한 경우, 낮은 물의 resuspension 볼륨 및 FACS 정렬 흐름 속도 (초 당 이벤트)에 따라 필요에 따라 조정 합니다. 가장 효율적인 정렬 밀도 (minimalizing 시간 동안 수집 된 셀의 수를 최대화) 컬렉션은 3000-7000 이벤트/초.
- 생존 염료를 사용 하는 경우에 흠 없는 컨트롤에 대 한 subaliquot 꺼내 주세요. 그런 다음 1:15,000 생존 염료를 추가 (재고: 20000 nM / µ L) 샘플 (1.3 nM / µ L 최종 농도)을 줄이기 위해 전단 넓은 구멍 팁을 사용 하 여.
참고: 각 조직 유형 또는 상태에 대 한 특정된 실험에서 세포 죽음의 정도 대 한 확인 하기 긴요 하다. 샘플 내에서 일부 세포 유형에 따라서 우선적으로 다운스트림 분석에서 제외 되 고, 다른 사람 보다 죽을 확률이 높다.- 어둠 속에서 얼음에 5-10 분 동안 생존 염료와 샘플을 품 어. 그런 다음 차가운 1%의 4 개 mL를 추가 BSA/HBSS 샘플. 8 분 초과 가능성 염료 제거 260 x g에서 원심. 치료 없이 생존으로 subaliquot 같은 방식으로 흠 없는 제어 염료.
2. 분리 가능한과 건강 한 세포 (주 1)
- FACS 시설 활성화 하는 적절 한 형광 셀 정렬 (FACS) 매개 변수를 다음과 같이 확인 합니다.
- 1 단계에서에서 마지막 원심 분리기가 완료 되 면 준비는 되도록 사전에 FACS 기계를 준비 하 고 컬렉션 구획 차가운 유지 되도록 얼음 블록을 사용 하 여.
- 다음 매개 변수를 사용 하 여: 유량: 1.0 (해당 대략 10 µ L/분); 필터: 1.5 ND; 노즐 크기: 100 µ m; 앞으로 분산형: 80-180 V (이벤트의 크기를 구분 하기 위해 필요에 따라 변경); 사이드 분산형: 150-220 V (단위/이벤트의 셰이프를 구분 하기 위해 필요에 따라 변경); 레이저: 100-400 V (구별 생존 염료 긍정적 vs 부정적 이벤트 & 생존 염료 통제에 대하여이 확인 하기 위해 필요에 따라 변경); 빌 게이츠: 모든 셀 수집 되도록 필요에 따라 변경 합니다. 그림 2-2 g을 참조 하십시오.
참고: FACS 매개 변수는 매우 셀 형식에 따라 정렬 고용 하 고 따라서 사용자가 최적화가 필요한.
- 준비 15 mL 8 mL 얼음 1%의 좁은 하단 튜브 BSA/HBSS 샘플 컬렉션. 튜브 및 표면 장력 내부 정적 컬렉션 효율성을 발생할 수 있습니다. 액체의 표면 사이의 인터페이스를 위해 컬렉션 전에 튜브를 반전 하 고 튜브의 내부는 촉 촉.
참고: 매우 낮은 셀 번호를 사용 하는 경우 적절 한 작은 컬렉션 선박을 조정 합니다. - 일단 모든 셀, 수집 샘플 8 분 260 x g에서 원심.
참고: 원심, 전에 추가 1 %BSA / 세척/밀어 셀 HBSS 측면 표면에서 아래로 및 반전/혼합 튜브 FACS 직후. - 1 %BSA / HBSS에 얼음에 계속 셀 펠 릿 resuspend 3 단계 처리와 호환 되는 샘플 당 최대 볼륨은 33.8 µ L, 그래서 마지막 셀 희석/물의 resuspension 볼륨은 33.8 µ L에 이상적인 셀 번호를 얻기 위해 적절 한 보장 합니다. 이 단계에 대 한 다른 희석 미디어 옵션 (및 모든 이전 희석 1 %BSA / HBSS) DMEM, 포함 및 혈 청 40%, 최대 하지만 칼슘, 마그네슘, 또는 시 약을 포함 하는 EDTA를 방지.
- 얼음에 시간의 최소 금액에 대 한 셀을 둡니다. 이상적으로 동료 준비 한다 모든 장비 및 시 약 다음 단계 (3 단계)에 대 한 단계 2의 최종 단계.
- 셀 준비 중요 한 검사
- FACS에서 얻은 세포 수의 견적을 확인 합니다. 조직 유형 및 분리에 따라 길이, 파편 및 셀 크기와 모양이 매우 유사 수 있습니다. 따라서, 형광 기자를 사용 하지 않으면 FACS 모든 파편을 제외할 수 없습니다. FACS 컬렉션 (FACS)에 따라 이벤트의 비율을 이해 하 수행 된 후 최종 세포 수는 주어진된 준비 (그림 2 g)에 대 한 셀 실제로 하는 것이 좋습니다. 세포 수 hemocytometer 또는 자동화 된 셀 카운터 (두번 반복)을 사용 하 여 수행 하 고 FACS 기계에 따라 수집 된 총 이벤트에 의해 표현 되는 실행 가능한 세포의 비율을 계산.
- 셀 준비를 확인 합니다. 확인 하는 더 큰 입자 (> 100 µ m) 다운스트림 단계에서 장비를 방해할 수 있습니다 그들은 있는지. 파편의 부적당 한 제거는 단 세포 미세 칩 막힘 위험 수 있습니다. 아무 큰 파편 파편은 존재를 보장 하기 위해 (위와) Nuc 파랑 나머지 플레이트. 이것 또한 세포는 단 수 확인 하면 (즉, 함께 고정 되지 않는) 여러 셀 대신 단일 셀 나타냅니다 그 다운스트림 단일 셀 유전자 분석 신뢰를 주는.
- 셀 번호 시퀀스를 결정: 최대 8 샘플을 한 번에 실행 될 수 있는 시스템으로 로드할 수 있는 샘플 당 성인 조직 파생 셀 숫자의 큰 범위가 있다. 저자는 샘플 당 500-50, 000 셀에서 어디에서 든 지 로드 하 고 좋은 품질 scRNA-Seq 데이터 집합을 얻은. 로드 하는 가장 적절 한 핸드폰 번호에 대 한 더 많은 토론 토론 섹션에서 찾을 수 있습니다. 시퀀스 된 핸드폰 번호의 최종 출력 무 겁 게 격리 단일 세포의 품질에 따라 달라 집니다. 로드 10000 성인 조직 유래 세포 돌아갈 수 있습니다 어디서 나 1, 000에서 4000 시퀀스 된 세포 (10-40% 반환). 높은 핸드폰 번호 (~ 10000 셀,이 시스템에 대 한 권장 하는 최대 수)를 시퀀싱에 관심이 있으면 다음 25000-100000 셀 로드 하셔야 합니다.
3. 보석 (유제에 젤 비드) 생성 및 바코드 (1 일)
참고:이 프로토콜의 3-6 단계는 가장 일반적인 microdroplet 기반 단일 셀 플랫폼, 10 X Genomics에 의해 제조와 함께 사용 하도록 설계 되었습니다. 자세한 지침 단계 3와 4는 제조 업체의에 설명 된 프로토콜 (크롬 단일 셀 3' 프로토콜 참조)11,12 와 함께이 프로토콜을 따라야 합니다. 최상의 결과 얻으려면 3 단계 하루에 1이이 프로토콜의 분리 (1 단계) 및 셀 절연 (2 단계) 단계 후 즉시 완료 해야 합니다.
- 제조 업체의 프로토콜 11,12따라 칩을 준비 합니다. 이 microdroplet 기반 단일 셀 플랫폼 별도로 각 셀의 transcriptome 색인 그 샘플 ~ 750000 바코드 기술을 사용 합니다. 이 유화 액 (보석) 생성 된 cDNA에서 일반적인 바코드를 공유 하는 어디에 젤 비드에 세포를 분할 하 여 이루어집니다. 보석 생성 하는 동안 생성 된 보석의 대부분 (90-99%) 포함 나머지, 셀, 대부분 세포 전달 됩니다, 그리고 단일 셀 포함.
- 칩 홀더에 칩을 놓습니다.
- 얼음에 셀 마스터 믹스를 준비 합니다.
- 사용 되지 않는 우물에 50% 글리세롤을 추가 하 고 잘 1 셀 마스터 믹스의 90 µ L를 추가 기름을 잘 3 분할의 잘 2, 및 270 µ L 젤 구슬의 90 µ L.
- 개 스와 함께 칩을 커버.
- 칩 로드 하 고 실행 하는 단일 셀 컨트롤러에서.
- 트레이 꺼내는, 칩 트레이, 트레이, 철회 놓고 플레이누릅니다. 단일 셀 3' 젤 구슬 보석에 뇌관 (읽기 1 시퀀싱 뇌관), 부분 Illumina R1 시퀀스가 포함 된 포함 16 뉴클레오티드 (nt) 10 x Barcode, 10 nt 독특한 분자 식별자 (우미), 그리고 폴 리-dT 뇌관 순서. 실행 하는 동안 컨트롤러에서 젤 비즈 출시 되며 세포 lysate 및 마스터 믹스와 함께 혼합.
- 샘플 및 PCR 튜브에서 100 µ L를 수집 합니다.
- 사전 설정된 PCR에 장소 PCR 튜브 기계 고 키트에 따라 PCR을 실행 합니다. 부 화, 다음 보석 포함 됩니다 폴 리 adenylated mRNA에서 전체 길이, 바코드 cDNA.
- 실행, 다음 장소-20 ° C에서 하룻밤 다음 단계로 이전 하기 전에 최대 1 주일.
4. 정리, 증폭, 도서관 건축 및 라이브러리 정량화 (2 일 이후)
참고: 단계 4에 대 한 자세한 지침 제조업체의 프로토콜 11,12 에서 설명 하 고 함께이 프로토콜을 따라야 합니다.
- Silane 자석 비즈를 사용 하 여 보석 반응 혼합물에서 남은 생 화 확 적인 시 약/뇌관을 제거.
- 증폭 전체 길이, 바코드 cDNA 도서관 건축을 위한 충분 한 질량을 생성.
- DNA 수율을 평가 합니다. 도서관 건축, 이전 샘플의 DNA 수율을 평가 합니다. 이 다운스트림 PCR 단계 (도서관 건설 기간 동안 샘플 인덱스 PCR)에서 사용 하 여 얼마나 많은 주기를 결정 합니다. 셀 유형과 셀 생산량 (예를 들어, 제어 대 부상, 등), 활성화 상태에 따라 달라질 수 있습니다, 주어진된 샘플의 RNA 내용에 따라 권장된 주기 수 변경할 수 있습니다.
- 셀에 대 한 시퀀싱 ~ 3000 조직 파생 (활성화 상태에 무관 한), 저자 발견 14 주기 (샘플: ~ 10-100 ng DNA) 표준입니다.
- Bioanalyzer를 사용 하 여 DNA 분석을 위해. 참조 사용자 가이드13.
- 샘플을 조각 하 고 DNA의 크기를 선택 합니다. 라이브러리 건설 하기 전에 적절 한 cDNA amplicon 크기를 효소 조각화 및 크기 선택 프로토콜을 사용 합니다.
- 라이브러리 건설에 대 한 샘플을 준비 합니다. R1 보석을 인큐베이션; 중 분자에 추가 됩니다 (1 뇌관 순서 읽기) 하는 동안 P5, P7 (샘플 지) r 2 라이브러리 생성 하는 동안 추가 됩니다 (읽기 2 뇌관 순서).
- DNA 수율을 평가 합니다. 대부분 시퀀싱 시설 DNA 수율 및 품질 정보를 포함 하는 최종 라이브러리의 제출이 필요 합니다. 따라서, 전체 프로토콜 및 시퀀싱 시설에 운반 하기 전에 완료 하는 bioanalyzer를 실행 합니다.
- 최대 2 개월까지-80 ° C에서 샘플을 저장 합니다.
- 시퀀싱 하기 전에 DNA 정량화 키트를 사용 하 여 샘플을 계량. 이 시퀀싱 시설에서 할 수 있습니다.
5. 도서관 시퀀싱 (3 일 이후)
참고:이 프로토콜에서 사용 하는 단일 셀 transcriptome barcoding 플랫폼 Illumina 호환 쌍-엔드 라이브러리 시작 하 고 끝나는 P5 및 P7 시퀀스를 생성 합니다. 최소 깊이 셀 형식 id 10000-50000 읽기/셀15,16몇 수를 해결 하는 데 필요한, 하지만 ~ 100000 읽기/셀 성인 vivo에서 세포 (염두에서에 두고 일부 셀에 대 한 최적의 비용 범위 무역으로 것이 좋습니다. 형식 또는 최소한 활성화 된 셀 상태 도달 한다 30000-50000 읽기/셀 채도).
- 갖춘 적절 한 Illumina 시퀀서는 시퀀싱 시설에 드라이 아이스에 cDNA 라이브러리 전송.
- 시퀀싱 시설에 다음 정보를 제공 합니다.
- 샘플 세부 정보 제공: 각 라이브러리;에 해당 하는 인덱스 Id 샘플 종; 기본 어셈블리 (즉, GRCm38 마우스에 대 한);에 대 한 게놈 데이터베이스 electropherogram (사이 200와 9000 bp); bioanalyzer에서 조각 크기를 보여주는 cDNA 농도 (ng / µ L) 및 총 도서관 농도 (총 수익률 범위 200-1400 ng); 샘플의 볼륨 (µ L)입니다.
- 제공 하는 시퀀싱 요청: DNA 정량화 키트;를 사용 하 여 샘플을 계량 어댑터/인덱스 유형 (DNA TruSeq); 판형 (Eppendorf twin.tec, 풀 스커트-DNA에 대 한 권장); 시퀀싱 기술/라이브러리 유형 (10 x, 전체 시퀀싱 지침 및 권장 사항을 주기)17.
- 얕은 시퀀싱 (선택적) 실행: 여러 생물 학적 샘플을 분석 하는 연구 샘플 (집계)에서 모든 샘플 데이터를 포함 하는 단일 유전자 바코드 매트릭스를 생성을 풀링에서 도움이 됩니다. 풀링 될 때 샘플 사이의 배치 효과 최소화 하기 위해 다른 라이브러리 간에 읽기 깊이 표준화 한다. 이 위해서 단일 셀 숫자의 정확한 근사는 필요 합니다. MiSeq 시퀀서 얕은 시퀀싱 고 정확한 셀 의견을 얻기 위해 비용 효율적이 고 실용적인 방법입니다.
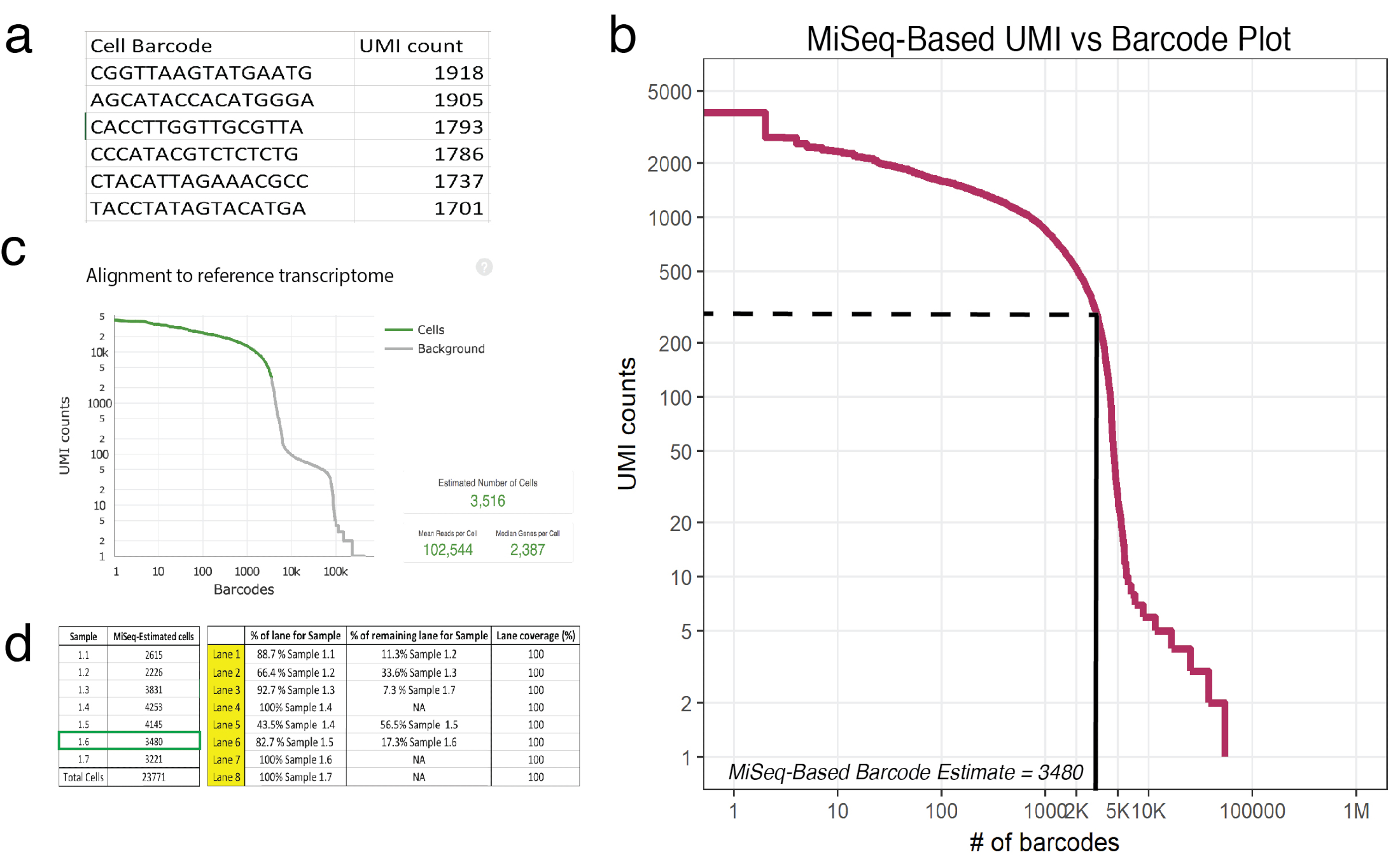
참고: MiSeq SR50 시퀀서를 사용 하 여 하나의 실행 정확 하 게 추정 약 20000 셀 충분 한 범위를 제공 합니다. 이 실행 우미 각 고유 바코드에 대 한 복구 수를 대략적인 것입니다. 그림 3a, 출력 예 (예제 1.6) (.csv) 헤더 바코드 표시 됩니다 하 고 자신 있게 매핑된 읽기에 의해 결정 되는 해당 우미 계산.- R 프로그래밍 언어에 익숙해질 수 bioinformatician와 상의 하십시오. 자세한 내용은18DataCamp 자습서를 참조 하십시오.
- 서식 파일19으로 제공 된 R 스크립트를 사용 하 여 sequencer에서 얻은 원시 데이터를 평가 합니다. 원시 데이터의 각 고유 셀 바코드에 매핑된 Umi 나타냅니다. 스크립트는 첫 번째 열은 바코드의 목록이 고 두 번째 열은 그 해당 우미.csv 파일 카운트 읽습니다. 이 스크립트는 각 샘플에 바코드 셀의 예상된 번호 뿐만 아니라 음모 (그림 3b)를 제공할 것입니다. 첫 번째 가파른 드롭의 1 / 3 지점에서 주어진된 샘플에 대 한 우미의 입력 된 번호 인지 확인 하는 스크립트를 조정 합니다. 그림 3b에서이 팔꿈치 3,480 바코드 셀에 해당 하는 약 225 Umi 떨어진다.
- HiSeq를 사용 하 여 전체 깊이 연속 비교 (3,516 셀 성공적으로 있던 시퀀싱, 그림 3 c), 얕은 시퀀싱 견적 예측 3,480 셀.
- (단계 5.3)에서 사용 하 여 셀 복구 근사 또는 복구 차트를 사용 하 여 깊은 시퀀싱 레인 배포를 계획 하는 제조업체의 프로토콜20 에서 발견. 그래서 얕은 시퀀싱 (이 종종 경우) 각 샘플에 있는 세포의 서로 다른 숫자는 계시 하는 경우 다음 레인 분포 계산 해야 그에 따라 각 샘플 비교 범위를 받아야 한다. 1 HiSeq 흐름 세포 (8 레인 구성) 최대 2.4 십억 사용자 지정 쌍된 끝 읽기 시퀀스 수 있습니다. 예 흐름 셀 설정 그림 3에 표시 됩니다.
6. 처리 파일 읽기
참고: 단일 셀 3'이 프로토콜을 사용 하 여 라이브러리를 시퀀싱 이진 기본 호출 (BCL) 형식의 원시 데이터를 생성 하 고. 셀 레인저 패키지는 BCL 파일에서 텍스트 기반 FASTQ 파일을 생성, 게놈 수행 하는 데 사용 됩니다 transcriptomic 정렬, 유전자 수, 디 멀티플렉싱, 그리고 샘플의 집계. 이 섹션에서는 사용자가 시퀀싱 시설에서 원시 BCL 데이터를 다운로드 하 여 필터링 된 유전자 바코드 매트릭스 다운스트림 생물 정보학에 대 한 준비를 생성할 수 있는 중요 한 단계는 제공 됩니다.
- 중앙된 서버를 사용 하 여 프로그램을 실행 하기 위한. BCL 파일, FASTQ 파일 및 처리 다운스트림 생물 정보학의 가장 중요 한 처리 능력을 요구 한다.
- 모든 원시 읽기 파일 서버 (또는 사용 가능한 경우 FASTQ 파일)를 다운로드 합니다.
- 서버 관리자는 중앙된 서버 또는 클러스터에 계정을 설정 하 고 유닉스21를 참조 하십시오.
- 서버 운영 체제에 대 한 적절 한 fetch 명령을 사용 하 여 시퀀싱 시설의 서버에서 모든 파일을 다운로드 하기.
- 명령줄에서 실행할 수 있는 보안 경로에서 파일을 다운로드 하는 명령을 제공 하는 대부분 시퀀싱 시설 (아래 예제 참조).
- "< 사용자 이름 >" 대체 및 제공 하는 자격 증명을 가진 명령줄 자리 표시자 "< 암호 >".
wget-O-"https://your_sequencing_facilitys_server.com/path_to_raw_read_files/-아니-쿠키-no-체크-인증서-후 데이터 ' j_username 사용자 이름 및 j_password = 비밀 번호 =' | wget-아니오-쿠키-no-체크-인증서-후 데이터 ' j_username 사용자 이름 및 j_password = 비밀 번호 ='-ci-
- 경우에 파일에 절대 경로 (예: https://your_sequencing_facilitys_server.com/path_to_raw_read_files/)를 제공 하 고, 반입 명령에이 경로 삽입 합니다.
- 압축 파일: ".gz" 확장명이 파일 끝을 다운로드, 그것은 압축 된 "gzip" 명령을 사용 하 여. , 실행 압축 명령줄에서 명령 (아래 예제 참조).
gunzip raw_read_files.gz - 자가.tar22로 서버에 셀 레인저의 최신 버전을 다운로드.
-
중요 한: 다운로드 전에 리눅스 시스템 만족23최소 요구 사항을 확인 합니다. 64 기가바이트 RAM과 1TB의 디스크 여유 공간 8 코어 인텔 프로세서의 최소한을 확인 합니다.
참고: 셀 레인저 미리 인간과 설치류 참조 transcriptomes을 제공합니다. 이 유전자에 GFP24같은 감지 cellranger mkref 명령을 사용 하 여 수정할 수 있습니다.
-
중요 한: 다운로드 전에 리눅스 시스템 만족23최소 요구 사항을 확인 합니다. 64 기가바이트 RAM과 1TB의 디스크 여유 공간 8 코어 인텔 프로세서의 최소한을 확인 합니다.
- 시퀀서의 기본 호출 파일 (Bcl) cellranger mkfastq 명령을 사용 하 여 FASTQ 파일을 생성 합니다.
참고: 프로그램 원시 읽기 (FASTQ 파일) 참조 게놈에 정렬 되며 다운스트림 분석을 위한 유전자 세포 매트릭스를 생성. 참조 게놈에 접합 인식 맞춤을 수행 하는 스타 동기 기를 사용 합니다. (즉, 단일 유전자 주석 호환 읽기)만 자신 있게 매핑된 읽기 사용 됩니다 우미 계산에 대 한.- 예를 들어 cellranger mkfastq 명령을 사용 하 여:
cellranger mkfastq -id = sample_name \
-= / 경로/로/샘플 실행 \
-csv=csv_file_containing_lane_sample_index.csv
- 예를 들어 cellranger mkfastq 명령을 사용 하 여:
- 단일 셀 유전자 수를 생성 하기 위해 mkfastq를 사용 하 여 생성 하는 FASTQ 파일에 cellranger 수를 실행 합니다.
- 예를 들어 cellranger count 명령을 사용 하 여:
cellranger 수 -id = sample_name \
-transcriptome = refdata-cellranger-mm10-1.2.0 \
-fastqs = / 절대/경로/로/fastq/파일 \
-샘플 same_sample_name_supplied_to_cellranger_mkfastq = \
-localcores = 30
- 예를 들어 cellranger count 명령을 사용 하 여:
- 다중 라이브러리 집합 (선택 사항): 샘플을 결합, cellranger aggr를 사용 하 여 cellranger 수 출력 하. 여러 라이브러리에서 풀링 하는 데이터를 포함 하는 단일 유전자 바코드 매트릭스에 결과. 예를 들어 cellranger aggr 명령:
cellranger aggr-id = sample_name \
-csv csv_with_libraryID_ & _path_to_molecule_h5.csv = \
-정상화 = 매핑된
참고: 라이브러리 집계할 수 있는 세 가지 정규화 모드를 사용 하 여 (매핑된, 원시, 없음). 매핑되는 것이 좋습니다 그것 샘플이 더 높은 깊이 라이브러리 모든 라이브러리 같은 시퀀싱 깊이25때까지. - 즉각적인 시각화/데이터의 분석을 위해 루 페 셀 브라우저26x 10로 (cellranger 수 또는 cellranger aggr를 사용 하 여 생성).cloupe 출력 파일을 가져옵니다.
7. scRNA-Seq 데이터 집합의 고급 분석
참고: 완전 한 scRNA-Seq 도구 데이터베이스 scRNA 도구3,27에서 찾을 수 있습니다. 아래는 쇠라2 를 사용 하 여 클러스터링 및 pseudotemporal 실험실된 셀에 대 한 프레임 워크 주문 단 안경6을 사용 하 여. 비록이 작품의 대부분은 로컬 컴퓨터에 행 해질 수 있다, 다음 단계는 계산 기관 서버를 사용 하 여 완료 됩니다 가정 합니다.
- 리눅스 플랫폼28를 사용 하 여 서버 계정에 Miniconda의 최신 버전을 다운로드.
- Conda29를 사용 하 여 R의 최신 버전을 설치 합니다.
- 서식 파일30제공된 Seurat R 스크립트를 사용 하 여 데이터를 플롯 합니다.
참고: Seurat는 품질 관리 검사, 클러스터링, 차동 유전자 표정 분석, 마커 유전자 식별, 차원 감소, 및 scRNA-Seq 데이터의 시각화를 가능 하 게 하는 R-기반 툴킷. Seurat 코딩 및 자습서의 포괄적인 설명31Satija 실험실 웹사이트 찾을 수 있습니다. - 서식 파일32로 제공된 단 안경 R 스크립트를 사용 하 여 데이터를 플롯 합니다.
참고: 단 안경 pseudotime 이상 식 변경의 시각화 및 기본 세포 운명 결정 하는 유전자를 식별 하는 또 다른 연구-기반 툴킷입니다. 단 안경 코딩 및 자습서의 포괄적인 설명33단 안경 웹사이트 찾을 수 있습니다. - KBET R-패키지 테스트 하 고 데이터 집합34풀링 결과로 배치 효과 수정 사용할 수 있습니다.
8. NCBI의 지리적 및 SRA 제출
참고: 원시 시퀀싱 파일에 쉽게 액세스할 수 있도록 재현성 및 해도, 이후 온라인 공개 저장소 accessioned 제출 권장 또는 원고 제출 전에 필요한. 생물 공학 정보 (NCBI)를 위한 국가 센터 진 식 옴니 버스 (지 오)와 시퀀스 읽기 아카이브 (SRA)는 높은 처리량 시퀀싱 데이터35,36공개적으로 액세스 가능한 데이터 저장소.
- NCBI의 지리적 제출자 계정37에 대 한 등록.
- 디렉토리/폴더 (GEO 제출자의 이름으로 라는 제목의)으로 세 가지 구성 요소를 포함 하는 완벽 한 지리적 제출: 1) 메타 데이터 기록 (프로젝트 제출 당 한 스프레드시트); 2) 원시 데이터 파일; 3) 데이터 파일을 처리.
- 다운로드 하 고 메타 데이터 스프레드시트38. 다음 공용 지리적 제출 가이드 (GSE100320)39로 사용할 수 있습니다. 디렉터리에 스프레드시트를 놓습니다.
- 장소 원시 데이터 파일은 디렉터리에 모든 라이브러리 cellranger 계산 스크립트에서 생성 된.
- 장소 처리 데이터 파일 (필터링 된 barcodes.tsv, genes.tsv, 및 matrix.mtx 파일) 디렉터리에 모든 라이브러리 cellranger 계산 스크립트에서 생성 된.
- GEO 제출자의 FTP 서버 자격 증명을 사용 하 여 모든 세 가지 구성 요소를 포함 하는 디렉터리를 전송. 리눅스/유닉스 사용자: ncftp, lftp, ftp, sftp, 및 ncftpput 사용할 수 있습니다.
- 모든 전송38지 오를 게 알립니다.
결과
ScRNA-Seq 데이터 집합을 분석 하도록 하는 오픈 소스 패키지의 레 퍼 토리는 이러한 패키지 사용 R 기반 언어3의 대다수와 가진40 극적으로 증가 했다. 여기, 두이 패키지를 사용 하 여 대표 결과 표시 됩니다: 평가의 자동된 그룹화 유전자 발현을 기반으로 셀을 단일 고 해결 궤적을 따라 하나의 셀을 순서이 세포 생물학 재조 처리합니다.
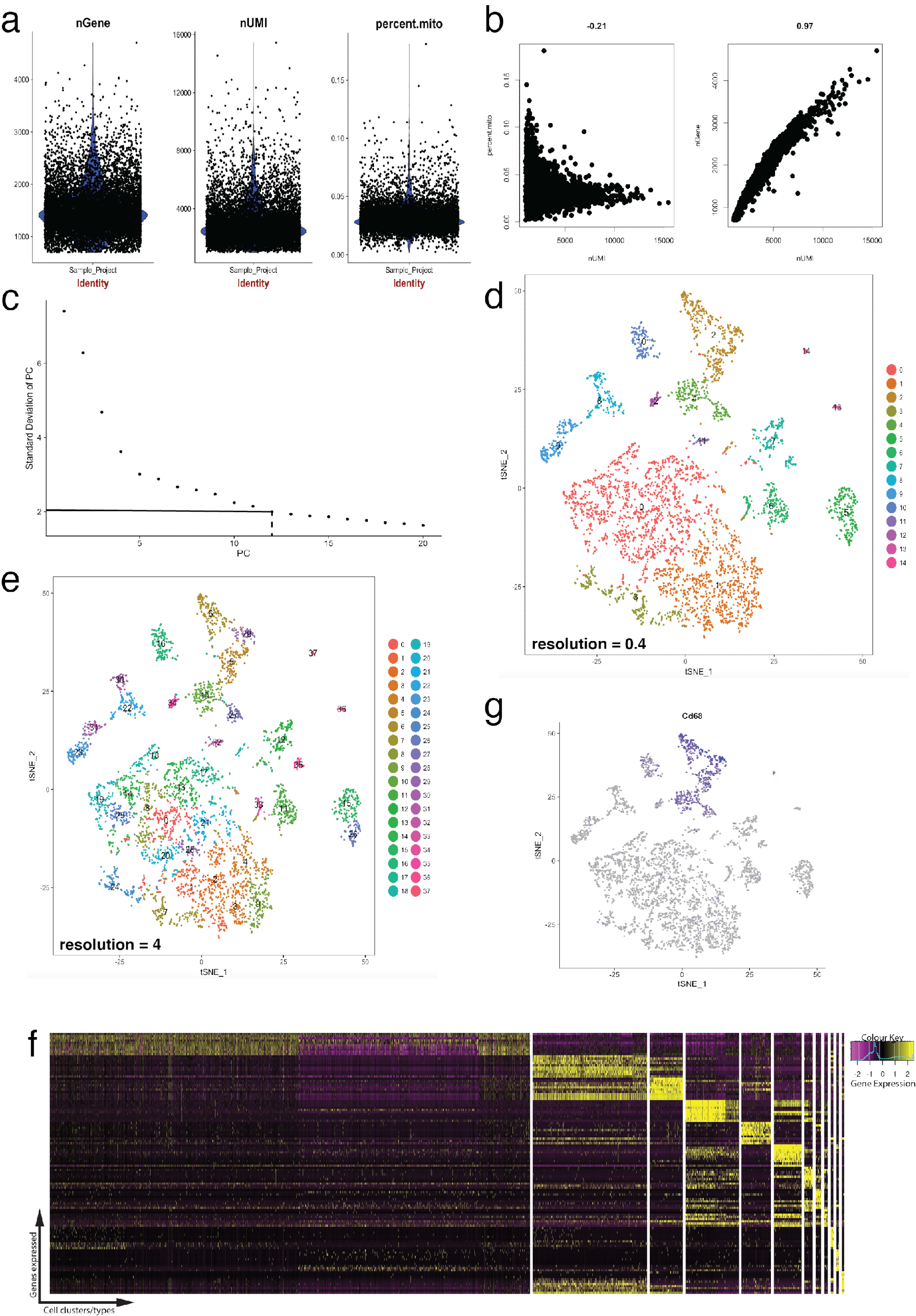
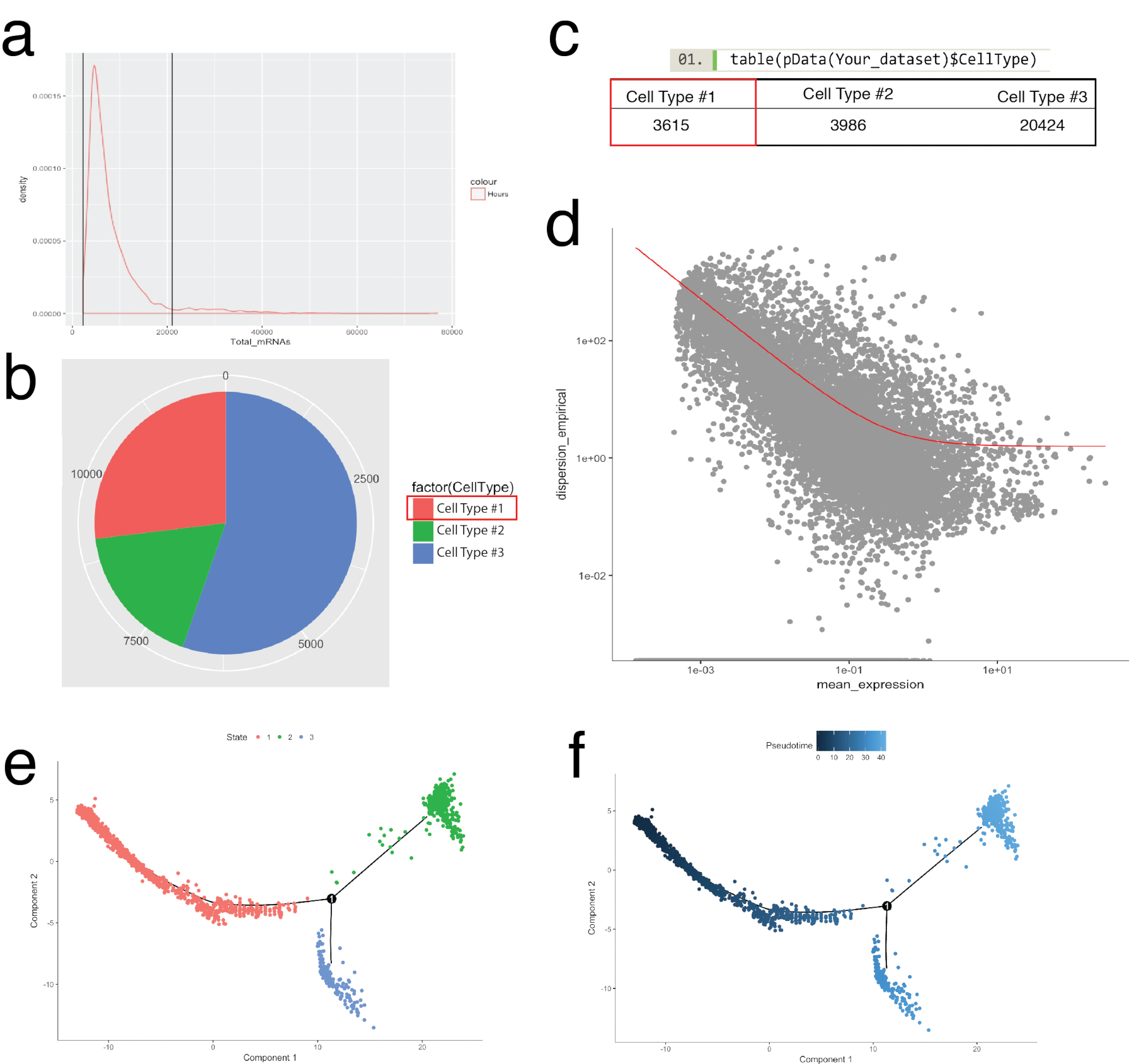
그림 4 는 미리 품질 검사 및 다운스트림 생물 정보학 분석을 처리 하기 위한 Seurat의 사용을 보여 줍니다. 첫째, 여과 및 분석에서 비정상적인 세포의 제거 품질 검사를 위해 필수적 이다. 이 미토 콘 드리 아 유전자, 유전자 (nGene)의 수 및 셀 남자와 outliers 식별 하 우미 (nUMI)의 수의 비율을 시각화 (그림 4b) 플롯 바이올린 (그림 4a)과 분산형을 사용 하 여. 분명 국외 자 수 유전자, 우미, 또는 미토 콘 드리 아 유전자의 비율의 어떤 셀 Seurat의 FilterCells 함수를 사용 하 여 제거 되었습니다. Seurat는 주성분 (PC)를 사용 하 여 이후 분석 점수 클러스터 셀, 통계적으로 중요 한 Pc 포함 하도록 결정에 중요 한 단계 이다. 팔꿈치 작 (그림 4c) PC 선택,' PC의 표준 편차 ' 고원 넘어 어떤 Pc에서 축 제외 했다 사용 되었다. 클러스터링의 해상도 또한 4 (높은 해상도 높은 세포 클러스터, 그림 4e 선도 하는 클러스터 수 변경할 수 있습니다, 0.4 (낮은 해상도 적은 셀 클러스터, 그림 4 d로 이어지는)에서 이르는 시연 조작 했다 ). 낮은 해상도, 높은 해상도에서 이것 또한 대표 있습니다 하위 또는 셀 인구의 과도 상태 반면 각 클러스터 정의 셀 형식을 나타내는 가능성이 높습니다. 이 인스턴스에서 저해상도 클러스터 설정은 특정된 클러스터 (그림 4 층)에서 가장 높은 표현된 유전자를 식별 하 식 heatmaps (Seurat의 DoHeatmap 함수를 사용 하 여) 추가 분석을 위해 사용 되었다. 이 경우 가장 높은 표현된 유전자는 각 클러스터를 고유 하 게 정의 된 유전자에 의해 표현 되었다 보여주는, 모든 다른 클러스터에 비해 특정된 클러스터에 차동 식 평가 하 여 확인 되었다. 또한, 개별 후보 유전자에 tSNE 음모에 Seurat의 FeaturePlot 기능 (그림 4 g)를 사용 하 여 구상 될 수 있다. 이 대 식 세포를 나타내는 클러스터 있었는지 해독에 대 한 허용. FeaturePlot를 사용 하 여, 우리 둘 다 2 클러스터 발견 4 Cd68-팬-대 식 세포 마커를 표현 했다.
단 안경 패키지 생물학 과정 (그림 5) 정리를 셀 클러스터에 Seurat, 식별을 확실 한지 알아보는 그리고 셀 궤도, 또는 pseudotemporal 주문, 건물 사용 되었다. Pseudotemporal 주문 사용할 수 있습니다 샘플에 대 한 단일 셀 식 프로필 생물 시간 코스를 따라 것으로 예상 된다. 셀 중간 상태, 두 대체 세포 운명의 분기 포인트를 해결 하기 위해 pseudotemporal 연속체를 따라 주문 될 수 있다 고 진 서명 인수 각 운명의 기본을 식별 합니다. 첫째, Seurat의 여과와 유사한 모든 셀에 걸쳐 mRNA의 분포는 로그 정상과 떨어진 사이 위와 더 낮은 그림 5a에서 식별 되는 가난한 품질 세포 제거 되었습니다. 단 안경의 newCellTypeHierarchy 함수를 사용 하 여 단일 세포 분류 되었다 그리고 알려진된 혈통 마커 유전자 (그림 5b, 5c)를 사용 하 여 계산 합니다. 예를 들어 PDGF 수용 체 알파 또는 섬유 특정 단백질 1을 표현 하는 세포를 만드는 섬유 아 세포를 정의 하기 위한 기준 셀 형식 # 1에 할당 했다. 다음,이 인구 (셀 타입 #1) 해독 구와 궤적을 평가 했다. 이 인구 내의 극단적인 상태를 나타내는 셀을 비교 하 고 (그림 5 d) 인구의 나머지 셀을 정렬 하기 위한 차동 유전자 발견 단 안경의 차동 GeneTest 함수 이용 되었다. 모든 셀에 걸쳐 매니폴드 학습 방법 (비-선형 차원 감소의 형식)을 적용 하 여 pseudotemporal 경로 따라 좌표 지정 되었다. 이 궤적은 다음 셀 주 (그림 5e) 및 pseudotime (그림 5 층)에 의해 시각.

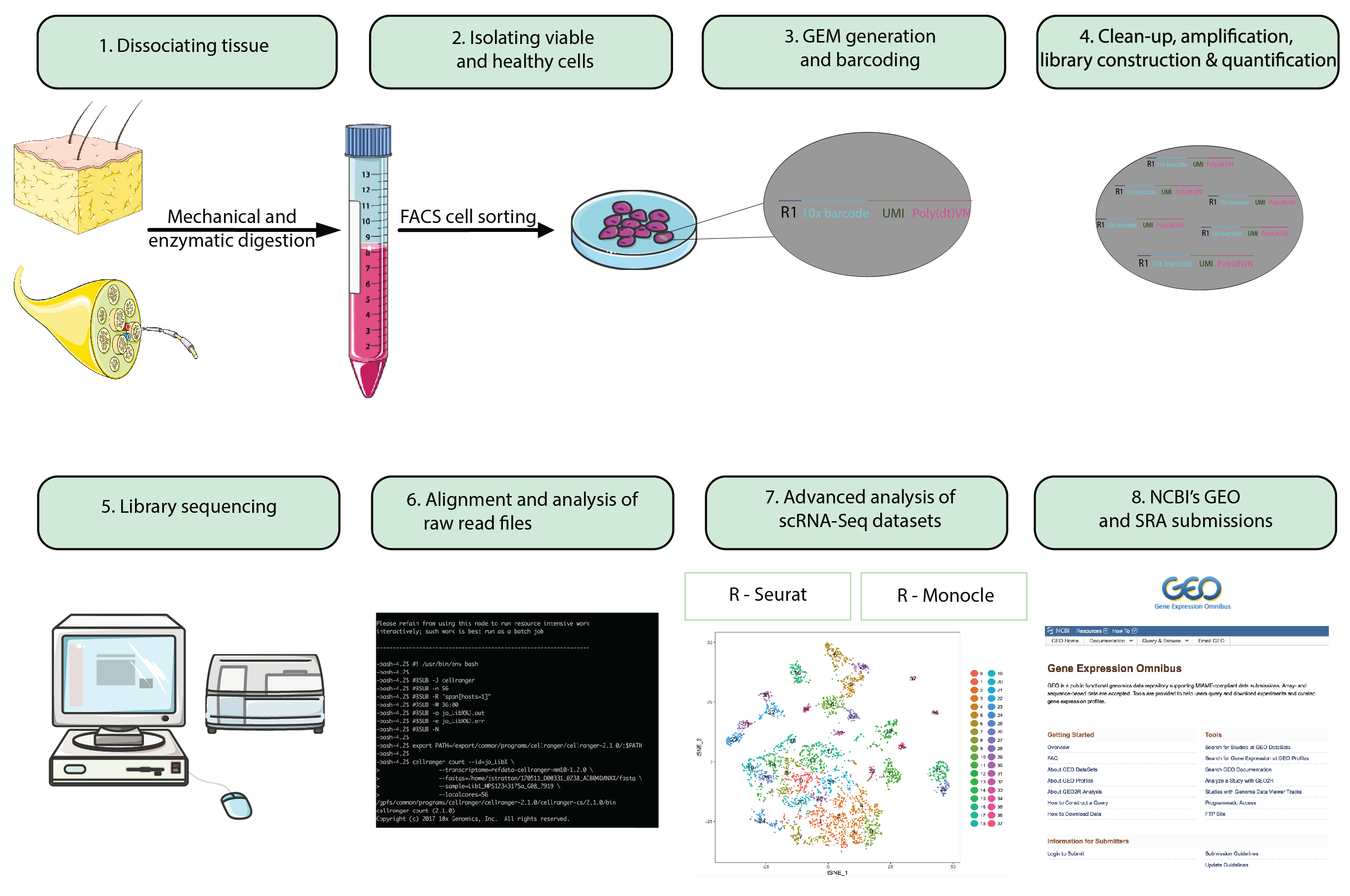
그림 1: 순서도. 단일 분석에서 전체 동물 준비 단계 세포 RNA-Seq 집합 공개적으로 사용 가능한 저장소에 마지막 데이터 집합을 제출. 유제 (보석)에 젤 구슬 구슬 수천 단일 세포의 캡슐화는 바코드 oligonucleotides와를 참조 하십시오. 이 그림의 더 큰 버전을 보려면 여기를 클릭 하십시오.
{kind=link}

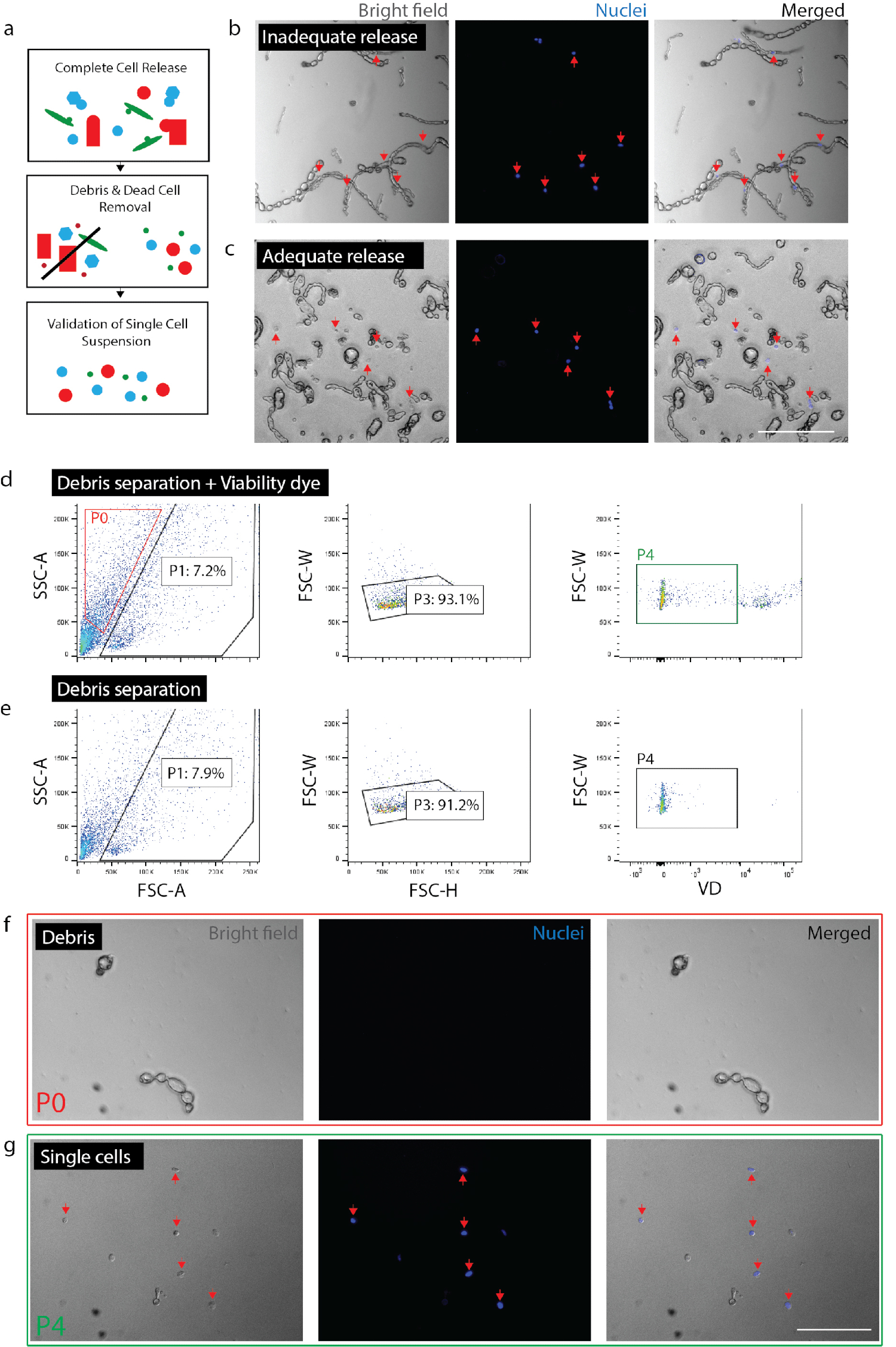
그림 2: 신경 조직에서 가능한 단일 세포 현 탁 액을 만들기. (a) 품질 관리 검사의 개요 만화. (b) 세포 및 세포 잔해는 아직도 파편 (빨간색 화살표)에 통합. (c) 세포 파편 (빨간색 화살표)에서 발표 했다. (FACS d) 세포 격리입니다. P0: 파편 분수; P1: 셀 같은 분수; Duplets;의 P3: 제외 P4: 생존 염료 (Sytox 오렌지) 부정적인 분수. (e) 생존 염료 컨트롤입니다. (f) 고립 된 파편을 대표 하는 P0 분수의 이미지. (g) 분리 가능한 셀 (빨간색 화살표)을 대표 하는 P4 분수의 이미지. (b) (c) (f) 및 (g) 핵 염료 이미징 하기 전에 20 분을 추가 했다. 스케일 바: 80 µ m. 이 그림의 더 큰 버전을 보려면 여기를 클릭 하십시오.
{kind=link}

그림 3: 처리 샘플 X 10에 복구 된 셀의 수를 예측 하는 얕은 시퀀싱. (a) 예 (예제 1.6) MiSeq에서 생성 된 csv 셀 바코드와 그 해당 우미의 자신 있게 매핑된 읽기에 따라 계산 됩니다. (b) 바코드 샘플 1.6에 대 한 순위 음모 셀 바코드의 기능으로 우미 수에 1 개의 중요 한 하락을 보여줍니다. 파선 및 고체 라인 셀과 검사에 의해 결정 되는 배경 간의 구분을 나타냅니다. (c) 셀 바코드 얕은 시퀀싱 정확 하 게 샘플 1.6에 대 한 셀의 개수를 계산 하는 것을 밝혀 셀 레인저 파이프라인 게시물-HiSeq를 사용 하 여 관찰. (d) 얕은 시퀀싱에 따라 흐름 셀 설정의 예는 셀 견적 파생. 1.6 샘플에 대 한 얕은 시퀀싱 예측 3480 셀 이후 1.17 차선 되도록 할당 된 > 셀 시퀀싱 범위 HiSeq에서 당 100000 읽습니다. 참고: 모든 차선은 100%를 추가 해야 합니다. 이 그림의 더 큰 버전을 보려면 여기를 클릭 하십시오.
{kind=link}

그림 4: 품질 관리 및 Seurat R 패키지를 사용 하 여 단일 셀 RNA-Seq dataset의 생물 정보학. (a) 유전자의, 독특한 분자 식별자 (Umi)의 수 및 미토 콘 드리 아 게놈에 성적 비율을 포함 하는 품질 관리 통계의 구획. (b) 샘플 유전자 감지 세포 미토 콘 드 리아 성적 및 Umi의 비정상적인 수준으로 플롯합니다. (c) 샘플 팔꿈치 줄거리 통계적으로 중요 한 Pc의 임시 결정에 사용. 대시 및 도트 점선 라인 그래프에서 분명 "팔꿈치"이 되는 컷오프를 나타냅니다. 이 팔꿈치 전에 PC 차원 다운스트림 분석에 포함 됩니다. (d, e) 그래프 기반 셀 클러스터 tSNE 플롯을 사용 하 여 저 차원 공간에서 두 개의 다른 해상도에서 시각. (f) 위쪽 표시자 유전자 (노란색) 각 클러스터에 대 한 Seurat의 DoHeatmap 함수를 사용 하 여 식 heatmap에 시각. (g)의 예, Cd68 유전자 세포 (보라색) Seurat의 FeaturePlot 함수를 사용 하 여 나타내는 마커 식 머릿속으로. 이 2-4 (패널 d)이 데이터이 집합의 해당 클러스터 세포를 나타냅니다 나왔다. 이 그림의 더 큰 버전을 보려면 여기를 클릭 하십시오.
{kind=link}

그림 5: 세포 분류 및 Monocle 툴킷을 사용 하 여 peudotemporal 궤적을 따라 주문. (a)는 샘플의 모든 셀에 걸쳐 mRNA (우미 카운트에서 유추)의 분포를 검사 합니다. 만 셀 0-사이 mRNA ~ 20000 다운스트림 분석을 위해 이용 되었다. (b의 c) 할당 하 고 셀 형식이 알려진된 계보 셀 표식에 따라 계산 합니다. 예를 들어 PDGF 수용 체 알파 또는 섬유 특정 단백질 1을 표현 하는 셀에 할당 된 셀 형식 #1 팬-섬유 아 세포를 나타내는 단 안경의 newCellTypeHierarchy 함수를 사용 하 여. 다른 세포 유형의 수 (b) 원형 차트 및 테이블 (c)으로 구상 될 수 있다. (d) 셀을 사용 하 여 형식 #1 (fibroblasts) 예를 들어, 셀 평균 식 대 진 분산을 보여 주는 산 점도 사용 하 여 구상 될 수 있다 주문에 사용 되는 유전자. 빨간 곡선 유전자 단 안경의 estimateDispersions 함수를 사용 하 여 평균-분산 모델에 의해 계산 된 주문에 대 한 사용에 대 한 구분을 보여 줍니다. 이 구분을 충족 하는 유전자는 다운스트림 pseudotime 주문 사용 되었다. (e, f) 셀의 "국가" (e)와 단 안경에 할당 된 "Pseudotime" (f)에 의해 감소 된 2 차원 공간에서 셀 궤적의 시각화. 이 그림의 더 큰 버전을 보려면 여기를 클릭 하십시오.
{kind=link}
토론
이 프로토콜 방법을 보여 줍니다 단 세포의 적절 한 준비 수 단일 세포의 수천의 transcriptional이 밝히기 차별 기능 상태 또는 조직 내에서 고유한 세포 id. 프로토콜과 형광 기자 단백질 또는 유전자 변형 도구를 필요로 하지 않는 인간;에서 그들을 포함 하는 관심의 다양 한 조직에서 단일 세포의 분리에 적용 될 수 있습니다. 각 조직 명심 독특한 이며이 프로토콜 조정/수정 어느 정도 필요 합니다.
세포 내에서 다양 하 고 높은 동적 transcriptional 프로그램 단일 셀 유전체학의 가치를 강조 했다. 높은-품질 RNA 분리, 이외에도 고품질 데이터 집합에 필요한 중요 한 샘플 준비 단계 세포 조직에서 발표 완전히 되 고 세포가 건강 하 고 그대로 하도록 이다. 이것은 상대적으로 곧장 앞으로 쉽게 세포를 수집 발표, 순환 셀 또는 셀 느슨하게 그대로 유지 하는 조직에 림프 조직 같이. 하지만이 다른 성인 조직, 세포 외 기질 및 자주 엄밀한 cytoskeletal 단백질 세포 구조를 유지에 관련 된 큰 거리에 걸쳐 선진국 세포질 건축 때문에 도전적 일 수 있다. 심지어 적절 한 분리에 대 한 기술로 셀의 전체 릴리스, 잠재적인 필요한 엄격 하 고 종종 긴 처리 mRNA 품질 및 셀 무결성을 변경할 것입니다. 또한, 효소를 이용한 분리에 사용 되는 높은 온도 transcriptional 서명29,30또한 영향을 미칩니다. 프로토콜의 현재 품질 관리 검사, 최적화 이러한 장애물을 극복 하는 데 도움이 수 어떻게 보여 조직 myelinated 성인 신경 및 세포 외 매트릭스 풍부한 성인 피부 등을 사용 하 여입니다.
어떤 scRNA-Seq 실험을 설계할 때 주요 고려 시퀀싱 깊이의 선택입니다. 시퀀싱을 매우 다중화 하 고 읽을 수 있는 깊이 최대 5 백만 읽기/셀14 스마트가 같은 전체 길이 RNA-Seq 방법을 사용 하 여 드롭-Seq2 를 사용 하 여 매우 낮은 것에서 변화할 수 있다 대부분 scRNA-Seq 실험은 일반적으로 셀 형식을 분류41,42에 대 한 충분 한 시퀀싱 10000 읽기/셀, 낮은과 중간에 높은 식 성적을 감지할 수 있습니다. 얕은 시퀀싱 깊이 복잡 한 조직 세포의 수천 드문 인구를 자신 있게 돌리다 필요할 어디에 걸쳐 희귀 세포 인구를 감지 하려는 경우 시퀀싱 비용에 저장 하는 값입니다. 하지만 얕은 깊이 시퀀싱 유전자 발현과 관련 된 미묘한 transcriptional 서명 프로세스에 대 한 자세한 내용은 필요한 경우 적합 하지 않습니다. 현재, 그것은 500000 읽기/셀, 셀에 유전자의 대다수는 발견 하지만이 프로토콜에 따라 다를 수 있습니다 및 조직 입력43,44추정. 전장 사본 시퀀싱 어셈블리에 대 한 필요를 circumvents 및 따라서, 소설 또는 드문 결합 이체를 검색할 수 있습니다, 하는 동안 자주 시퀀싱 비용 스케일링 복잡 한 조직 시스템을 구성 하는 세포의 수천 검토를 같은 접근 제한. 반면, 3' 태그가 단일 셀 라이브러리와 같은 일반적으로이 프로토콜에서 설명 하는 것 들 낮은 복잡도 얕은 시퀀싱을 필요로. 5 지원된 시퀀서 중 하나에 설명 된 프로토콜을 사용 하 여 생성 하는 라이브러리의 순차를 설정할 수는 주의 하는 것이 중요 하다: 1) NovaSeq, 2) HiSeq 3000/4000, 3) HiSeq 2500 빠른 실행 및 높은 출력, 4) NextSeq 500/550, 및 5) MiSeq.
섬세 한 조직 및 세포 아직 처리를 위한 필요를 감소 시키는 다른 접근 방법 단일 셀 RNA-Seq, 단일 셀 RNA-Seq의 혜택의 일부를 유지, 단일 핵45에서 RNA의 분석 이다. 이 이렇게는 RNA 저하, 그리고 더 많은 극단적인 조치, 핵의 적절 한 릴리스를 줄이고 더 빠른 처리를 허용 하 고 따라서 가능성이 해당된 조직 내에서 모든 셀을 나타내는 transcriptional 프로필의 자부 캡처 수 있습니다. 이 물론,만 따라서 따라 어떤 실험 목표가이 접근 하거나 적합 하지 않을 수 있습니다 관심의 특정된 셀 내에서 현재 transcriptional 활동의 일부를 제공할 것 이다.
특정된 조직 내에서 세포 id의 완전 한 특성, 외 scRNA-Seq 데이터 집합에 대 한 가장 중요 한 분석 중 하나는 '정의' 세포 인구에 걸쳐 중간 transcriptional 상태의 평가 이다. 이러한 중간 상태 전통적인 대량 RNA-Seq 접근 가능 했던 식별된 인구 내의 셀 간의 혈통 관계에 대 한 통찰력을가 르 친다 수 있습니다. 여러 scRNA-Seq bioinformatic 공구 지금이 명료 하 게 개발 되었습니다. 이러한 도구는, 예를 들어 암 세포 종양/전이성 상태로 전환 다양 한 터미널 운명 또는 상태와 무부하 상태 사이 왕복 하는 면역 세포로 성숙 하는 줄기 세포 관련 프로세스를 평가할 수 있습니다. 셀에 미묘한 transcriptome 차이 계보 편견, FateID, 같은 최근에 개발한 bioinformatic 도구47유추할 수 표시 될 수 있습니다. 이후 세포 전환 사이의 구분은 어려울 수 있습니다, 그리고 깊은 시퀀싱 필요한46수 transcriptional 차이 주어진 확인 미묘한 수 있습니다. 다행히, 다른 흐름 셀에 라이브러리를 다시 실행 하 여 데이터 집합을 추가 조사에 관심이 얕게 시퀀스 된 도서관의 범위를 늘릴 수 있습니다.
함께 찍은,이 프로토콜 transcriptionally 프로 한 실험에서 단일 세포의 수천 수백 사용자 수 있게 하는 적응 하기 쉬운 워크플로우를 제공 합니다. ScRNA-Seq 데이터 집합의 마지막 품질 최적화 된 세포 격리, cytometry, cDNA 라이브러리 생성 및 원시 유전자 바코드 매트릭스의 해석에 의존합니다. 이 위해,이 프로토콜에는 다양 한 조직 유형의 연구를 사용 하도록 쉽게 수정할 수 있는 모든 주요 단계에 대 한 포괄적인 개요를 제공 합니다.
공개
아무 공개
감사의 말
우리는 캘거리 대학에서 동물 관리 시설 직원 뿐만 아니라 UCDNA 서비스 시설에서 지원 직원을 인정합니다. 우리는 그의 생물 정보학 지원에 대 한 매트 Workentine와 옌스 Durruthy 그의 기술 지원에 대 한 감사합니다. 이 작품은 jb에 요, 및 친교 (대표)는 알버타 어린이 건강 연구 연구소 CIHR 새로운 탐정 수상 CIHR 그랜트 (R.M. 그리고 jb에 요)에 의해 자금.
자료
| Name | Company | Catalog Number | Comments |
| Products | |||
| RNAse out | Biosciences | 786-70 | |
| Pentobarbital sodium | Euthanyl | 50mg/kg | |
| HBSS | Gibco | 14175-095 | |
| Dispase 5U/ml | StemCell Technologies | 7913 | 5 mg/ml |
| Collagenase-4 125 CDU/mg | Sigma-Aldrich | C5138 | 2 mg/ml |
| DNAse | Sigma-Aldrich | DN25 | 10mg/ml |
| BSA | Sigma-Aldrich | A7906 | |
| 15 ml Narrow bottom tube VWR® High-Performance Centrifuge Tubes | VWR | 89039-666 | |
| Sytox Orange Viability Dye | Molecular Probes | 11320972 | 1.3 nM/µl |
| Nuc Blue Live ReadyProbes | Invitrogen | R37605 | |
| Agilent 2100 Bioanalyzer High senitivity DNA Reagents | Agilent | 5067-4626 | |
| Kapa DNA Quantification Kit | Kapa Biosystems | KK4844 | |
| Chromium Single Cell 3' reagents | 10x Genomics | ||
| Equipment | |||
| BD FACSAria III | BD Biosciences | ||
| Agilent 2100 Bioanalyzer Platform | Agilent | ||
| Illumina® HiSeq 4000 | Illumina | ||
| Illumina® MiSeq SR50 | Illumina | ||
| 10X Controller + accessories | 10x Genomics | ||
| Software | |||
| The Cell Ranger | 10x GENOMICS | support.10xgenomics.com/single-cell-gene-expression/software/overview/welcome | |
| Loupe Cell Browser | 10x GENOMICS | support.10xgenomics.com/single-cell-gene-expression/software/downloads/latest | |
| R | https://anaconda.org/r/r |
참고문헌
- Shalek, A. K., et al. Single-cell RNA-seq reveals dynamic paracrine control for cellular variation. Nature. 510, 363-369 (2014).
- Macosko, E. Z., et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell. 161, 1202-1214 (2015).
- Zappia, L., Phipson, B., Oshlack, A. Exploring the single-cell RNA-seq analysis landscape with the scRNA-tools database. bioRxiv:206573. , (2018).
- Dulken, B. W., Leeman, D. S., Boutet, S. C., Hebestreit, K., Brunet, A. Single cell transcriptomic analysis defines heterogeneity and transcriptional dynamics in the adult neural stem cell lineage. Cell Reports. 18 (3), 777-790 (2017).
- Llorens-Bobadilla, E., et al. Single-Cell Transcriptomics Reveals a Population of Dormant Neural Stem Cells that Become Activated upon Brain Injury. Cell Stem Cell. 17 (3), 329-340 (2015).
- Trapnell, C., et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nature Biotechnology. 32, 381-386 (2014).
- Aibar, S., et al. SCENIC: single-cell regulatory network inference and clustering. Nature Methods. 14, 1083-1086 (2017).
- Mayer, C., et al. Developmental diversification of cortical inhibitory interneurons. Nature. 555 (7697), 457-462 (2018).
- Stratton, J. A., et al. Purification and Characterization of Schwann Cells from Adult Human Skin and Nerve. eNeuro. 4 (3), (2017).
- Biernaskie, J. A., McKenzie, I. A., Toma, J. G., Miller, F. D. Isolation of skin-derived precursors (SKPs) and differentiation and enrichment of their Schwann cell progeny. Nature Protocols. 1 (6), 2803-2812 (2007).
- 10X Genomics. User Guides. , Available from: https://www.10xgenomics.com/resources/user-guides/ (2018).
- 10X Genomics. Chromium Single Cell 3' Training Module. , Available from: http://go.10xgenomics.com/training-modules/single-cell-gene-expression (2018).
- Agilent. , Available from: https://www.agilent.com/en-us/library/usermanuals?N=135 (2018).
- Kolodziejczyk, A. A. Single Cell RNA-Sequencing of Pluripotent States Unlocks Modular Transcriptional Variation. Cell Stem Cell. 17, 471-485 (2015).
- Jaitin, D. A., et al. Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types. Science. 343, 776-779 (2014).
- Pollen, A. A., et al. Low-coverage single-cell mRNA sequencing reveals cellular heterogeneity and activated signaling pathways in developing cerebral cortex. Nature Biotechnology. 32, 1053-1058 (2014).
- 10X Genomics. Sequencing Requirements for Single Cell 3'. , Available from: https://support.10xgenomics.com/single-cell-gene-expression/sequencing/doc/specifications-sequencing-requirements-for-single-cell-3 (2018).
- Datacamp. Introduction to R. , Available from: https://www.datacamp.com/courses/free-introduction-to-r (2018).
- Droplet-based, high-throughput single cell transcriptional analysis of adult mouse tissue using 10X Genomics#39; Chromium Single Cell 3' (v2) system: From tissue preparation to bioinformatic analysis. , Available from: https://figshare.com/s/97b83e649e5eefd01357 (2018).
- 10X Genomics. User Guides. , Available from: https://www.10xgenomics.com/resources/user-guides/ (2018).
- UNIX Tutorial for Beginners. , Available from: http://www.ee.surrey.ac.uk/Teaching/Unix/ (2018).
- 10X Genomics. Creating a Reference Package with cellranger mkref. , Available from: https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/advanced/references (2018).
- 10X Genomics. System Requirements. , Available from: https://support.10xgenomics.com/single-cell-gene-expression/software/overview/system-requirements (2018).
- 10X Genomics. Software Downloads. , Available from: https://support.10xgenomics.com/single-cell-gene-expression/software/downloads/latest (2018).
- 10X Genomics. Aggregating Multiple Libraries with cellranger aggr. , Available from: https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/using/aggregate#depth_normalization (2018).
- 10X Genomics. Loupe Cell Browser Gene Expression Tutorial. , Available from: https://support.10xgenomics.com/single-cell-gene-expression/software/visualization/latest/tutorial (2018).
- scRNA-tools. A table of tools for the analysis of single-cell RNA-seq data. , Available from: https://www.scrna-tools.org/ (2018).
- Conda. Downloading conda. , Available from: https://conda.io/docs/user-guide/install/download.html (2018).
- Anaconda. r / packages / r 3.5.1. , Available from: https://anaconda.org/r/r (2018).
- Droplet-based, high-throughput single cell transcriptional analysis of adult mouse tissue using 10X Genomics' Chromium Single Cell 3' (v2) system: From tissue preparation to bioinformatic analysis. , Available from: https://figshare.com/s/97b83e649e5eefd01357 (2018).
- Satija Lab. Seurat - Guided Clustering Tutorial. , https://satijalab.org/seurat/pbmc3k_tutorial.html (2018).
- Droplet-based, high-throughput single cell transcriptional analysis of adult mouse tissue using 10X Genomics' Chromium Single Cell 3' (v2) system: From tissue preparation to bioinformatic analysis. , Available from: https://figshare.com/s/97b83e649e5eefd01357 (2018).
- Monocle. , Available from: http://cole-trapnell-lab.github.io/monocle-release/docs/#constructing-single-cell-trajectories (2018).
- Github. An R package to test for batch effects in high-dimensional single-cell RNA sequencing data. , (2018).
- Edgar, R. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Research. 30, 207-210 (2002).
- Leinonen, R., Sugawara, H., Shumway, M. The sequence read archive. Nucleic Acids Research. 39, D19-D21 (2011).
- NIH. GenBank Submission Portal Wizards. , Available from: https://www.ncbi.nlm.nih.gov/account/register/?back_url=/geo/submitter/ (2018).
- NIH. Submitting data. , Available from: https://submit.ncbi.nlm.nih.gov/geo/submission/ (2018).
- Shah, P. T., et al. Single-Cell Transcriptomics and Fate Mapping of Ependymal Cells Reveals an Absence of Neural Stem Cell Function. Cell. 173, 1045-1057 (2018).
- Anon, Method of the Year 2013. Nature Methods. 11, 1(2013).
- Adam, M., Potter, A. S., Potter, S. S. Psychrophilic proteases dramatically reduce single-cell RNA-seq artifacts: a molecular atlas of kidney development. Development. 144, 3625-3632 (2017).
- Wu, Y. E., Pan, L., Zuo, Y., Li, X., Hong, W. Detecting activated cell populations using single-cell RNA-seq. Neuron. 96, 313-329 (2017).
- Zeigenhain, C., et al. Comparative Analysis of Single-Cell RNA Sequencing Methods. Molecular Cell. 65 (4), 631-643 (2017).
- Wu, A. R., et al. Quantitative assessment of single-cell RNA-sequencing methods. Nature Methods. 11 (1), 41-46 (2014).
- Habib, N., et al. Div-Seq: Single-nucleus RNA-Seq reveals dynamics of rare adult newborn neurons. Science. 353 (6302), 925-928 (2016).
- Janes, K. A. Single-cell states versus single-cell atlases - two classes of heterogeneity that differ in meaning and method. Current Opinions in Biotechnology. 39, 120-125 (2016).
- Herman, J. S., Sagar,, Grün, D. FateID infers cell fate bias in multipotent progenitors from single-cell RNA-seq data. Nature Methods. 15 (5), 379-386 (2018).
재인쇄 및 허가
JoVE'article의 텍스트 или 그림을 다시 사용하시려면 허가 살펴보기
허가 살펴보기더 많은 기사 탐색
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. 판권 소유