
Method Article
Transcriptomics de célula única baseada em código de barras da gota de tecidos de mamíferos adultos
* Estes autores contribuíram igualmente
Neste Artigo
Resumo
Este protocolo descreve os processos gerais e controle de qualidade verifica necessário para a preparação de células simples mamíferos adultas saudáveis para preparações de RNA-Seq de célula única baseada em gotículas, alta taxa de transferência. Parâmetros de sequenciamento, alinhamento de leitura e análise de bioinformatic a jusante de célula única também são fornecidos.
Resumo
A análise da expressão gênica de célula única através de milhares de células individuais dentro de um tecido ou microambiente é uma ferramenta valiosa para identificar composição celular, discriminação dos Estados funcionais e vias moleculares subjacentes observado tecido funções e comportamentos animais. No entanto, o isolamento de células única intactas e saudáveis de tecidos de mamíferos adultos para análise molecular subsequente a jusante única célula pode ser um desafio. Este protocolo descreve os processos gerais e controle de qualidade verifica necessário para obter preparações de alta qualidade adulto única célula do sistema nervoso ou pele que permitiu análise e sequenciamento de célula única imparcial subsequentes do RNA. Diretrizes para a análise de bioinformatic jusante também são fornecidas.
Introdução
Com o desenvolvimento da alta taxa de transferência única célula tecnologia1,2 e avanços em ferramentas de Bioinformática amigável ao longo da última década3, surgiu-se um novo campo de análise de expressão do gene de alta resolução – sequenciamento de RNA de célula única (scRNA-Seq). O estudo da expressão gênica de célula única foi desenvolvido para identificar heterogeneidade dentro de populações de células definidos, tais como em células estaminais ou células cancerosas, ou para identificar raras populações de células4,5, que estavam usando inatingível técnicas de sequenciamento de RNA em massa tradicional. Ferramentas de Bioinformatic permitiram a identificação de sub-populações romance (Seurat)2, visualização da ordem de células ao longo de um psuedotime espacial (monóculo)6, definição das redes de sinalização ativas dentro ou entre populações ( CÊNICA)7, predição da Assembleia do único-células em um espaço 3D artificial (Seurat e muito mais)8. Com estas análises novas e excitantes disponíveis para a comunidade científica, scRNA-Seq é rápido, tornando-se a nova abordagem padrão para análise de expressão do gene.
Apesar do vasto potencial de scRNA-Seq, skillsets técnicos necessários para produzir um conjunto de dados limpo e com precisão, interpretar resultados pode ser um desafio para os recém-chegados. Aqui, um protocolo básico, mas abrangente, a partir do isolamento de células únicas de tecidos todo primários para visualização e apresentação de dados para publicação é apresentada (Figura 1). Em primeiro lugar, o isolamento de células saudáveis único pode ser considerado desafiador, como tecidos diferentes variam em seu grau de sensibilidade à digestão enzimática e subsequente dissociação mecânica. Este protocolo fornece orientação nessas etapas de isolamento e identifica os pontos de verificação de controle de qualidade importante durante todo o processo. Em segundo lugar, a compreensão da compatibilidade e requisitos entre tecnologia única célula e sequenciamento de próxima geração pode ser confuso. Este protocolo fornece diretrizes para implementar uma plataforma amigável, baseada em gotículas barcoding monocelulares e realizar o sequenciamento. Finalmente, programação de computadores é um requisito importante para a análise de conjuntos de dados transcriptomic de célula única. Este protocolo fornece recursos para começar com a linguagem de programação R e fornece orientação sobre como implementar dois pacotes de R populares scRNA Seq-específicos. Juntos, este protocolo pode orientar os recém-chegados na realização de análises scRNA-Seq para a obtenção de resultados claros, interpretáveis. Este protocolo pode ser ajustado para a maioria dos tecidos no mouse e importante poderia ser modificado para uso com outros organismos, incluindo o tecido humano. Ajustes dependendo do tecido e do usuário serão necessários.
Há várias considerações a ter em mente ao seguir este protocolo; incluindo, 1) seguir todas as diretrizes de controle de qualidade nas etapas 1 e 2 do presente protocolo é recomendado para garantir uma suspensão de célula única viável de todas as células dentro da amostra de interesse, assegurando a contagem de número de células de total exato (resumidas em Figura 2 ). Uma vez que este é alcançado, e se forem seguidas todas as condições otimizadas, as etapas de controle de qualidade podem ser descartadas (para economizar tempo - preservar a qualidade do RNA e redução de células perda). Confirmando o sucesso isolamento de células únicas de alta viabilidade do tecido de interesse é altamente recomendo antes de qualquer um jusante de processamento. 2) uma vez que alguns tipos de células são mais sensíveis do que outros ao estresse, técnicas de dissociação excessiva podem inadvertidamente viés da população, confundindo, portanto, a análise a jusante. Dissociação suave sem ruptura celular desnecessários e digestão é fundamental para alcançar altos rendimentos de celulares e uma representação exata da composição do tecido. Forças de cisalhamento ocorrem durante as etapas de trituração, FACS e ressuspensão. 3) como com qualquer trabalho de RNA, é melhor introduzir como RNase adicional pequeno da amostra quanto possível durante a preparação. Isto ajudará a manter a qualidade do RNA. Utilizar soluções de inibidor do ribonuclease com lavagem de ferramentas limpas e qualquer equipamento que não é livre de RNase mas Evite produtos DEPC tratada. 4) realize preparações tão rapidamente quanto possível. Isto ajudará a manter a qualidade do RNA e reduzir a morte celular. Dependendo do comprimento de dissecação de tecido e o número de animais, considere começar várias dissecações ou preparações ao mesmo tempo. 5) prepare células no gelo quando possível para manter a alta qualidade do RNA, reduzir a morte celular e lenta atividade transcricional e sinalização de células. Embora, processamento gelado é ideal para a maioria dos tipos de células, alguns tipos de células (por exemplo, os neutrófilos) melhor desempenho quando processado à temperatura ambiente. 6) Evite o cálcio, magnésio, EDTA e tratada com DEPC produtos durante a preparação da célula.
Protocolo
Todos os protocolos aqui descritos estão em conformidade com e aprovaram pelo Comitê de cuidado Animal da Universidade de Calgary.
1. desassociar o tecido (dia 1)
- Eutanásia em ratos com uma overdose de pentobarbital de sódio (i.p., 50 mg/kg) ou conforme o caso, de acordo com o protocolo de ética animal. Em seguida remover pelos indesejados de costas e pernas do mouse e etanol-esterilizar a região de dissecação.
- Disse o tecido ou microambiente de interesse. Para este protocolo, usamos os tecidos da pele e nervos para demonstrar a generalização de gotículas célula única baseada em código de barras transcriptomics seguindo a dissociação de tecido adulto.
- Para o nervo ciático, usam o protocolo detalhado encontrado em Stratton et al . 9. brevemente, cortar a pele da região posterior das costas/pernas do rato. Fazer uma incisão ao longo do comprimento da coxa com uma lâmina de bisturi estéril. Use a pinça fina e tesoura para expor e remover o nervo ciático.
- Para a pele de volta, usam o protocolo detalhado encontrado em Biernaskie et al 10. brevemente, dissecar a pele dorsal de costas fazendo incisões de ombro a ombro, entre a alcatra e nas costas, usando a pinça fina e tesoura. Corte a pele em fatias finas (0,5 cm de espessura), usando uma lâmina de bisturi estéril.
- Lave o tecido 2 vezes com HBSS gelada e remover indesejados do tecido conjuntivo, os depósitos de gordura ou detritos sob um microscópio de dissecação.
- Para a derme da pele somente, flutuar as fatias em dispase (5 mg/mL, 5 U/mL) em HBSS por 30-40 min a 37 ° C. Cirurgicamente, separe a epiderme da derme. Descarte a epiderme ou dissociar ainda mais usando a tripsina se de interesse.
- Picar a amostra em pedaços de 1-2 mm, usando uma lâminas de bisturi estéril e colocar em enzima colagenase frio-IV de recém descongelado 2 mg/mL (2 mg/mL, 125 CDU/mg, nos meios de comunicação F12).
- Para o nervo, use ~ 500 µ l por 2 nervos ciático de x. Para a pele, use ~ 8 mL por rato 1 x volta da pele.
Nota: Os tecidos devem ser totalmente submerso na solução de colagenase-IV. É fundamental que qualquer enzimas de digestão são tratadas, armazenadas e preparadas adequadamente. Se enzimas são deixadas à temperatura ambiente por longos períodos de tempo, o isolamento de célula única requerem trituração mecânica excessiva e reduzir a viabilidade celular. Colagenase-IV também pode ser feita em meios de cultura celular, onde a viabilidade celular é mais ideal. No entanto, isso pode alterar a atividade enzimática ou assinatura transcriptional então deve ser otimizada pelo usuário.
- Para o nervo, use ~ 500 µ l por 2 nervos ciático de x. Para a pele, use ~ 8 mL por rato 1 x volta da pele.
- Incube a amostra na enzima em um banho de 37 ° C por 30 min com agitação suave cada 10 min. Um shaker colocado a 37 ° C é também uma alternativa apropriada.
- Triture com uma pipeta P1000 20 - 30 vezes em adição de pós-enzima 30 min.
- Repita a trituração cada 30 min até solução aparece nublada e pedaços de tecido são em grande parte dissociados.
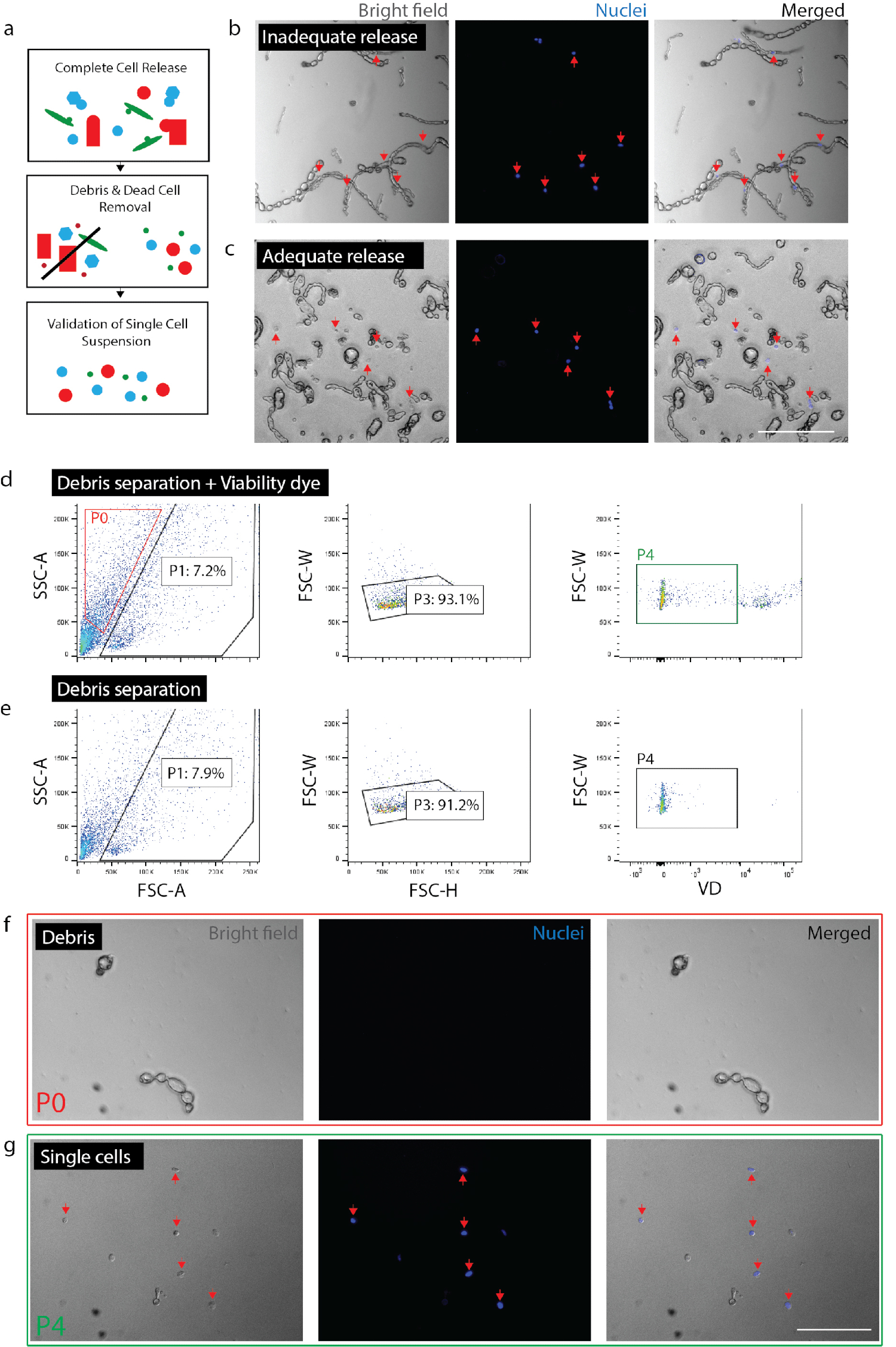
Nota: Certifique-se de liberação cheia de células (Figura 2b, 2C). Para confirmar a liberação completa, placa células com Nuc azul (2 gotas por 1 mL) e depois de 20 min, verifique sob o microscópio para garantir que todos os núcleos estão associados com células únicas, em vez de detritos. É fundamental para verificar o grau de liberação de células dentro de um determinado experimento para cada tipo de tecido ou condição. No tecido fibrótico (ou seja, lesão crônica) ou tecido adulto ileso, a liberação de células irá variar drasticamente de lesão aguda ou tecido embrionário. Isto é especialmente importante porque certos tipos de células são menos propensos a liberação do tecido do que outros, excluindo preferencialmente as células da análise a jusante.- Para o nervo, dissociar o tecido para 0,5-1,5 horas no total. Para a pele, dissociar o tecido por 2 horas no total (na última hora de incubação, adicionar DNase (1 mg/mL) para a amostra de pele).
- Filtrar com um filtro de 40 µm. Lave o filtro com gelada 1% BSA/HBSS.
- Centrifugar a 260 x g por 8 min. Em seguida retire o sobrenadante.
- Resuspenda o pellet de células em HBSS contendo 1% de BSA usando uma ponta de todo o furo e colocar no gelo. O volume de ressuspensão é baseado no volume de tecido (peso molhado de 800 mg para pele = 800 µ l volume; 10 mg molhado peso para nervos = 100 µ l volume).
- Opcionalmente, comece com um volume de ressuspensão de baixo e ajustar conforme necessário com base na taxa de fluxo (eventos por segundo) no classificador FACS. A densidade de tipo mais eficiente (maximizar o número de células coletadas durante a minimalizing tempo) para coleções é 7.000-3.000 eventos por segundo.
- Se usar tintura de viabilidade, tire um subaliquot para um controle imaculado. Em seguida, adicione corante de viabilidade 1:15,000 (estoque: 20.000 nM / µ l) de amostra (concentração final de 1,3 nM / µ l) usando uma dica de largo diâmetro para reduzir a distorção.
Nota: É fundamental para verificar o grau de morte celular dentro de um determinado experimento para cada tipo de tecido ou condição. Alguns tipos de células dentro de uma amostra são mais propensos a morrer do que outros, assim, preferencialmente sendo excluídos da análise a jusante.- Incube a amostra com tintura de viabilidade para 5-10 min no gelo no escuro. Em seguida, adicione 4 mL de gelado 1% BSA/HBSS a amostra. Centrifugar a 260 x g, durante 8 min remover o corante em excesso de viabilidade. Tratar a subaliquot com nenhuma viabilidade tingir controle imaculado da mesma maneira.
2. isolar as células viáveis e saudáveis (dia 1)
- Certifique-se de que as instalações de FACS segue célula apropriada fluorescência ativada classificação parâmetros (FACS).
- Preparar a máquina de FACS com antecedência para garantir que ele está pronto, uma vez concluída a centrifugação final na etapa 1 e certifique-se de que o compartimento de coleta é mantido frio usando blocos de gelo.
- Use os seguintes parâmetros: taxa de fluxo: 1.0 (corresponde aproximadamente a 10 µ l/min); Filtro: 1.5 ND; Tamanho do bocal: 100 µm; Dispersão para a frente: 80-180 V (alterar, se necessário, a fim de distinguir o tamanho de eventos); Dispersão de lado: 150-220 V (alterar, se necessário, a fim de distinguir a granularidade/forma de eventos); Laser: 100-400 V (alterar, se necessário, a fim de distinguir a viabilidade tintura positivos vs negativos eventos & saca contra nenhum controle de tintura de viabilidade); Gates: Alterar conforme necessário para garantir que todas as células são coletadas. Ver Figura 2d-2 g.
Nota: Os parâmetros de FACS são altamente dependentes dos tipos de célula e classificador de empregado e, portanto, precisa ser otimizado pelo usuário.
- Prepare-se 15 mL estreito-tubos com 8 mL de gelado 1% BSA/HBSS para coletas de amostra. Estática no interior do tubo e a tensão de superfície pode afetar a eficiência de coleta. Inverter os tubos antes de coleções para garantir a interface entre a superfície do líquido e do interior do tubo é úmido.
Nota: Se trabalhar com números de telemóvel muito baixo, ajuste a embarcação pequena coleção conforme apropriado. - Uma vez que todas as células são coletadas, centrifugar a amostra a 260 x g por 8 min.
Nota: Antes da centrifugação, adicionar 1% BSA/HBSS para lavagem/empurrar as células abaixo da superfície lateral e invertido/mistura do tubo imediatamente após FACS. - Resuspenda o pellet celular em 1% BSA/HBSS e manter no gelo. O volume máximo por exemplo que é compatível com o processamento de etapa 3 µ l 33.8, então certifique-se que volume de diluição/ressuspensão de final de célula é apropriado para obter o número de célula ideal em 33.8 µ l. Outras opções de mídia de diluição para esta etapa (e todas as diluições anteriores em 1% BSA/HBSS) incluem DMEM e até 40% de soro, mas evite o cálcio, magnésio ou EDTA contendo reagentes.
- Embora as células no gelo para uma quantidade mínima de tempo. Idealmente cooperador deve preparar todos os equipamentos e reagentes para a etapa seguinte (etapa 3) durante as etapas finais da etapa 2.
- Célula preparação críticas cheques
- Confirme as estimativas do número de células Obtida de FACS. Dependendo do tipo de tecido e dissociação comprimentos, detritos e células podem ser muito semelhantes em tamanho e forma. Assim, a menos que um repórter fluorescente é usado, FACS não pode excluir todos os detritos. É recomendável que uma contagem de células final após coleta de FACS é realizada para compreender qual é a porcentagem de eventos (de acordo com FACS) são, de facto, as células de uma determinada preparação (Figura 2 g). Realizar a contagem de células usando um hemocytometer ou contador automatizado de células (repetir duas vezes) e calcular a percentagem de células viáveis é representado pelos acontecimentos totais coletados de acordo com a máquina de FACS.
- Valide a preparação da pilha. Validar que não há partículas grandes (> 100 µm) estão presentes como eles podem entupir o equipamento em etapas a jusante. Inadequada remoção de detritos correm o risco de entupimento o chip microfluidic monocelulares. Placa de células restantes com Nuc azul (como acima) para garantir que não há fragmentos grandes destroços estão presentes. Isso também permitirá a confirmação de que as células são singular (i.e., não colem) dar confiança essa análise genética a jusante única célula representa células únicas, em vez de várias células.
- Decidir sobre números de telemóvel à sequência: há uma grande variedade de números de telemóvel de derivado de tecido adulto por exemplo que pode ser carregado no sistema com até 8 amostras que podem ser executados simultaneamente. Autores têm carregado em qualquer lugar a partir de 500 – 50.000 células por amostra e obtido boa qualidade scRNA-Seq datasets. Mais discussão sobre os números de celular mais adequados para carregar pode ser encontrado na seção de discussão. A saída final dos números de telemóvel sequenciado depende muito da qualidade do único-células isoladas. Carregamento de 10.000 células adultas de tecidos derivados pode retornar em qualquer lugar de 1.000 a 4.000 células sequenciadas (10-40% de retorno). Se estiver interessado em sequenciamento celulares alta (~ 10.000 células, o número máximo recomendado para este sistema), então a 25.000-100.000 células de carga serão necessárias.
3. GEM (grânulo de Gel em emulsão) geração e código de barras (dia 1)
Nota: As etapas 3 a 6 do presente protocolo são projetadas para ser usado em conjunto com o mais comum microdroplet plataforma baseada em célula única, fabricado pela 10 X Genomics. Orientações detalhadas para etapas 3 e 4 estão descritas do fabricante (Veja o protocolo de cromo única célula 3')11,12 do protocolo e devem ser seguidas em conjunto com este protocolo. Para melhores resultados, etapa 3 devem ser preenchidos imediatamente após dissociação (etapa 1) e as etapas de isolamento (etapa 2) célula no dia 1 do presente protocolo.
- Prepare a microplaqueta de acordo com protocolo 11,12 do fabricante. Esta plataforma de célula única baseada em microdroplet usa a tecnologia que amostras ~ 750.000 códigos de barras para indexar o transcriptoma da cada célula separadamente. Isto é conseguido através de células de particionamento em grânulo de Gel em emulsões (gemas), onde o cDNA gerado compartilham um código de barras comum. Durante a geração do GEM, as células são entregues para que a maioria (90-99%) das gemas geradas não conter nenhuma célula, enquanto o restante, em sua maior parte, contêm uma única célula.
- Coloque o chip no suporte do chip.
- Prepare a mistura mestre de célula no gelo.
- Adicionar 50% glicerol poços não utilizados e adicionar 90 µ l do mix principal célula 1 bem, 90 µ l dos grânulos do gel para bem 2 e 270 µ l de óleo para 3 bem de particionamento.
- Cobrir o chip com a junta.
- Carregar o chip e executado em um controlador de célula única.
- Ejetar a bandeja, coloque o chip na bandeja, retire a bandeja e pressione Play. Uma única célula 3' gel de pérola em uma GEM inclui cartilhas contendo uma sequência parcial de Illumina R1 (leia 1 cartilha de sequenciamento), um 16 nucleotídeos (nt) 10 x Barcode, um 10 nt identificador exclusivo Molecular (UMI) e uma sequência de primeira demão poli-dT. Durante a execução, gel de grânulos no controlador são liberados e misturados com mistura de lisado e mestre de célula.
- Colete 100 µ l da amostra e coloque em um tubo PCR.
- Lugar da polimerase em PCR pré-definida da máquina e executar o PCR de acordo com o kit. Após incubação, gemas incluirá o cDNA completos, código de barras do mRNA poli-adenylated.
- Após a execução, coloque a-20 ° C durante a noite para até 1 semana antes anterior para a próxima etapa.
4. limpar, amplificação, construção de biblioteca e quantificação de biblioteca (a partir do dia 2)
Nota: Orientações detalhadas para 4 etapas estão descritas no protocolo 11,12 do fabricante e devem ser seguidas em conjunto com este protocolo.
- Use grânulos magnético de silano para remover restos reagentes bioquímicas/primeiras demão da mistura de reação de GEM.
- Amplificar o cDNA completos, código de barras para gerar massa suficiente para construção de biblioteca.
- Avalie o rendimento do ADN. Antes da construção da biblioteca, avalie o rendimento do ADN da amostra. Isto determinará quantos ciclos para usar na etapa PCR a jusante (índice de amostra PCR durante a construção da biblioteca). Dependendo do conteúdo de RNA de uma determinada amostra, que pode variar dependendo dos Estados de ativação (por exemplo, controle vs feridos, etc.), tipo de célula e o rendimento da célula, o ciclo recomendado número pode mudar.
- Para sequenciamento ~ 3.000 derivado de tecido as células (irrelevantes para Estados de ativação), os autores encontraram que 14 ciclos (exemplos: ~ 10-100 ng de DNA) é o padrão.
- Use um Bioanalyzer para análise de DNA. Referem-se ao usuário guia13.
- Amostra do fragmento e selecione o tamanho do DNA. Antes da construção da biblioteca, use protocolos de seleção de tamanho e fragmentação enzimática para obter tamanho de amplicons do cDNA apropriado.
- Prepare a amostra para a construção da biblioteca. Enquanto R1 (ler 1 cartilha de sequência) é adicionado para as moléculas durante a incubação GEM; P5, P7 (um índice de amostra), e R2 (Leia 2 sequência da primeira demão) são adicionados durante a construção da biblioteca.
- Avalie o rendimento do ADN. A maioria das instalações de sequenciamento exigem apresentação de bibliotecas finais que incluem informações de rendimento e qualidade do DNA. Assim, execute o bioanalyzer após a conclusão do protocolo todo e antes de transportar para as instalações de sequenciamento.
- Armazenar as amostras a-80 ° C por até 2 meses.
- Antes de sequenciamento, quantificar as amostras usando um kit de quantificação de DNA. Isso pode ser feito na instalação de sequenciamento.
5. Biblioteca sequenciamento (dia 3 em diante)
Nota: A plataforma de código de barras monocelulares transcriptome utilizada neste protocolo gera bibliotecas emparelhado-fim de Illumina-compatível começando e terminando com sequências P5 e P7. Apesar de profundidade mínima necessária para resolver o tipo de célula de identidade pode ser sómente 10.000-50.000 leituras/célula15,16, ~ 100.000 leituras/célula é recomendada como um trade-off custo-cobertura ideal para adultas células in vivo (tendo em mente um celular tipos ou Estados minimamente ativado celular chegará a saturação em 30.000-50.000 leituras/célula).
- Transporte de bibliotecas de cDNA em gelo seco para uma instalação de sequenciamento, equipado com um sequenciador Illumina apropriado.
- Forneça as seguintes informações para as instalações de sequenciamento:
- Fornecer detalhes de amostra: amostra índice identificações correspondentes para cada biblioteca; espécies; banco de dados genômico para Primária assembly (isto é, GRCm38 para mouse); electroferograma mostrando fragmento tamanhos desde o bioanalyzer (entre 200 e 9.000 bp); cDNA concentração (ng / µ l) e a concentração total de biblioteca (rendimentos totais variam de 200 – 1400 ng); volume (µ l) da amostra.
- Fornecer as solicitações da sequenciamento: quantificar as amostras usando um kit de quantificação de DNA; adaptador/tipo de índice (DNA TruSeq); tipo da placa (Eppendorf twin.tec, saIão - recomendado para DNA); sequenciamento de tecnologia/biblioteca tipo (10 x, sequenciação completa instruções e recomendações do ciclo)17.
- Executar sequenciamento superficial (opcional): estudos analisando várias amostras biológicas irão beneficiar de pool de amostras (agregação) para gerar um único gene-código de barras matriz que contém os dados de todas as amostras. Para minimizar efeitos de lote entre amostras quando o pool, a profundidade de leitura entre diferentes bibliotecas deve ser padronizada. Para fazer isso, uma aproximação precisa de números única célula é necessária. O sequencer MiSeq permitirá sequenciamento superficial e é uma maneira cost-effective, prática para obter estimativas de célula precisos.
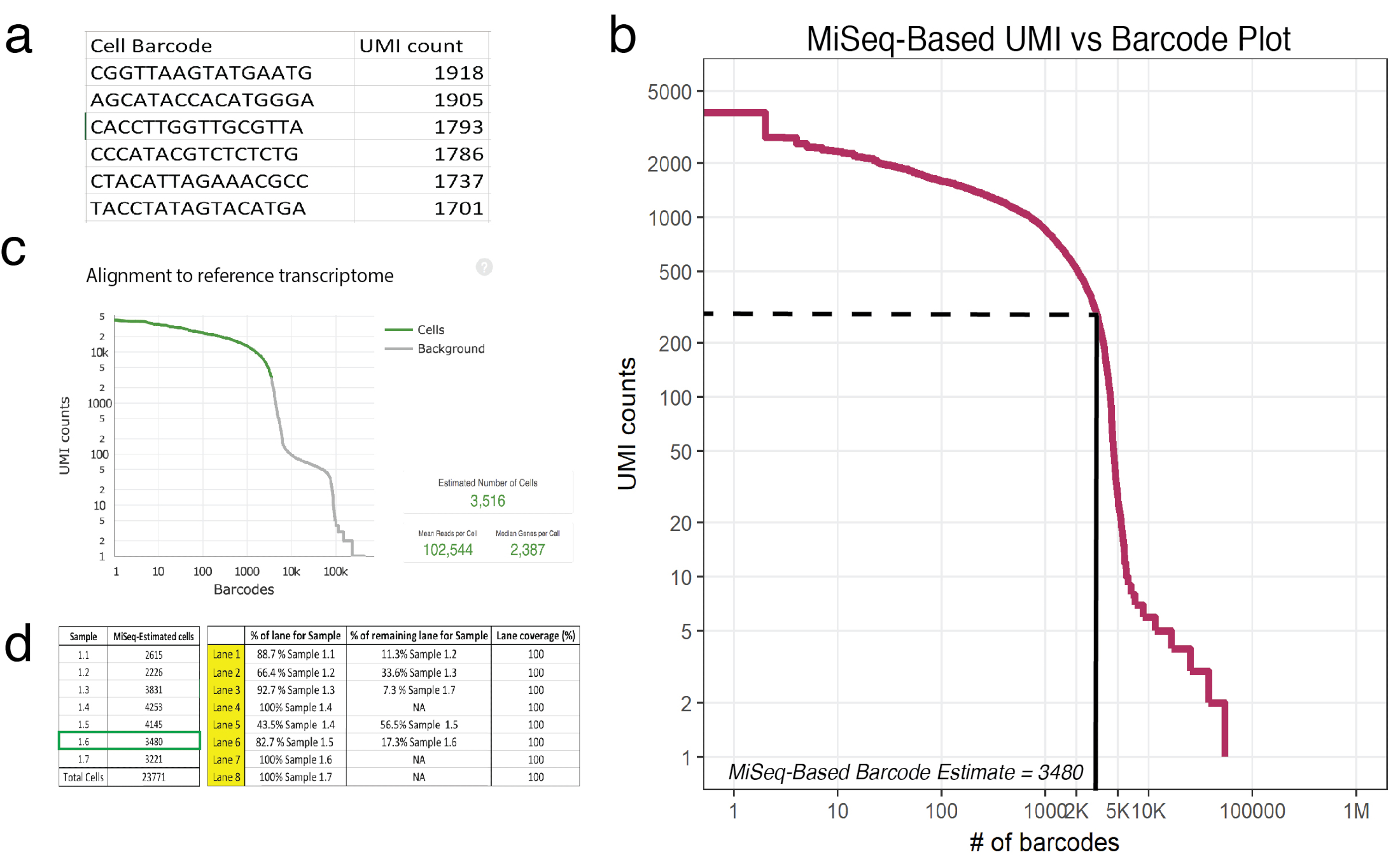
Nota: Uma corrida com um sequenciador MiSeq SR50 fornece cobertura suficiente para estimar com precisão cerca de 20.000 células. Esta corrida será aproximar o número de UMI recuperado para cada código de barras único. Na Figura 3a, é mostrado o cabeçalho de uma saída de exemplo (exemplo 1.6) (CSV), listagem de códigos de barras e sua correspondente UMI conta conforme determinado pela leituras confiàvel mapeadas.- Consulte com um bioinformático para se familiarizar com a linguagem de programação de R. Consulte DataCamp tutoriais para mais informações18.
- Avalie os dados brutos obtidos a partir do sequencer usando o script fornecido de R como um modelo19. Dados brutos refere-se ao número de UMIs mapeado para cada código de barras exclusivos de célula. O script lê um arquivo CSV, onde a primeira coluna é uma lista de códigos de barras e a segunda coluna são seu correspondente UMI conta. Este script irá fornecer uma trama (Figura 3b) bem como a estimativa do número de células de código de barras em cada amostra. Ajuste o script para garantir que o número introduzido de UMI contagens para uma determinada amostra é o ponto de um terço da primeira gota íngreme. Na Figura 3b, esse cotovelo cai cerca de 225 UMIs correspondente a 3.480 células de código de barras.
- Comparável ao sequenciamento completo-profundidade usando HiSeq (onde 3.516 células foram com sucesso sequenciado, Figura 3C), estimativas de sequenciamento superficial previram 3.480 células.
- Uso celular aproximações de recuperação (da etapa 5.3) ou usar o gráfico de recuperação encontrado no protocolo20 para planejar a distribuição de pista para o sequenciamento de mais profundo do fabricante. Cada amostra deve receber cobertura comparável, então se sequenciamento superficial revela que existem diferentes números de células em cada amostra (que é frequentemente o caso) em seguida distribuição de pista deve ser calculada em conformidade. Uma célula de fluxo de HiSeq (que compreende 8 pistas) pode sequenciar até 2,4 bilhões leituras personalizado final emparelhados. Configuração de célula de fluxo de exemplo é apresentada na Figura 3d.
6. processamento de ler arquivos
Nota: Uma única célula 3' biblioteca usando esse protocolo de sequenciamento gera dados brutos em formato binário chamada base (BCL). A célula Ranger pacote é usado para gerar arquivos FASTQ baseado em texto de arquivos BCL, executar genômica e alinhamentos de transcriptomic, gene contagens, demultiplexação e agregação de amostras. Nesta seção, os principais passos que permitem que os usuários transfiram dados brutos BCL de uma instalação de sequenciamento e gerar matrizes gene-barcode filtrado prontas para bioinformática a jusante é apresentado.
- Use um servidor centralizado para executar o programa. BCL arquivos FASTQ e maior parte a jusante bioinformática processamento exige capacidade de processamento significativo.
- Baixe todos os arquivos raw de leitura no servidor (ou arquivos FASTQ se estiverem disponíveis).
- Consulte o administrador do servidor para configurar uma conta em um servidor centralizado ou cluster e para familiarizar-se com Unix21.
- Use um comando de busca apropriado para o sistema operacional do servidor para baixar todos os arquivos do servidor a facilidade sequenciamento.
- A maioria das instalações de sequenciamento fornecem um comando para baixar arquivos de um caminho seguro que pode ser executado a partir da linha de comando (Veja exemplo abaixo).
- Substituir o "< username >" e "< senha >" espaços reservados na linha de comando com credenciais fornecidas.
wget - O - "https://your_sequencing_facilitys_server.com/path_to_raw_read_files/..--não-cookies-não-verificação-certificado - pós-dados ' j_username = username & j_password = senha ' | wget - não-cookies-não-verificação-certificado - pós-dados ' j_username = username & j_password = a senha ' - ci -
- Se apenas um caminho absoluto para arquivos é fornecido (ou seja, https://your_sequencing_facilitys_server.com/path_to_raw_read_files/), insira um comando de busca esse caminho.
- Descompacte arquivos: se baixou final de arquivos com a extensão ". gz", foi compactada usando o comando "gzip". Para descompactar, executar Descompacte o comando na linha de comando (Veja exemplo abaixo).
gunzip raw_read_files.gz - Baixe a última versão da Ranger de célula para o servidor como uma auto-contido. tar22.
-
Crítica: Antes de baixar, certifique-se de que sistema Linux atende a requisitos mínimos de23. Assegure um mínimo de 8-core processador Intel com 64 GB de RAM e 1 TB de espaço livre em disco.
Nota: Ranger célula fornece pré-construídos humanos e roedores referência transcriptomes. Estas podem ser modificadas usando o comando cellranger mkref para detectar genes como GFP24.
-
Crítica: Antes de baixar, certifique-se de que sistema Linux atende a requisitos mínimos de23. Assegure um mínimo de 8-core processador Intel com 64 GB de RAM e 1 TB de espaço livre em disco.
- Gera arquivos FASTQ de arquivos de base chamada do Sequenciador (BCLs) usando o comando de mkfastq de cellranger.
Nota: O programa irá alinhar crus leituras (a partir de arquivos FASTQ) para um genoma de referência e gerar matrizes gene-célula para análise a jusante. Ele usa o alinhador de estrela que executa o alinhamento de emenda-ciente das leituras de um genoma de referência. Só lê confiàvel mapeada (i.e., leituras compatíveis com anotação de um único gene) é usados para a contagem de UMI.- Por exemplo, use o comando de mkfastq de cellranger:
cellranger mkfastq ..--id = sample_name \
... = / caminho/para/exemplo executar \
..--csv=csv_file_containing_lane_sample_index.csv
- Por exemplo, use o comando de mkfastq de cellranger:
- Execute o Conde de cellranger em arquivos FASTQ gerados usando mkfastq para gerar contagens de célula única gene.
- Por exemplo, use o comando do Conde de cellranger:
contagem de cellranger ..--id = sample_name \
-transcriptoma = refdata-cellranger-mm10-1.2.0 \
-fastqs = / absoluto/caminho/para/fastq/files \
-amostra = same_sample_name_supplied_to_cellranger_mkfastq \
-localcores = 30
- Por exemplo, use o comando do Conde de cellranger:
- Agregação de biblioteca multi (opcional): para combinar as amostras, saídas de contagem cellranger usando cellranger agregação da piscina. Isso resulta em uma matriz de gene único-código de barras que contém dados agrupados de várias bibliotecas. Comando de agregação de cellranger exemplo:
agregação de cellranger..--id = sample_name \
-csv = csv_with_libraryID_ & _path_to_molecule_h5.csv \
-normalizar = mapeado
Nota: As bibliotecas podem ser agregadas usando três modos de normalização (mapeado, cru, nenhum). Mapeado é recomendada, pois isso subamostras bibliotecas de maior profundidade, até que todas as bibliotecas têm igual sequenciamento profundidade25. - Para imediata visualização/análise de dados, importe o arquivo de saída de .cloupe (gerado com o Conde de cellranger ou cellranger agregação) para 10 x lupa celular navegador26.
7. avançada análise de conjuntos de dados scRNA-Seq
Nota: Um banco de dados de ferramentas scRNA-Seq completo pode ser encontrado em ferramentas scRNA3,27. Abaixo está um quadro para a célula sem supervisão clustering usando Seurat2 e pseudotemporal ordenação usando monóculo6. Embora muito deste trabalho pode ser feito em um computador local, as seguintes etapas assumem que computação será concluída usando um servidor institucional.
- Baixe a versão mais recente do Miniconda na conta do servidor usando o Linux plataforma28.
- Instale a versão mais recente do R usando conda29.
- Plotar os dados usando o script de Seurat R fornecido como um modelo30.
Nota: Seurat é um toolkit do R-baseado que permite verificações de controle de qualidade, clustering, diferencial análise da expressão de gene, identificação do gene marcador, redução de dimensionalidade e visualização de dados de scRNA-Seq. Encontram-se uma descrição detalhada de Seurat codificação e tutoriais sobre o site de laboratório de Jaqueline Silva31. - Plotar os dados usando o script fornecido do monóculo R como um modelo32.
Nota: O monóculo é outro toolkit R-baseado que permite a visualização de alterações de expressão sobre pseudotime e identifica genes célula destino decisões. Uma descrição abrangente de monóculo codificação e tutoriais pode ser encontrada sobre o monóculo site33. - R-pacotes como o kBET podem ser empregadas para testar e corrigir efeitos de lote como resultado da conjugação de conjuntos de dados34.
8. do NCBI GEO e SRA submissões
Nota: Desde que o fácil acesso aos arquivos de sequenciamento bruto garantir reprodutibilidade e reanálise, submissões adquiridas para repositórios publicamente disponíveis on-line são recomendadas ou necessárias antes da submissão do manuscrito. National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) e arquivo de leitura da sequência (SRA) são repositórios de dados publicamente acessíveis para arranjar em sequência do elevado-throughput dados35,36.
- Registre-se para GEO Submitter conta37 do NCBI.
- Apresentação de GEO completo que inclui três componentes compilados em uma diretório/pasta (intitulada como username do submitter GEO): 1) registro de metadados (uma folha de cálculo por submissão de projeto); 2) arquivos de dados brutos; 3) transformados em arquivos de dados.
- Baixar e preencher a planilha de metadados38. A seguinte apresentação de GEO pública pode ser usada como um guia (GSE100320)39. Coloque a folha de cálculo no diretório.
- Lugar arquivos de dados Raw gerados a partir de script de contagem de cellranger para todas as bibliotecas no diretório.
- Lugar processados dados (arquivos filtrados barcodes.tsv, genes.tsv e matrix.mtx) gerados a partir de script de contagem de cellranger para todas as bibliotecas no diretório.
- Use as credenciais do servidor FTP do apresentador GEO para transferir o diretório que contém todos os três componentes. Para usuários de Linux/Unix: ncftp, lftp, ftp, sftp e ncftpput podem ser usados.
- Informe GEO para todas as transferências38.
Resultados
O repertório de pacotes de código aberto projetado para analisar conjuntos de dados scRNA-Seq aumentou dramaticamente a40 , com a maioria de uso esses pacotes de idiomas baseados em R3. Aqui, apresentam-se resultados representativos usando dois desses pacotes: avaliar agrupamento sem supervisão de único baseadas na expressão gênica de células e ordenação de células únicas ao longo de uma trajetória para resolver heterogeneidade de célula e desconstruir biológico processos.
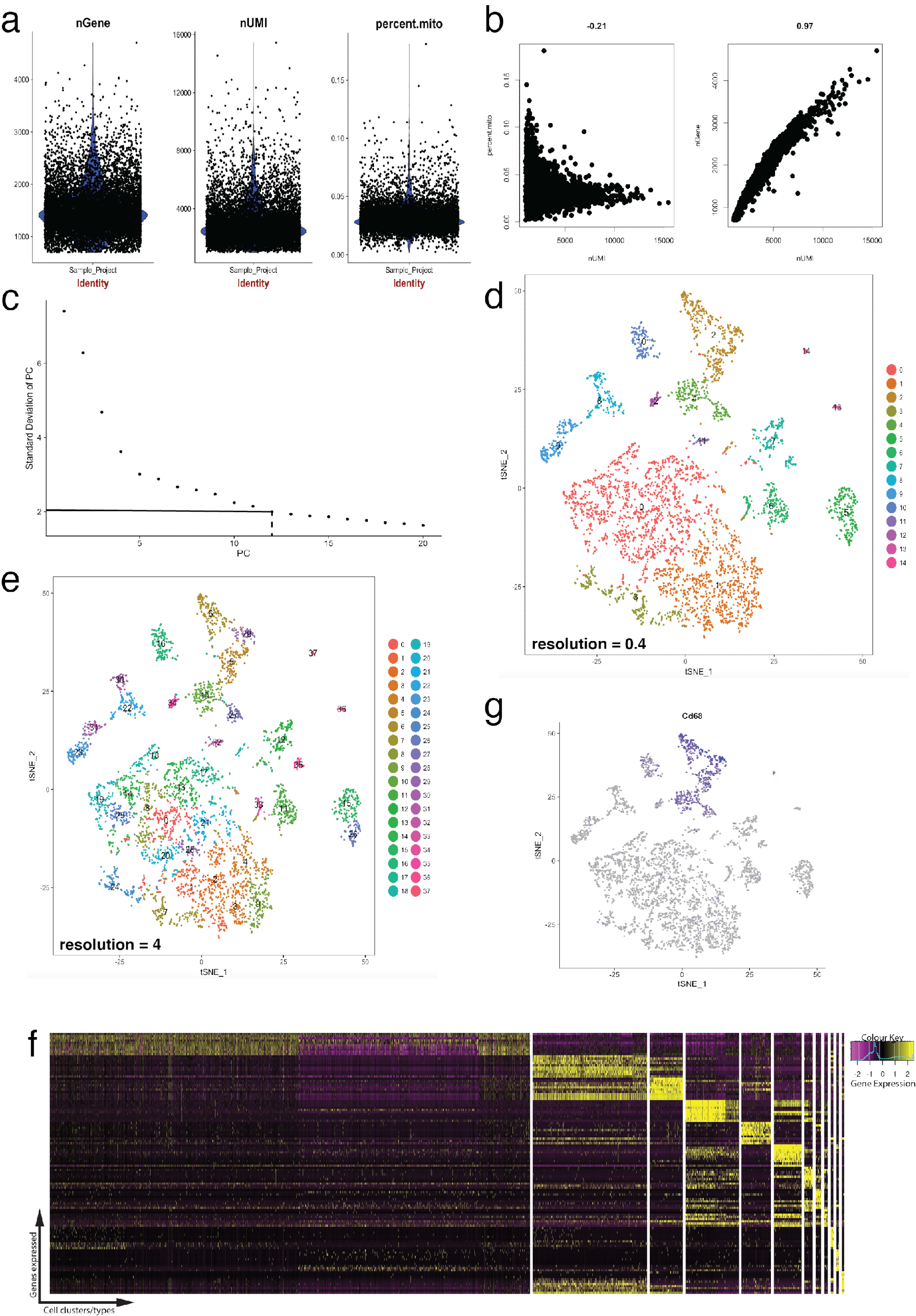
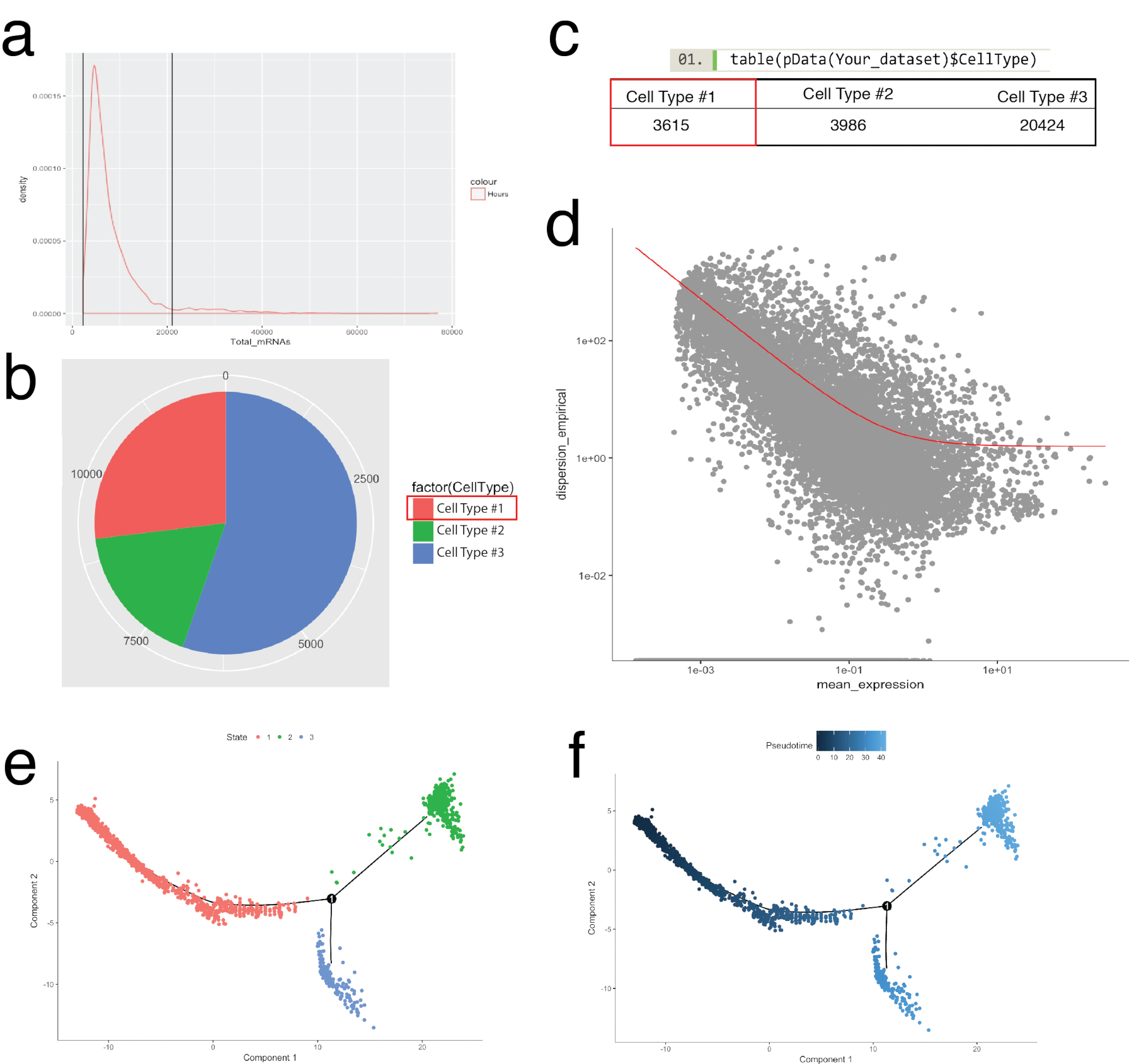
A Figura 4 ilustra o uso de Seurat para pré-processamento de controlos de qualidade e análise de Bioinformática a jusante. Primeiro, filtragem e remoção de células desviantes de análise é essencial para o controlo de qualidade. Isto foi feito usar o violino (figura 4a) e dispersão parcelas (figura 4b) para visualizar a porcentagem de genes mitocondriais e número de genes (nGene) número de UMI (nUMI) para identificar parelhas de célula e exceções. Qualquer célula com um número de outlier clara de genes, UMI ou a porcentagem de genes mitocondriais foi removida usando a função FilterCells de Seurat. Desde que Seurat usa o componente principal (PC) resultados de análise de aglomerados de células, determinando estatisticamente significativas PCs para incluir é uma etapa crítica. Parcelas de cotovelo (Figura 4C) foram usadas para a seleção de PC, no quais PCs além do planalto do 'desvio padrão de PC' eixo foram excluídos. A resolução de cluster foi manipulada também demonstrando que o número de clusters pode ser alterado, variando de 0,4 (baixa resolução, levando a menos aglomerados de células, Figura 4D) a 4 (resolução alta, levando a maiores aglomerados de células, Figura 4e ). Em baixa resolução, é provável que cada cluster representa um tipo de célula definida, Considerando que, em alta resolução isso também pode representar subtipos ou Estados de transição de uma população celular. Nesta instância, configurações de baixa resolução cluster foram utilizadas para analisar mais expressão heatmaps (usando a função DoHeatmap de Seurat) para identificar os genes mais altamente expressos em um determinado cluster (Figura 4f). Neste caso, os mais altamente expressos genes foram identificados através da avaliação de expressão diferencial em um determinado cluster contra todos os outros grupos combinados, demonstrando que cada cluster foi exclusivamente representada por genes definidos. Além disso, genes candidatos individuais podem ser visualizados nas parcelas de tSNE usando a função FeaturePlot de Seurat (Figura 4 g). Isto permitiu decifrar se houve clusters que representou os macrófagos. Usando FeaturePlot, descobrimos que ambos cluster 2 e 4 foram expressando Cd68 - um marcador de pan-macrófago.
O pacote de monóculo foi usado para corroborando aglomerados de células identificados em Seurat e para a construção de trajetórias de célula, ou encomendar pseudotemporal, recapitular os processos biológicos (Figura 5). Ordenação pseudotemporal pode ser usado para amostras onde perfis de expressão de célula única são esperados para seguir um curso de tempo biológico. Células podem ser encomendadas ao longo de um continuum pseudotemporal para resolver Estados intermediários, pontos de bifurcação de dois destinos alternativos da célula e identificam as assinaturas de gene subjacente a aquisição de cada destino. Em primeiro lugar, semelhante à filtragem de Seurat, células de má qualidade foram removidas de tal forma que a distribuição do mRNA através de todas as células foi log normal e caiu entre os limites superiores e inferiores, conforme identificado na Figura 5a. Em seguida, usando a função de newCellTypeHierarchy do monóculo, células únicas foram classificadas e contados usando genes marcadores de linhagem conhecida (Figura 5b, 5C). Por exemplo, células expressando PDGF receptor alfa ou fibroblasto específico da proteína 1 foram atribuídas a célula tipo #1 para criar um critério para a definição de fibroblastos. Em seguida, esta população (célula tipo #1) foi avaliada para decifrar as trajetórias de fibroblasto. Para fazer isso, a função de GeneTest diferencial do monóculo foi utilizada, que comparou as células que representam os Estados extremos no seio da população e encontrado genes diferenciais para ordenar as células restantes na população (Figura 5D). Através da aplicação de métodos de aprendizagem múltipla (um tipo de redução de dimensionalidade não-linear) em todas as células, foi atribuída uma coordenada ao longo do caminho pseudotemporal. Esta trajetória foi depois visualizada pelo estado da célula (Figura 5e) e pseudotime (Figura 5f).

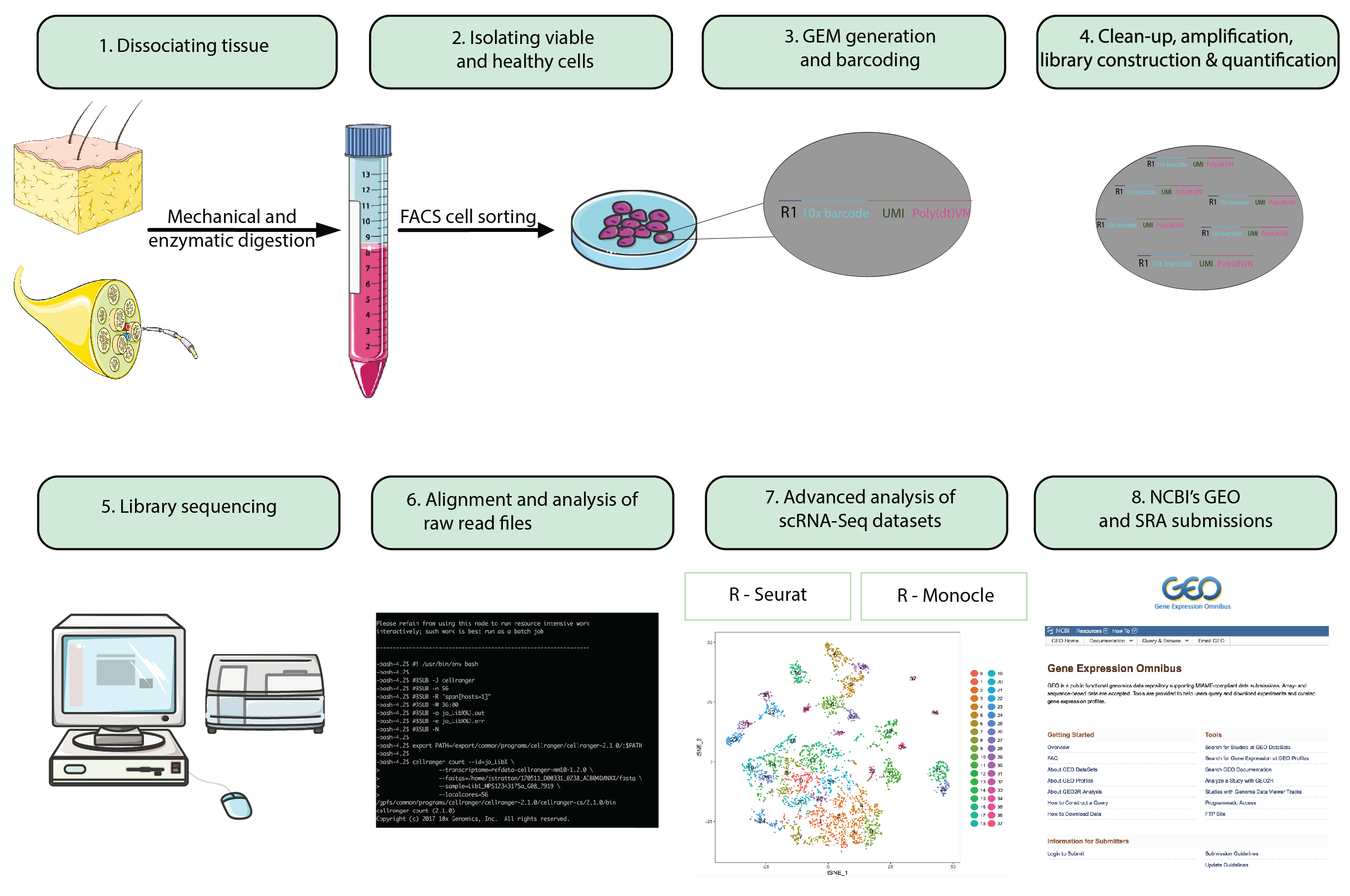
Figura 1: fluxograma. Etapas de preparação todo animal para analisar uma única célula RNA-Seq datasets para apresentação de conjuntos de dados finais para um repositório disponível publicamente. Grânulos do gel em emulsão (gemas) consulte contas com código de barras oligonucleotides que encapsulam milhares de células únicas. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 2: criando suspensão viável única célula do tecido nervoso. (a) desenho animado visão geral dos controlos do controlo de qualidade. (b) células e restos com células ainda incorporados em escombros (setas vermelhas). (c) células lançadas de detritos (setas vermelhas). (d) célula isolamento pela FACS. P0: fração de detritos; P1: célula-como fração; P3: exclusão de duplets; P4: negativo fração viabilidade tintura (Sytox Orange). (e) não há controle de tintura de viabilidade. (f) a imagem da fração P0 representando fragmentos isolados. (g) imagem da fração de P4, que representa células viáveis isoladas (setas vermelhas). (b) (c) (f) e (g) tinha corante nuclear adicionado 20 minutos antes de imagem. Barras de escala: 80 µm. clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 3: sequenciamento superficial prevê o número de células recuperadas em 10 X de amostras processadas. um exemplo (1.6 de amostra) de csv gerado pelo MiSeq listando célula códigos de barras e sua correspondente UMI conta conforme determinado pela leituras confiàvel mapeadas. (b) enredo de classificação do código de barras para amostra 1.6 mostra uma significativa queda na contagem UMI em função de códigos de barras do celular. As linhas tracejadas e sólidas representam o corte entre as células e fundo, conforme determinado pela inspeção visual. (c) códigos de barras célula observaram usando a célula Ranger gasoduto post-HiSeq revela raso sequenciamento aproximados com precisão o número de células por exemplo 1.6. (d) um exemplo de uma configuração de célula de fluxo baseado no sequenciamento superficial derivada estimativas de célula. Por exemplo 1.6, desde sequenciamento superficial previu 3480 células, 1,17 pistas foram designadas para assegurar > 100.000 leituras por cobertura de sequenciamento de celular em HiSeq. Nota: Todas as faixas devem adicionar a 100%. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 4: controle de qualidade e bioinformática de célula única RNA-Seq dataset usando o pacote de Seurat R. (a) parcelas de métricas de controle de qualidade que incluem o número de genes, o número de identificadores exclusivos moleculares (UMIs) e a porcentagem de transcrições de mapeamento de genoma mitocondrial. (b) gene amostra parcelas detectar células com níveis desviantes de transcrições mitocondriais e UMIs. (c) enredo de cotovelo amostra utilizado para determinação de ad hoc dos PCs estatisticamente significativas. As linhas tracejadas e ponto-tracejado representam o corte onde um claro "cotovelo" torna-se evidente no gráfico. Dimensões do PC antes esse cotovelo são incluídos na análise a jusante. (d, e) Aglomerados de células gráfico baseado visualizados em duas resoluções diferentes em um baixo-dimensional espaço usando um enredo tSNE. (f) superior genes marcadores (amarelos) para cada cluster visualizados em uma expressão heatmap usando a função DoHeatmap de Seurat. (g) visualizando a expressão do marcador de, por exemplo, Cd68 gene representando os macrófagos (roxos) usando a função FeaturePlot de Seurat. Isto sugere que o cluster 2 e 4 (no painel d) deste dataset representa os macrófagos. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 5: categorização e ordenando ao longo da trajetória de peudotemporal usando o toolkit de monóculo celular. (a) fiscalizar a distribuição de mRNA (inferido UMI contagens) em todas as células em uma amostra. Somente as células com mRNA entre 0 - ~ 20.000 foram utilizados para análise a jusante. (b, c) Atribuindo e contando os tipos de células com base em marcadores de células de linhagem conhecida. Por exemplo, células expressando PDGF receptor alfa ou fibroblasto específico da proteína 1 foram atribuídas a célula tipo #1 representando panfibroblastos usando a função de newCellTypeHierarchy do monóculo. Número de diferentes tipos de células pode ser visualizado como um gráfico de pizza (b) e como uma tabela (c). (d) usando a célula tipo #1 (fibroblastos), por exemplo, os genes usados para ordenar as células podem ser visualizadas usando um gráfico de dispersão que demonstra a dispersão de gene vs expressão significa. A curva vermelha mostra o corte para genes usados para requisitar calculado pelo modelo de média-variância usando a função de estimateDispersions do monóculo. Genes que atendam este corte foram usados para requisitar pseudotime a jusante. (e, f) Visualização trajectórias de célula em um espaço reduzido bidimensional colorido por "Estado" do célula (e) e pelo monóculo atribuído "Pseudotime" (f). Clique aqui para ver uma versão maior desta figura.
{kind=link}
Discussão
Este protocolo demonstra como a preparação adequada de células únicas pode descobrir a heterogeneidade transcriptional de milhares de células únicas e discriminar Estados funcionais ou identidades únicas de celulares dentro de um tecido. O protocolo não exige ferramentas transgénicas ou proteínas fluorescentes repórter e pode ser aplicado para o isolamento de células únicas de diversos tecidos de interesse, incluindo os de seres humanos; tendo em mente cada tecido é único e este protocolo exigirá algum grau de adaptação/modificação.
Os programas transcriptional diversificados e altamente dinâmicos dentro de células têm enfatizado o valor da célula única genómica. Além de isolar o RNA de alta qualidade, uma etapa de preparação de amostra crítico necessária para conjuntos de dados de alta qualidade é garantir que as células são completamente liberadas do tecido e que as células são saudáveis e intacta. Isto é relativamente direta para coleta de células que são facilmente lançou, como circulantes de células ou tecidos onde as células são frouxamente retidas, como em tecidos linfoides. Mas isso pode ser um desafio para outros tecidos adultos, devido à arquitetura celular altamente desenvolvidos, abrangendo grandes distâncias, em torno da matriz extracelular e as proteínas do citoesqueleto rígida frequentemente envolvidos na manutenção da estrutura celular. Mesmo com técnicas de dissociação apropriado para a versão completa de células, há um potencial que o rigoroso e muitas vezes demorado processamento necessário alteraria a integridade de qualidade e celular mRNA. Além disso, as altas temperaturas, utilizadas para dissociação enzima assistida também afetam assinaturas transcriptional29,30. A intenção do protocolo é apresentar controle de qualidade verifica, usando tecidos tais como o nervo adulto mielinizado e a pele de adulta ricos em matriz extracelular, para demonstrar como a otimização pode ajudar a superar esses obstáculos.
Uma consideração importante ao projetar qualquer experimento scRNA-Seq é a escolha de profundidade de sequenciamento. Sequenciamento pode ser altamente multiplexado e ler profundidade pode variar sendo muito baixo usando Drop-Seq2 para até 5 milhões de leituras/célula14 usando um método de RNA-Seq completo como Smart-Seq A maioria dos experimentos de scRNA-Seq podem detectar transcrições de moderada a alta expressão com sequenciamento tão baixo quanto 10.000 leituras/célula, que é geralmente suficiente para4241,classificação do tipo celular. Sequenciamento superficial profundidade é de valor para economizar em custos de sequenciamento ao tentar detectar populações de células raras em tecidos complexos, onde milhares de células podem ser necessários para confiantemente atribuem populações raras. Mas o sequenciamento de profundidade não é adequado quando são necessárias informações detalhadas na expressão dos genes e processos associados com assinaturas transcriptional sutis. Atualmente, estima-se que a grande maioria dos genes em uma célula é detectada com 500.000 leituras/célula, mas isso pode variar dependendo do protocolo e tecido tipo43,44. Enquanto transcrição completos no sequenciamento contorna a necessidade de montagem e pode, portanto, detectar romance ou variantes raras da tala, custos de sequenciamento frequentemente limitam a escala tais abordagens para examinar milhares de células que inclui um sistema complexo de tecido. Em contraste, 3' com a tag bibliotecas unicelulares tais como as descritas neste protocolo normalmente têm baixa complexidade e exigem mais raso sequenciamento. É importante notar que bibliotecas geradas usando o protocolo descrito podem ser sequenciadas em uma das cinco sequenciadores suportados: 1) NovaSeq, 2) HiSeq 3000/4000, 2500 3) HiSeq execução rápida e alta saída, 4) NextSeq 500/550 e MiSeq 5).
Uma abordagem alternativa a única célula RNA-Seq, que reduz a necessidade de tecidos delicados e célula manipulação ainda mantém alguns dos benefícios da única célula RNA-Seq, é a análise do RNA do núcleo único45. Esta abordagem permite o processamento mais rápido, reduzindo a degradação do RNA e tomar medidas mais drásticas para garantir a adequada liberação de núcleos e assim provavelmente permite uma captura mais confiante dos perfis transcriptional que representa todas as células dentro de um determinado tecido. Isto, claro, só forneceria uma porção da atividade transcricional presente dentro de uma determinada célula, assim, dependendo de quais objetivos experimentais são de interesse, esta abordagem pode ou não ser adequado.
Além a caracterização completa de identidades celulares dentro de um determinado tecido, uma das mais valiosas análises para conjuntos de dados scRNA-Seq é a avaliação de Estados intermediários transcriptional em populações de células 'definido'. Estes Estados intermediários podem dar insights sobre as relações de linhagem entre células dentro das populações identificadas, que não foi possível com a tradicional massa que se aproxima do RNA-Seq. Várias ferramentas de bioinformatic scRNA-Seq agora foram desenvolvidas para elucidar isso. Tais ferramentas podem avaliar os processos envolvidos em, por exemplo, as células cancerosas em transição para um estado oncogênica/metastático, células-tronco amadurecimento em diversos destinos terminais ou células imunitárias vaivém entre os Estados ativos e inativo. Transcriptoma sutis diferenças nas células também podem ser indicativas de enviesamentos de linhagem que, ferramentas de bioinformatic recentemente desenvolvidos como FateID, pode inferir47. Uma vez que as distinções entre as células de transição podem ser difícil verificar tendo em conta as diferenças transcriptional pode ser sutil, sequenciamento mais profundo pode ser necessário46. Felizmente, a cobertura de uma biblioteca superficialmente sequenciada pode ser aumentada se interessado na investigação do conjunto de dados mais re-executando a biblioteca na outra célula de fluxo.
Tomados em conjunto, este protocolo fornece um fácil-para-adaptar-se de fluxo de trabalho que permite aos usuários transcricionalmente perfil centenas ou milhares de single-células dentro de um experimento. A qualidade final de um conjunto de dados de scRNA-Seq baseia-se no isolamento de célula otimizado, citometria de fluxo, geração de biblioteca de cDNA e interpretação de matrizes gene cru-código de barras. Para este fim, este protocolo fornece uma visão abrangente de todas as etapas-chave que pode ser facilmente modificado para permitir estudos de tipos diversos de tecido.
Divulgações
Sem divulgações
Agradecimentos
Reconhecemos que o pessoal de apoio à instalação de serviços de UCDNA, bem como o pessoal de instalação Cuidado Animal na Universidade de Calgary. Agradecemos a Matt Workentine para seu apoio de bioinformática e Jens Durruthy para seu suporte técnico. Este trabalho foi financiado por um subsídio CIHR (R.M. e J.B.), um CIHR New Investigator Award, J.B., e saúde Research Institute Fellowship (J.S. uma infantil de Alberta).
Materiais
| Name | Company | Catalog Number | Comments |
| Products | |||
| RNAse out | Biosciences | 786-70 | |
| Pentobarbital sodium | Euthanyl | 50mg/kg | |
| HBSS | Gibco | 14175-095 | |
| Dispase 5U/ml | StemCell Technologies | 7913 | 5 mg/ml |
| Collagenase-4 125 CDU/mg | Sigma-Aldrich | C5138 | 2 mg/ml |
| DNAse | Sigma-Aldrich | DN25 | 10mg/ml |
| BSA | Sigma-Aldrich | A7906 | |
| 15 ml Narrow bottom tube VWR® High-Performance Centrifuge Tubes | VWR | 89039-666 | |
| Sytox Orange Viability Dye | Molecular Probes | 11320972 | 1.3 nM/µl |
| Nuc Blue Live ReadyProbes | Invitrogen | R37605 | |
| Agilent 2100 Bioanalyzer High senitivity DNA Reagents | Agilent | 5067-4626 | |
| Kapa DNA Quantification Kit | Kapa Biosystems | KK4844 | |
| Chromium Single Cell 3' reagents | 10x Genomics | ||
| Equipment | |||
| BD FACSAria III | BD Biosciences | ||
| Agilent 2100 Bioanalyzer Platform | Agilent | ||
| Illumina® HiSeq 4000 | Illumina | ||
| Illumina® MiSeq SR50 | Illumina | ||
| 10X Controller + accessories | 10x Genomics | ||
| Software | |||
| The Cell Ranger | 10x GENOMICS | support.10xgenomics.com/single-cell-gene-expression/software/overview/welcome | |
| Loupe Cell Browser | 10x GENOMICS | support.10xgenomics.com/single-cell-gene-expression/software/downloads/latest | |
| R | https://anaconda.org/r/r |
Referências
- Shalek, A. K., et al. Single-cell RNA-seq reveals dynamic paracrine control for cellular variation. Nature. 510, 363-369 (2014).
- Macosko, E. Z., et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell. 161, 1202-1214 (2015).
- Zappia, L., Phipson, B., Oshlack, A. Exploring the single-cell RNA-seq analysis landscape with the scRNA-tools database. bioRxiv:206573. , (2018).
- Dulken, B. W., Leeman, D. S., Boutet, S. C., Hebestreit, K., Brunet, A. Single cell transcriptomic analysis defines heterogeneity and transcriptional dynamics in the adult neural stem cell lineage. Cell Reports. 18 (3), 777-790 (2017).
- Llorens-Bobadilla, E., et al. Single-Cell Transcriptomics Reveals a Population of Dormant Neural Stem Cells that Become Activated upon Brain Injury. Cell Stem Cell. 17 (3), 329-340 (2015).
- Trapnell, C., et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nature Biotechnology. 32, 381-386 (2014).
- Aibar, S., et al. SCENIC: single-cell regulatory network inference and clustering. Nature Methods. 14, 1083-1086 (2017).
- Mayer, C., et al. Developmental diversification of cortical inhibitory interneurons. Nature. 555 (7697), 457-462 (2018).
- Stratton, J. A., et al. Purification and Characterization of Schwann Cells from Adult Human Skin and Nerve. eNeuro. 4 (3), (2017).
- Biernaskie, J. A., McKenzie, I. A., Toma, J. G., Miller, F. D. Isolation of skin-derived precursors (SKPs) and differentiation and enrichment of their Schwann cell progeny. Nature Protocols. 1 (6), 2803-2812 (2007).
- 10X Genomics. User Guides. , Available from: https://www.10xgenomics.com/resources/user-guides/ (2018).
- 10X Genomics. Chromium Single Cell 3' Training Module. , Available from: http://go.10xgenomics.com/training-modules/single-cell-gene-expression (2018).
- Agilent. , Available from: https://www.agilent.com/en-us/library/usermanuals?N=135 (2018).
- Kolodziejczyk, A. A. Single Cell RNA-Sequencing of Pluripotent States Unlocks Modular Transcriptional Variation. Cell Stem Cell. 17, 471-485 (2015).
- Jaitin, D. A., et al. Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types. Science. 343, 776-779 (2014).
- Pollen, A. A., et al. Low-coverage single-cell mRNA sequencing reveals cellular heterogeneity and activated signaling pathways in developing cerebral cortex. Nature Biotechnology. 32, 1053-1058 (2014).
- 10X Genomics. Sequencing Requirements for Single Cell 3'. , Available from: https://support.10xgenomics.com/single-cell-gene-expression/sequencing/doc/specifications-sequencing-requirements-for-single-cell-3 (2018).
- Datacamp. Introduction to R. , Available from: https://www.datacamp.com/courses/free-introduction-to-r (2018).
- Droplet-based, high-throughput single cell transcriptional analysis of adult mouse tissue using 10X Genomics#39; Chromium Single Cell 3' (v2) system: From tissue preparation to bioinformatic analysis. , Available from: https://figshare.com/s/97b83e649e5eefd01357 (2018).
- 10X Genomics. User Guides. , Available from: https://www.10xgenomics.com/resources/user-guides/ (2018).
- UNIX Tutorial for Beginners. , Available from: http://www.ee.surrey.ac.uk/Teaching/Unix/ (2018).
- 10X Genomics. Creating a Reference Package with cellranger mkref. , Available from: https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/advanced/references (2018).
- 10X Genomics. System Requirements. , Available from: https://support.10xgenomics.com/single-cell-gene-expression/software/overview/system-requirements (2018).
- 10X Genomics. Software Downloads. , Available from: https://support.10xgenomics.com/single-cell-gene-expression/software/downloads/latest (2018).
- 10X Genomics. Aggregating Multiple Libraries with cellranger aggr. , Available from: https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/using/aggregate#depth_normalization (2018).
- 10X Genomics. Loupe Cell Browser Gene Expression Tutorial. , Available from: https://support.10xgenomics.com/single-cell-gene-expression/software/visualization/latest/tutorial (2018).
- scRNA-tools. A table of tools for the analysis of single-cell RNA-seq data. , Available from: https://www.scrna-tools.org/ (2018).
- Conda. Downloading conda. , Available from: https://conda.io/docs/user-guide/install/download.html (2018).
- Anaconda. r / packages / r 3.5.1. , Available from: https://anaconda.org/r/r (2018).
- Droplet-based, high-throughput single cell transcriptional analysis of adult mouse tissue using 10X Genomics' Chromium Single Cell 3' (v2) system: From tissue preparation to bioinformatic analysis. , Available from: https://figshare.com/s/97b83e649e5eefd01357 (2018).
- Satija Lab. Seurat - Guided Clustering Tutorial. , https://satijalab.org/seurat/pbmc3k_tutorial.html (2018).
- Droplet-based, high-throughput single cell transcriptional analysis of adult mouse tissue using 10X Genomics' Chromium Single Cell 3' (v2) system: From tissue preparation to bioinformatic analysis. , Available from: https://figshare.com/s/97b83e649e5eefd01357 (2018).
- Monocle. , Available from: http://cole-trapnell-lab.github.io/monocle-release/docs/#constructing-single-cell-trajectories (2018).
- Github. An R package to test for batch effects in high-dimensional single-cell RNA sequencing data. , (2018).
- Edgar, R. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Research. 30, 207-210 (2002).
- Leinonen, R., Sugawara, H., Shumway, M. The sequence read archive. Nucleic Acids Research. 39, D19-D21 (2011).
- NIH. GenBank Submission Portal Wizards. , Available from: https://www.ncbi.nlm.nih.gov/account/register/?back_url=/geo/submitter/ (2018).
- NIH. Submitting data. , Available from: https://submit.ncbi.nlm.nih.gov/geo/submission/ (2018).
- Shah, P. T., et al. Single-Cell Transcriptomics and Fate Mapping of Ependymal Cells Reveals an Absence of Neural Stem Cell Function. Cell. 173, 1045-1057 (2018).
- Anon, Method of the Year 2013. Nature Methods. 11, 1(2013).
- Adam, M., Potter, A. S., Potter, S. S. Psychrophilic proteases dramatically reduce single-cell RNA-seq artifacts: a molecular atlas of kidney development. Development. 144, 3625-3632 (2017).
- Wu, Y. E., Pan, L., Zuo, Y., Li, X., Hong, W. Detecting activated cell populations using single-cell RNA-seq. Neuron. 96, 313-329 (2017).
- Zeigenhain, C., et al. Comparative Analysis of Single-Cell RNA Sequencing Methods. Molecular Cell. 65 (4), 631-643 (2017).
- Wu, A. R., et al. Quantitative assessment of single-cell RNA-sequencing methods. Nature Methods. 11 (1), 41-46 (2014).
- Habib, N., et al. Div-Seq: Single-nucleus RNA-Seq reveals dynamics of rare adult newborn neurons. Science. 353 (6302), 925-928 (2016).
- Janes, K. A. Single-cell states versus single-cell atlases - two classes of heterogeneity that differ in meaning and method. Current Opinions in Biotechnology. 39, 120-125 (2016).
- Herman, J. S., Sagar,, Grün, D. FateID infers cell fate bias in multipotent progenitors from single-cell RNA-seq data. Nature Methods. 15 (5), 379-386 (2018).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoExplore Mais Artigos
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados