
Method Article
Transcriptomique cellule unique axée sur le codage à barres de gouttelettes de tissus de mammifères adultes
* Ces auteurs ont contribué à parts égales
Dans cet article
Résumé
Ce protocole décrit les processus générales et contrôle qualité vérifie nécessaire pour la préparation de cellules unique chez les mammifères adultes saines pour les préparations de RNA-Seq cellule unique axée sur la gouttelette, haut débit. Séquençage des paramètres, l’alignement de lecture et l’analyse bioinformatique de cellule unique en aval sont également fournis.
Résumé
L’analyse de l’expression génique cellulaire unique sur des milliers de différentes cellules dans un tissu ou un micro-environnement est un outil précieux pour identifier la composition cellulaire, la discrimination des États fonctionnels et les voies moléculaires qui sous-tendent les tissus observés fonctions et les comportements animaux. Toutefois, l’isolement des monocellules intacts, sains provenant de tissus de mammifères adultes pour l’analyse moléculaire ultérieures unicellulaire en aval peut être difficile. Ce protocole décrit les processus générales et contrôle qualité vérifie nécessaires pour obtenir des préparations de qualité adult unicellulaire du système nerveux ou de la peau qui activé l’analyse et le séquençage d’ultérieures unicellulaire impartiale RNA. Lignes directrices pour l’analyse bioinformatique en aval sont également fournis.
Introduction
Avec le développement du haut débit unicellulaire technologie1,2 et les avancements dans les outils de bio-informatique conviviale au cours de la dernière décennie3, un nouveau champ d’analyse de l’expression génique à haute résolution est apparu – séquençage de RNA unicellulaires (scRNA-Seq). L’étude de l’expression génique cellulaire unique est d’abord développée pour identifier l’hétérogénéité au sein de populations de cellules définis, tels que dans les cellules souches ou cellules cancéreuses, ou pour identifier les rares populations de cellules4,5, qui étaient inaccessibles à l’aide techniques de séquençage de RNA traditionnelles en vrac. Outils bioinformatiques ont permis l’identification de nouveaux sous-groupes (Seurat)2, visualisation de l’ordre de cellules le long d’un psuedotime espace (Monocle)6, définition des réseaux de signalisation actifs au sein ou entre populations ( SCENIC)7, prédiction de l’Assemblée de cellules unique dans un espace 3D artificiel (Seurat, etc.)8. Avec ces analyses nouvelles et passionnantes dont dispose la communauté scientifique, scRNA-Seq rapide-devient la nouvelle approche standard pour l’analyse de l’expression génique.
Malgré l’énorme potentiel de scRNA-Seq, les niveaux de compétences techniques nécessaire pour produire un ensemble de données propre à interpréter correctement les résultats peut être difficile aux nouveaux arrivants. Ici, un protocole de base, mais complet, à partir de l’isolement des cellules individuelles de tissus ensemble primaires à la visualisation et la présentation des données pour publication est présenté (Figure 1). Tout d’abord, l’isolement de cellules individuelles en bonne santé peut être réputé difficile, puisque les différents tissus varient dans leur degré de sensibilité à la digestion enzymatique et dissociation mécanique ultérieure. Ce protocole donne des indications à ces mesures d’isolement et identifie les points de contrôle importants de contrôle de qualité tout au long du processus. En second lieu, il peut être déroutant de comprendre la compatibilité et les exigences entre la technologie de cellule unique et séquençage de prochaine génération. Ce protocole fournit des directives pour mettre en œuvre une plate-forme conviviale, axée sur les gouttelettes barcoding unicellulaires et effectuez le séquençage. Enfin, la programmation informatique est une condition préalable importante pour l’analyse des ensembles de données transcriptomiques unicellulaires. Ce protocole fournit des ressources pour débuter avec le langage de programmation R et fournit des conseils sur la mise en œuvre de deux paquets de R scRNA-Seq-specific populaires. Ensemble, ce protocole peut guider les nouveaux arrivants en analyse scRNA-Seq pour obtenir des résultats clairs et interprétables. Ce protocole peut être ajusté à la plupart des tissus chez la souris et ce qui est important, pourrait être modifié pour être utilisé avec d’autres organismes, y compris les tissus humains. Ajustements selon le tissu et l’utilisateur sera nécessaires.
Il y a plusieurs considérations à garder à l’esprit tout en suivant ce protocole ; y compris, 1) à la suite toutes les directives de contrôle de la qualité aux étapes 1 et 2 du présent protocole est recommandée pour assurer une suspension monocellulaire viable de toutes les cellules de l’échantillon d’intérêt tout en assurant la précision totale numéros globules (résumées dans Figure 2 ). Une fois que cela est possible, et si toutes les conditions optimisées sont suivies, les mesures de contrôle de la qualité peuvent être déposés (pour gagner du temps - préserver la qualité de RNA réduisant cellulaires et perte). Confirmant la réussite de l’isolement de cellules uniques de grande viabilité des tissus d’intérêt est hautement recommander avant tout en aval de traitement. 2) étant donné que certains types de cellules sont plus sensibles que d’autres au stress, techniques de dissociation excessive peuvent biaiser par inadvertance de la population, donc confusion analyse en aval. Dissociation douce sans cisaillement cellulaires inutiles et la digestion est essentielle pour atteindre des rendements élevés de cellulaires et une représentation exacte de la composition du tissu. Forces de cisaillement se produisent au cours des étapes de trituration, FACS et la remise en suspension. 3) comme avec n’importe quel travail d’ARN, est-il préférable d’introduire comme peu RNase supplémentaire dans l’échantillon que possible au cours de la préparation. Cela aidera à maintenir la qualité RNA. Utiliser des solutions d’inhibiteur de la ribonucléase avec rinçage à nettoyer l’outillage et tout matériel qui n’est pas exempte de RNase mais éviter les produits traités DEPC. 4) effectuer des préparations aussi rapidement que possible. Cela aidera à maintenir la qualité RNA et réduire la mort cellulaire. Selon la longueur de la dissection des tissus et plusieurs animaux, considère la création de multiples dissections/préparations en même temps. 5) préparer les cellules sur la glace si possible de maintenir la haute qualité RNA, réduire la mort cellulaire et ralentir l’activité transcriptionnelle et de signalisation de cellules. Quoique, traitement glacée est idéal pour la plupart des types de cellules, certains types de cellules (par exemple, neutrophiles) performants lors du traitement à température ambiante. 6) éviter le calcium, magnésium, EDTA et les produits traités DEPC au cours de la préparation de cellules.
Protocole
Tous les protocoles décrits ici sont conformes et approuvé par le Comité de protection des animaux de l’Université de Calgary.
1. séparer les tissus (jour 1)
- Euthanasier la souris avec une surdose de pentobarbital de sodium (i.p., 50 mg/kg) ou le cas échéant selon le protocole de l’éthique animale. Enlever les poils indésirables de la dos et les jambes de la souris, puis la région de dissection de l’éthanol-stériliser.
- Disséquer le tissu ou le microenvironnement d’intérêt. Pour ce protocole, les tissus de la peau et du système nerveux nous permet de démontrer la généralisabilité des gouttelettes cellule unique axée sur le codage à barres de transcriptomique après dissociation tissu adulte.
- Pour le nerf sciatique, utilisent le protocole détaillé à Stratton et al. 9. brièvement, couper la peau de la région postérieure des jambes/dos de souris. Faites une incision le long de la cuisse avec une lame de bistouri stérile. Utiliser les ciseaux et pinces fines pour exposer et enlever le nerf sciatique.
- Pour la peau du dos, utilisent le protocole détaillé à Biernaskie et al. 10. en bref, disséquer la peau dorsale arrière en faisant des incisions d’une épaule à l’autre, dans l’ensemble de la croupe et à l’arrière avec des ciseaux et des pinces fines. Couper la peau en tranches fines (0,5 cm d’épaisseur) à l’aide d’une lame de bistouri stérile.
- Laver le tissu 2 fois avec HBSS glacee et enlever les indésirables du tissu conjonctif, des dépôts de graisse ou des débris sous un microscope à dissection.
- Pour le derme de la peau seulement, flotter les tranches dans a (5 mg/mL, 5 U/mL) en HBSS pendant 30-40 min à 37 ° C. Séparer chirurgicalement l’épiderme du derme. Jeter l’épiderme ou dissocier plus loin à l’aide de la trypsine si cela présente un intérêt.
- Émincer l’échantillon en morceaux de 1 à 2 mm à l’aide d’une lames de bistouri stériles et mis en enzyme collagénase froid-IV de fraîchement décongelés 2 mg/mL (2 mg/mL, 125 CDU/mg, dans les médias de F12).
- Pour le nerf, utilisez ~ 500 µL par 2 nerfs sciatiques de x. Pour la peau, utiliser environ 8 mL par souris 1 x retour peau.
Remarque : Les tissus doivent être complètement submergées dans la solution de collagènase-IV. Il est essentiel que des enzymes de digestion sont manipulés, entreposés et préparés convenablement. Si les enzymes sont laissées à la température ambiante pendant de longues périodes de temps, isolement unicellulaires exigera trituration mécanique excessive et réduire la viabilité des cellules. Collagènase-IV peut également être composé dans les milieux de culture cellulaire où la viabilité cellulaire est plus optimale. Toutefois, cela pourrait altérer l’activité enzymatique ou transcriptionnelle signature donc doit être optimisé par l’utilisateur.
- Pour le nerf, utilisez ~ 500 µL par 2 nerfs sciatiques de x. Pour la peau, utiliser environ 8 mL par souris 1 x retour peau.
- Incuber l’échantillon de l’enzyme dans un bain de 37 ° C pendant 30 min avec agitant doucement toutes les 10 min. Un shaker placé à 37 ° C est aussi une alternative appropriée.
- Broyer avec un pipetteur P1000 20 - 30 fois à ajout d’enzyme après 30 min.
- Répéter la trituration toutes les 30 minutes jusqu'à ce que la solution a un aspect trouble et de morceaux de tissus sont en grande partie dissociées.
Remarque : Assurez-vous que la version complète de cellules (Figure 2 b, 2C). Pour confirmer le communiqué complet, la plaque cellules bleu Nuc (2 gouttes par mL 1) et après 20 min, vérifier au microscope pour s’assurer que tous les noyaux sont associés à des cellules individuelles plutôt que des débris. Il est essentiel de vérifier le degré de la cellule libération dans une expérience donnée pour chaque type de tissu ou de la condition. En tissu fibreux (par exemple, une blessure chronique) ou tissu adulte blessé, la libération des cellules variera considérablement d’une blessure aiguë ou de tissu embryonnaire. Ceci est particulièrement important parce que certains types de cellules sont moins susceptibles de libérer des tissus que d’autres, excluant ainsi préférentiellement les cellules de l’analyse en aval.- Pour le nerf, dissocier le tissu pour 0,5 à 1,5 heures au total. Pour la peau, se dissocient tissu pour 2 heures au total (dans la dernière heure d’incubation, ajouter DNase (1 mg/mL) à l’échantillon de peau).
- Filtrer deux fois avec un filtre de 40 µm. Rincer le filtre avec glacee 1 % BSA/HBSS.
- Centrifuger à 260 x g pendant 8 min. Retirez ensuite le surnageant.
- Resuspendre le culot dans HBSS contenant 1 % de BSA en utilisant un embout large-alésage et placez sur la glace. Le volume de la remise en suspension est basé sur le volume tissulaire (poids humide 800 mg pour peau = 800 µL volume ; 10 mg poids pour nerfs humide = 100 µL volume).
- Éventuellement, commencez avec un volume faible remise en suspension et ajustez si nécessaire basée sur la vitesse d’écoulement (événements par seconde) sur trieuse de FACS. La densité de tri plus efficace (maximiser le nombre de cellules recueillies alors que réduire le temps) pour les collections est 3 000-7 000 événements par seconde.
- Si à l’aide de colorant de viabilité, retirez un subaliquot pour un contrôle sans coloration. Puis ajoutez le colorant de viabilité 1:15,000 (stock : 20 000 nM/µL) d’échantillon (concentration finale de 1,3 nM/µL) en utilisant un embout large calibre pour réduire la tonte.
Remarque : Il est essentiel de vérifier le degré de la mort des cellules dans une expérience donnée pour chaque type de tissu ou de la condition. Certains types de cellules au sein d’un échantillon sont plus susceptibles de mourir que les autres, étant ainsi préférentiellement exclus de l’analyse en aval.- Incuber l’échantillon avec de la teinture de viabilité pendant 5 à 10 minutes sur la glace dans l’obscurité. Puis ajouter 4 mL de glacé 1 % BSA/HBSS à échantillon. Centrifuger à 260 x g pendant 8 min enlever le colorant excédentaire de viabilité. Traiter le subaliquot avec aucune viabilité teindre contrôle non colorée de la même manière.
2. isoler les cellules viables et en bonne santés (jour 1)
- S’assurer que l’installation de FACS suit cellule à fluorescence appropriée activée (FACS) paramètres de tri.
- Préparer la machine de FACS à l’avance pour s’assurer qu’elle est prête lorsque la centrifugeuse finale à l’étape 1 est terminée et faire en sorte que le compartiment de la collection est conservé au froid à l’aide de blocs de glace.
- Utilisez les paramètres suivants : débit : 1.0 (correspond grosso modo à 10 µL/min) ; Filtre : 1,5 ND ; Taille de la buse : 100 µm ; Dispersion vers l’avant : 80-180 V (changement si nécessaire afin de distinguer la taille des événements) ; Diffusion latérale : 150-220 V (changement si nécessaire afin de distinguer la granularité/forme d’événements) ; Laser : 100-400 V (changement si nécessaire afin de distinguer les événements négatifs positifs vs colorant de viabilité & vérifier ce contre aucun contrôle de colorant de viabilité) ; Gates : Modifier au besoin pour que toutes les cellules sont obtenus. Voir Figure 2d-2 g.
Remarque : Les paramètres FACS dépendent fortement les types de cellules et la trieuse employées et doit donc être optimisé par l’utilisateur.
- Préparer 15 mL et de base étroite tubes 8 ml de glacée 1 % BSA/HBSS pour les collections de l’échantillon. Statique à l’intérieur du tube et la tension superficielle peut affecter l’efficacité de collecte. Inverser les tubes avant collections afin d’assurer l’interface entre la surface du liquide et l’intérieur du tube est humide.
Remarque : Si vous travaillez avec un nombre très faible de cellules, ajuste pour petit récipient selon le cas. - Une fois que toutes les cellules sont recueillies, centrifuger échantillon à 260 x g pendant 8 min.
Remarque : Avant la centrifugation, ajouter 1 % BSA/HBSS lavage/pousser les cellules vers le bas de la surface latérale et inverti/mélanger le tube immédiatement après les FACS. - Resuspendre le culot dans 1 % BSA/HBSS et garder sur la glace. Le volume maximal par exemple qui est compatible avec le traitement de l’étape 3 est 33,8 µL, donc vous assurer que le volume de dilution/remise en suspension cellulaire finale a lieu d’obtenir nombre de cellule idéal à 33,8 µL. Autres options de support de dilution pour cette étape (et toutes les dilutions précédentes dans 1 % BSA/HBSS) incluent DMEM et jusqu'à 40 % de sérum, mais éviter de calcium, de magnésium ou EDTA contenant des réactifs.
- Laissez les cellules sur la glace pour un minimum de temps. Idéalement collègue doit préparer tous les matériels et réactifs pour l’étape suivante (étape 3) pendant les étapes finales de l’étape 2.
- Cellule préparation contrôles critiques
- Confirmer les estimations du nombre de cellules provenant des FACS. Selon le type de tissu et de dissociation, longueurs, de débris et de cellules peuvent être très semblables en taille et en forme. Ainsi, sauf si un journaliste fluorescent est utilisé, les FACS ne peut exclure tous les débris. Il est recommandé qu’un nombre de cellules final après que collection FACS est effectuée afin de comprendre quel est le pourcentage d’événements (selon les FACS) sont en fait des cellules pour une préparation donnée (Figure 2 g). Effectuer le nombre de cellules à l’aide d’un hémocytomètre ou compteur de cellules automatisées (répéter deux fois) et calculer le pourcentage de cellules viables qui est représenté par le nombre total d’événements collecté selon machine FACS.
- Valider la préparation de cellules. Valider le fait que sans les grosses particules (> 100 µm) sont présents car ils peuvent boucher les équipements en étapes en aval. Perturbation dans l’évacuation des débris risquent d’obstruer la puce microfluidique de cellule unique. Plaque de cellules restantes avec le fil bleu Nuc (voir ci-dessus) pour vous assurer qu'aucun fragment de gros débris n’est présents. Cela permettra également la confirmation que les cellules sont au singulier (c.-à-d.ne pas collées entre elles) donnant confiance cette analyse génétique de la cellule unique en aval représente les cellules individuelles plutôt que plusieurs cellules.
- Décider sur le nombre de cellules à séquence : il y a une large gamme de nombres de cellules adultes dérivés de tissus par exemple qui peut être chargé dans le système avec jusqu'à 8 échantillons qui peuvent être exécutées simultanément. Auteurs ont chargé n’importe où à partir de 500 – 50 000 cellules par échantillon et obtenu de bonne qualité scRNA-Seq datasets. On trouvera une discussion plus approfondie concernant le nombre de cellules plus approprié pour charger dans la section « Discussion ». La sortie finale du nombre de cellules séquencée dépend fortement de la qualité de simple-cellules isolées. Chargement de 10 000 cellules tissu adultes peut retourner n’importe où de 1 000 à 4 000 cellules séquencés (retour 10-40 %). Si vous êtes intéressé dans la séquence des numéros de cellulaire élevée (~ 10 000 cellules, le nombre maximal recommandé pour ce système), puis à charger 25 000-100 000 cellules sera nécessaire.
3. GEM (perle de Gel en émulsion) génération et codes à barres (jour 1)
NOTE : Les étapes 3 à 6 du présent protocole sont conçus pour être utilisé en conjonction avec la plus courante microgouttelettes cellule unique plate-forme, fabriquée par 10 X Genomics. Des lignes directrices détaillées pour les étapes 3 et 4 sont décrites dans les directives du fabricant du protocole (voir protocole 3' Chrome unicellulaire)11,12 et doivent être suivies en conjonction avec ce protocole. Pour de meilleurs résultats, étape 3 doit être rempli immédiatement après dissociation (étape 1) et les mesures d’isolement (étape 2) cellule le jour 1 du présent protocole.
- Préparer la puce selon protocole 11,12 les directives du fabricant. Cette plate-forme unicellulaires microgouttelettes utilise la technologie qu’échantillons ~ 750 000 codes à barres pour indexer séparément du transcriptome de chaque cellule. Ceci est réalisé en partitionnant les cellules en Gel talon en émulsion (GEMs) où les ADNc généré partagent un code à barres commun. Pendant la génération du GEM, les cellules sont fournis afin que la majorité (90-99 %) des gemmes générés ne contiennent aucune cellule, alors que le reste, la plupart du temps, contiennent une seule cellule.
- Mettre la puce dans le support de la puce.
- Préparer le mélange maître cellule sur la glace.
- Ajouter 50 % de glycérol à puits inutilisés et 90 µL du mélange maître cellule bien 1, 90 µL de billes de gel à bien 2 et 270 µL de partitionnement d’huile pour bien 3.
- Couvrir la puce avec le joint.
- Charger la puce et exécuter dans un contrôleur de cellule unique.
- Éjecter le tiroir, placer la puce dans plateau, fermer le tiroir et appuyez sur Play. Une perle dans un joyau unique cellule 3' gel comprend amorces contenant une séquence partielle de Illumina R1 (lire 1 apprêt de séquençage), un 16 nucléotides (nt) 10 x Barcode, une 10 nt identificateur moléculaire Unique (UMI) et une séquence d’apprêt poly-dT. Pendant la course, billes de gel dans le contrôleur sont libérés et mélangés avec mélange de maître et de lysat cellulaire.
- Collecter 100 µL de l’échantillon et les place dans un tube de PCR.
- Tubes PCR lieu prédéterminé par PCR en machine et exécutent la PCR selon le kit. Après incubation, les gemmes comprendra des ADNc barcoded complet, de l’ARNm poly-adenylated.
- Après la course, placez à-20 ° C durant la nuit jusqu'à 1 semaine avant précédant à l’étape suivante.
4. nettoyage, Amplification, la Construction de la bibliothèque et bibliothèque Quantification (jour 2)
NOTE : Des lignes directrices détaillées pour les 4 étapes sont décrites dans protocole 11,12 les directives du fabricant et doivent être suivies en conjonction avec ce protocole.
- Billes magnétiques de silane permet de retirer les restes réactifs biochimiques/amorces du mélange réactionnel GEM.
- Amplifier les ADNc barcoded complet, pour générer une masse suffisante pour la construction de la bibliothèque.
- Évaluer le rendement d’ADN. Avant la construction de la bibliothèque, évaluer le rendement d’ADN de l’échantillon. Ceci déterminera le nombre de cycles à utiliser dans l’étape de l’ACP en aval (échantillon Index PCR pendant la construction de la bibliothèque). Selon la teneur en ARN d’un échantillon donné, qui peut varier selon les États d’activation (par exemple, contrôle vs blessés, etc.), type de cellules et le rendement de la cellule, le nombre de cycle recommandé peut changer.
- Pour le séquençage ~ 3 000 tissus cellules dérivées (rien à voir avec les États d’activation), les auteurs ont constaté que 14 cycles (exemples : ~ 10-100 ng d’ADN) est la norme.
- Utiliser un Bioanalyzer pour l’analyse de l’ADN. Se référer à l’utilisateur Guide13.
- Fragment d’échantillon, puis sélectionnez la taille de l’ADN. Avant la construction de la bibliothèque, utilisez fragmentation enzymatique et protocoles de sélection de taille pour obtenir taille amplicon ADNc approprié.
- Préparation des échantillons pour la construction de la bibliothèque. Tandis que R1 (lire 1 séquence d’amorçage) est ajouté aux molécules durant l’incubation GEM ; P5, P7 (un indice de l’échantillon), et R2 (lire 2 séquence d’amorce) sont ajoutés au cours de la construction de la bibliothèque.
- Évaluer le rendement d’ADN. La plupart des installations de séquençage nécessitent des bibliothèques finales qui incluent des informations de qualité et de rendement ADN. Ainsi, exécutez le bioanalyzer après l’achèvement de l’ensemble du protocole et avant de les transporter à la facilité de séquençage.
- Conserver les échantillons à-80 ° C pendant 2 mois.
- Avant de séquençage, quantifier les échantillons à l’aide d’un kit de quantification de l’ADN. Cela peut être fait à l’installation de séquençage.
5. bibliothèque de séquençage (jour 3)
Remarque : La plate-forme de barcoding transcriptome unicellulaires utilisée dans le présent protocole génère les bibliothèques jumelé-fin compatible Illumina commençant et finissant par séquences P5 et P7. Bien que la profondeur minimale nécessaire pour résoudre l’identité type cellulaire peut être aussi peu que 10 000-50 000 lectures/cellule15,16, ~ 100 000 lectures/cellule est recommandée comme un compromis optimal de couverture des coûts pour les cellules adultes in vivo (en gardant à l’esprit certaines cellules types ou États de cellules activées minimalement atteindra saturation à 30 000-50 000 lectures/cellule).
- Transport de banques d’ADNc sur glace sèche à une installation de séquençage équipée d’un séquenceur Illumina approprié.
- Fournir les renseignements suivants à la facilité de séquençage :
- Fournir les détails de l’échantillon : l’échantillon ID d’index correspondant à chaque bibliothèque ; espèces ; base de données génomique pour l’Assemblée primaire (c.-à-d., GRCm38 pour souris) ; électrophorégramme montrant la taille de fragment de le bioanalyzer (entre 200 et 9 000 bp) ; ADNc (ng/µL) concentration et en bibliothèque total (total des rendements vont de 200 à 1400 ng) ; volume (µL) de l’échantillon.
- Fournir des demandes de séquençage : quantifier les échantillons à l’aide d’un kit de quantification de l’ADN ; type d’adaptateur/index (TruSeq ADN) ; type de plaque (Eppendorf twin.tec, jupe - recommandé pour l’ADN) ; le séquençage de technologie/bibliothèque type (10 x, instructions de séquençage complet et recommandations cycle)17.
- Courir peu profond séquencement (facultatif) : études analysant plusieurs échantillons biologiques bénéficieront de la mise en commun des échantillons (agrégation) pour générer une matrice de gène-code à barres unique contenant des données provenant de tous les échantillons. Pour minimiser les effets lot entre échantillons lorsque la mise en commun, la profondeur entre différentes bibliothèques devrait être normalisée. Pour ce faire, il faut une approximation précise du nombre de cellules simples. Le séquenceur MiSeq permettra de séquençage peu profond et est un moyen rentable et pratique d’obtenir des estimations exactes cellule.
Remarque : Une seule frappe à l’aide d’un séquenceur MiSeq SR50 offre une couverture suffisante pour estimer avec précision environ 20 000 cellules. Cette course sera approximativement le nombre de UMI récupéré pour chaque code à barres unique. Dans la Figure 3 a, l’en-tête d’une sortie de l’exemple (échantillon 1.6) (.csv) est affichée, énumérant les codes à barres et ses correspondant UMI compte tel que déterminé par lectures mappés en toute confiance.- Consulter un bioinformaticien afin de vous familiariser avec le R de langage de programmation. Se référer à DataCamp tutoriels pour plus d’informations,18.
- Évaluer les données brutes obtenues depuis le séquenceur en utilisant le script R fourni comme un modèle19. Données brutes désigne nombre de UMIs mappé à chaque code à barres unique cellule. Le script lit un fichier .csv, où la première colonne est une liste de codes à barres et la deuxième colonne sont ses correspondant UMI compte. Ce script fournira une parcelle (Figure 3 b) ainsi que le nombre de cellules avec code à barres dans chaque échantillon. Ajuster le script pour faire en sorte que le nombre de saisies d’UMI pour un échantillon donné est au point d’un tiers de la première descente raide. Dans la Figure 3 b, ce coude tombe environ 225 UMIs correspondant à 3 480 cellules avec code à barres.
- Comparable au séquençage de toute l’épaisseur à l’aide de HiSeq (où 3 516 cellules ont été séquencés, Figure 3C), estimations peu profond séquencement prédit 3 480 cellules.
- Utilisation cellulaire des approximations de récupération (de l’étape 5.3) ou utilisez le tableau de récupération trouvés dans protocole20 au plan de distribution de voie pour l’ordonnancement de la plus profonde du fabricant. Chaque échantillon doit recevoir une couverture comparable, donc si peu profond séquencement révèle qu’il existe différents nombres de cellules dans chaque échantillon (qui est souvent le cas) puis distribution de voie doit être calculée en conséquence. Une cellule d’écoulement HiSeq (qui comprend 8 voies) pouvez séquencer jusqu'à 2,4 milliards fin appariés personnalisé lectures. Exemple flux cellule mise en place est présenté en Figure 3d.
6. traitement lire fichiers
Remarque : Séquençage d’une seule cellule 3' bibliothèque utilisant ce protocole génère des données brutes en format binaire appel de base (BCL). La cellule Ranger package est utilisé pour générer des fichiers FASTQ basé sur du texte de fichiers BCL, effectuez génomique et transcriptomique alignements, comtes de gène, démultiplexage et agrégation des échantillons. Dans cette section, les principales étapes qui permettent aux utilisateurs de télécharger les données brutes de BCL provenant d’une installation de séquençage et de générer filtrée gène-barcode matrices prêtes pour la bioinformatique en aval est présenté.
- Utiliser un serveur centralisé pour l’exécution du programme. BCL fichiers de FASTQ et la plupart de la bioinformatique en aval de traitement exige une puissance de traitement importante.
- Télécharger tous les fichiers raw de lecture sur le serveur (ou des fichiers FASTQ si elles sont disponibles).
- Consulter avec l’administrateur du serveur pour configurer un compte sur un serveur centralisé ou un cluster et de se familiariser avec Unix21.
- Utilisez une commande fetch appropriée pour le système d’exploitation du serveur pour télécharger tous les fichiers du serveur de la facilité de séquençage.
- La plupart des installations de séquençage fournissent une commande permettant de télécharger des fichiers depuis un chemin sûr qui peut être exécuté depuis la ligne de commande (voir exemple ci-dessous).
- Remplacer le « < nom d’utilisateur > » et « mot de passe < > » espaces réservés dans la ligne de commande avec les informations d’identification fournies.
wget - O - « https://your_sequencing_facilitys_server.com/path_to_raw_read_files/--non-cookies--non-cocher-certificat--post-Data "j_username = nom d’utilisateur & j_password = mot de passe ' | wget--non-cookies--non-cocher-certificat--post-Data "j_username = nom d’utilisateur & j_password = mot de passe ' - ci -
- Si seul un chemin absolu aux fichiers est fourni (c.-à-d. https://your_sequencing_facilitys_server.com/path_to_raw_read_files/), insérez ce chemin d’accès dans une commande fetch.
- Décompressez les fichiers : si téléchargé fin de fichiers avec l’extension « .gz », il a été compressé à l’aide de la commande « gzip ». Pour décompresser, run décompressez commande dans la ligne de commande (voir exemple ci-dessous).
gunzip raw_read_files.gz - Télécharger la dernière version de cellule Ranger sur le serveur comme un.tar autonome22.
-
Critique : Avant de télécharger, s’assurer que le système Linux répond aux exigences minimales de23. Garantir un minimum de processeur 8-core Intel avec 64 Go de RAM et 1 to d’espace disque libre.
Remarque : Cellule Ranger offre transcriptomes pré-construit référence humaine et les rongeurs. Ceux-ci peuvent être modifiés à l’aide cellranger mkref commande pour détecter les gènes comme GFP24.
-
Critique : Avant de télécharger, s’assurer que le système Linux répond aux exigences minimales de23. Garantir un minimum de processeur 8-core Intel avec 64 Go de RAM et 1 to d’espace disque libre.
- Générer des fichiers FASTQ de fichiers de base appel du séquenceur (BCLs) à l’aide de la commande de mkfastq de cellranger.
NOTE : Le programme va aligner lectures brutes (à partir de fichiers FASTQ) à un génome de référence et générer des matrices de gène-cellule d’analyse en aval. Il utilise l’aligneur STAR qui effectue l’alignement épissage prenant en charge les lectures et un génome de référence. Seul avec confiance mappés lectures (c.-à-d.lectures compatible avec une annotation de gène unique) sont utilisés pour le comptage des UMI.- Par exemple, utilisez la commande de mkfastq de cellranger :
cellranger mkfastq --id = sample_name \
--courir = / chemin/de/échantillon \
--csv=csv_file_containing_lane_sample_index.csv
- Par exemple, utilisez la commande de mkfastq de cellranger :
- Cellranger nombre d’exécutions sur FASTQ fichiers générés à l’aide de mkfastq pour générer des chefs de cellule unique gène.
- Par exemple, utilisez la commande de comte de cellranger :
cellranger comte --id = sample_name \
transcriptome--= refdata-cellranger-mm10-1.2.0 \
--fastqs = / absolu/chemin/vers/fastq/les fichiers \
--exemple = same_sample_name_supplied_to_cellranger_mkfastq \
--localcores = 30
- Par exemple, utilisez la commande de comte de cellranger :
- La multi-bibliothèque agrégation (facultatif) : pour combiner les échantillons, piscine cellranger comte de sorties à l’aide de cellranger aggr. Cela se traduit par une matrice de gène-code à barres unique contenant des données regroupées à partir de plusieurs bibliothèques. Dans l’exemple cellranger aggr commande :
cellranger aggr--id = sample_name \
--csv = csv_with_libraryID_ & _path_to_molecule_h5.csv \
--normaliser = mappé
Remarque : Les bibliothèques peuvent être agrégés à l’aide de trois modes de normalisation (mappé, raw, none). Mappée est recommandé car il souséchantillons bibliothèques profondeur supérieure jusqu'à ce que toutes les bibliothèques ont séquençage égale profondeur25. - Pour une visualisation immédiate/analyse de données, importer le fichier de sortie .cloupe (généré à l’aide de nombre de cellranger ou de cellranger aggr) dans 10 x Loupe cellule navigateur26.
7. avancées d’analyse des ensembles de données scRNA-Seq
NOTE : Une base de données des outils scRNA-Seq complète se trouvent à scRNA-outils3,27. Ci-dessous est un cadre pour une cellule sans surveillance clustering à l’aide de Seurat2 et pseudotemporal vous passez votre commande à l’aide de Monocle6. Bien qu’une grande partie de ce travail peut se faire sur un ordinateur local, les étapes suivantes supposent que calcul sera complété à l’aide d’un serveur institutionnel.
- Télécharger la version la plus récente sur le compte du serveur à l’aide de la plate-forme de Linux28de Miniconda.
- Installez la dernière version de R en utilisant conda29.
- Tracer des données en utilisant le script de Seurat R fourni comme un modèle de30.
NOTE : Seurat est une boîte à outils axés sur la R qui permet aux contrôles de qualité, clustering, différentiel analyse d’expression de gène, identification de gènes marqueurs, réduction dimensionnelle et visualisation de données scRNA-Seq. On trouvera une description complète de Seurat de codage et de tutoriels sur le site Web de Siva Lab31. - Tracer des données en utilisant le script de Monocle R fourni comme un modèle32.
NOTE : Monocle est un autre outils axés sur la R qui permet la visualisation des modifications de l’expression plus pseudotime et identifie les gènes qui sous-tendent les décisions destin cellulaire. On trouvera une description complète de Monocle de codage et de tutoriels sur le site de Monocle33. - R-paquets tels que kBET peuvent être utilisées pour tester et corriger les effets du lot en raison de la mise en commun des datasets34.
8. de NCBI GEO et présentations de SRA
Remarque : Puisqu’un accès facile aux fichiers raw séquençage garantir la reproductibilité et réanalyse, présentations aliénées dans les référentiels en ligne accessibles au public sont recommandées ou requis avant la soumission du manuscrit. National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) et lecture de séquence Archive (SRA) sont des référentiels de données accessibles au public pour le séquençage haut débit données35,36.
- Registre de NCBI GEO Submitter compte37.
- Présentation de GEO complète qui comprend trois composants compilés dans un répertoire/dossier (intitulé comme nom d’utilisateur de l’émetteur de la GEO) : 1) enregistrement de métadonnées (un tableur par soumission du projet) ; 2) fichiers de données brutes ; 3) fichiers de données traitées.
- Téléchargez et remplissez la feuille de calcul de métadonnées38. La présentation de GEO publique suivante peut être utilisée comme un guide (GSE100320)39. Placez la feuille de calcul dans le répertoire.
- Fichiers de données brutes place générée du comte cellranger script pour toutes les bibliothèques dans le répertoire.
- Place traitée données fichiers (filtrée barcodes.tsv, genes.tsv et matrix.mtx) généré du comte cellranger script pour toutes les bibliothèques dans le répertoire.
- Références de serveur FTP de l’émetteur GEO permet de transférer le répertoire contenant les trois éléments. Pour les utilisateurs de Linux/Unix : ncftp, lftp, ftp, sftp et ncftpput peuvent être utilisés.
- Notifier les GEO pour les transferts38.
Résultats
Le répertoire des paquets open source conçu pour analyser des ensembles de données scRNA-Seq a considérablement augmenté40 avec la majorité de ces utilisation de paquets langages basés sur R3. Ici, les résultats représentatifs à l’aide de deux de ces paquets sont présentés : évaluer groupement sans surveillance de simple basés sur l’expression des gènes des cellules et ordonnant des cellules isolées le long d’une trajectoire afin de résoudre l’hétérogénéité de cellules et déconstruire biologique processus.
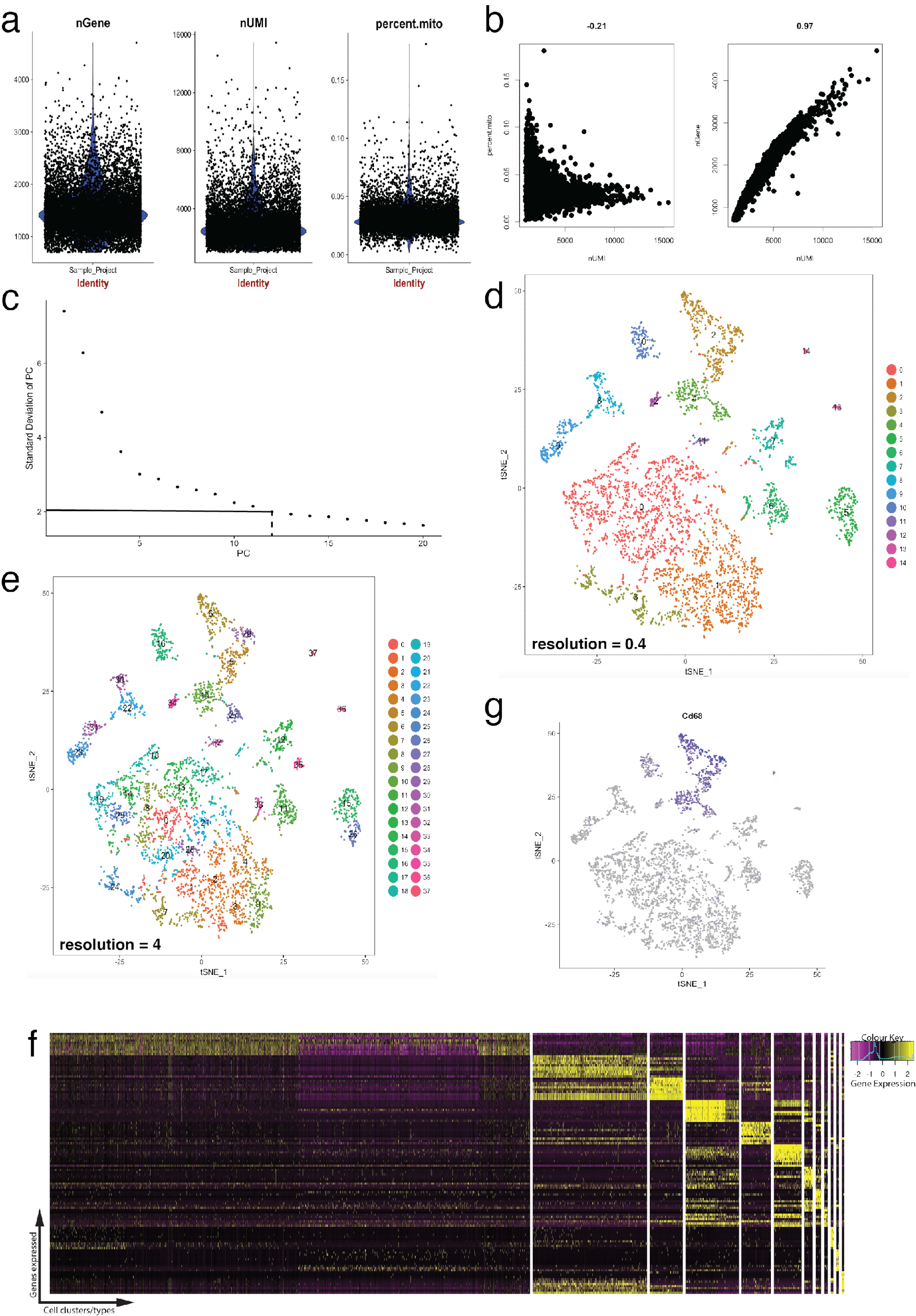
La figure 4 illustre l’utilisation de Seurat pour pré-traitement des contrôles de qualité et analyse bioinformatique en aval. Tout d’abord, filtration et élimination des cellules déviantes de l’analyse est essentielle pour le contrôle de la qualité. Cela a été fait à l’aide de violon (Figure 4 a) et nuages de points XY complote (Figure 4 b) pour visualiser le pourcentage de gènes mitochondriaux, nombre de gènes (nGene) et nombre de UMI (nUMI) pour identifier les doublets de cellule et de valeurs aberrantes. N’importe quelle cellule avec un nombre de claire aberrantes de gènes, UMI ou le pourcentage de gènes mitochondriaux a été supprimé à l’aide de la fonction FilterCells de Seurat. Depuis Seurat utilise des composantes principales (PC) analyse scores aux cellules de grappes, déterminer PCs statistiquement significatifs à inclure est une étape cruciale. Parcelles de coude (Figure 4C) ont été utilisés pour la sélection de PC, dans lequel PCs au-delà du plateau de le « écart-type de PC » axe ont été exclus. La résolution de clustering a été également manipulée ce qui démontre que le nombre de clusters peut être modifié, allant de 0,4 (basse résolution conduisant à moins agrégats cellulaires, Figure 4D) à 4 (haute résolution conduisant à la plus élevées agrégats cellulaires, Figure 4e ). À basse résolution, il est probable que chaque cluster représente un type de cellule définie, tandis qu’à haute résolution, cela peut aussi représenter sous-types ou États transitoires d’une population de cellules. Dans ce cas, des paramètres de cluster basse résolution ont été utilisés pour analyser les heatmaps expression (à l’aide de la fonction DoHeatmap Seurat) pour identifier les gènes exprimés plus fortement dans un cluster donné (Figure 4f). Dans ce cas, les gènes plus fortement exprimés ont été identifiés en évaluant l’expression différentielle dans un cluster donné par rapport à tous les autres groupes combinés, démontrant que chaque grappe était particulièrement bien représenté par gènes définis. En outre, les gènes candidats individuels peuvent être visualisées tSNE parcelles à l’aide de la fonction FeaturePlot de Seurat (Figure 4 g). Ceci permettait à déchiffrer s’il existait des clusters qui représentaient des macrophages. À l’aide de FeaturePlot, nous avons constaté que les deux cluster 2 et 4 exprimaient Cd68 - un marqueur de pan-macrophage.
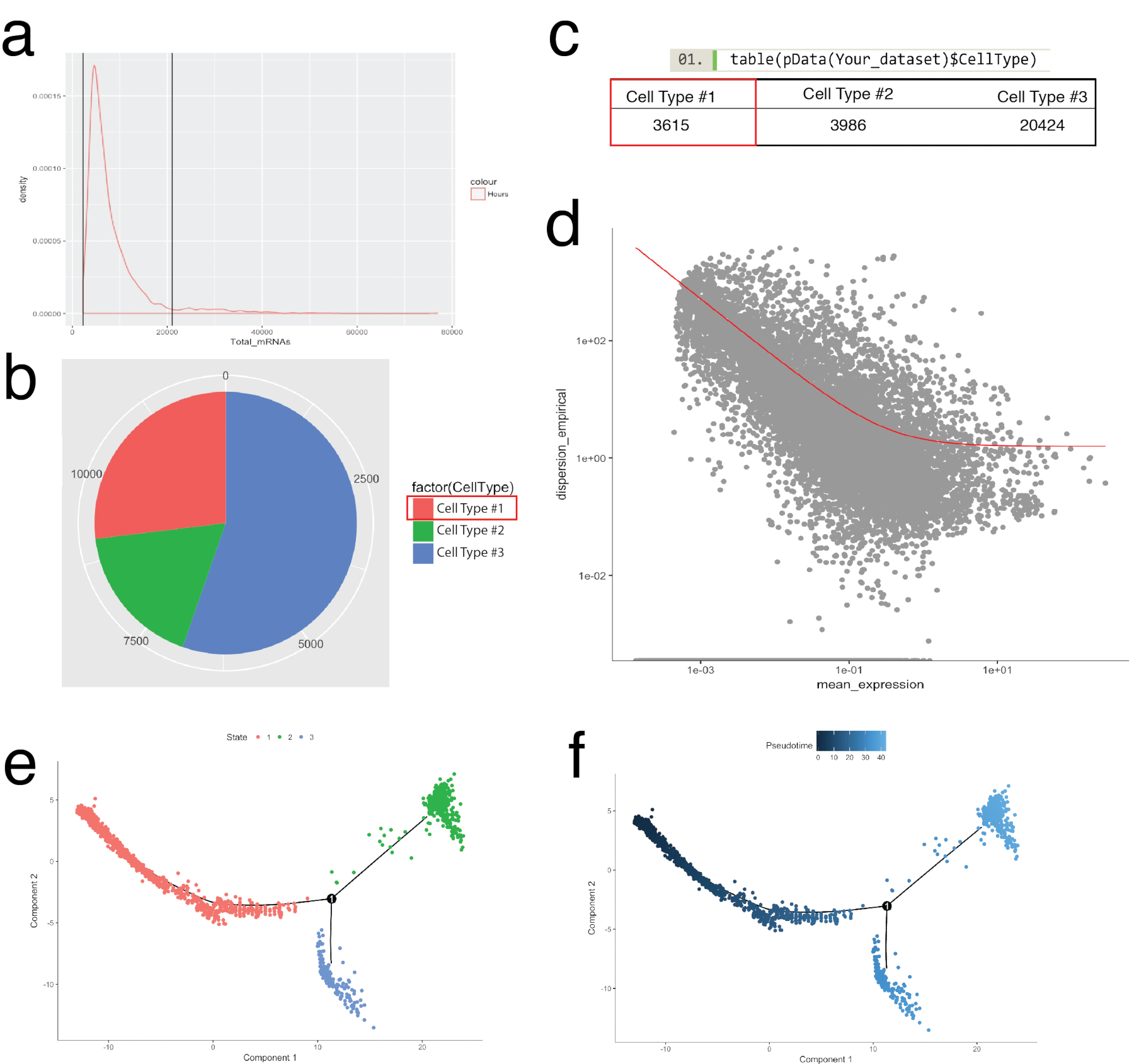
Le paquet de Monocle servait pour corroborer des agrégats cellulaires identifiés à Seurat et créer des trajectoires de la cellule, ou une commande pseudotemporal, à récapituler les processus biologiques (Figure 5). Commande pseudotemporal peut être utilisé pour les échantillons où les profils d’expression unicellulaires sont censées suivre une évolution biologique. Les cellules peuvent être commandés selon un continuum pseudotemporal pour résoudre les États intermédiaires, des points de bifurcation des deux destins cellulaires alternatifs et identifient les signatures de gène qui sous-tendent l’acquisition de chaque sort. Tout d’abord, semblable à la filtration de Seurat, cellules de mauvaise qualité ont été retirés de telle sorte que la répartition des ARNm entre toutes les cellules était log normale et est tombé entre les limites supérieures et inférieures, tel qu’indiqué dans la Figure 5 a. Ensuite, les cellules individuelles en utilisant la fonction newCellTypeHierarchy de Monocle, ont été classés et compté à l’aide de gènes marqueurs de la lignée connue (Figure 5 b, 5C). Par exemple, les cellules exprimant le PDGF récepteurs alpha ou fibroblastes spécifiques Protein 1 ont été assignés à cellule Type #1 pour créer un critère de définition des fibroblastes. Ensuite, cette population (cellule Type #1) a été évaluée pour déchiffrer les trajectoires des fibroblastes. Pour ce faire, fonction de GeneTest différentielle de Monocle a été utilisée, qui comparé les cellules représentant les États extrêmes au sein de la population et trouvé les gènes différentiels pour commander les cellules restantes dans la population (Figure 5 d). En appliquant des méthodes d’apprentissage multiples (un type de réduction dimensionnelle non-linéaire) dans l’ensemble de toutes les cellules, une coordonnée le long d’un chemin de pseudotemporal a été affectée. Cette trajectoire a été visualisée ensuite par la cellule état (Figure 5e) et pseudotime (Figure 5f).

Figure 1 : organigramme. Étapes de préparation animale à analyser seule cellule RNA-Seq datasets à la présentation finale datasets vers un référentiel accessible au public. Billes de gel en émulsion (GEMs) se référer aux billes avec des oligonucléotides avec code à barres qui encapsulent des milliers de cellules individuelles. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
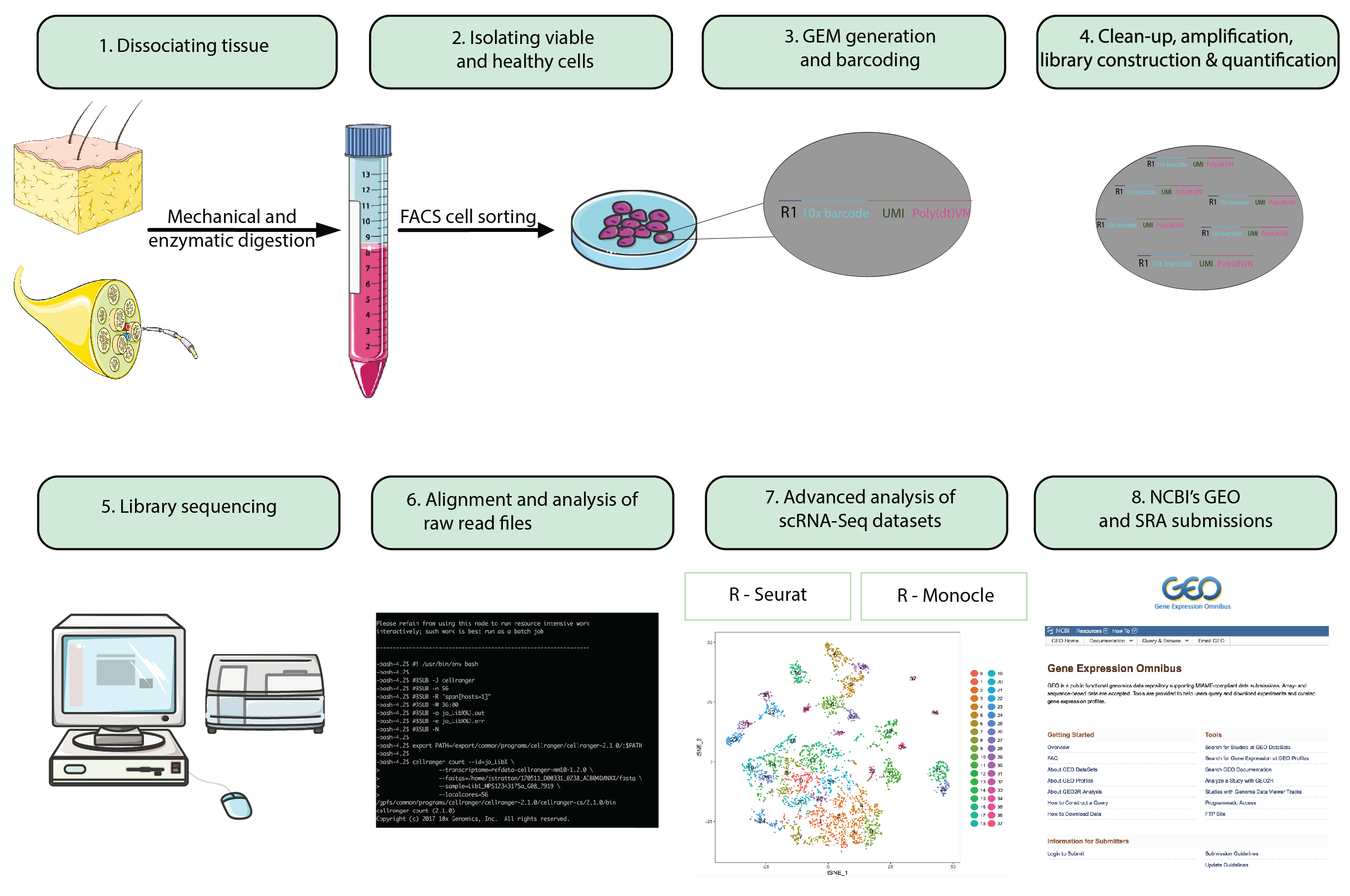{kind=link}

Figure 2 : création de suspension monocellulaire viable du tissu nerveux. (a) dessin animé vue d’ensemble des contrôles de qualité. (b) les cellules et les débris de cellules encore incorporés dans les débris (flèches rouges). (c) cellules libérés de débris (flèches rouges). d cellule isolation par FACS. P0 : fraction de débris ; P1 : fraction de cellules ; P3 : exclusion des duolets ; P4 : fraction de négatif viabilité colorant (Sytox Orange). e aucun contrôle de colorant de viabilité. (f) image de P0 fraction représentant isolé des débris. (g) l’image de P4 fraction représentant isolé des cellules viables (flèches rouges). (b) (c) (f) et (g) avait nucléaire colorant ajouté 20 minutes avant l’imagerie. Barreaux de l’échelle : 80 µm. s’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
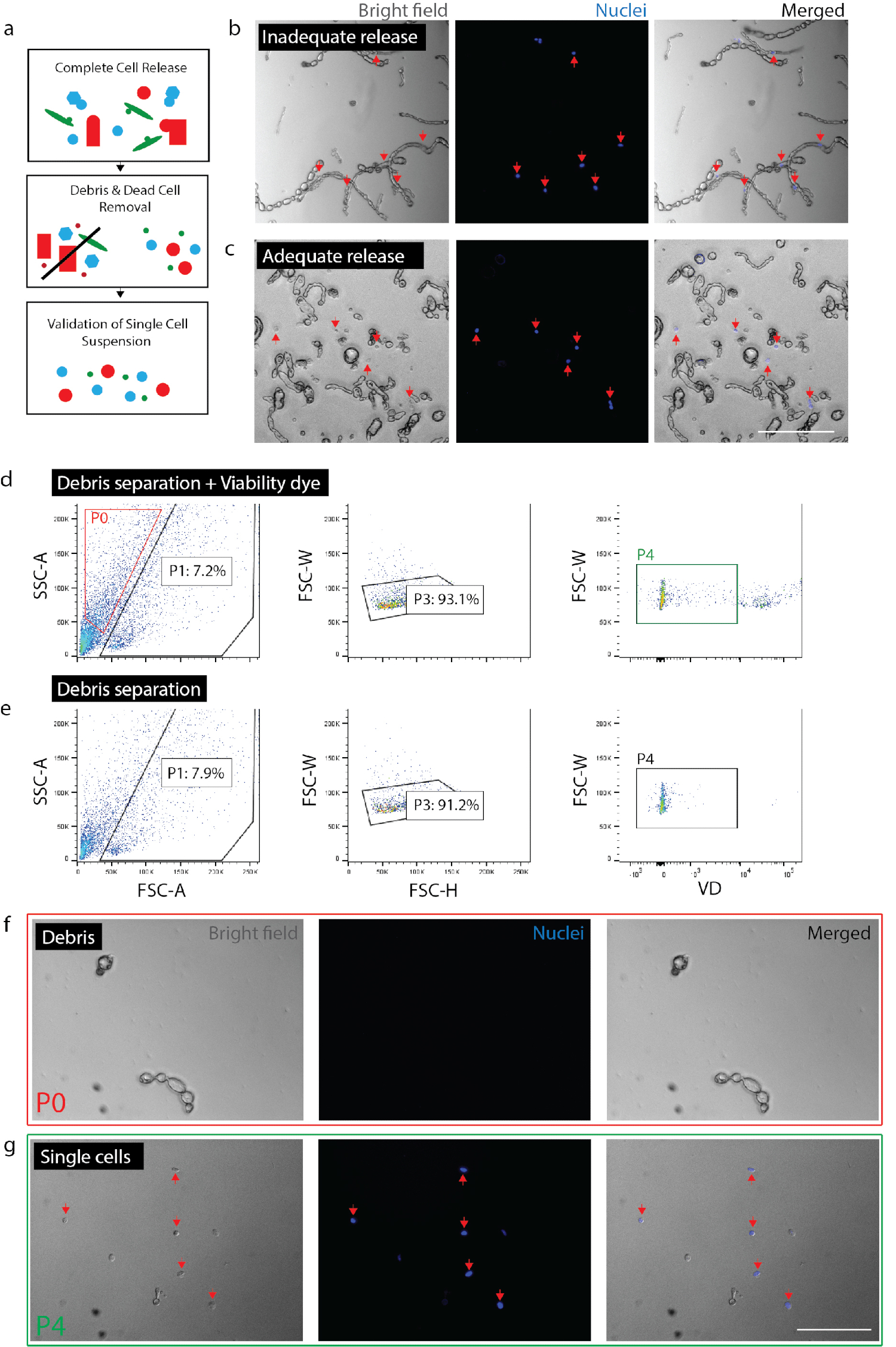{kind=link}

Figure 3 : séquençage peu profond prédit le nombre de cellules récupérées dans 10 X les échantillons traités. un exemple (échantillon 1.6) du csv générés par MiSeq liste cellule codes-barres et ses correspondant UMI compte tel que déterminé par lectures mappés en toute confiance. (b) code à barres rang tracé pour échantillon 1.6 montre une baisse significative au comte UMI en fonction de la cellule des codes à barres. Les lignes en pointillés et solides représentent le niveau de la coupure entre les cellules et fond tel que déterminé par inspection visuelle. (c) codes à barres cellules observées à l’aide de la cellule Ranger pipeline post-HiSeq révèle peu profond séquencement approximée avec précision le nombre de cellules pour échantillon 1.6. (d) un exemple d’une mise en place de flux-cellule basée sur le séquençage peu profond dérivé estimations de la cellule. Pour échantillon 1.6, car peu profond séquencement prédit 3480 cellules, 1,17 voies ont été assignés pour assurer > 100 000 lectures par la couverture de séquençage de cellule en HiSeq. Remarque : Toutes les voies doivent ajouter à 100 %. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
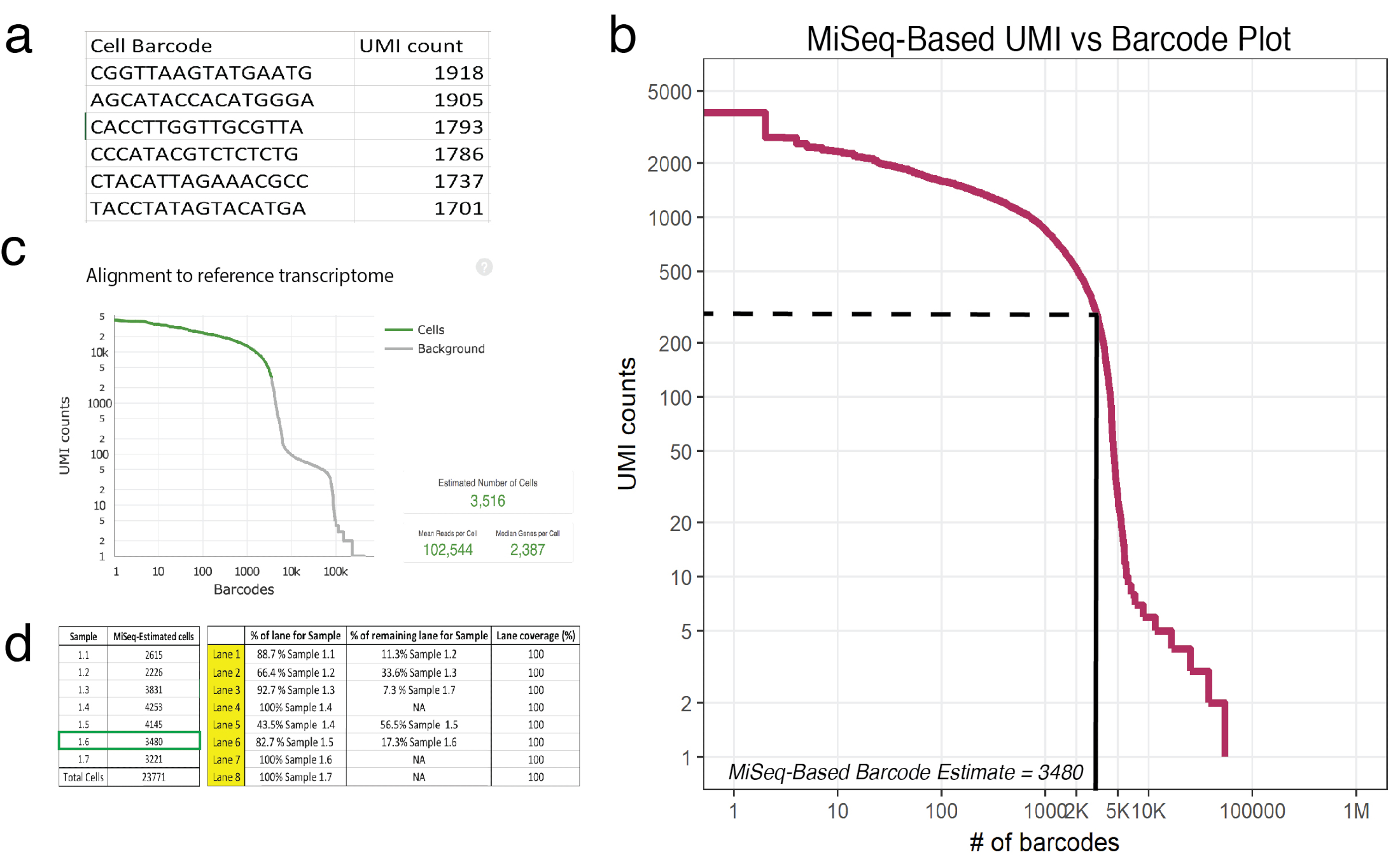{kind=link}

Figure 4 : contrôle de la qualité et de la bioinformatique des unicellulaires RNA-Seq dataset à l’aide de progiciel Seurat R. b les parcelles de paramètres de contrôle de la qualité incluent le nombre de gènes, nombre d’identificateurs moléculaires uniques (UMIs) et le pourcentage des transcriptions mappage sur le génome mitochondrial. b gène de l’échantillon parcelles detecter des cellules avec des niveaux déviants des transcriptions mitochondriales et UMIs. (c) coude placette utilisé pour ad hoc détermination du PCs statistiquement significatives. Les lignes en pointillés et discontinue dot représentent le seuil où un « coude » net apparaît dans le graphique. Dimensions de PC avant ce coude sont incluses dans l’analyse en aval. (d, e) Amas de cellule basée sur les graphiques visualisés à deux résolutions différentes dans un espace de faibles dimensions à l’aide d’un tracé tSNE. (f) top gènes marqueurs (jaunes) pour chaque cluster visualisées sur un heatmap d’expression à l’aide de la fonction DoHeatmap de Seurat. (g) visualiser l’expression du marqueur de, par exemple, les gènes Cd68 représentant des macrophages (violets) à l’aide de la fonction FeaturePlot de Seurat. Ceci suggère que ce cluster 2 et 4 (en panneau d) de ce groupe de données représente des macrophages. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}

Figure 5 : cellule de catégorisation et ordonnant le long de la trajectoire de peudotemporal à l’aide de Monocle toolkit. b inspecter la distribution de l’ARNm (déduite des comtes UMI) dans l’ensemble de toutes les cellules dans un échantillon. Seulement les cellules avec l’ARNm entre 0 - ~ 20 000 ont été utilisées pour l’analyse en aval. (b, c) L’assignation et comptage des types cellulaires issus des marqueurs de cellules de lignée connue. Par exemple, les cellules exprimant le PDGF récepteurs alpha ou fibroblastes spécifiques Protein 1 ont été assignés à la cellule Type #1 représentant pan-fibroblastes en utilisant la fonction newCellTypeHierarchy de Monocle. Nombre de différents types de cellules peut être visualisée sous forme de diagramme circulaire (b) et sous forme de tableau (c). (d) à l’aide de cellules Type #1 (fibroblastes) à titre d’exemple, les gènes utilisés pour commander les cellules peuvent être visualisées à l’aide d’un diagramme de dispersion qui montre la dispersion des gènes vs expression moyenne. La courbe rouge montre la coupure pour gènes utilisées pour le classement calculé par le modèle moyenne-variance à l’aide de la fonction estimateDispersions de Monocle. Les gènes qui répondent à ce seuil de décision ont été utilisés pour la commande pseudotime en aval. (e, f) Visualisation des trajectoires de cellule dans un espace réduit bidimensionnel colorée par « État » de la cellule (e) et attribué Monocle « Pseudotime » (f). S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}
Discussion
Ce protocole montre comment la préparation adéquate des cellules simples peut découvrir l’hétérogénéité transcriptionnelle de milliers de cellules individuelles et discriminer les États fonctionnels ou identités cellulaires uniques au sein d’un tissu. Le protocole n’exige pas de protéines fluorescentes journaliste ou outils transgéniques et peut être appliqué à l’isolement de cellules isolées de divers tissus d’intérêt, y compris ceux de l’homme ; gardant à l’esprit de chaque tissu est unique et ce protocole exigera une certaine adaptation ou la modification.
Les programmes de transcriptional diversifiés et très dynamiques au sein de cellules ont souligné la valeur de la cellule unique génomique. En dehors d’isoler l’ARN de haute qualité, une étape de préparation d’échantillon critique nécessaire pour les ensembles de données de haute qualité est de s’assurer que les cellules sont complètement libérés du tissu et que les cellules sont sains et intacts. C’est relativement simple pour prélever des cellules qui sont facilement mises, comme les cellules circulantes ou dans les tissus où les cellules sont plus ou moins conservés, comme dans les tissus lymphoïdes. Mais cela peut être difficile pour les autres tissus adultes, en raison de l’architecture cellulaire hautement développée, s’étendant sur grandes distances, entourant la matrice extracellulaire et les protéines du cytosquelette souvent rigides impliqués dans le maintien de la structure cellulaire. Même avec des techniques de dissociation appropriées pour la libération totale des cellules, il est possible que le traitement souvent long et rigoureux nécessaire modifierait intégrité de qualité et de la cellule de mRNA. En outre, les hautes températures utilisées pour la dissociation assistée par enzyme affectent également la transcription signatures29,30. Le but du protocole est de présenter le contrôle de la qualité vérifie, à l’aide de tissus tels que le nerf adult myélinisée et la peau adulte riches en matrice extracellulaire pour démontrer comment l’optimisation peut aider à surmonter ces obstacles.
Une considération importante lors de la conception de toute expérience de scRNA-Seq est le choix de la profondeur de séquençage. Séquençage peut être hautement multiplexé et lu profondeur peut varier d’une très faible à l’aide de Drop-Seq2 et jusqu'à 5 millions de lectures/cellule14 en utilisant une méthode de RNA-Seq longue tels que Smart-suiv. Plupart des expériences de scRNA-Seq peuvent détecter des transcriptions de modérée à forte expression avec le séquençage aussi bas que 10 000 lectures/cellule, qui est généralement suffisant pour cellule type classification41,,42. Profondeur de séquençage peu profonde est précieux pour économiser sur les coûts de séquençage en essayant de détecter des populations cellulaires rares à travers les tissus complexes où des milliers de cellules peuvent être nécessaire d’attribuer avec certitude les populations rares. Mais le séquençage de faible profondeur n’est pas suffisant lorsque des informations détaillées sur l’expression des gènes et des processus associées aux signatures de transcriptionnelles subtiles sont nécessaires. Actuellement, on estime que la grande majorité des gènes dans une cellule est détectée avec 500 000 lectures/cellule, mais cela peut varier selon le protocole et le tissu type43,44. Tandis que la transcription du séquençage contourne la nécessité pour l’Assemblée et peut, par conséquent, détecter roman ou épissure rares variantes, coûts de séquençage limitent souvent mise à l’échelle de telles approches afin d’examiner des milliers de cellules comprenant un système complexe de tissus. En revanche, 3' tag libraries unicellulaires tels que ceux décrits dans le présent protocole en général ont plus faible complexité et requièrent moins profond séquencement. Il est important de noter que bibliothèques générés à l’aide du protocole décrit peuvent être séquencés sur l’un des cinq séquenceurs supportés : 1) NovaSeq, 2) HiSeq 3000/4000, 3) HiSeq 2500 course rapide et High Output, 4) NextSeq 500/550 et MiSeq 5).
Une approche alternative à cellule unique RNA-Seq, qui réduit la nécessité pour les tissus délicats et cellule gère encore conserve certains des avantages d’une seule cellule RNA-Seq, est l’analyse de l’ARN de noyaux unique45. Cette approche permet un traitement plus rapid, réduire la dégradation de l’ARN et plus des mesures extrêmes pour assurer une diffusion adéquate des noyaux et donc probablement permet une capture plus confiante des profils transcriptionnels qui représente toutes les cellules dans un tissu donné. Ceci, bien sûr, seulement fournirait une partie de l’activité transcriptionnelle présente dans une cellule donnée, donc selon quels objectifs expérimentaux sont d’intérêt de cette approche peut ou peut ne pas convenir.
Sans compter que la caractérisation complète des identités cellulaires au sein d’un tissu donné, une des analyses plus précieux pour les ensembles de données scRNA-Seq est l’évaluation des États intermédiaires de transcriptional parmi les populations cellulaires « définie ». Ces États intermédiaires peuvent donner un aperçu des relations de lignée entre cellules au sein de populations identifiées, ce qui n’était pas possible avec la traditionnelle en vrac que RNA-Seq s’approche. Plusieurs outils bioinformatiques de scRNA-Seq ont maintenant été développés afin d’élucider ce. Ces outils peuvent évaluer les processus impliqués dans, par exemple, les cellules cancéreuses la transition vers un état oncogènes ou métastatique, venant à échéance dans divers destins terminales ou navette entre États quiescentes et actives les cellules immunitaires des cellules souches. Transcriptome de subtiles différences dans les cellules peuvent également être indicatives des biais de lignage, bioinformatique récemment mis au point des outils tels que FateID, peut déduire47. Étant donné que les distinctions entre les cellules de transition peuvent être difficiles à déterminer compte tenu des différences de transcriptionnelles peut être subtile, séquençage plus profond peut être nécessaire de46. Heureusement, la couverture d’une bibliothèque très superficiellement séquencée peut être augmentée si vous êtes intéressé dans la détection de l’ensemble de données plus loin en exécutant à nouveau la bibliothèque sur une autre cellule de flux.
Globalement, ce protocole prévoit un flux de travail facile-à-adaptation qui permet aux utilisateurs de profil transcriptionnellement des centaines de milliers de cellules au sein d’une expérience unique. La qualité finale d’un dataset scRNA-Seq repose sur l’isolement cellulaire optimisée, la cytométrie en flux, cDNA bibliothèque génération et interprétation des matrices de gène-code à barres brutes. À cette fin, le présent protocole donne un aperçu détaillé de toutes les étapes clés qui peuvent être facilement modifiées pour permettre des études de types de tissus différents.
Déclarations de divulgation
Aucune divulgation
Remerciements
Nous reconnaissons que le personnel de soutien à l’installation de Services de UCDNA, ainsi que le personnel de l’établissement animalier à l’Université de Calgary. Nous remercions Matt Workentine pour son soutien de bio-informatique et Jens Durruthy pour son soutien technique. Ce travail a été financé par une subvention des IRSC (R.M. et J.B.), une bourse de nouveau chercheur des IRSC à J.B. et l’Alberta Children Health Research Institute Fellowship (J.S.).
matériels
| Name | Company | Catalog Number | Comments |
| Products | |||
| RNAse out | Biosciences | 786-70 | |
| Pentobarbital sodium | Euthanyl | 50mg/kg | |
| HBSS | Gibco | 14175-095 | |
| Dispase 5U/ml | StemCell Technologies | 7913 | 5 mg/ml |
| Collagenase-4 125 CDU/mg | Sigma-Aldrich | C5138 | 2 mg/ml |
| DNAse | Sigma-Aldrich | DN25 | 10mg/ml |
| BSA | Sigma-Aldrich | A7906 | |
| 15 ml Narrow bottom tube VWR® High-Performance Centrifuge Tubes | VWR | 89039-666 | |
| Sytox Orange Viability Dye | Molecular Probes | 11320972 | 1.3 nM/µl |
| Nuc Blue Live ReadyProbes | Invitrogen | R37605 | |
| Agilent 2100 Bioanalyzer High senitivity DNA Reagents | Agilent | 5067-4626 | |
| Kapa DNA Quantification Kit | Kapa Biosystems | KK4844 | |
| Chromium Single Cell 3' reagents | 10x Genomics | ||
| Equipment | |||
| BD FACSAria III | BD Biosciences | ||
| Agilent 2100 Bioanalyzer Platform | Agilent | ||
| Illumina® HiSeq 4000 | Illumina | ||
| Illumina® MiSeq SR50 | Illumina | ||
| 10X Controller + accessories | 10x Genomics | ||
| Software | |||
| The Cell Ranger | 10x GENOMICS | support.10xgenomics.com/single-cell-gene-expression/software/overview/welcome | |
| Loupe Cell Browser | 10x GENOMICS | support.10xgenomics.com/single-cell-gene-expression/software/downloads/latest | |
| R | https://anaconda.org/r/r |
Références
- Shalek, A. K., et al. Single-cell RNA-seq reveals dynamic paracrine control for cellular variation. Nature. 510, 363-369 (2014).
- Macosko, E. Z., et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell. 161, 1202-1214 (2015).
- Zappia, L., Phipson, B., Oshlack, A. Exploring the single-cell RNA-seq analysis landscape with the scRNA-tools database. bioRxiv:206573. , (2018).
- Dulken, B. W., Leeman, D. S., Boutet, S. C., Hebestreit, K., Brunet, A. Single cell transcriptomic analysis defines heterogeneity and transcriptional dynamics in the adult neural stem cell lineage. Cell Reports. 18 (3), 777-790 (2017).
- Llorens-Bobadilla, E., et al. Single-Cell Transcriptomics Reveals a Population of Dormant Neural Stem Cells that Become Activated upon Brain Injury. Cell Stem Cell. 17 (3), 329-340 (2015).
- Trapnell, C., et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nature Biotechnology. 32, 381-386 (2014).
- Aibar, S., et al. SCENIC: single-cell regulatory network inference and clustering. Nature Methods. 14, 1083-1086 (2017).
- Mayer, C., et al. Developmental diversification of cortical inhibitory interneurons. Nature. 555 (7697), 457-462 (2018).
- Stratton, J. A., et al. Purification and Characterization of Schwann Cells from Adult Human Skin and Nerve. eNeuro. 4 (3), (2017).
- Biernaskie, J. A., McKenzie, I. A., Toma, J. G., Miller, F. D. Isolation of skin-derived precursors (SKPs) and differentiation and enrichment of their Schwann cell progeny. Nature Protocols. 1 (6), 2803-2812 (2007).
- 10X Genomics. User Guides. , Available from: https://www.10xgenomics.com/resources/user-guides/ (2018).
- 10X Genomics. Chromium Single Cell 3' Training Module. , Available from: http://go.10xgenomics.com/training-modules/single-cell-gene-expression (2018).
- Agilent. , Available from: https://www.agilent.com/en-us/library/usermanuals?N=135 (2018).
- Kolodziejczyk, A. A. Single Cell RNA-Sequencing of Pluripotent States Unlocks Modular Transcriptional Variation. Cell Stem Cell. 17, 471-485 (2015).
- Jaitin, D. A., et al. Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types. Science. 343, 776-779 (2014).
- Pollen, A. A., et al. Low-coverage single-cell mRNA sequencing reveals cellular heterogeneity and activated signaling pathways in developing cerebral cortex. Nature Biotechnology. 32, 1053-1058 (2014).
- 10X Genomics. Sequencing Requirements for Single Cell 3'. , Available from: https://support.10xgenomics.com/single-cell-gene-expression/sequencing/doc/specifications-sequencing-requirements-for-single-cell-3 (2018).
- Datacamp. Introduction to R. , Available from: https://www.datacamp.com/courses/free-introduction-to-r (2018).
- Droplet-based, high-throughput single cell transcriptional analysis of adult mouse tissue using 10X Genomics#39; Chromium Single Cell 3' (v2) system: From tissue preparation to bioinformatic analysis. , Available from: https://figshare.com/s/97b83e649e5eefd01357 (2018).
- 10X Genomics. User Guides. , Available from: https://www.10xgenomics.com/resources/user-guides/ (2018).
- UNIX Tutorial for Beginners. , Available from: http://www.ee.surrey.ac.uk/Teaching/Unix/ (2018).
- 10X Genomics. Creating a Reference Package with cellranger mkref. , Available from: https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/advanced/references (2018).
- 10X Genomics. System Requirements. , Available from: https://support.10xgenomics.com/single-cell-gene-expression/software/overview/system-requirements (2018).
- 10X Genomics. Software Downloads. , Available from: https://support.10xgenomics.com/single-cell-gene-expression/software/downloads/latest (2018).
- 10X Genomics. Aggregating Multiple Libraries with cellranger aggr. , Available from: https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/using/aggregate#depth_normalization (2018).
- 10X Genomics. Loupe Cell Browser Gene Expression Tutorial. , Available from: https://support.10xgenomics.com/single-cell-gene-expression/software/visualization/latest/tutorial (2018).
- scRNA-tools. A table of tools for the analysis of single-cell RNA-seq data. , Available from: https://www.scrna-tools.org/ (2018).
- Conda. Downloading conda. , Available from: https://conda.io/docs/user-guide/install/download.html (2018).
- Anaconda. r / packages / r 3.5.1. , Available from: https://anaconda.org/r/r (2018).
- Droplet-based, high-throughput single cell transcriptional analysis of adult mouse tissue using 10X Genomics' Chromium Single Cell 3' (v2) system: From tissue preparation to bioinformatic analysis. , Available from: https://figshare.com/s/97b83e649e5eefd01357 (2018).
- Satija Lab. Seurat - Guided Clustering Tutorial. , https://satijalab.org/seurat/pbmc3k_tutorial.html (2018).
- Droplet-based, high-throughput single cell transcriptional analysis of adult mouse tissue using 10X Genomics' Chromium Single Cell 3' (v2) system: From tissue preparation to bioinformatic analysis. , Available from: https://figshare.com/s/97b83e649e5eefd01357 (2018).
- Monocle. , Available from: http://cole-trapnell-lab.github.io/monocle-release/docs/#constructing-single-cell-trajectories (2018).
- Github. An R package to test for batch effects in high-dimensional single-cell RNA sequencing data. , (2018).
- Edgar, R. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Research. 30, 207-210 (2002).
- Leinonen, R., Sugawara, H., Shumway, M. The sequence read archive. Nucleic Acids Research. 39, D19-D21 (2011).
- NIH. GenBank Submission Portal Wizards. , Available from: https://www.ncbi.nlm.nih.gov/account/register/?back_url=/geo/submitter/ (2018).
- NIH. Submitting data. , Available from: https://submit.ncbi.nlm.nih.gov/geo/submission/ (2018).
- Shah, P. T., et al. Single-Cell Transcriptomics and Fate Mapping of Ependymal Cells Reveals an Absence of Neural Stem Cell Function. Cell. 173, 1045-1057 (2018).
- Anon, Method of the Year 2013. Nature Methods. 11, 1(2013).
- Adam, M., Potter, A. S., Potter, S. S. Psychrophilic proteases dramatically reduce single-cell RNA-seq artifacts: a molecular atlas of kidney development. Development. 144, 3625-3632 (2017).
- Wu, Y. E., Pan, L., Zuo, Y., Li, X., Hong, W. Detecting activated cell populations using single-cell RNA-seq. Neuron. 96, 313-329 (2017).
- Zeigenhain, C., et al. Comparative Analysis of Single-Cell RNA Sequencing Methods. Molecular Cell. 65 (4), 631-643 (2017).
- Wu, A. R., et al. Quantitative assessment of single-cell RNA-sequencing methods. Nature Methods. 11 (1), 41-46 (2014).
- Habib, N., et al. Div-Seq: Single-nucleus RNA-Seq reveals dynamics of rare adult newborn neurons. Science. 353 (6302), 925-928 (2016).
- Janes, K. A. Single-cell states versus single-cell atlases - two classes of heterogeneity that differ in meaning and method. Current Opinions in Biotechnology. 39, 120-125 (2016).
- Herman, J. S., Sagar,, Grün, D. FateID infers cell fate bias in multipotent progenitors from single-cell RNA-seq data. Nature Methods. 15 (5), 379-386 (2018).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationExplorer plus d’articles
This article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.