
Method Article
Bestimmende 3'-Termini und Sequenzen der im Entstehen begriffenen einsträngige virale DNA-Moleküle während HIV-1 Reverse Transkription in infizierten Zellen
In diesem Artikel
Zusammenfassung
Hier präsentieren wir einen Tiefe Sequenzierung Ansatz, der eine objektive Bestimmung der im Entstehen begriffenen 3'-Termini sowie mutagenen Profile der einzelsträngigen DNA-Moleküle enthält. Die Hauptanwendung ist die Charakterisierung der im Entstehen begriffenen retrovirale ergänzende DNAs (cDNAs), die Zwischenprodukte, die während des Prozesses der retroviralen reversen Transkription generiert.
Zusammenfassung
Überwachung von Nukleinsäure-Zwischenprodukte während der Replikation des Virus gibt Einblicke in die Wirkungen und Wirkmechanismen von antiviralen Substanzen und Host Zellproteinen auf virale DNA-Synthese. Hier gehen wir auf das Fehlen eines Zell-basierte, reichweitenstarken und hochauflösende Assays, die retrovirale reversen Transkription Zwischenprodukte im physiologischen Rahmen einer Virusinfektion zu definieren vermag. Das beschriebene Verfahren erfasst die 3'-Termini, der im Entstehen begriffenen komplementäre DNA (cDNA) Moleküle in HIV-1 infizierten Zellen mit Einzel-Nukleotid-Auflösung. Das Protokoll beinhaltet Ernte der ganzen Zelle DNA, gezielte Anreicherung der viralen DNA über Hybrid Capture, Adapter Ligation, Größe Fraktionierung von Gel Reinigung, PCR-Amplifikation, Tiefe Sequenzierung und Datenanalyse. Ein wichtiger Schritt ist die effiziente und unvoreingenommene Ligatur der Adapter Moleküle, 3'-DNA Termini zu öffnen. Anwendung des beschriebenen Verfahrens bestimmt die Fülle der umgekehrte Abschriften jeder bestimmten Länge in einer Probe. Darüber hinaus Informationen über die (interne) Sequenz Variation in umgekehrter Transkripte und damit jede möglichen Mutationen. Der Test eignet sich im Allgemeinen für alle Fragen rund um DNA-3'-Erweiterung, unter der Voraussetzung, dass die Vorlage-Sequenz bekannt ist.
Einleitung
Um zu sezieren und zu verstehen, dass die virale Replikation vollständig, zunehmend Techniken verfeinert, die Replikation zu erfassen sind Zwischenprodukte erforderlich. Insbesondere kann die genaue Definition der viralen Nukleinsäure-Arten im Rahmen der infizierten Zellen liefern neue Erkenntnisse, da viele virale Replikationsmechanismen bisher wurde in isolierten in Vitro Reaktionen untersucht. Ein Paradebeispiel ist der Prozess der reversen Transkription in Retroviren, wie humane Immundefizienz-Virus (HIV-1) 1. Die verschiedenen Schritte der reversen Transkription HIV-1, bei denen das virale Enzym Reverse Transkriptase (RT) die einsträngige RNA-Genom in doppelsträngige DNA kopiert wurden in erster Linie im Primer Extension Assays mit gereinigten Proteinen und Nukleinsäuren Säuren,1,2,3,4,5. Während grundlegende Prinzipien hergestellt wurden, solche Tests nicht zu übernehmen alle virale und zelluläre Komponenten und nicht unbedingt biologisch relevante Zusammensetzungen der beteiligten Faktoren. Daher haben wir eine leistungsstarke Technik, die Spektren der reversen Transkription Zwischenprodukten mit ihren exakten cDNA 3'-Termini (d. h. Ermittlung ihrer genauen Längen) und Nukleotidsequenzen im Zusammenhang mit Infektionen des Lebens bestimmen entwickelt Zellen-6. Sammlung von Daten aus Zeit Kurs Experimente genutzt werden, um das Profil der Transkripte unter verschiedenen Bedingungen, wie z. B. das Vorhandensein von antiviralen Moleküle oder Proteine, die Einfluss auf die Effizienz und Processivity der DNA-Synthese zu vergleichen und Akkumulation. Dies ermöglicht ein genaueres Verständnis des natürlichen Erreger Lebenszyklus, die oft die Basis für gezielte Drug Design und erfolgreiche therapeutische Intervention.
HIV-1 reverse Transkription umfasst eine Reihe von aufeinander folgenden Veranstaltungen initiiert durch Tempern eine tRNA-Grundierung, die genomische RNA-Vorlage, welche dann durch RT, ein kurzen einzelsträngigen cDNA-Protokoll namens eine Minus-Strang stark-Stop zu produzieren verlängert wird (-Sss) (siehe ( Abbildung 1). Anschließend wird die cDNA - Sss vom 5'-Long Terminal wiederholen (LTR), die 3' LTR der genomischen RNA, wo es glüht und dient als Grundierung für anhaltende RT Dehnung von Minus vermittelte DNA Strang übertragen (siehe Bewertungen auf reverse Transkription1 , 2 , 3 , ( 4). diese ersten Strang zählt die Bandbreitenbegrenzung Schritte der reversen Transkription; Daher ist die cDNA - Sss bekannt zu akkumulieren. Die Grundkonstruktion für Workflow und Bibliothek der reversen Transkription Produkte in infizierten Zellen zu erfassen ist in Abbildung 2adargestellt. Die spezifische Primer und Analyse, die Einstellungen, die in das Protokoll verwendet und in Tabelle 1 aufgeführten Zielen alle früh reverse Transkription cDNA Zwischenprodukte im Längenbereich von 23 bis ~ 650 nt, das 180-182-nt - Sss-DNA enthält. Jedoch werden geeignete geringfügige Anpassungen der Strategie ermöglichen Anwendung nicht nur spät reversen Transkription Produkte, aber auch andere Viren und Systeme, wo ist das Ziel, 3'-OH enthaltende DNA-enden zu erkennen. Wichtige Einschränkungen berücksichtigen umfassen die Länge des PCR-Produktes in der Bibliothek; insbesondere werden Vorlagen, in denen der Abstand zwischen den Adapter auf der offenen 3'-Terminus und die vorgelagerten Grundierung Website überschreiten ~ 1000 NT, wahrscheinlich weniger effizient sequenziert werden, potentiell Einführung technische Vorurteile während Bibliothek Vorbereitung (irreführend, siehe Diskussion für mehr Details und Anpassung Vorschläge).
Bisher konzentrierten gemeldeten Techniken für die systematische Ermittlung der 3'-Termini der Nukleinsäure-Stränge auf RNA, nicht DNA-Moleküle. Ein Beispiel ist 3' Rennen (schnelle Verstärkung der cDNA enden)7, die stützt sich auf die Polyadenylation der mRNA. Darüber hinaus wurden Adapter Ligatur-basierte Strategien beschäftigen RNA Ligases entwickelt, die RLM-Rennen (RNA-Ligase-vermittelten)8 oder Spitze (Ligatur-basierte Amplifikation der cDNA enden)9einbezogen. Es ist wichtig zu betonen, dass die Ligatur-basierte Erweiterungen empfindlich auf jede Bias durch die Ligatur Reaktion selbst eingeführt werden. Z. B. möglicherweise Ligatur je nach einer bestimmten Nukleotid in der 3'-Position, die Sequenz, total Molekül Länge oder lokale Struktur mehr oder weniger effizient. Solche Präferenzen Ligase führen zu unvollständigen Erfassung von Molekülen und falsche Angaben in der Anzeige, die wir und andere9,10beobachtet haben. Um während der Adapter zusätzlich Schritte im Protokoll beschriebenen Ligatur Voreingenommenheit zu minimieren, wir eine Reihe von Ligatur Strategien getestet und fand die Verwendung von T4 DNA-Ligase mit einer Haarnadel einzelsträngigen DNA-Adapter (wie von Kwok Et al.beschrieben. 11) das einzige Verfahren mit in der Nähe von quantitativen Ligatur, die nicht signifikante Unterschiede in der Ligatur Effizienz mit einem speziell ausgewählten Steuerelement Oligonukleotide6Beurteilung geführt hat. Die Wahl dieser Ligatur-Strategie ist daher ein wesentliches Merkmal für den Erfolg dieses Protokolls.
Bis heute, Überwachung der HIV-1-RT-Progression in infizierten Zellen in erster Linie erreicht wurde durch die Messung der reversen Transkription Produkte verschiedener Länge mit quantitativen PCR (qPCR) mit Primer-Sonden-Sets, die eindeutig kürzer oder länger zu messen (frühe und spät, beziehungsweise) cDNA Produkte12,13,14. Während dieser Ansatz qPCR intrinsischen Effizienz des Prozesses reverse Transkription in zellulären Systemen zu bestimmen ist, ist die Ausgabe der relativ geringen Auflösung ohne Reihenfolge Informationen abgeleitet wird. Unser neue Ansatz, basierend auf optimierten Adapter Ligation, PCR-vermittelten Bibliothek Generation und Tiefe Sequenzierung Adressen die Technologielücke und bietet die Möglichkeit, reverse Transkription während HIV-1 Infektion quantitativ und bei Einzel-Nukleotid zu überwachen Auflösung.
Wir haben den Nutzen dieser Methode in einer Studie, die unterschieden zwischen zwei vorgeschlagenen Modelle für die Kapazität des Faktors HIV-1 Einschränkung APOBEC3G (Apolipoprotein B mRNA editing Enzym katalytische Polypeptid-wie 3 G) zu stören illustriert, mit der Produktion von viralen reverse Transkripte6.
Protokoll
Hinweis: Lesen Sie bitte die Tabelle der Materialien für bestimmte Reagenzien und Geräte, die in diesem Protokoll verwendeten.
1. Virus Produktions- und Zelle Infektion
Achtung: Infektiöse HIV-1 sollte nur in zugelassenen Biosafety Containment Laboratorien gehandhabt werden.
Hinweis: Produktion von HIV-1-Partikeln durch transiente Transfektion von menschlichen embryonalen Niere (HEK) 293T Zellen, wie in beschrieben Schritt 1.1, ist ein standard-Verfahren und wurde zuvor beschriebenen15,16. Zuvor beschriebenen allgemeinen Zellkultur, die Verfahren sind17.
- Virus HIV-1-Produktion.
- Pflegen Sie 293T Zellen in Dulbecco den Eagle Medium (DMEM) ergänzt mit 10 % fetalen bovine Serum (FBS) und 1 % Penicillin/Streptomycin (volle DMEM) in eine Standardzelle Kultur Inkubator bei 37 ° C und 5 % CO2 wie oben beschrieben17geändert.
- Entfernen Sie in einem standard Laminar-Flow Gewebekultur Haube die Wachstumsmedien und 3 mL vorgewärmten (37 ° C) Trypsin in einer in der Nähe von Zusammenfluss 10 cm Zelle Kulturschale (~1.2 X 107 Zellen) von 293T Zellen. Setzen Sie die Schale wieder in den Inkubator für 2-3 min.
- Nimmt das Gericht aus dem Inkubator zurück in der Gewebekultur-Haube und 7 mL voll Medium. Pipette rauf und runter in die Schale mehrmals auf die Zellen aufzuwirbeln. Teilen Sie der Zellen 1:4 durch Zugabe von 2,5 mL Zellsuspension auf eine neue 10 cm Schüssel und füllen Sie es mit 7,5 mL voll Medium.
- Am nächsten Tag mischen Sie 10 µg der proviralen HIV-1-Plasmid DNA (z. B. pNL4.3) mit 1 mL serumfreien minimal wesentliche Medium und fügen Sie Polyethylenimine (PEI) Lösung (25.000 mW, 1 mg/mL pH 7) 4,5 µL pro 1 µg DNA. Die 293T Zellen tropfenweise hinzufügen und 10 min bei RT inkubieren.
- 24 h nach Transfektion, das Medium zu entfernen und ersetzen Sie es mit 6 mL volle DMEM mit RNase-freie DNase bei 20 U/mL Medium. Ersetzen Sie nach 6 h das Medium mit 10 mL der vollen DMEM.
- 48 h nach Transfektion, den Überstand zu ernten und durch einen 0,22 µm-Filter, mit einer 10 mL Spritze, in ein 15 mL Polypropylen Röhrchen zu filtern.
- Einem offenen dünnwandigen Ultrazentrifugen Rohr 2 mL steril 20 % Saccharose in 1 x Phosphat gepufferte Kochsalzlösung (PBS) hinzufügen. Überlagern Sie langsam die Saccharose mit der gefilterten Zelle überstand.
- Zentrifuge für 1 h 15 min bei 134.000 x g bei 4 ° C mit einer Ultrazentrifuge.
- Entfernen Sie vorsichtig die Röhren aus der Ultrazentrifuge. Langsam ausziehen der Überstand und Saccharose mit einer Absaugung oder einer Pipette. Verwenden Sie eine kleinere Pipette und kippen Sie das Rohr zu, wenn die letzte Saccharoselösung herausnehmen. Lassen Sie das gebeizte Virus in den Boden des Röhrchens.
Hinweis: Das Diabolo wird nicht sichtbar sein. - Fügen Sie 200 µL 1 X PBS, lassen in den Kühlschrank für 4 bis 12 h, Aufschwemmen und Einfrieren in 20 µL-Aliquots bei-80 ° C.
- Bestimmen, p24Gag Inhalte über die eine HIV-1-Antigen p24 ELISA kit (folgende Angaben des Herstellers).
- T-Zell-Linie-Infektion.
- Kultur eine immortalisierte T-Zell-Linie (zB., CEM-SS-Zellen) in Roswell Park Memorial Institute (RPMI) 1640 Medium mit 10 % FBS und 1 % Penicillin/Streptomycin (volle RPMI) ergänzt. Zählen Sie die Zellen mit einem Hemocytometer18 und Samen 1 gut pro Probe mit 1 mL voll RPMI mit 2 x 106 Zellen/mL in einer 12-Well-Format-Zelle-Kultur-Platte.
- Hinzufügen von HIV-1-Teilchen entspricht 150 ng p24Gag und Stelle die Platte in ein Schwingen Zentrifugieren Eimer mit Deckel Biocontainment Spin infizieren-durch Zentrifugieren in einem Benchtop-Zentrifuge für 2 h bei 2.000 x g bei 30 ° C.
- Platte aus der Zentrifuge zu entfernen und in einem standard Gewebekultur Inkubator bei 37 ° C und 5 % CO21 h ruhen lassen.
- Zum Abwaschen Eingabe Virus sammeln Sie Zellen durch Übertragung der Zellsuspensionen für Mikrozentrifugenröhrchen und in einem Microcentrifuge bei RT (RT) bei 500 X g für 2 min zentrifugieren. Der Überstand ohne zu stören die Zelle Pellet ausziehen.
- Aufschwemmen der Zelle Pellets in 1 mL vorgewärmten (37 ° C), steril 1 X PBS. Wiederholen Sie das Zentrifugieren, überstand entfernen und Wiederfreisetzung Schritte zwei weitere Male.
- Erneut Zentrifugieren Sie, entfernen Sie überstand zu und Aufschwemmen Sie der Zelle Pellets in 1 mL voll RPMI. Fügen Sie jede Aussetzung zu einem Brunnen in einem neuen 12-well-Platte.
- 6 h nach dem ersten Zusatz der Virus (4 h nach Zentrifugation) ernten Sie die Zellen durch Zentrifugation wie in Schritt 1.2.4 getan. Entfernen und entsorgen des Überstands. Die Zelle-Pellets können bei-80 ° C eingefroren oder verarbeitet direkt für DNA-Extraktion.
(2) DNA-Extraktion, HIV-1 DNA Quantifizierung und Anreicherung von Hybrid Capture
- Extrahieren Sie ganze Zelle DNA mit den Blut- und Gewebeproben total DNA-Extraktion Kit durch Anschluss an das Kit-Handbuch für Gewebekultur Zellen. Die einzige Änderung ist Elution in 200 µL der Nuklease-freie H2O anstelle der mitgelieferten Elution Buffer.
Hinweis: Nach Zugabe von chaotropen Lyse Puffer (das Kit "AL Puffer") und Proteinase, Proben können aus dem Biosafety-Containment-Labor entfernt und behandelt in einem standard-Sicherheits-Niveau-Labor für den Rest des Protokolls. - Bestimmen Sie die Kopienzahl des HIV-1 cDNA von qPCR.
- Das Eluat von Schritt 2.1 17 µL und fügen Sie 2 µL 10 X Restriktionsenzym Puffer zusammen mit 1 µL Restriktionsenzym DpnI. Inkubation für 1 h bei 37 ° C, jede mögliche verbleibende Eingabe Plasmid DNA aus Transfektion zu entfernen.
- QPCR für Minus-Strang stark-Stop cDNA anhand der folgenden Grundierung Sonde durchzuführen: oHC64 (5′-Taactagggaacccactgc-3 ') und oHC65 (5′-Gctagagattttccacactg-3 ') und Sonde oHC66 (5′-FAM-Acacaacagacgggcacacacta-TAMRA-3′). qPCR Setup und genauen Bedingungen finden Sie in den Referenzen6,13. Proben mit einer seriellen Verwässerung des pNL4.3 proviralen Plasmid als eine Standardkurve Heftnummern cDNA Moleküle bestimmen mitnehmen.
Hinweis: Siehe Diskussion zur erwarteten Mengen.
- HIV-1 DNA-Anreicherung durch Hybrid Capture.
Hinweis: Aus diesen Schritt nach vorn ist es vorzuziehen, Mikrozentrifugenröhrchen mit niedrigen Nukleinsäure Bindungseigenschaften sowie Aerosol-Filter-Pipettenspitzen für alle DNA-Proben zu verwenden. Wenn möglich, arbeiten Sie in einer PCR-Workstation. Alle Schritte und Reagenzien sind bei RT (RT), sofern nicht anders angegeben.- Um ein master-Mix von magnetischen Streptavidin Perlen vorzubereiten, pipette 100 µL Perlen pro Probe zu einem einzigen Microcentrifuge Schlauch. Legen Sie das Rohr auf ein Magnet für Mikrozentrifugenröhrchen geeignet.
- Nachdem die Perlen in Richtung der Magnet-Seite des Rohres (~ 1 min) niedergelassen haben, nehmen Sie den Puffer speichern, entfernen Sie den Schlauch aus der Magnet Aufschwemmen Sie Perlen in 500 µL von Bind und waschen Sie Puffer (BW Puffer, 5 mM Tris-HCL pH 7.5, 0,5 mM EDTA 1 M NaCl) zu waschen.
- Setzen Sie den Schlauch wieder auf Magnet entfernen des Überstands und 500 µL Kasein-Lösung. Nehmen des Magneten, Aufschwemmen und 10 min bei RT inkubieren, dann waschen mit BW-Puffer.
Hinweis: Eine Wäsche bezieht sich auf das Rohr auf den Magneten zu platzieren, überstand abnehmen, das Rohr aus dem Magneten, Hinzufügen des Puffers und resuspending. - Platzieren Sie Schlauch wieder auf den Magneten zu, überstand nehmen Sie ab und Aufschwemmen Sie Perlen in 500 µL Puffer BW. Fügen Sie 50 Pmol jedes aufzeichnende biotinylierte Oligonukleotide (siehe Tabelle 1, drei Oligos in diesem Fall) pro Probe. (Wenn 5 DNA-Proben bearbeitet werden sollen, z. B. 500 µL des magnetischen Beads aus Schritt 2.3.1 und 250 Pmol von jedem Oligonukleotid).
- 30 min bei RT beim Schaukeln in einer durchgängigen über Mixer inkubieren.
- Perlen mit den immobilisierten Oligonukleotiden zweimal mit 500 µL 1 X 10-Puffer (10 mM Tris HCl pH 8.0, 1 mM EDTA, 100 mM NaCl) zu waschen.
- Aufschwemmen Sie Perlen in 10 µL 1 X zehn Puffer pro Probe.
- Für jede Probe ein Microcentrifuge Schlauch beschriften und fügen Sie 10 µL Perlen Suspension, 170 µL DNA (aus Schritt 2.1) und 90 µL 3 x 10 Puffer. Brüten Sie in einem Block von trockener Hitze bei 92 ° C für 2 min um die DNA zu denaturieren.
- Eine andere trockene Hitze-Block, der auf 52 ° C eingestellt ist, die Rohre nach und inkubieren 1H invertieren, regelmäßig zu mischen (~ alle 10 min) während diese Inkubation.
- Einmal waschen mit 500 µL 1 x zehn Puffer und Aufschwemmen in 35 µL Nuklease-freie H2O.
- Um eluieren, brüten Sie die Rohre bei 92 ° C in einem trockenen Hitze Block 2 min. lang. Verschieben Sie dann schnell die Rohre auf den Magneten (ein Rohr zu einem Zeitpunkt). Übertragen Sie Perlen an der Seite des Rohres gebunden sind, den Überstand mit der HIV-1 DNA zu einem frischen Schlauch.
- Optional: Wiederholen Sie, qPCR (wie in Schritt 2.2.2) um die wiederhergestellten HIV-1-cDNA festzustellen.
Hinweis: Siehe Diskussion zur erwarteten Mengen.
3. Adapter Ligation
- Vorbereitung des Adapters
- Aufschwemmen lyophilisierter Adapter (siehe Tabelle 1 "voll Kwok + MiSeq") bei 100 µM im Nuklease-freie H2O.
- Pro Probe plus einer Kontrollprobe kombinieren 0,45 µL 10 x T4 DNA-Ligase Puffer, 4 µL des Adapters und 0,05 µL Nuklease-freie H2O. Hitze bis 92 ° C für 2 min und lassen Sie abkühlen langsam.
Hinweis: Wenn die Option verfügbar ist, verwenden Sie eine PCR-Maschine mit einstellbarer Abkühlgeschwindigkeit (Nutzung 2 % Rate). Dies dauert etwa 30 Minuten von 92 ° C bis 16 ° C. Alternativ verwenden Sie einen trockene Hitze-Block bei 92 ° C und ausschalten. Herausnehmen Sie der Adapter Mastermix, wenn die Heizblock zurück bei RT ist Das ist den Adapter eine Haarnadel-Struktur zu bilden zu lassen (siehe Abbildung 2b).
- Bereiten Sie eine Kontrollreaktion mit einer Reihe von synthetisierte Oligonukleotide (siehe Tabelle 2) anstelle von DNA aus Zellen extrahiert.
- Aktien von 100 µM jedes Oligonukleotid zu machen. 1 µL der einzelnen 17 Oligonucelotides und 8 µL H2O für ein äquimolaren Verhältnis in einem Endvolumen von 25 µL.
- Verdünnen Sie die Mischung 1: 2.500 Nuklease-freie h2O in eine serielle Verdünnung. Kombinieren 1 µL des Mixes mit 17,3 µL Nuklease-freie H2O, in der Kontrolle Probe Ligatur im Schritt 3.3.1 zu verwenden, so dass jedes Oligonukleotid bei 1,6 Fmol vorhanden ist (gleichbedeutend mit 0.026 nM 60 µL-Reaktion).
- Einrichten von Verbindlichkeiten
- Für 60 µL Endvolumen Reaktionen in PCR-Rohre verbinden 6 µL 10 x T4 DNA-Ligase Puffer, 24 µL 40 % PEG, 6 µL 5 M Betain, 4,5 µL (400 Pmol) des Adapters (Pre-Annelead wie in Schritt 3.1.2), 1,2 µL T4 DNA-Ligase (2.000.000 Einheiten/mL) und 18,3 µL DNA (von Schritt 2.3.11)
Hinweis: Besondere Vorsicht bei viskosen Lösungen wie 40 % PEG, genaue Mengen beizubehalten. Machen Sie kein Mastermix. - Richten Sie die gleiche Reaktion wie in Schritt 3.3.1, sondern mit dem Steuerelement Oligonukleotid-Mix in Schritt 3.2.2 vorbereitet durchgeführt.
- Die Reaktionen gut mischen und über Nacht in einer PCR-Maschine bei 16 ° C inkubieren.
- Für 60 µL Endvolumen Reaktionen in PCR-Rohre verbinden 6 µL 10 x T4 DNA-Ligase Puffer, 24 µL 40 % PEG, 6 µL 5 M Betain, 4,5 µL (400 Pmol) des Adapters (Pre-Annelead wie in Schritt 3.1.2), 1,2 µL T4 DNA-Ligase (2.000.000 Einheiten/mL) und 18,3 µL DNA (von Schritt 2.3.11)
4. Adapter Entfernung und Größe Trennung
- Denaturierung Gelelektrophorese
- Fügen Sie 30 µL Formamid-haltigen DNA-Gel Ladepuffer zu jeder Ligatur-Reaktion. Mischen Sie gut durch pipettieren.
- Für 2 min bei 94 ° C im PCR-Maschine erhitzen, dann sofort auf Eis gelegt.
- Platzieren Sie eine Fertigteile 6 % Tris/Borat/EDTA (TBE) denaturierenden Harnstoff Polyacrylamid Gel (10-Well-Kamm) in einem entsprechenden Gel-Tank. 1 X TBE (89 mM Tris-Base, 89 mM Borsäure, 2 mM EDTA) laufenden Puffer und vorab das Gel für 20 min bei 250 V/Max konstanten laufen.
- Waschen Sie die Geltaschen mit fließendem Puffer mit einer Spritze und Nadel 21G.
- Last jedes 90 µL Probe in drei Brunnen (30 µL pro Well) und Lauf für 20 min (250 V/Max) bis dunkelblaue Färbung vorne auf halbem Wege durch das Gel geht.
- Färben und schneiden Nukleinsäuren aus dem gel
- Bereiten Sie 3 kleine Mikrozentrifugenröhrchen (0,5 mL vor) pro Probe durch stossen Löcher in den Boden mit einer 21 G Spritzennadel (nehmen Sie Vorsicht beim Arbeiten mit Kleie). Legen Sie jedes vorbereitete Rohre zu einem 2,0 mL Microcentrifuge Schlauch und beschriften Sie sie mit dem Beispielnamen plus "niedrig", "mid" oder "high".
- Herausnehmen und Hebeln öffnen die Gel-Kassette. Schneiden Sie das Gel vertikal mit einer Rasierklinge zu großzügig den Streifen mit der 3 Brunnen der geladenen Samples Verbrauchssteuern. Fügen Sie die Gel-Streifen in einen Behälter mit 1 x TBE (ca. 30 mL) und 5 µL Cyanin Nukleinsäure-Fleck. 3-5 min inkubieren.
Hinweis: Das Gel Extraktionsschritt reagiert besonders empfindlich auf Cross-Kontamination. Es ist ratsam, nur 1 Exemplar pro Gel und mit einem separaten, saubere Behälter für jedes Gel Färbung führen. Handschuhe sollten geändert werden, wenn Gel Partikel behandschuhte Fingern kontaktieren. - Reinigen Sie die Oberfläche von einem blauen Licht Transilluminator gründlich mit DdH2O. Nehmen Sie das Gel-Stück aus dem Färbung Container und fügen Sie es dem Lichtkasten.
- Schalten Sie den Leuchtkasten und inspizieren Sie die gefärbten Nukleinsäuren durch die orange-Filter.
Hinweis: Der Adapter in der Regel scheint überlastet und läuft als große "blob" mit aufgespaltenen HIV-1 DNA als einen Streifen oben ausgeführt. - Mit einer neuen Rasierklinge, schneiden Sie die Seiten des Gels gibt es Bereiche mit keine Probe geladen noch vorhanden. Weiter, nur über den Adapter zu entfernen den Adapter und niedriger gel Teile schneiden. Zu guter Letzt Taschen wegschneiden ganz oben auf dem Gel einschließlich ca. 1 mm des Gels, die haben oft eine scharfe intensiven Signal des höheren Molekulargewichts DNA.
- Teilen Sie das Gel-Reststück mit der Probe, die in der Regel ~ 2 x 3 cm groß, horizontal in drei sogar Stücke: "niedrig", "mid" und "hochmolekularen" Bereiche.
Hinweis: Jedes Stück wird jetzt separat behandelt [dh., es werden drei Rohre (niedrig, Mitte und hoch)] pro Originalprobe. - Jedes der drei Gel Fragmente in kleinere Stücke (~ 2 x 2 mm Partikel) schneiden und in die vorbereitet 0,5 mL Mikrozentrifugenröhrchen (Schritt 4.2.1) überführen.
- Drehen Sie bei Höchstgeschwindigkeit mit offenen Deckel für 1 min, die Gel-Stücke durch das Loch in der 2 mL Tube erstelle ich eine Gel-Matsch zu pressen. Wenn keine Gel-Partikel im unteren Teil der 0,5 mL Tube bleiben, zu der 2 mL Tube manuell mit einer Nadel oder Pipette Spitze übertragen.
- DNA-Extraktion
- Fügen Sie 1 mL Harnstoff Gel Extraktionspuffer (0,5 M NH4CH3CO2, 1 mM EDTA, 0,2 % SDS) zu den Gel-Matsch. Rohre für ein Minimum von 3 h drehen (über Nacht ist zulässig) bei RT mit einem durchgängigen über Mixer.
- Verwenden Sie einen sauberen Satz Pinzette Zentrifuge Spalten mit Cellulose-Acetat-Membranfilter (0,2 µm), die Membran zu Verstopfung vermeidet ein kleines rundes Glasfaser Filter hinzufügen. Setzen Sie den Filter mit einer invertierten Pipettenspitze.
- Kurz drehen Sie die 2 mL-Röhrchen mit Gel Matsch und Extraktion-Puffer in einem Microcentrifuge und übertragen Sie 700 µL des Überstands an die vorbereitete Filter Spalten. Halten Sie das Gel Matsch und verbleibende Überstand.
- Zentrifugieren Sie die Filter-Spalten in einem Microcentrifuge Höchstgeschwindigkeit für 1 min. Transfer der Bestrahlungskammer in einen neuen 2,0 mL Microcentrifuge Schlauch.
- Laden Sie die Spalten mit der restlichen überstand. Versuchen Sie, so viel Flüssigkeit wie möglich aus der Extraktion Matsch zu erhalten. Übertragung von Gel Stücke ist nicht von Belang. Wieder drehen und Flowthroughs der gleichen Extraktion Proben zu kombinieren.
- DNA-Fällung
- Fügen Sie 3 µL PolyA RNA (1 µg/µL; als Träger), 1 µL von Glykogen und 0,7 mL Isopropanol in der Bestrahlungskammer aus Schritt 4.3.5. Vortex kurz und Einfrieren bei-80 ° C über Nacht.
- Proben aus dem-80 ° C Gefrierschrank nehmen und kurz auftauen lassen. Setzen Sie sie in einem Microcentrifuge gekühlt (4 ° C) und Spin für 30 min bei Höchstgeschwindigkeit.
- Entfernen und entsorgen des Überstands. Seien Sie vorsichtig, nicht die Kugel zu entfernen. 30 bis 50 µL Flüssigkeit zu verlassen, wenn es ist ungewiss, dass Pellet sonst entfernt werden würde.
Hinweis: In der Regel alle "hoch" Beispiele zeigen einen sichtbareren Pellet als "mid" und "low" Proben. - Fügen Sie 800 µL von 80 % Ethanol. Rohre und Spin wieder für 1 min bei höchster Geschwindigkeit zu invertieren. Die Mehrheit von Ethanol mit einer Pipette, kurz Spinnen Sie die Rohre wieder zu, und entfernen Sie mehr Ethanol mit einem kleineren Volumen-Pipette.
- Lassen Sie alle restlichen Ethanol, indem man die Rohre mit einem geöffnetem Deckel in einen Block 55 ° C trockene Hitze verdampfen. Wenn Proben sind trocken (ca. 2-4 min) hinzufügen 20 µL der Nuklease-freie H2O und Verbreitung rund um den Boden des Rohres um sicherzustellen das DNA-Pellet wird aufgelöst. Die DNA Probe kann bei-20 ° c gelagert werden
(5) PCR-Amplifikation und Bibliothek-Vorbereitung
- Richten Sie ein 40 µL-PCR-Reaktion mit 20 µL der DNA-Polymerase Premix, 18 µL ausgefällt und Bodensatz DNA aus Schritt 4.4.5, 1 µL vorwärts Grundierung "MP1.0 + 22HIV" (10µM) (siehe Tabelle 1), und 1 µL der Multiplex-Oligo-Primer (Index Primer 1 bis 24) (siehe Tabelle der Ma Materialien).
Hinweis: Führen Sie die drei Reaktionen (Low, mid, High) jeder Probe in separaten PCR-Reaktionen, aber mit der gleichen Grundierung indiziert. Verwenden Sie einen anderen Index für jede der ursprünglichen Infektion Proben.- Führen Sie die PCR-Reaktionen unter den folgenden Bedingungen: 2 min bei 94 ° C Denaturierung, dann 18 Zyklen der 3-Stufen-PCR; 15 s bei 94 ° C Denaturierung 15 s Glühen bei 55 ° C und 30 s-Erweiterung bei 68 ° C.
- Als Option zur Qualitätskontrolle PCR-Reaktionen mit hoher Empfindlichkeit automatisiert Gel-Elektrophorese System zu analysieren. Nehmen Sie 2 µL einer niedrigen, mittleren und hohen Probe gemäß Anweisungen des Herstellers ausgeführt.
Hinweis: Die beiden Primer soll sichtbar und oft laufen auf eine berechnete Länge von etwa 45 bis 95 nt (tatsächliche Länge abweicht). Darüber hinaus sollte DNA erkannt werden, zwischen 150 bis 500 nt. Wenn kein Signal vorhanden ist, ist es ratsam, zusätzliche PCR-Zyklen, zwischen 2 und 10 zusätzliche Zyklen hinzuzufügen. Fügen Sie keine zusätzlichen Zyklen für die Oligonukleotid-Kontrollproben im Schritt 3.3.2 erstellt. - Zum Entfernen verwenden die Primer ein paramagnetischen Wulst-PCR Aufräum System.
- Nehmen Sie 20 µL jeder PCR-Reaktion und bündeln Sie die Proben zusammen (mischen Sie alle Beispiele zu diesem Zeitpunkt). Frieren Sie die restlichen 20 µL Reaktionen als Backup bei-20 ° C.
- Lassen Sie die paramagnetischen Perlen ans RT und Mischen der gepoolten PCR-Reaktionen mit 1,8 x Volumen der Wulst Lösung. Durch Pipettieren mischen und 5 min inkubieren.
Hinweis: Als ein Beispiel, wenn 4 Proben vorbereitet waren und niedrige, mittleren und hohen Reaktionen haben, das Volumen wäre 4 x 3 x 20 µL = 240 µL PCR-Reaktionen mit einer 432 µL-Perle-Lösung. - Setzen Sie die Rohre auf einem Microcentrifuge Schlauch Magnet, lassen Sie die Perlen für ~ 1 min zu binden, und Ausziehen des Überstands verwerfen. Lassen Sie die Rohre auf den Magneten und fügen Sie 500 µL von 80 % Ethanol.
- Lassen Sie das Ethanol für 30 s, dann gründlich ausziehen und lassen die Perlen Airdry für ~ 5 min. hinzufügen 40 µL Nuklease-freie H2O, nehmen Sie die Rohre aus den Magneten und pipette nach oben und unten mehrmals.
- Verlassen Sie Aufhängung für 5 min. legte das Rohr zurück auf den Magneten zu, lassen Sie die Perlen auf die Seite zu begleichen und überstand an einen neuen Schlauch übertragen. Dies ist die Bibliothek. Nehmen Sie 10 µL aliquoten für Qualitätskontrollen und frieren Sie den Rest bei-20 ° C.
6. Bewertung der Bibliothek
- Bestimmen Sie die Bibliothek Qualität, Konzentration und Molarity.
- Verwenden Sie eine fluorometrisch Quantifizierungsmethode. Messen Sie 1 µL und 3 µL der Bibliothek mit einem hochempfindlichen DsDNA-Assay Kit gemäß Herstellervorschrift.
Hinweis: Typische Konzentrationen liegen zwischen 1 und 10 ng/µL. - Maßnahme durch eine hohe Empfindlichkeit DNA Molekulargewicht Bibliotheksspektrum automatisiert Gelelektrophorese, wie oben (Schritt 5.2) beschrieben.
- Einsatz der automatisierten gel-Elektrophorese-Analyse zur Bestimmung der durchschnittlichen Molekulargewicht der Bibliothek und berechnen, um die Bibliothek in Nuklease-freie H verdünnen2O 4 nM. Mehrere Bibliotheken können kombiniert werden, solange alle Indizes einzigartig sind.
- Verwenden Sie eine fluorometrisch Quantifizierungsmethode. Messen Sie 1 µL und 3 µL der Bibliothek mit einem hochempfindlichen DsDNA-Assay Kit gemäß Herstellervorschrift.
- Optionale niedriger Durchsatz Qualitätskontrolle
- Vorbehaltlich der DNA Bibliothek TA Klonen19 Bibliothek Moleküle in Vektoren für Verstärkung einfügen. Befolgen Sie das Kit, wachsen, ~ 10-20 Kolonien und Extrakt DNA über Miniprep Protokolle, wie hier beschrieben20.
- Sequenzen und Bibliothek-spezifische Adapter abgeleitet Sequenz Vektor mit lokalen Sequenzierung Services und überprüfen Sie, dass die Einsätze der gewünschten HIV-1 enthalten.
(7) Hochdurchsatz-Sequenzierung Run
- Erstellen Sie eine Sequenzierung Musterblatt mit kommerzieller Software, die mit der Sequenzierung-Plattform zur Verfügung gestellt.
- Geben Sie das ausgewählte Sequencing Kit. Wählen Sie in der Regel eine 150-Zyklus-Kit, aber andere sind abhängig von der gewünschten lesen Sie Länge geeignet.
- Wählen Sie "Nur Fastq" als die Anwendung Workflow. Wählen Sie eine der Vorlagen, die die 24 Indizes vorhanden in den Multiplex-Oligonukleotid-Kits (im Kit-Handbuch angegeben) enthält.
- Wählen Sie "25 nt" für Read1 und "125 nt" für Read2. Halten Sie 6 nt für einzelnen Index lesen.
Hinweis: Bei der internen Analyse ist nur Read2 in der Analyse verwendet. Halten Sie Read1 auf ein Minimum von 25 nt für die Sequenzierung Plattform Algorithmus Zwecke.
- Befolgen Sie die Anweisungen des Herstellers genau für die Vorbereitung vor dem Rechenlauf Bibliothek und das Setup. Entscheiden Sie sich für maximal 20 Uhr Konzentration und Einsatz eine 15 % PhiX Spitze, als die Bibliothek von sehr geringer Komplexität ist.
8. die Datenanalyse
- Überprüfen Sie, ob der Pass Filter Prozentsatz und durchschnittliche Q30 Qualitätsfaktor nach Sequenzierung Plattform Herstellerrichtlinien akzeptabel sind.
Hinweis: Pass-Filter ist in der Regel > 90 % und Q30 Partituren sind in der Regel > 80 %. - Download der. fastq.gz Dateien des Herstellers Sequenzierung Hub.
- Einrichten der Sequenzierung Skript
- Erstellen Sie ein neues Verzeichnis (Ordner) namens "AnalysisXYZ" und gehen Sie zu https://github.com/malimlab/seqparse zum download alle Quellcode-Dateien (parse_sam.pl, rc_extract.pl, parse.sh) in dieses Verzeichnis.
- Die kurze lesende Aligner Bowtie, Version 1.1.2, von http://bowtie-bio.sourceforge.net/index.shtml in das gleiche Verzeichnis herunterladen.
- Der Download wird ein Unterverzeichnis innerhalb von "AnalysisXYZ" namens "Bowtie-1.1.2" erstellt. Öffnen Sie in diesem Verzeichnis Unterverzeichnis "Indizes" und laden Sie die mitgelieferten Template-Sequenzen, bestehend aus 6 Dateien mit den Erweiterungen .ebwt.
- Laden Sie die FASTQ/A kurze Lesevorgänge Pre-processing Toolkit Fastx-0.0.13 von http://hannonlab.cshl.edu/fastx_toolkit/download.html in das Verzeichnis "AnalysisXYZ".
- Download Samtools (https://sourceforge.net/projects/samtools/files/) und Bam-Readcount (https://github.com/genome/bam-readcount) in das Verzeichnis "Dokumente".
- Verschieben der. fastq.gz, im Schritt 8.2, aller heruntergeladenen Dateien, lesen 2 s (Ende... _R2_001.fastq.gz) in das Verzeichnis "AnalysisXYZ".
- Öffnen Sie das Befehl Konsole/Terminal. Nach der "AnalysisXYZ" als das aktuelle Verzeichnis mit cd Befehlen bewegen Geben Sie "./parse.sh.", führen Sie die Skripts.
- Finden die CSV-Dateien mit Zusammenfassungen für alle Proben auf insgesamt lesen Sie zählt, Länge angepasst zählt, zu lesen und lesen Sie zählt normalisiert, sowie Dateien mit der Basis-Variante für jede Probe in einem Verzeichnis namens Parse_results im Verzeichnis "Analyse-XYZ".
Hinweis: Siehe die Diskussion für weitere Informationen zu den Analyseprozess. Das Skript gibt Csv-Dateien mit insgesamt liest für jedes Nukleotid entlang der HIV-1-NL4.3 -starke-Stop-Sequenzen und ersten Strang Transfer bis Nukleotid 635 zurück. Als Orientierungshilfe werden 50.000 bis 100.000 einzigartige liest in der Regel in Proben von Infektionen mit den angegebenen Zellzahlen und virale Inokula und ohne antivirale Proteine oder Verbindungen beobachtet. Die Oligonukleotid-Kontrollprobe produziert in der Regel 100.000 bis 200.000 mal gelesen.
Ergebnisse
In diesem Artikel beschriebene Verfahren angewendet wurde, auf eine breitere Studie an Mechanismen, die Hemmung der reversen Transkription HIV-1 durch die antiretrovirale menschliche Protein APOBEC3G (A3G)6. Abbildung 3 zeigt repräsentative Ergebnisse nach Einsatz des Protokolls in Proben von CEM-SS-T-Zellen infiziert mit Vif-mangelhaft HIV-1 in das Fehlen oder Vorhandensein von A3G. Die Gesamtzahl der einzigartigen liest erhalten von jeder Probe nach Dubletten PCR, die die gleichen 6 herausfiltern nt Barcode und die gleiche Länge (durchgeführt durch die Analysesoftware zur Verfügung gestellt) werden dargestellt in Abbildung 3ein. Zunehmende Verringerung der A3G die Gesamtzahl lesen Sie reflektieren die inhibitorische Wirkung des A3G RT vermittelte cDNA Synthese zuvor beschrieben und gemessen an qPCR6,13,21,22. In Abbildung 3bder Anteil der Moleküle auf jede mögliche Länge innerhalb der ersten 182 nt werden angezeigt. Für HIV-1 Infektion in Ermangelung von A3G, ist die häufigste Art der wichtigsten 180 nt stark-Stop Molekül selbst, mit einigen Anhäufung der Lesevorgänge in die kürzere Reichweite (23 bis 40 nt) (top-Grafik, blauen Histogramme). Die Zugabe von A3G Änderungen, die dieses Profil da ein starker Anstieg der kürzere, abgeschnitten cDNA Moleküle an ein paar ganz bestimmte, reproduzierbare Positionen ist erkannt (mittlere und untere Graphen). Da A3G ein Cytidin-Deaminase, Cytosin-Uridin ist (gekennzeichnet als C-to-Tape) auftreten Mutationen in der cDNA, wenn A3G in der infizierenden Virionen21,23,24vorhanden ist. Mit den erhaltenen Sequenzierungsinformationen, war der Anteil der C-to-Tape-Mutationen im selben Diagramm (rote gestrichelte Linie) aufgetragen. Es sei darauf hingewiesen, dass die Mutationszucht Profil sich aus alle einzigartig liest kombiniert ergibt und Berichterstattung über jedes Nukleotid variiert. Jedoch bei Bedarf Sequenzinformation kann im Zusammenhang mit jedem Molekül zurück und korreliert mit einer spezifischen 3'-Terminus. Die Angaben stammen aus Pollpeter Et al. 6 und die Korrelation zwischen mutagenen und cDNA Länge Profile wurde nachgewiesen durch Erkennung und Spaltung von deaminated cDNA durch die zelluläre DNA-Reparatur Maschinen.
Eine positive-Kontrolle für die 3'-Mapping-Ansatz kann leicht durch die Verarbeitung eines Pools von synthetischen Oligonukleotiden bekannten Sequenz, Länge und Konzentration hergestellt werden. Dieses Steuerelement an der Adapter Ligatur im Schritt 3.3.2 hinzugefügt, wird empfohlen, in allen Multiplex Bibliotheken aufgenommen werden. Daten aus einer Kontrollprobe sollte auf den erwarteten Eingabe Verhältnissen, mit nur sehr geringen Hintergrund liest die Oligonucleotides haben. Abbildung 4 zeigt die Ergebnisse einer Positivkontrolle Reihe von 17 chemisch synthetisierte Oligonukleotide (Sequenzen, siehe Tabelle 2), die bei äquimolaren Verhältnisse gemischt wurden. Wie erwartet, erscheinen alle Moleküle in fast gleicher Fülle mit nur geringe Schwankungen (obere Grafik). Während die meisten Positionen innerhalb der Sss - DNA-Sequenz, die nicht durch ein Oligonukleotid vertreten waren null lesen Sie zählt zurückgeben, beobachteten wir kleinere Arten, die 1 oder 2 nt kürzer als die eigentliche Steuerung Oligonucleotides sind. Wir haben diese kleine Arten nicht weiter untersucht, aber davon ausgehen, dass sie abgebaut oder unvollständige Produkte eventuell vorhandene in der zur Verfügung gestellten Oligonukleotid-Aktien beim Kauf darstellen (Oligonukleotide wurden bestellt, wie HPLC, für die gereinigt die Hersteller zeigt Reinheit von > 80 %). Die untere Grafik zeigt die Kontrollprobe aus einer anderen Bibliothek wo Variante ist etwas höher, zwischen den 17 Oligonukleotide und korreliert mit Gesamtlänge, dass längere Kontrolle Moleküle effizienter erkannt werden dann kürzere laufen. Dies kann durch eine geringfügige Verzerrung im PCR-Reaktionen oder beim clustering während der MiSeq-Sequenzierung, die optimale Größe einfügen hat und auftreten mit Bibliotheken mit besonders breiten Einsatz reicht. Eine einfache Methode zum Adressieren diese Vorspannung ist die Anwendung der einen Normalisierungsfaktor basierend auf der Piste, die angibt, die Vorspannung Molekül Länge (rosa Linie) korrelieren. Die erforderlichen Berechnungen sind in der Analyse-Programm enthalten (siehe Punkt 8.3 im Protokoll).

Abbildung 1: das Diagramm zeigt die ersten Schritte der HIV-1 reverse Transkription. Der Prozess beginnt mit der tRNA(Lys,3) (Orange), die Grundierung-Bindungsstelle (PBS) Glühen in der genomischen viralen RNA (Schritt 1), wodurch die Initiierung und Dehnung der virale cDNA (blau, Schritt 2). Begleitend wird die Vorlage genomische RNA durch RNaseH Aktivität der RT (Schritt 3) abgebaut. Die erste vollständige Zwischenprodukt bei der reversen Transkription ist der Minus-Strang stark-Stopp (-) Sss cDNA, die ist abgeschlossen, wenn die RT katalysierten Polymerisation der 5'-Terminus des gRNA (R) Wiederholbereichs (Schritt 3) erreicht. (-) Sss Zwischenprodukt wird an den 3'-Terminus der genomischen RNA-Vorlage durch Tempern, ergänzende terminal 3'-lange (LTR) R Wiederholbereich übertragen. Von hier aus weiter Polymerisation (Schritt 4). In dem beschriebenen Verfahren wird das Fortschreiten der reversen Transkription durch Zuordnung die genaue Länge der im Entstehen begriffenen virale cDNA (blau) bestimmt. PPT, Polypurine-Darm-Trakt; U5, einzigartige 5'-Sequenz; U3, einzigartige 3'-Sequenz. Diese Zahl ist aus einer früheren Veröffentlichung6neu aufgelegt. Bitte klicken Sie hier für eine größere Version dieser Figur.
{kind=link}

Abbildung 2 : Workflow Gliederung und schematische Darstellungen der PCR Verstärkung Strategie und Adapter Ligation. (a) Workflow skizziert die wichtigsten Schritte von der beschriebenen Technik festzustellen, 3'-Termini von HIV-1 umkehren Transkripte in infizierten Zellen. Die Figur ist aus einer früheren Veröffentlichung6angepasst. (b) schematische Darstellung der PCR Verstärkung Strategie und Adapter Ligatur. Im Entstehen begriffenen cDNA Moleküle unterschiedlicher Länge, die in den vorherigen Schritten gereinigt worden sind, eine einsträngige DNA-Adapter mit T4 DNA-Ligase ligiert. Die Haarnadel-Adapter (benannte "volle Kwok + MiSeq", siehe Tabelle 1) Design wurde inspiriert von Kwok Et Al. 11. Adapter führt eine zufällige 6 nt Barcode-Sequenz, die für die Basis-Kopplung zur Unterbindung Erleichterung erlaubt und dient gleichzeitig als Bezeichner für einzigartige liest. Die 3'-Termini des Adapters trägt ein Distanzstück (SpC3) selbst Ligatur zu verhindern. Ligiertes Produkte sind aus überschüssigen Adapter durch denaturierenden Polyacrylamid-Gelelektrophorese (PAGE) getrennt. Nukleinsäuren im Gel sind gefärbt und geschnitten in drei separate, gleich große Gel-Stücke in der Gegend von oben den Adapter zum Brunnen wie in25. Die Produkte sind nach der Elution, Niederschlag und Wiederfreisetzung, PCR verstärkt mit Zündkapseln Tempern der bekannten Sequenz des Adapters (Primer 1, Multiplex-Oligonukleotid-Kit, siehe Tabelle of Materials) und eine Grundierung, die Durchführung der ersten 22 nt des HIV-1 5'-LTR-Sequenz unmittelbar nach der tRNA (Primer 2, MP1.0 + 22HIV). Die 5'-Termini der gewählten Primer tragen Adapter für die gewählten Sequenzierung Plattform (P5 und P7) sowie eine Index-Sequenz, einzelne Proben laufen in derselben Bibliothek zu unterscheiden. Ausgangspunkte für die Sequenzierung lesen Sie Primer angegeben sind. Das blaue Feld zeigt die Region von Interesse für die ursprüngliche 3'-Termini des aufgenommenen Moleküls bestimmen. Diese Zahl wird von einer früheren Veröffentlichung6angepasst. Bitte klicken Sie hier für eine größere Version dieser Figur.
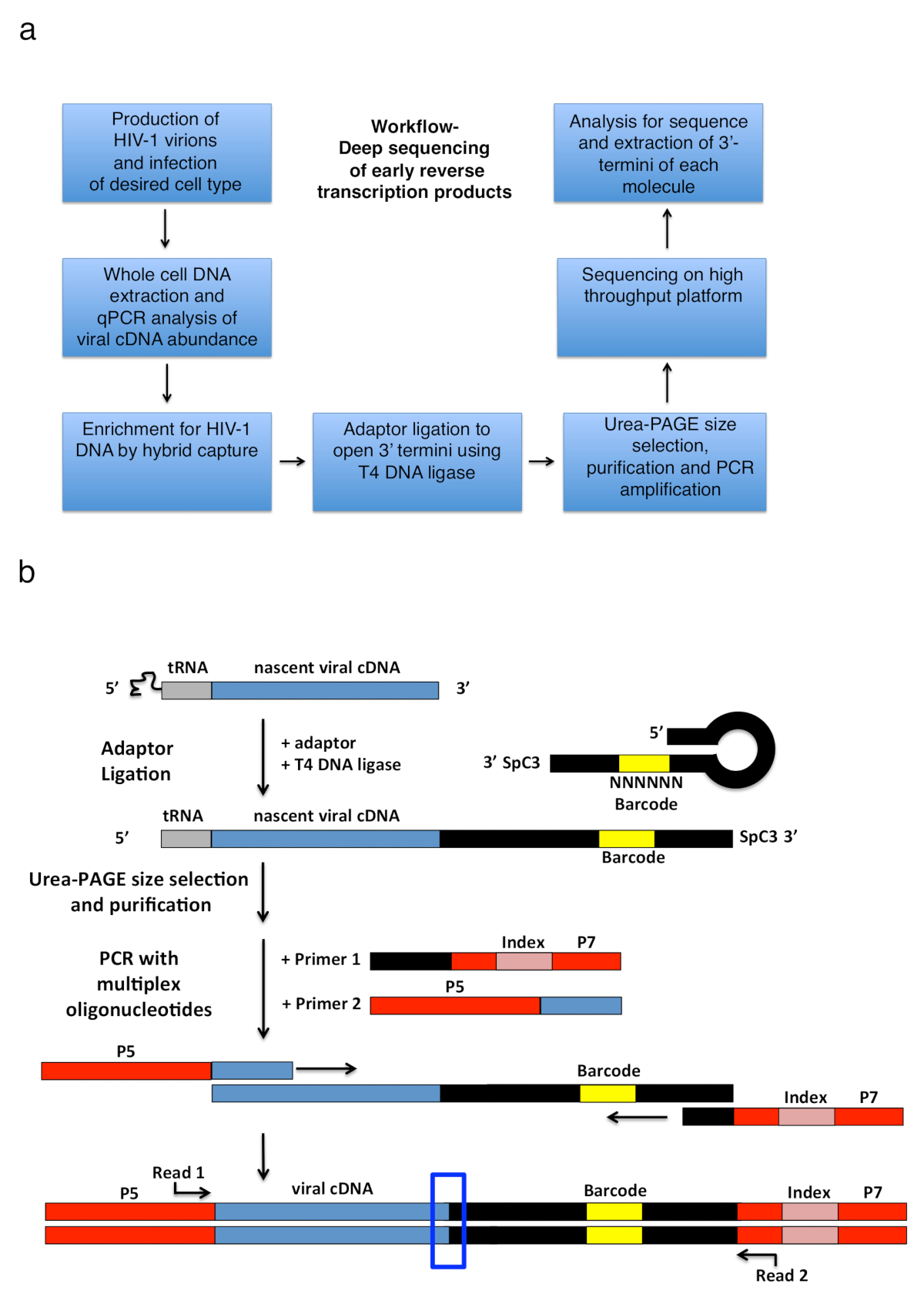{kind=link}

Abbildung 3 : Repräsentative Ergebnisse. (a) die Gesamtzahl lesen Sie von repräsentativen Stichproben mit dem beschriebenen Protokoll verarbeitet. Hierunter fallen alle Sequenzen, die als einzigartige liest der HIV-1-Moleküle mit ihren 3'-Termini innerhalb der ersten 635 identifiziert wurden nt Minus Strang cDNA (bis die PPT, siehe Abbildung 1). Infektion mit HIV-1 nicht tragen A3G ergibt die höchste Anzahl der Lesevorgänge, während A3G cDNA Synthese hemmt und dadurch reduziert sich die Anzahl insgesamt. Nicht infizierte Zellen diente als Negativkontrolle, während eine Reihe von synthetischen Oligonukleotiden Positivkontrolle bietet. (b) die relative Häufigkeit der cDNAs für jede Länge zwischen nt Position 23 und 182 (Full-length - Sss cDNA ist 180 bis 182 nt) von HIV-1-NL4.3 ist in blauen Histogramme (Skala auf der linken y-Achse) Ablauf (x-Achse) dargestellt. Die relative Häufigkeit der cDNA wurde aus der absoluten Anzahl der Sequenzen Abbruch an einen bestimmten Nukleotid innerhalb der Sss - cDNA-Sequenz dividiert durch die Summe der alle Lesevorgänge Messung 182nt oder weniger berechnet. In rot gestrichelt dargestellt, sind die Prozentsätze der Lesevorgänge mit C-T/U Mutationen an der jeweiligen Position (Skala auf der rechten y-Achse–). Abbildung 3 b ist von einer früheren Veröffentlichung6neu aufgelegt. Bitte klicken Sie hier für eine größere Version dieser Figur.
{kind=link}

Abbildung 4 : Repräsentative Ergebnisse der Kontrollproben. Gezeigt werden zwei Profile für Pools mit äquimolaren Mengen von 17 verschiedenen Länge synthetische Oligonukleotide. Diese Oligonukleotide Sequenzen von HIV-1-NL4.3 und wurden ausgewählt, um verschiedene Längen zu decken und alle 4 Basen als eine 3'-Nukleotid (siehe Tabelle 2). Die obere Grafik zeigt die Positivkontrolle Probe aus Abbildung 3ein. Keine signifikante Tendenz zu Molekül Länge oder die offene 3'-Termini wird erkannt. Die untere Grafik zeigt eine andere Bibliothek ausführen, die produziert einer kleinere Länge Vorspannung in der Sequenzierung. In diesem Fall ist es ratsam, einen Normalisierungsfaktor anzuwenden, die von der Piste (in rosa dargestellt) abgeleitet wird, die die Größe der Vorspannung darstellt. Diese Zahl ist aus einer früheren Veröffentlichung6neu aufgelegt. Bitte klicken Sie hier für eine größere Version dieser Figur.
{kind=link}
| Oligo-name | Länge im nt | Sequenz | Zweck | Hersteller (Reinigung) | ||||||||||||||
| voll Kwok + MiSeq | 61 | 5'-PHO-Tgaagagcctagtcgctgttcannnnnnctgcccatagagagatcggaagagcacacgtct-SpC3-3 " | Adapter | IDT DNA-Technologien (HPLC) | ||||||||||||||
| 2xBiotin SS Köder | 40 | 5'-Biotin-Cagtgtggaaaatctctagcagtggcgcccgaacagggac-Biotin-3 " | Hybrid-Capture | Eurofins MWG (HPLC) | ||||||||||||||
| Biotin 1-16 ss | 22 | 5'-Cagtgtggaaaatctctagcag-BiTEG-3 " | Hybrid-Capture | Eurofins MWG (HPLC | ||||||||||||||
| Biotin-tRNA + CTG | 16 | 5'-Cagtggcgcccgaaca-BITEG-3 " | Hybrid-Capture | Eurofins MWG (HPLC) | ||||||||||||||
| MP1.0 + 22HIV | 82 | 5'-Aatgatacggcgaccaccgagatctacactctttccctacacgacgctcttccgatctcactgctagagattttccacactg-3 " | PCR-Amplifikation | Eurofins MWG (HPLC | ||||||||||||||
Tabelle 1: Tabelle der Oligonukleotide einschließlich der Länge, Sequenzen und Änderungen, die verwendet werden in der beschriebenen Protokoll. Der Tisch ist aus einer früheren Veröffentlichung6angepasst. Bitte klicken Sie hier, um diese Tabelle als Excel-Datei herunterladen.
| Oligo-name | Länge im nt | Sequenz | Hersteller (Reinigung) | |||||||||||||
| HTP con lange C | 120 | 5'-Ctgctagagattttccacactgactaaaagggtctgagggatctctagttaccagagtcacacaacagacgggcacacactactttgagcactcaaggcaagctttattgaggcttaagc-3 " | Eurofins MWG (HPLC) | |||||||||||||
| HTP con lange G | 119 | 5'-Ctgctagagattttccacactgactaaaagggtctgagggatctctagttaccagagtcacacaacagacgggcacacactactttgagcactcaaggcaagctttattgaggcttaag-3 " | Eurofins MWG (HPLC) | |||||||||||||
| HTP con lange T | 116 | 5'-Ctgctagagattttccacactgactaaaagggtctgagggatctctagttaccagagtcacacaacagacgggcacacactactttgagcactcaaggcaagctttattgaggctt-3 " | Eurofins MWG (HPLC) | |||||||||||||
| HTP Con lange A | 118 | 5'-Ctgctagagattttccacactgactaaaagggtctgagggatctctagttaccagagtcacacaacagacgggcacacactactttgagcactcaaggcaagctttattgaggcttaa-3 " | Eurofins MWG (HPLC) | |||||||||||||
| HTP Con Mitte C | 76 | 5'-Ctgctagagattttccacactgactaaaagggtctgagggatctctagttaccagagtcacacaacagacgggcac-3 " | Eurofins MWG (HPLC) | |||||||||||||
| HTP Con Mitte G (a) | 71 | 5'-Ctgctagagattttccacactgactaaaagggtctgagggatctctagttaccagagtcacacaacagacg-3 " | Eurofins MWG (HPLC) | |||||||||||||
| HTP Con Mitte G (b) | 72 | 5'-Ctgctagagattttccacactgactaaaagggtctgagggatctctagttaccagagtcacacaacagacgg-3 " | Eurofins MWG (HPLC) | |||||||||||||
| HTP Con Mitte A | 69 | 5'-Ctgctagagattttccacactgactaaaagggtctgagggatctctagttaccagagtcacacaacaga-3 " | Eurofins MWG (HPLC) | |||||||||||||
| HTP Con Mitte T | 85 | 5'-Ctgctagagattttccacactgactaaaagggtctgagggatctctagttaccagagtcacacaacagacgggcacacactactt-3 " | Eurofins MWG (HPLC) | |||||||||||||
| HTP Con kurzen A | 40 | 5'-Ctgctagagattttccacactgactaaaagggtctgaggga-3 " | Eurofins MWG (HPLC) | |||||||||||||
| HTP con kurze T | 33 | 5'-Ctgctagagattttccacactgactaaaagggt-3 " | Eurofins MWG (HPLC) | |||||||||||||
| HTP con kurzen G | 41 | 5'-Ctgctagagattttccacactgactaaaagggtctgaggg-3 " | Eurofins MWG (HPLC) | |||||||||||||
| HTP con kurzen C | 34 | 5'-Ctgctagagattttccacactgactaaaagggtc-3 " | Eurofins MWG (HPLC) | |||||||||||||
| HTP Con 46 (T) | 46 | 5'-Ctgctagagattttccacactg Actaaaagggtctgagggatctct-3 " | Eurofins MWG (HPLC) | |||||||||||||
| HTP-Con 83 (C) | 83 | 5'-Ctgctagagattttccacactg Actaaaagggtctgagggatctctagttaccagagtcacacaacagacgggcacacactac-3 " | Eurofins MWG (HPLC) | |||||||||||||
| HTP-Con 103 (C) | 103 | 5'-Ctgctagagattttccacactg Actaaaagggtctgagggatctctagttaccagagtcacacaacagacgggcacacactactttgagcactcaaggcaagc-3 " | Eurofins MWG (HPLC) | |||||||||||||
| HTP Con 107 (A) | 107 | 5'-Ctgctagagattttccacactg Actaaaagggtctgagggatctctagttaccagagtcacacaacagacgggcacacactactttgagcactcaaggcaagcttta-3 " | Eurofins MWG (HPLC) | |||||||||||||
Tabelle 2: Tabelle 17 synthetische Kontrolle Oligonukleotide als eine positive Kontrollprobe. Die Top 13-Oligonukleotide wurden ausgewählt, basierend auf der Größe [lange (116 bis 120 nt), Mitte (69 bis 85 nt), kurze (33 bis 41 nt)] sowie deren 3'-Termini. Der Tisch ist aus einer früheren Veröffentlichung6angepasst. Bitte klicken Sie hier, um diese Tabelle als Excel-Datei herunterladen.
Diskussion
Die Verfügbarkeit für die schnelle, zuverlässige und kostengünstige Tiefe Sequenzierung revolutioniert hat viele Aspekte auf dem Gebiet der Life Sciences, großen Tiefe Sequenzierung-basierte Analysen ermöglicht. Eine verbleibende Herausforderung liegt in der innovativen Gestaltung und Erstellung des Vertreters Sequenzierung Bibliotheken. Hier beschreiben wir ein Protokoll, um entstehende virale cDNA Molekülen, speziell die Zwischenprodukte des HIV-1 reverse Transkription Prozesses zu erfassen.
Der wichtigste Schritt in dieser Strategie ist die Unterbindung eines Adapters offen 3'-Termini in einer quantitativen und unvoreingenommene Weise. Wirkungsgrade von grundsätzlich zwischen zwei SsDNA Termini, beide inter- und intramolekulare, untersucht und für verschiedene Anwendungen11,26,27,28,29optimiert wurden. Die Möglichkeit zur Nutzung eines Haarnadel-Adapters mit T4 DNA-Ligase unter den in Schritt 3.3 beschriebenen Bedingungen ist das Ergebnis der empirischen Optimierung in dem wir verschiedene Ligases, Adaptern und Reagenzien für die Unterbindung der synthetischen Oligonukleotiden vertreten ausgewertet HIV-1 Sequenzen (Tabelle 2) (Daten nicht gezeigt). In dieser in-Vitro -Test-Reaktionen bestätigt wir, dass die T4-DNA-Ligase Ligatur der Haarnadel-Adapter vermittelt, wie von Kwok Et al.beschrieben. 11, hat eine sehr geringe Vorspannung und in der Nähe von vollständigen Unterbindung der Akzeptor Moleküle erreicht, wenn der Adapter im Überschuss eingesetzt wird. Die Ligatur-Effizienz war unberührt durch die Zugabe von Nukleotidsequenz, die den Adapter für die Multiplex-Primer-System in Einklang zu bringen (siehe Abbildung 4). Im Vergleich dazu fanden wir, dass eine thermostabile 5' DNA/RNA-Ligase ("Ligase A", siehe Tabelle der Materialien für genaue Ligases im Vergleich hier), das ist ein veränderter RNA Ligase, die teilweise entwickelt wurde, Ligation Effizienz mit SsDNA als den Akzeptor zu verbessern 27, war in der Tat viel effektiver Ligation zwei SsDNA Moleküle als RNA-Ligase ("Ligase B"), aber eine deutliche Tendenz mit starke Unterschiede in der Effizienz der Ligatur sogar zwischen Oligonukleotide mit einzelnen Basislänge unterschieden [Tabelle 2 hatte ; HTP con Mitte G (a) und (b)]. Darüber hinaus fanden wir nur eine minimale Verzerrung in Reaktionen mit "Ligase C" kombiniert mit einem Adapter tragen eine randomisierte 5'-Termini (eine Strategie verwendet, um bekannte Nukleotid-Bias "Ligase c" versetzen; siehe z. B. Ding Et Al. 30). aber das "Ligase C"-vermittelte intermolekulare Verbindlichkeiten waren unvollständig, Rendern dem T4 DNA-Ligase-System die bessere Wahl.
Mehrstufig Qualitätskontrolle im Laufe des Protokolls und die Aufnahme von positiven und negativen Kontrollen ermöglichen die Erkennung potenzieller Probleme vor Assay Fortsetzung und Orientierungshilfen für die Problembehandlung Bemühungen. Die qPCR Quantifizierungen in Schritten 2.2.2 und 2.3.12 sicherstellen, dass die Menge an input-Material ausreicht. Typische cDNA Kopie Zahlen im Bereich von 200 µL Elution (aus Schritt 2.1) von etwa 10.000 bis 300.000 pro Mikroliter. Der Hybrid-Capture-Schritt kann dazu führen, dass einige Verlust von HIV-1-Allgemeine cDNA Menge aber sollte eine starke Anreicherung von bestimmten HIV-1 cDNA führen, über zelluläre DNA, die mit entsprechenden Grundierungen genomischen DNA vor und nach Anreicherung durch Quantifizierung bestimmt werden kann qPCR oder DNA-Konzentration zu messen. Wiederhergestellte HIV-1 cDNA nach die Hybrid capture Schritte sollte mindestens 10 % des Eingangs. Low Ausgangsmaterial kann sonst erklären, dass eine erfolgreiche Oligonukleotid-positiv-Kontrolle (siehe Punkt 3.3.2) aber nur begrenzte liest in den Proben erreicht. Niedrige lesen Sie zahlen insgesamt auch Überschätzung der Bibliothek Konzentration aufgrund des Vorhandenseins von irrelevanten DNA-Arten ohne MiSeq Adapter erklärt werden könnte. Dies ergäbe sich niedrige Cluster Dichte und kann durch Bestimmung der Konzentration von HIV-1 Sequenzen in der Bibliothek von qPCR neben den Gesamtbetrag der DNA durch fluorometrischen Assays verbessert werden. Aufgrund der hochsensiblen Natur der Methode sollte besondere Vorsicht geboten, auch Low-Level-Kontamination, sowohl von anderen Proben (insbesondere durch die hohe Konzentration Kontrolle Oligonukleotid Bestände) sowie von Laborgeräten zu vermeiden. Arbeiten in einer UV-Sterilisation PCR-Workstation ist in dieser Hinsicht vorteilhaft. Die automatisierte Gelelektrophorese der endgültigen Bibliothek (Schritt 6.1.2) ist eine weitere Maßnahme der Qualitätskontrolle. Nukleinsäure-liegt Größe in der Regel beobachtet zwischen 150 bis 500 nt. Primer, die in die optionale Steuerung erkannt werden können, nach der PCR und vor Reinigung (siehe Hinweis im Schritt 5.2) jetzt nicht vorhanden sein sollte. In einem repräsentativen Ergebnis die Probe Intensitätskurve hat eine Spitze rund 160 bis 170 nt und eine zweite schärfer Spitze rund 320 bis 350 nt. Dies spiegelt wahrscheinlich die oft gesehen höheren Fülle in relativ kurz (1 bis 20 nt Einsatzlänge) reverse Transkripte und in voller Länge stark-Stop (180 bis 182 nt Einsatzlänge) (Abbildung 3b).
Während die vorgestellten Protokoll und ausgewählten Primer spezifisch für frühe HIV-1 reverse Transkription Konstrukte sind, ist die Methode generell für jede Studie mit dem Ziel, offene 3'-Termini der DNA zu bestimmen. Die wichtigsten Modifikationen in anderen Zusammenhängen werden die Methode für Hybrid Capture und die Grundierung-Design-Strategie sein. Z. B. wenn das Ziel ist es zu spät HIV-1-Transkripte angepasst werden, eine größere Anzahl von verschiedenen aufzeichnende biotinylierte Oligonukleotide Glühen über die gesamte Länge der cDNA wäre ratsam und wird wahrscheinlich den Verlust in der Hybrid-Capture-Schritt abnehmen. Wie in der Einleitung erwähnt, ist es wichtig, Grenzen zu berücksichtigen, wenn den Bereich entwerfen, über die 3'-Termini erkannt werden, um unterschiedliche Quellen der Befangenheit zu vermeiden. Zunächst möglicherweise eine Vorspannung in die PCR-Reaktionen wenn die Vorlagen mit dem Adapter von stark unterschiedlicher Länge sind. Zweitens die Sequenzierung Plattform hier verwendet (z.B. MiSeq) hat eine bevorzugte einfügen Längenbereich für optimale clustering und deutlich kürzere und längere Produkte dürfen nicht mit der gleichen Effizienz sequenziert werden. Teilweise kann dies rechnerisch, wie durch die Berechnung eines Korrekturfaktors für lineare Länge Bias berücksichtigt werden (siehe Abbildung 4, untere Grafik). Jedoch wenn die Region wo die 3'-Termini mapping erwünscht ist lang ist (> 1000 nt), ist es ratsamer, die Reaktionen mit den aufgespaltenen Transkripte aufgeteilt und mehrere upstream Primer verwenden, um 3'-Termini in Abschnitten zu beurteilen.
Das Auswertungsprogramm geschrieben wurde intern für den spezifischen Zweck der Analyse der beiden letzten Nukleotid der HIV-1 Sequenz angrenzend an die feste Adapter-Sequenz sowie die base Variation der alle Basen, Mutationen zu identifizieren. Die einzelnen Schritte umfassen Folgendes: Erstens sind die Adapter-Sequenzen getrimmt, mit dem Fastx-0.0.13 Toolkit; dann werden alle Sequenzen, die dupliziert werden (d.h. identische Sequenzen einschließlich des Barcodes) entfernt. Alle übrigen einzigartige Lesevorgänge werden dann auf die HIV-1 Sequenz mit Fliege (http://bowtie-bio.sourceforge.net/index.shtml) mit der maximalen Missverhältnis auf drei Basen festgelegt ausgerichtet. Die Vorlage-Sequenz besteht aus den ersten 635 nt von HIV-1 cDNA (NL4.3 Stamm), umfasst die Sss - Sequenz und der erste Strang Transfer Produkt bis zu der Polypurine Strecke (U5-R-U3-PPT; siehe Abbildung 1). Dabei sind nur die mitgelieferte Software und Vorlagen direkt geeignet, wenn die Methode für die gleiche Anwendung (Erkennung von frühen reverse Transkriptionen von HIV-1-NL4.3) verwendet wird. Anpassungen müssen für andere Zielsequenzen erfolgen. Die Positionen der 3'-Termini für jede Leseoperation wurden durch die Position auf der Achse bestimmt. Base Anrufe für jede Position werden aufgezeichnet und Mutationsraten aus die Gesamtabdeckung der jeder Basis, die variiert, wie Lesevorgänge unterschiedlich lang sind und lange Einsätze nicht vollständig durch die 125-Base Sequenzierung in Read2 abgedeckt werden können, berechnet werden.
Abschließend möchte ich sagen, glauben wir das beschriebene Verfahren zu einem wertvollen Werkzeug für viele Arten von Studien. Offensichtliche Anwendungen umfassen Untersuchungen von Mechanismen, die Hemmung der reversen Transkription durch antiretrovirale Medikamente oder zellulären Einschränkung Faktoren. Allerdings sollte nur relativ geringfügige Anpassungen notwendig sein, passen Sie das System auf 3'-Termini mapping innerhalb von anderen einzelsträngiger DNA virale Zwischenprodukte, die beispielsweise in Parvovirus Replikation vorhanden sind. Darüber hinaus bieten das Prinzip der Methode, vor allem seine optimierte Ligatur Schritt, ein zentraler Bestandteil der Vorbereitung von Bibliotheksentwurf zur Charakterisierung der 3'-DNA-Erweiterungen, einschließlich Dehnungen katalysiert durch zelluläre doppelsträngige DNA Polymerasen.
Offenlegungen
Die Autoren erklären, dass sie nichts preisgeben.
Danksagungen
Die Autoren erkennen die Unterstützung von Mitgliedern der Malim-Labor, Luis Apolonia, Jernej Ule und Rebecca Oakey. Die Autoren danken Matt Arno in die Kings College London genomische Centre und Debbie Hughes am University College London (UCL), Institut für Neurologie nächste Generation Sequencing Facility, um Hilfe mit MiSeq Sequenzierung ausgeführt wird. Die Arbeit wurde von der UK Medical Research Council (G1000196 und Herr/M001199/1, M.M.), vom Wellcome Trust (106223/Z/14/Z, M.M.), der Europäischen Kommission siebten Rahmenprogramms (RP7/2007-2013) unter Finanzhilfevereinbarung unterstützt keine. PIIF-GA-2012-329679 (, D.P.) und der Department of Health über eine National Institutes for Health Research umfassende Biomedical Research Center Award an Guy es and St. Thomas' NHS Foundation Trust in Partnerschaft mit Kings College London des Königs College Hospital NHS Foundation Trust.
Materialien
| Name | Company | Catalog Number | Comments |
| 293T cells | ATCC | CRL-3216 | |
| Dulbecco's Modified Eagle's Medium | Gibco | 31966-021 | |
| Penicillin/Streptomycin | Gibco | 15150-122 | |
| Fetal Bovine Serum | Gibco | 10270-106 | |
| HeraCell Vios 250i CO2 Incubator | Thermo Scientific | 51030966 | |
| Laminar flow hood - CAS BioMAT2 | Wolflabs | CAS001-C2R-1800 | |
| 10mm TC-treated culture dish | Corning | 430167 | |
| TrypLE™ Express (1x), Stable Trypsin Replacement Enzyme | Gibco | 12605-010 | |
| OptiMEM® (Minimal Essential Medium) | Gibco | 31985-047 | |
| HIV-1 NL4-3 Infectious Molecular Clone (pNL4-3) | NIH Aids reagent program | 114 | |
| Polyethylenimine (PEI) - MW:25000 | PolySciences Inc | 23966-2 | dissolved at 1mg/ml and adjusted to pH7 |
| RQ1- Rnase free Dnase | Promega | M6101 | |
| Filter 0.22 μm | Triple Red Limited | FPE404025 | |
| 15 mL polypropylene tubes | Corning | CLS430791 | |
| Sucrose | Calbiochem | 573113 | |
| Phosphate Buffered Saline (1x) | Gibco | 14190-094 | |
| Ultracentrifuge tubes | Beckman Coulter | 344060 | |
| Ultracentrifuge | Sorval | WX Ultra Series | Th-641 Rotor |
| Alliance HIV-1 p24 antigen ELISA kit | Perkin Elmer | NEK050001KT | |
| CEM-SS cells | NIH Aids reagent program | 776 | |
| Roswell Park Memorial Institute Medium | Gibco | 31870-025 | |
| CoStar® TC treated multiple well plates | Corning | CLS3513-50EA | |
| Benchtop centrifuge: Heraus™ Multifuge™ X3 FR | Thermo Scientific | 75004536 | |
| TX-1000 Swinging Bucket Rotor | Thermo Scientific | 75003017 | |
| Microcentrifuge: 5424R | Eppendorf | 5404000060 | |
| Total DNA extraction kit (DNeasy Blood and Tissue kit) | Qiagen | 69504 | |
| Nuclease free H2O | Ambion | AM9937 | |
| Cutsmart buffer | New England Biolabs (part of DpnI enzyme) | R0176S | |
| DpnI restriction enzyme | New England Biolabs | R0176S | |
| Oligonucleotides for qPCR | MWG Eurofins | N/A | HPSF purification |
| TaqMan PCR Universal Mastermix | Thermo | 4304437 | |
| LoBind Eppendorf® tubes | Eppendorf | 30108078 | |
| Axygen™ aerosol filter pipette tips, 1000 μL | Fisher Scientific | TF-000-R-S | |
| Axygen™ aerosol filter pipette tips, 200 μL | Fisher Scientific | TF-200-R-S | |
| Axygen™ aerosol filter pipette tips, 20 μL | Fisher Scientific | TF-20-R-S | |
| Axygen™ aerosol filter pipette tips, 10 μL | Fisher Scientific | TF-10-R-S | |
| PCR clean hood | LabCaire | Model PCR-62 | |
| DynaMag™2-magnet | Thermo | 12321D | |
| Streptavidin MagneSphere® paramagnetic particles | Promega | Z5481 | |
| Casein | Thermo Scientific | 37582 | |
| End over end rotator, Revolver™ 360° | Labnet | H5600 | |
| Tris-Base | Fisher Scientific | BP152-5 | |
| Hydrochloric Acid | Sigma | H1758-100ML | |
| EDTA disodium salt dihydrate | Electran (VWR) | 443885J | |
| Sodium Chloride | Sigma | S3014 | |
| Dri-Block® Analog Block Heater | Techne | UY-36620-13 | |
| PCR tubes and domed caps | Thermo Scientific | AB0266 | |
| PCR machine | Eppendorf | Mastercycler® series | |
| T4 DNA ligase | New England Biolabs | M0202M | |
| 40% Polyethylene glycol solution (PEG) in H2O, MW: 8000 | Sigma | P1458-25ML | |
| Betaine solution, 5M | Sigma | B0300-1VL | |
| Gel loading buffer II (formamide buffer) | Thermo Scientific | AM8546G | |
| Precast 6% TBE urea gels | Invitrogen | EC6865BOX | |
| Mini cell electrophoresis system | Invitrogen, Novex | XCell SureLock™ | |
| Tris/Borate/EDTA solution (10x) | Fisher Scientific | 10031223 | |
| Needle 21 G x1 1/2 | VWR | 613-2022 | |
| SYBR Gold nucleic acid stain (10000x) | Life Technologies | S11494 | |
| Dark Reader DR46B transilluminator | Fisher Scientific | NC9800797 | |
| Ammonium acetate | Merck | 101116 | |
| SDS solution 20% (w/v) | Biorad | 161-0418 | |
| Centrifuge tube filter | Appleton Woods | BC591 | |
| Filter Glass Fibre Gf/D 10mm | Whatman (VWR) | 512-0427 | |
| polyadenylic acid (polyA) RNA | Sigma | 10108626001 | |
| Glycogen, molecular biology grade | Thermo Scientific | R0561 | |
| Isopropanol (2-propanol) | Fisher Scientific | 15809665 | |
| Ethanol, molecular biology grade | Fisher Scientific | 10041814 | |
| Accuprime™ Supermix I (DNA polymerase premix) | Life Technologies | 12342-010 | |
| NEBNext® Multiplex Oligo for Illumina (Index Primer Set 1 and 2) | New England Biolabs | E7335S; E7500S | |
| Tapestation D1000 Screentape High sensitivity | Agilent Technologies | 5067- 5584 | |
| Tapestation D1000 Reagents | Agilent Technologies | 5067- 5585 | |
| 2200 Tapestation - automated gel electrophoresis system | Agilent Technologies | G2965AA | |
| Agencourt® AMPure® beads XP | Beckman Coulter | A63880 | |
| Qubit™ dsDNA HS Assay Kit | Invitrogen | Q32851 | |
| Qubit™ 2.0 Fluorometer | Invitrogen | Q32866 | |
| Topo™ TA cloning Kit | Invitrogen | 450071 | |
| Sequencing platform: MiSeq System | Illumina | ||
| Experiment Manager (Sample sheet software) | Illumina | Note: Use TruSeq LT as a template | |
| Miseq™ Reagent kit V3 (150 cycle) | Illumina | MS-102-3001 | |
| Sequencing hub: Basespace | Illumina | https://basespace.illumina.com | |
| Ligase A: Thermostable 5’ App DNA/RNA ligase | NEB | M0319S | Not used in this protocol, but tested in optimization process with results described in the discussion. |
| Ligase B: T4 RNA ligase 1 | NEB | M0204 | Not used in this protocol, but tested in optimization process with results described in the discussion. |
| Ligase C: CircLigase | Epicentre | CL4111K | Not used in this protocol, but tested in optimization process with results described in the discussion. |
Referenzen
- Herschhorn, A., Hizi, A. Retroviral reverse transcriptases. Cellular and Molecular Life Sciences. 67 (16), 2717-2747 (2010).
- Hu, W. S., Hughes, S. H. HIV-1 reverse transcription. Cold Spring Harbor Perspectives in Medicine. 2 (10), (2012).
- Levin, J. G., Mitra, M., Mascarenhas, A., Musier-Forsyth, K. Role of HIV-1 nucleocapsid protein in HIV-1 reverse transcription. RNA Biology. 7 (6), 754-774 (2010).
- Menendez-Arias, L., Sebastian-Martin, A., Alvarez, M. Viral reverse transcriptases. Virus Research. , (2016).
- Telesnitsky, A., Goff, S. P., Coffin, J. M., Hughes, S. H., Varmus, H. E. . Retroviruses. , (1997).
- Pollpeter, D., et al. Deep sequencing of HIV-1 reverse transcripts reveals the multifaceted antiviral functions of APOBEC3G. Nature Microbiology. 3 (2), 220-233 (2018).
- Frohman, M. A., Dush, M. K., Martin, G. R. Rapid production of full-length cDNAs from rare transcripts: amplification using a single gene-specific oligonucleotide primer. Proceedings of the National Academy of Sciences of the United States of America. 85 (23), 8998-9002 (1988).
- Liu, X., Gorovsky, M. A. Mapping the 5' and 3' ends of Tetrahymena thermophila mRNAs using RNA ligase mediated amplification of cDNA ends (RLM-RACE). Nucleic Acids Research. 21 (21), 4954-4960 (1993).
- Ince, I. A., Ozcan, K., Vlak, J. M., van Oers, M. M. Temporal classification and mapping of non-polyadenylated transcripts of an invertebrate iridovirus. Journal of General Virology. 94, 187-192 (2013).
- Hafner, M., et al. RNA-ligase-dependent biases in miRNA representation in deep-sequenced small RNA cDNA libraries. RNA. 17 (9), 1697-1712 (2011).
- Kwok, C. K., Ding, Y., Sherlock, M. E., Assmann, S. M., Bevilacqua, P. C. A hybridization-based approach for quantitative and low-bias single-stranded DNA ligation. Analytical Biochemistry. 435 (2), 181-186 (2013).
- Abram, M. E., Tsiang, M., White, K. L., Callebaut, C., Miller, M. D. A cell-based strategy to assess intrinsic inhibition efficiencies of HIV-1 reverse transcriptase inhibitors. Antimicrobial Agents and Chemotherapy. 59 (2), 838-848 (2015).
- Bishop, K. N., Verma, M., Kim, E. Y., Wolinsky, S. M., Malim, M. H. APOBEC3G inhibits elongation of HIV-1 reverse transcripts. PLoS Pathogens. 4 (12), 1000231 (2008).
- Zack, J. A., Haislip, A. M., Krogstad, P., Chen, I. S. Incompletely reverse-transcribed human immunodeficiency virus type 1 genomes in quiescent cells can function as intermediates in the retroviral life cycle. Journal of Virology. 66 (3), 1717-1725 (1992).
- Adachi, A., et al. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. Journal of Virology. 59 (2), 284-291 (1986).
- Shah, V. B., Aiken, C. In vitro uncoating of HIV-1 cores. Journal of Visualized Experiments. (57), (2011).
- JoVE Science Education Database. Science Education Database: Basic Methods in Cellular and Molecular Biology: Passaging Cells. Journal of Visualized Experiments. , (2018).
- JoVE Science Education Database. JoVE Science Education Database: Basic Methods in Cellular and Molecular Biology: Using a Hemocytometer to Count Cells. Journal of Visualized Experiments. , (2018).
- Zhou, M. Y., Gomez-Sanchez, C. E. Universal TA cloning. Current Issues in Molecular Biology. 2 (1), 1-7 (2000).
- Zhang, S., Cahalan, M. D. Purifying plasmid DNA from bacterial colonies using the QIAGEN Miniprep Kit. Journal of Visualized Experiments. (6), 247 (2007).
- Mangeat, B., et al. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature. 424 (6944), 99-103 (2003).
- Gillick, K., et al. Suppression of HIV-1 infection by APOBEC3 proteins in primary human CD4(+) T cells is associated with inhibition of processive reverse transcription as well as excessive cytidine deamination. Journal of Virology. 87 (3), 1508-1517 (2013).
- Harris, R. S., et al. DNA deamination mediates innate immunity to retroviral infection. Cell. 113 (6), 803-809 (2003).
- Zhang, H., et al. The cytidine deaminase CEM15 induces hypermutation in newly synthesized HIV-1 DNA. Nature. 424 (6944), 94-98 (2003).
- Konig, J., et al. iCLIP--transcriptome-wide mapping of protein-RNA interactions with individual nucleotide resolution. Journal of Visualized Experiments. (50), (2011).
- Troutt, A. B., McHeyzer-Williams, M. G., Pulendran, B., Nossal, G. J. Ligation-anchored PCR: a simple amplification technique with single-sided specificity. Proceedings of the National Academy of Sciences of the United States of America. 89 (20), 9823-9825 (1992).
- Zhelkovsky, A. M., McReynolds, L. A. Structure-function analysis of Methanobacterium thermoautotrophicum RNA ligase - engineering a thermostable ATP independent enzyme. BMC Molecular Biology. 13 (24), (2012).
- Li, T. W., Weeks, K. M. Structure-independent and quantitative ligation of single-stranded DNA. Analytical Biochemistry. 349 (2), 242-246 (2006).
- Gansauge, M. T., et al. Single-stranded DNA library preparation from highly degraded DNA using T4 DNA ligase. Nucleic Acids Research. 45 (10), 79 (2017).
- Ding, Y., Kwok, C. K., Tang, Y., Bevilacqua, P. C., Assmann, S. M. Genome-wide profiling of in vivo RNA structure at single-nucleotide resolution using structure-seq. Nature Protocols. 10 (7), 1050-1066 (2015).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten