
Method Article
Sequenze di molecole nascente Single-Stranded DNA virale durante HIV-1 e 3'-Termini determinante retrotrascrizione in cellule infettate
In questo articolo
Riepilogo
Qui presentiamo un metodo di sequenziamento profondo che fornisce una determinazione obiettiva della nascente 3'-termini nonché profili mutational di molecole di DNA single-stranded. L'applicazione principale è la caratterizzazione del nascente retrovirali DNAs complementare (cDNA), gli intermedi generati durante il processo di trascrizione inversa retrovirale.
Abstract
Monitoraggio di acido nucleico intermedi durante la replicazione del virus fornisce gli effetti e meccanismi d'azione di composti antivirali e proteine della cellula ospite: approfondimenti sulla sintesi del DNA virale. Qui affrontiamo la mancanza di un'analisi basata sulle celle, ad alta copertura e ad alta risoluzione che è in grado di definire la trascrizione inversa retrovirali intermedi all'interno del contesto fisiologico dell'infezione del virus. Il metodo descritto consente di acquisire le 3'-estremità delle molecole complementari nascente di DNA (cDNA) all'interno delle cellule di infezione da HIV-1 a risoluzione di singolo nucleotide. Il protocollo coinvolge raccolta dell'intera cellula del DNA, arricchimento mirato del DNA virale tramite ibrido cattura, legatura di adattatore, frazionamento di dimensione di purificazione del gel, amplificazione PCR, sequenziamento profondo e analisi dei dati. Un passo fondamentale è la legatura efficiente e imparziale delle molecole di adattatore per aprire termini 3'-DNA. Applicazione del metodo descritto determina l'abbondanza delle trascrizioni inversione di ogni lunghezza particolare in un dato campione. Fornisce anche informazioni circa la variazione di sequenza (interna) di trascrizioni inversione e quindi qualsiasi potenziali mutazioni. In generale, il dosaggio è adatto a tutte le domande relative al DNA 3'-estensione, purché la sequenza di modello è noto.
Introduzione
Per sezionare e capire la replicazione virale completamente, sempre più raffinate tecniche che catturano replica intermedi sono necessari. In particolare, la definizione precisa delle specie dell'acido nucleico virale all'interno del contesto delle cellule infettate può fornire nuove intuizioni, poiché molti meccanismi di replicazione virale hanno fino ad oggi è stato esaminato nelle reazioni isolate in vitro . Un primo esempio è il processo di trascrizione inversa nei retrovirus, come il virus dell'immunodeficienza umana 1 (HIV-1). I vari passaggi di trascrizione inversa di HIV-1, durante il quale il virale enzima transcriptase (RT) copia del genoma di single-stranded RNA in DNA double-stranded, sono stati studiati principalmente in saggi di estensione di primer con proteine purificate e acido nucleico acidi1,2,3,4,5. Mentre sono stati stabiliti i principi fondamentali, tali dosaggi non incorporano tutti i componenti virali e cellulari e potrebbero non riflettere stoichiometries biologicamente rilevanti dei fattori coinvolti. Pertanto, abbiamo progettato una tecnica potente per determinare gli spettri di trascrizione inversa intermedi con il loro preciso cDNA 3'-termini (cioè, determinare la loro esatte lunghezze) e sequenze del nucleotide nel contesto delle infezioni di vita le cellule6. Raccolta di dati da tempo esperimenti in corso possono essere utilizzati per confrontare il profilo di trascrizioni in varie condizioni, come la presenza di molecole antivirali o proteine, che possono influenzare l'efficienza e il processivity della sintesi del DNA e accumulo. Questo consente una comprensione più dettagliata del ciclo di vita naturale dell'agente patogeno, che è spesso la base per la progettazione di farmaci mirati e riuscito intervento terapeutico.
La trascrizione inversa di HIV-1 comprende una serie di eventi successivi iniziata da ricottura di un primer di tRNA per il modello di RNA genomico, che si è poi esteso da RT per produrre una breve filamento singolo cDNA trascrizione chiamata un filo meno forte-stop (-sss) (Vedi Figura 1). Successivamente, il cDNA - sss è trasferito dal terminal 5'-lungo ripetere (LTR) a 3'-LTR del RNA genomico, dove accoppia e serve come il primer per continuato RT mediato allungamento di meno filo del DNA (Vedi recensioni su trascrizione inversa1 , 2 , 3 , 4). questo primo trasferimento di filo è uno dei passaggi tasso-di limitazione di trascrizione d'inversione; quindi, il cDNA - sss è noto ad accumularsi. Il disegno di base del flusso di lavoro e biblioteca per catturare i prodotti di trascrizione inversa nelle cellule infette è descritta nella Figura 2a. I primers specifici e analisi le impostazioni che vengono utilizzate nel protocollo ed elencate nella tabella 1 destinazione tutti presto inversa trascrizione cDNA intermedi all'interno della gamma di lunghezza di 23 a ~ 650 nt, che comprende 180-182 nt - sss del DNA. Tuttavia, gli opportuni adattamenti minori alla strategia consentirà applicazione non solo fine trascrizione inversa prodotti, ma anche altri virus e sistemi, dove l'obiettivo è quello di rilevare le estremità del DNA contenente 3'-OH. Importanti limitazioni da considerare comprendono la gamma di lunghezza del prodotto PCR finale nella libreria; in particolare, modelli in cui la distanza tra l'adattatore sull'aperta 3'-terminale e il sito di primer a Monte superare ~ 1000 nt saranno probabilmente meno efficientemente sequenziati, introducendo potenzialmente ingannevoli pregiudizi tecnici durante libreria preparazione ( vedere la discussione per ulteriori dettagli e suggerimenti di adattamento).
Precedentemente segnalati tecniche per la determinazione sistematica di 3'-termini dei acidi nucleici sono concentrati su RNA e DNA, non molecole. Un esempio è 3' gara (amplificazione veloce delle estremità del cDNA)7, che si basa sulla poliadenilazione del mRNA. Inoltre, le strategie basate su legatura adattatore che impiegano ligasi di RNA sono state sviluppate, che hanno incluso RLM-RACE (gara di ligasi-mediata RNA)8 o pizzo (basato su legatura amplificazione del cDNA ends)9. È importante sottolineare che basati su legatura amplificazioni sono sensibili a qualsiasi distorsione introdotta dalla reazione di legatura si. Ad esempio, la legatura può essere più o meno efficiente a seconda di un particolare nucleotide in posizione 3', la sequenza, lunghezza totale molecola o struttura locale. Tali preferenze ligasi portano cattura incompleta delle molecole e travisamento nella lettura, che noi ed altri abbiamo osservato9,10. Per minimizzare il bias della legatura durante la procedura di aggiunta di adattatore nel protocollo descritto nel presente documento, abbiamo testato una serie di strategie di legatura e trovato l'uso di ligasi del DNA T4 con un adattatore di DNA single-stranded tornante (come descritto da Kwok et al. 11) di essere l'unica procedura con vicino la legatura quantitativa che non ha provocato le differenze significative in termini di efficienza legatura quando valutati con una serie di controllo oligonucleotidi6appositamente selezionati. La scelta di questa strategia di legatura è, dunque, una caratteristica fondamentale nel successo di questo protocollo.
Ad oggi, monitoraggio della progressione di RT di HIV-1 in cellule infettate principalmente è stata compiuta misurando prodotti trascrizione d'inversione, di varia lunghezza con PCR quantitativa (qPCR), utilizzo di set di primer-sonda che misura in modo univoco più o meno lunghi (precoce e tardi, rispettivamente) cDNA prodotti12,13,14. Mentre questo approccio qPCR è opportuno determinare l'efficienza intrinseca del processo di trascrizione inversa in sistemi cellulari, l'output è relativamente bassa risoluzione, senza informazioni di sequenza essendo derivata. Il nostro nuovo approccio, basato su adattatore ottimizzato la legatura, generazione della libreria PCR-mediata e indirizzi di sequenziamento profondo il divario tecnologico e offre la possibilità di monitorare la trascrizione d'inversione durante l'infezione da HIV-1 quantitativamente e a singolo nucleotide ad alta risoluzione.
Abbiamo illustrato l'utilità di questo metodo in uno studio che distingue tra due modelli proposti per la capacità del fattore di restrizione APOBEC3G di HIV-1 (enzima modifica catalitica polipeptide-come 3G del mRNA di apolipoproteina B) interferire con il produzione di trascrizioni virali inversa6.
Protocollo
Nota: Consultare la Tabella materiali per reagenti specifici e delle attrezzature utilizzate nel presente protocollo.
1. produzione virus e l'infezione delle cellule
Attenzione: Infettive HIV-1 deve essere gestita solo in laboratori di contenimento di biosicurezza approvato.
Nota: La produzione di particelle di HIV-1 mediante trasfezione transitoria di rene embrionale umano (HEK) 293T cellule, come descritto nel passo 1.1, sono una procedura standard ed sono stato descritto in precedenza15,16. Coltura cellulare generale le procedure sono descritte precedentemente17.
- Produzione di virus HIV-1.
- Mantenere le cellule 293T in di Dulbecco per volta medium di Eagle (DMEM) completati con 10% siero bovino fetale (FBS) e 1% di penicillina/streptomicina (DMEM completo) in un'incubatrice di coltura cellulare standard a 37 ° C e 5% CO2 come descritto in precedenza17.
- In un cappuccio di flusso laminare standard coltura tissutale è necessario rimuovere il supporto di crescita e aggiungere 3 mL di tripsina pre-riscaldata (37 ° C) a una piastra di coltura cellulare di vicino-confluenti 10cm (~1.2 x 107 cellule) delle cellule 293T. Mettere il piatto indietro nell'incubatrice per 2-3 min.
- Prendere il piatto dall'incubatrice torna nella cappa di coltura del tessuto e aggiungere 7 mL di terreno completo. Pipettare su e giù all'interno il piatto più volte per risospendere le cellule. Dividere le celle 1:4 con l'aggiunta di 2,5 mL di sospensione cellulare per un nuovo piatto di 10 cm e riempirlo con 7,5 mL di terreno completo.
- Il giorno successivo, mescolare 10 µ g di proviral HIV-1 DNA plasmidico (ad esempio pNL4.3) con 1 mL di terreno privo di siero minimo essenziale e aggiungere soluzione polyethylenimine (PEI) (25.000 mW, 1 mg/mL pH 7) a 4,5 µ l a 1 µ g di DNA. Incubare per 10 min a RT e aggiungere goccia a goccia per le cellule 293T.
- 24 ore dopo la trasfezione, rimuovere il mezzo e sostituirlo con 6 mL di DMEM completo contenente dnasi RNAsi-libera a 20 U/mL di terreno. Dopo 6 h, sostituire il mezzo con 10 mL di DMEM completo.
- 48 ore dopo la trasfezione, raccogliere il surnatante e filtrarlo attraverso un filtro da 0,22 µm, utilizzando una siringa da 10 mL, in una provetta da 15 mL in polipropilene.
- Aggiungere 2 mL di saccarosio al 20% sterile in 1x tamponato fosfato salino (PBS) in una provetta di tetto aperto sottili ultracentrifuga. Lentamente si sovrappongono il saccarosio con la cellula filtrato del surnatante.
- Centrifugare per 1 h e 15 min a 134.000 x g a 4 ° C, utilizzando un'ultracentrifuga.
- Rimuovere con attenzione i tubi dall'ultracentrifuga. Togliere lentamente il surnatante e il saccarosio mediante un'aspirazione o una pipetta. Utilizzare una pipetta più piccola e inclinare il tubo quando stipula l'ultima soluzione di saccarosio. Lasciare il virus pellettato nella parte inferiore del tubo.
Nota: È possibile che il pellet non sarà visibile. - Aggiungere 200 µ l di PBS 1X, lasciare in frigo per 4 a 12 h, risospendere e congelare in 20 aliquote µ l a-80 ° C.
- Determinare p24Gag contenuto utilizzando l'un antigene di HIV-1 p24 ELISA kit (istruzioni del produttore riportato di seguito).
- Infezione di linea di T-cellula.
- Cultura di una linea di T-cellule immortalizzata (ad es., cellule di CEM-SS) in medium di Roswell Park Memorial Institute (RPMI) 1640 supplementato con 10% FBS e 1% penicillina/streptomicina (RPMI completo). Contare le celle utilizzando un emocitometro18 e seme 1 pozzetto per esempio con 1 mL di RPMI completo con 2 x 106 cellule/mL in una piastra di coltura cellulare formato 12-pozzetti.
- Aggiungere particelle di HIV-1 equivalente a 150 ng p24Gag e posizionare la piastra in un'oscillazione centrifugare secchio con biocontenimento ClickSeal per spin-infettare tramite centrifugazione in una centrifuga da banco per 2 h a 2.000 x g a 30 ° C.
- Rimuovere la piastra dalla centrifuga e lasciarlo riposare per 1 ora in un incubatore di coltura del tessuto standard a 37 ° C e 5% CO2.
- Per lavare il virus in ingresso, è possibile raccogliere le cellule trasferendo le sospensioni cellulari per microcentrifuga e centrifugazione in una microcentrifuga a RT (RT) a 500 x g per 2 min. Decollare il sopranatante senza disturbare il pellet cellulare.
- Risospendere il pellet di cellule in 1 mL di pre-riscaldata (37 ° C), PBS 1X sterile. Ripetere la centrifugazione, rimozione del surnatante e risospensione passi due volte.
- Centrifugare nuovamente, rimuovere il supernatante e risospendere il pellet di cellule in 1 mL di RPMI completo. Aggiungere ogni sospensione ad un pozzo in una nuova piastra di ben 12.
- In aggiunta post-iniziale di 6 h di virus (post-centrifugazione 4h), raccogliere le cellule mediante centrifugazione come fatto nel passaggio 1.2.4. Rimuovere e scartare il surnatante. Il pellet di cellule può essere congelato a-80 ° C o trattato direttamente per estrazione del DNA.
2. DNA estrazione, la quantificazione del DNA di HIV-1 e arricchimento di Hybrid Capture
- Estrarre il DNA di cellule intere con un kit di estrazione di DNA sangue e tessuto totale seguendo il manuale kit per cellule di coltura del tessuto. L'unico cambiamento è eluizione in 200 µ l di nucleasi-free H2O invece che il buffer di eluizione fornito.
Nota: Dopo l'aggiunta del buffer di lisi caotropici (del kit "AL buffer") e proteinasi, campioni possono essere rimosso dal laboratorio di contenimento di biosicurezza e gestiti in un laboratorio di livello di sicurezza standard per il resto del protocollo. - Determinare il numero di copie di HIV-1 cDNA di qPCR.
- Prendere 17 µ l di eluato dal punto 2.1 e aggiungere 2 µ l di tampone di enzima di restrizione x 10 insieme a 1 µ l di enzima di restrizione DpnI. Incubare per 1h a 37 ° C per rimuovere qualsiasi potenziale residuo input DNA plasmidico dalla transfezione.
- Svolgere qPCR per meno-filo forte-stop cDNA utilizzando il seguente set di primer sonda: oHC64 (5 ′-taactagggaacccactgc-3 ') e oHC65 (5 ′-gctagagattttccacactg-3 ') e la sonda oHC66 (5 ′-FAM-acacaacagacgggcacacacta-TAMRA-3 ′). installazione di qPCR e condizioni esatte si trovano nei riferimenti6,13. Portare con sé i campioni con una diluizione seriale del plasmide proviral pNL4.3 come una curva standard per determinare il numero di copie di molecole di cDNA.
Nota: Vedi discussione per quantitativi attesi.
- Arricchimento di HIV-1 DNA di cattura di ibrido.
Nota: Da questo passo in avanti, è preferibile utilizzare per microcentrifuga con proprietà di associazione a basso tenore di acido nucleico, nonché puntali aerosol filtro per tutti i campioni di DNA. Se possibile, lavorare in una workstation PCR. Tutti i passaggi e i reagenti sono a RT (RT) salvo diversa indicazione.- Per preparare un mix master dei branelli magnetici streptavidina, pipettare 100 perline µ l per campione in una provetta da microcentrifuga singolo. Posizionare il tubo su un magnete adatto per microcentrifuga.
- Dopo che le perle sono stabiliti verso il lato del magnete del tubo (~ 1 min), togliere il buffer di memoria, rimuovere il tubo dal magnete e risospendere perline in 500 µ l di binding e lavare buffer (buffer di BW, 5 mM Tris-HCL pH 7.5, 0,5 mM EDTA NaCl di 1m) per lavare.
- Riposizionare il tubo sul magnete, rimuovere il supernatante e aggiungere 500 µ l di soluzione di caseina. Prendere del magnete, risospendere e incubare per 10 min a RT, quindi lavare con tampone di BW.
Nota: Un lavaggio si riferisce al posizionamento del tubo sul magnete, prendendo il sovranatante, prendendo il tubo fuori il magnete, aggiungendo il buffer e risospendere. - Riposizionare il tubo sul magnete, togliere il supernatante e risospendere perline in 500 µ l di tampone di BW. Aggiungere 50 pmoli di ogni acquisizione oligonucleotidi biotinilati (Vedi tabella 1, i tre oligos in questo caso) per campione. (Ad esempio, se 5 campioni di DNA sono da elaborare, utilizzare 500 µ l di biglie magnetiche da passo 2.3.1 e 250 pmol di ogni oligonucleotide).
- Incubare per 30 min a RT mentre a dondolo in un mixer più fine.
- Lavare perline con gli oligonucleotidi immobilizzati due volte con 500 µ l di tampone dieci 1x (10 mM Tris-HCl pH 8.0, 1 mM EDTA, NaCl 100 mM).
- Risospendere perline in 10 µ l di tampone dieci 1x per campione.
- Per ogni campione etichettare un tubo del microcentrifuge e aggiungere 10 µ l di sospensione di perline, 170 µ l di DNA (dal punto 2.1) e 90 µ l di tampone dieci: 3x. Incubare in un blocco di calore secco a 92 ° C per 2 min denaturare il DNA.
- Spostare i tubi in un blocco diverso calore secco, che è impostato a 52 ° C, e incubare per 1 h. Inverti per mescolare regolarmente (~ ogni 10 min) durante questa incubazione.
- Lavare una volta con 500 µ l di tampone di dieci 1x e risospendere in 35 µ l privo di nucleasi H2O.
- Per eluire, incubare le provette a 92 ° C in un blocco di calore secco per 2 min. Quindi, spostare rapidamente i tubi sul magnete (un tubo alla volta). Una volta perline sono vincolati al lato del tubo, trasferire il surnatante contenente il DNA di HIV-1 in una nuova provetta.
- Opzionale: Ripetere qPCR (come fatto al punto 2.2.2) per determinare il cDNA recuperato di HIV-1.
Nota: Vedi discussione per quantitativi attesi.
3. adattatore legatura
- Preparare l'adattatore
- Risospendere adattatore liofilizzato (Vedi tabella 1 "completo Kwok + MiSeq") a 100 µM a nucleasi-free H2O.
- Per esempio, più un controllo campione, combinare 0,45 µ l di 10 x T4 DNA ligasi buffer, 4 µ l di adattatore e 0,05 µ l di nucleasi-free H2O. Heat a 92 ° C per 2 min e lasciare ogni raffreddare lentamente.
Nota: Se l'opzione è disponibile è possibile utilizzare una macchina PCR con velocità di raffreddamento regolabile (2% tasso di utilizzo). Questo richiede circa 30 min da 92 ° C a 16 ° C. In alternativa, utilizzare un blocco di calore secco a 92 ° C e spegnere. Estrarre l'adattatore mastermix quando il blocco di calore è tornato a TA. Questo è di lasciare l'adattatore a formare una struttura di tornante (Vedi Figura 2b).
- Preparare una reazione di controllo con un set di oligonucleotidi sintetizzati (Vedi tabella 2) invece di DNA estratto dalle cellule.
- Fanno scorte di 100 µM di ogni oligonucleotide. Mescolare 1 µ l di ciascuno del 17 oligonucelotides e aggiungere 8 µ l di H2O per un rapporto equimolare in un volume finale di 25 µ l.
- Diluire il 1:2,500 mix a nucleasi-free H2O a una diluizione seriale. Unire 1 µ l della miscela con 17,3 µ l di nucleasi-free H2O utilizzare nella legatura del campione di controllo nel passaggio 3.3.1 in modo che ogni oligonucleotide è presente al fmol 1,6 (equivalente a 0,026 nM nella reazione 60 µ l).
- Configurazione di legature
- Per combinano reazioni di volume finale 60 µ l in provette PCR 6 µ l di 10 x T4 DNA ligasi del buffer, 24 µ l di 40% PEG, 6 µ l di 5m betaina, 4,5 µ l (400 pmol) dell'adattatore (pre-cotto come descritto al punto 3.1.2), 1,2 µ l di T4 DNA ligasi (2.000.000 unità/mL) e 18,3 µ l di DNA (da Passo 2.3.11)
Nota: Prestare particolare attenzione con soluzioni viscose quali 40% PEG per mantenere accurati volumi. Non fare un mastermix. - Impostare la stessa reazione come eseguita nel passaggio 3.3.1 ma con il controllo del oligonucleotide mix preparata al punto 3.2.2.
- Mescolare bene le reazioni e incubare in una macchina PCR a 16 ° C durante la notte.
- Per combinano reazioni di volume finale 60 µ l in provette PCR 6 µ l di 10 x T4 DNA ligasi del buffer, 24 µ l di 40% PEG, 6 µ l di 5m betaina, 4,5 µ l (400 pmol) dell'adattatore (pre-cotto come descritto al punto 3.1.2), 1,2 µ l di T4 DNA ligasi (2.000.000 unità/mL) e 18,3 µ l di DNA (da Passo 2.3.11)
4. adattatore rimozione e separazione dimensioni
- Denaturare Elettroforesi del gel
- Aggiungere 30 µ l di buffer per ogni reazione di legatura di caricamento del gel del DNA contenente formammide. Mescolare bene il pipettaggio.
- Scaldare per 2 min a 94 ° C in macchina PCR, poi immediatamente mettere il ghiaccio.
- Collocare un prefabbricato 6% Tris/Borato/EDTA (TBE) urea poliacrilammide gel denaturante (10 pozzetti pettine) in un carro armato del gel appropriato. Aggiungere 1 tampone di corsa di x TBE (89 mM di Tris-base, acido borico 89 mM, 2 mM EDTA) e pre-corsa il gel per 20 min a costante di 250 V/max.
- Lavare le tasche di gel con tampone utilizzando una siringa ed un ago 21G di corsa.
- Carico ogni 90 µ l di campione in tre pozzi (30 µ l per pozzetto) ed eseguire per 20 min (250 V/max) fino alla parte anteriore di colorante blu scuro è a metà strada attraverso il gel.
- Colorazione e taglio degli acidi nucleici dal gel
- Preparare 3 microcentrifuga piccolo (0,5 mL) per campione da frugando fori nella parte inferiore utilizzando un ago di siringa di 21 G (prendere attenzione mentre si lavora con diesis). Inserire ciascuna delle provette preparate in un tubo del microcentrifuge 2,0 mL e con l'etichetta con il nome di campione più "basso", "medio" o "alto".
- Estrarre e aprire il vassoio del gel. Tagliare il gel verticalmente con una lama di rasoio generosamente per asportare la striscia con i 3 pozzi di campioni caricati. Aggiungi la striscia di gel a un contenitore con 1 x TBE (circa 30 mL) e 5 µ l di macchia di acido nucleico cianina. Incubare per 3-5 min.
Nota: La fase di estrazione del gel è particolarmente sensibile alla contaminazione. Si consiglia di eseguire solo 1 campione al gel e utilizzando un contenitore separato, pulito per ogni gel di colorazione. Guanti dovrebbero essere cambiati se particelle di gel contattare le dita guantate. - Pulire la superficie di un transilluminatore luce blu accuratamente con ddH2O. Prendere il pezzo di gel dal contenitore della colorazione e aggiungerlo alla casella di luce.
- Accendere la luce box e ispezionare gli acidi nucleici macchiati attraverso il filtro arancio.
Nota: L'adattatore in genere appare overload e viene eseguito come grande "blob" con dei DNA di HIV-1 che corre sopra come una striscia. - Utilizzando una nuova lama di rasoio, tagliata via i lati del gel, se ci sono aree con nessun campione caricato ancora presenti. Quindi, tagliare appena sopra l'adattatore per rimuovere l'adattatore e parti inferiori del gel. Infine, tagliare via cima del gel compreso circa 1mm del gel tasche, che spesso hanno un forte segnale intenso di più alto peso molecolare del DNA.
- Dividere la parte restante di gel contenente il campione, che è in genere ~ 2 x 3 cm nel formato, orizzontalmente in tre pezzi anche: "bassa", "media" e "alto" peso molecolare aree.
Nota: Ogni pezzo sarà ora trattato separatamente [cioè., ci saranno tre tubi (bassa, media e alta)] al campione originale. - Ciascuno dei tre gel frammenti tagliati in piccoli pezzi (~ 2 x 2 particelle mm) e trasferirli nelle provette microcentrifuga prepped 0,5 mL (punto 4.2.1).
- Girare alla massima velocità con coperchi aperti per 1 min spremere i pezzi gel attraverso il foro nel tubo 2ml per creare una fanghiglia di gel. Se eventuali particelle di gel rimangono nella parte inferiore del tubo 0,5 mL, trasferirli al tubo 2ml manualmente utilizzando una punta di spillo o pipetta.
- Estrazione del DNA
- Aggiungere 1 mL di tampone di estrazione del gel di urea (0,5 M NH4CH3CO2, 1 mM EDTA, 0,2% SDS) lo Slusho gel. Ruotare i tubi per un minimo di 3 ore (durante la notte è accettabile) presso RT con un mixer più fine.
- Utilizzare un set di pinzette puliti per aggiungere un filtro di fibra di vetro rotondo piccolo a centrifuga colonne con filtri a membrana in acetato di cellulosa (0,2 µm), che evita l'intasamento della membrana. Mettere il filtro in luogo con una pipetta invertito.
- Brevemente spin le provette da 2 mL con buffer di fanghiglia ed estrazione di gel in una microcentrifuga e trasferire 700 µ l del surnatante le colonne filtro preparato. Tenere il gel di fanghiglia e surnatante rimanenti.
- Centrifugare le colonne di filtro in una microcentrifuga alla massima velocità per 1 minuto l'attraversamento da trasferire in un nuovo tubo del microcentrifuge 2,0 mL.
- Ricaricare le colonne con il surnatante rimanente. Cercare di ottenere quanto più liquido possibile dall'estrazione di fanghiglia. Trasferimento di pezzi gel non è una preoccupazione. Girare di nuovo e combinare flowthroughs degli stessi campioni di estrazione.
- Precipitazione del DNA
- 3 µ l di polyA RNA (1 µ g / µ l; come un elemento portante), 1 µ l di glicogeno e 0,7 mL di isopropanolo per l'attraversamento del passaggio 4.3.5. Vortex brevemente e congelare a-80 ° C durante la notte.
- Prelevare campioni fuori dal congelatore a-80 ° C e lasciarli scongelare brevemente. Metterli in una microcentrifuga raffreddato (4 ° C) e spin per 30 min a velocità massima.
- Rimuovere e scartare il surnatante. Essere molto attenti a non rimuovere il pellet. Se è incerto che pellet sarebbe stato rimosso in caso contrario, lasciare 30-50 µ l di liquido.
Nota: In genere tutti gli esempi di "alti" mostrano un pellet più visibile rispetto a campioni di "medio" e "bassi". - Aggiungere 800 µ l di etanolo di 80%. Invertire i tubi e spin ancora per 1 min alla massima velocità. Rimuovere la maggior parte dell'etanolo con una pipetta, brevemente girare di nuovo i tubi e rimuovere più etanolo con una pipetta a volume più piccola.
- Evaporare qualsiasi residuo etanolo inserendo i tubi con un coperchio aperto in un blocco di calore secco di 55 ° C. Quando i campioni sono a secco (2-4 min) aggiungere 20 µ l di nucleasi-free H2O e diffusione intorno alla parte inferiore del tubo per garantire il pellet di DNA è dissolto. Il campione di DNA può essere conservato a-20 ° C.
5. PCR amplificazione e preparazione di biblioteca
- Impostare una reazione di PCR di 40 µ l con 20 µ l di DNA polimerasi pre-mix, 18 µ l di precipitato e ridisciolto DNA dal passaggio 4.4.5, 1 µ l di primer forward "MP1.0 + 22" (10 µm) (Vedi tabella 1) e 1 µ l di primer oligo multiplex (primer indice 1-24) (Vedi tabella di Ma teriali).
Nota: Eseguire le tre reazioni (basso, medio, alto) di ciascun campione in reazioni di PCR separate, ma con lo stesso indicizzata primer. Utilizzare un indice diverso per ciascuno dei campioni originali di infezione.- Eseguire le reazioni di PCR nelle seguenti condizioni: 2 min a denaturazione 94 ° C, quindi 18 cicli della PCR 3-passo; 15 s a denaturazione 94 ° C, 15 s ricottura a 55 ° C e 30 s estensione a 68 ° C.
- Come opzione di controllo di qualità, analizzare le reazioni di PCR con sistema di elettroforesi del gel di alta sensibilità automatizzato. Prendere 2 µ l di un campione di basso, media e alto per l'esecuzione come da istruzioni del produttore.
Nota: I due primer devono essere visibile e spesso vengono eseguiti con una lunghezza calcolata di circa 45 e 95 nt (lunghezza effettiva è diversa). Inoltre, il DNA dovrebbe essere rilevato tra 150 e 500 nt. Se non è presente alcun segnale, si consiglia di aggiungere ulteriori cicli PCR, tra 2 e 10 ulteriori cicli. Non aggiungere ulteriori cicli per i campioni di controllo del oligonucleotide creati al punto 3.3.2. - Per rimuovere i primer utilizzano un sistema di pulizia PCR basato su tallone paramagnetico.
- Prendere 20 µ l di ogni reazione di PCR e la piscina i campioni insieme (mescolare a questo punto tutti i campioni). Congelare le reazioni di µ l 20 restanti come backup a-20 ° C.
- Lasciate che i branelli paramagnetici raggiungere RT e mescolare le reazioni di PCR in pool con 1.8 x il volume della soluzione della perla. Mescolare pipettando e incubare per 5 min.
Nota: Come un esempio, se 4 campioni sono stati preparati e ciascuno hanno reazioni di basse, media e alte, il volume sarebbe 4 x 3 x 20 µ l = 240 reazioni di PCR µ l con una soluzione di perlina di 432 µ l. - Mettere i tubi su un magnete di tubo del microcentrifuge, lasciate le perline si legano per ~ 1 min e prendere il sovranatante di disfarsi. Lasciare le provette sul magnete e aggiungere 500 µ l di etanolo di 80%.
- Lasciare l'etanolo per 30 s, poi togliere accuratamente e lasciare il airdry perline per ~ 5 min aggiungere 40 µ l di nucleasi-free H2O, prendere i tubi fuori il magnete e pipettare su e giù più volte.
- Lasciare la sospensione per 5 min, mettere il tubo indietro sul magnete, lasciate le perline e trasferire il surnatante in una provetta nuova trasferirsi al lato. Questa è la libreria. Prendere un 10 µ l aliquota per controlli di qualità e congelare il resto a-20 ° C.
6. valutare la libreria
- Determinare la qualità di biblioteca, la concentrazione e la molarità.
- Utilizzare un metodo di Quantificazione fluorometrica. Misurare 1 µ l e 3 µ l della libreria con un kit di test ad alta sensibilità dsDNA seguendo le istruzioni del produttore.
Nota: Le concentrazioni tipiche sono tra 1 e 10 ng / µ l. - Lo spettro di peso molecolare di DNA biblioteca dall'alta sensibilità di misura automatizzato elettroforesi del gel come descritto in precedenza (punto 5.2).
- Uso l'automatizzato gel analisi elettroforesi per determinare il peso molecolare medio della biblioteca e calcola per diluire la biblioteca di nucleasi-free H2O a 4 nM. Diverse librerie possono essere combinate, purché tutti gli indici sono unici.
- Utilizzare un metodo di Quantificazione fluorometrica. Misurare 1 µ l e 3 µ l della libreria con un kit di test ad alta sensibilità dsDNA seguendo le istruzioni del produttore.
- Opzionale e bassa velocità effettiva di controllo di qualità
- La libreria di DNA in base alla clonazione19 TA inserire molecole libreria vettori per l'amplificazione. Seguire le istruzioni del kit, crescere fuori ~ 10-20 colonie ed Estratto di DNA tramite miniprep protocolli, come descritto qui20.
- Vettore di sequenza utilizzando servizi di sequenziamento locale e verificare che gli inserti contengono il desiderato HIV-1 derivato sequenze e libreria specifici adattatori.
7. high-Throughput sequenziamento Run
- Creare un foglio di esempio di sequenziamento con il software commerciale fornito con la piattaforma di sequenziamento.
- Indicare il kit selezionato sequenziamento. In genere, scegli un kit 150-ciclo, ma altri sono adatti a seconda della lunghezza di lettura desiderata.
- Selezionare "Fastq solo" come il flusso di lavoro di applicazione. Scegli uno dei modelli che contiene gli 24 indici presenti nei kit multiplex del oligonucleotide (indicato nel manuale kit).
- Selezionare "25 nt" per Read1 e "125 nt" per Read2. Tenere 6 nt per singolo indice leggere.
Nota: L'analisi in loco solo Read2 viene utilizzato nell'analisi. Tenere Read1 a un minimo di 25 nt per scopi di algoritmo di piattaforma di sequenziamento.
- Seguire le istruzioni del produttore proprio per la preparazione di libreria pre-esecuzione e installazione. Optare per il massimo 20 pM concentrazione e uso un picco di PhiX 15%, come la biblioteca è molto bassa complessità.
8. analisi dei dati
- Verifica se la percentuale di filtro di pass e Punteggio di qualità medio Q30 è accettabile secondo le istruzioni del produttore del piattaforma di sequenziamento.
Nota: Filtro passa in genere è > 90% e Q30 punteggi sono in genere > 80%. - Scarica il. file fastq.gz da hub di sequenziamento del produttore.
- Impostare lo script di sequenziamento
- Creare una nuova directory (cartella) denominata "AnalysisXYZ" e di andare a https://github.com/malimlab/seqparse per scaricare tutti i file di codice sorgente (parse_sam.pl, rc_extract.pl, parse.sh) in questa directory.
- Scarica il breve lettura aligner Bowtie, versione 1.1.2, da http://bowtie-bio.sourceforge.net/index.shtml nella stessa directory.
- Il download verrà creata una sottodirectory all'interno di "AnalysisXYZ" denominato "Papillon-1.1.2". All'interno di questa directory aprire la sottodirectory "indici" e scaricare le sequenze di modello fornito che consiste di 6 file con estensioni di .ebwt.
- Scarica il FASTQ/A brevi letture di pre-processing toolkit fastx-0.0.13 da http://hannonlab.cshl.edu/fastx_toolkit/download.html nella directory "AnalysisXYZ".
- Scaricare sia Samtools (https://sourceforge.net/projects/samtools/files/) e bam-readcount (https://github.com/genome/bam-readcount) nella directory "Documenti".
- Spostare il. file fastq.gz, scaricati al punto 8.2, di tutte le leggi 2s (che termina in... _R2_001.fastq.gz) nella directory "AnalysisXYZ".
- Aprire una console/terminale di comando. Spostare il "AnalysisXYZ" come directory corrente utilizzando i comandi cd. Tipo "./parse.sh." per eseguire gli script.
- Trovare i file CSV con riepiloghi per tutti i campioni sul totale lettura conta, lunghezza regolata leggere conteggi e normalizza la lettura conta, così come i file con la variazione di base, per ciascun campione in una directory denominata parse_results all'interno della directory "Analisi XYZ".
Nota: Vedere la discussione per ulteriori informazioni sul processo di analisi. Lo script restituisce i file csv con totale letture per ogni nucleotide lungo le sequenze di HIV-1NL4.3 forte-stop e il primo filo trasferimento fino al nucleotide 635. Come una guida, 50.000 a 100.000 uniche letture sono tipicamente osservate in campioni da infezioni con i numeri di cella indicata e inoculi virali e senza proteine antivirali o composti. Il campione di controllo del oligonucleotide di solito produce 100.000 a 200.000 letture.
Risultati
La tecnica descritta in questo articolo è stata applicata ad uno studio più ampio per affrontare i meccanismi di inibizione della trascrizione inversa di HIV-1 di proteina umana antiretrovirale APOBEC3G (A3G)6. La figura 3 Mostra i risultati rappresentativi ottenuti dopo che impiegano il protocollo in campioni da CEM-SS T-cellule infettate con vif-carenti HIV-1 in assenza o in presenza di A3G. Il numero totale di letture unici ottenuti da ogni campione dopo il filtraggio eventuali duplicati PCR che hanno la stessa 6 tracciati in Figura 3unnt codice a barre e la stessa lunghezza (cantata dal software di analisi fornito). I livelli aumentanti di A3G riducono il numero totale di lettura che riflette l'effetto inibitorio di A3G sulla RT mediato sintesi di cDNA precedentemente descritti e misurati da qPCR6,13,21,22. Nella Figura 3b, la frazione di molecole ad ogni possibile lunghezza all'interno del primo 182 nt sono mostrati. Infezione da HIV-1 in assenza di A3G, la specie più abbondante è il principale 180 nt forte-stop molecola stessa, con qualche accumulo di letture nella gamma più corta (23 a 40 nt) (istogrammi grafico, blu in alto). L'aggiunta di modifiche A3G che questo profilo come un forte incremento della più breve, troncato molecole di cDNA a poche posizioni molto specifici, riproducibile è rilevato (grafici di medio e Bassi). Poiché A3G è una citidina deaminasi, citosina-a-uridina (identificata come C-a-T) mutazioni nel cDNA verificano quando A3G è presente nelle infettante virioni21,23,24. Utilizzando le informazioni di sequenza ottenuta, la percentuale di mutazioni di C-a-T è stata tracciata sul grafico stesso (linea rossa tratteggiata). Dovrebbe essere notato che il profilo mutazionale deriva da letture tutti uniche combinate e copertura di ogni nucleotide varierà. Tuttavia, se richiesto informazioni di sequenza possono essere legati indietro ad ogni molecola e correlati con un specifico 3'-terminale. Sono stati considerati i dati forniti da Pollpeter et al. 6 e la correlazione tra mutazionale e profili di lunghezza di cDNA è stata dimostrata per essere dovuto il rilevamento e la fenditura di deaminated cDNA da cellulare macchine di riparazione del DNA.
Un controllo positivo per l'approccio di 3'-mappatura possa essere prodotte facilmente dalla lavorazione di un pool di oligonucleotidi sintetici di sequenza nota, la lunghezza e la concentrazione. Questo controllo viene aggiunto a legatura adattatore nel passaggio 3.3.2 e consigliato di essere incluso in tutte le librerie "multiplex". I dati ottenuti da un campione di controllo dovrebbero avere tutti gli oligonucleotidi ai rapporti di input previsti, con letture di sfondo solo molto minori. La figura 4 Mostra i risultati di un gruppo di controllo positivo di 17 oligonucleotidi sintetizzati chimicamente (per sequenze, vedere tabella 2), che sono stati miscelati a rapporti equimolari. Come previsto, tutte le molecole appaiono nel vicino uguale abbondanza con solo piccole variazioni (grafico in alto). Mentre la maggior parte delle posizioni all'interno della sequenza di DNA - sss che non erano rappresentate da un oligonucleotide restituire zero lettura conta, abbiamo osservato le specie minori che sono 1 o 2 nt inferiore i oligonucleotides di effettivo controllo. Ci non sono ulteriormente studiati queste specie minori ma si supponga che rappresentano prodotti degradati o incompleti potenzialmente presenti nelle scorte del oligonucleotide fornito al momento dell'acquisto (oligonucleotidi sono state ordinate come HPLC purificato, per il quale il produttore indica > 80% di purezza). Il grafico inferiore mostra il campione di controllo da un'altra libreria esegue, dove la variazione è leggermente più alto tra i 17 oligonucleotides e correla con la lunghezza totale in quanto più molecole di controllo vengono rilevati in modo più efficiente quindi più brevi. Questo può essere dovuto a una distorsione minore in reazioni di PCR o nel clustering durante la sequenza di MiSeq, che ha un optimum di dimensione inserto e può verificarsi con le librerie che trasportano gli intervalli particolarmente ampio inserto. Un modo di base per affrontare questa polarizzazione è l'applicazione di un fattore di normalizzazione basato sul pendio che indica il bias correlare alla lunghezza della molecola (linea rosa). I calcoli necessari sono inclusi nel programma di analisi (Vedi punto 8.3 nel protocollo).

Figura 1: diagramma che mostra i primi passi di HIV-1 retrotrascrizione. Il processo inizia con ricottura di tRNA(Lys,3) (arancione) per il sito di legame del primer (PBS) nel RNA genomico virale (passaggio 1), che permette l'avvio e l'allungamento del cDNA virale (blu, passaggio 2). Simultaneamente, il RNA genomico modello è degradato da RNaseH attività di RT (passaggio 3). Il primo intermedio completo nel processo di trascrizione inversa è la meno-filo forte fermata (-) sss cDNA, che è completa quando la polimerizzazione catalizzata RT raggiunge il 5'-capolinea dell'area (R) ripetuta gRNA (passaggio 3). Sss (-) intermedio viene trasferito al 3'-terminale del modello RNA genomico di ricottura per la regione (LTR) R di ripetizione terminale lunga 3' complementare. Da qui, polimerizzazione continua (passaggio 4). Nel metodo descritto la progressione di trascrizione d'inversione è determinata dal mapping la lunghezza esatta del cDNA virale nascente (blu). PPT, tratto polipurinico; U5, 5'-sequenza univoca; U3, 3'-sequenza unica. Questa figura è ripubblicata da una precedente pubblicazione6. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}

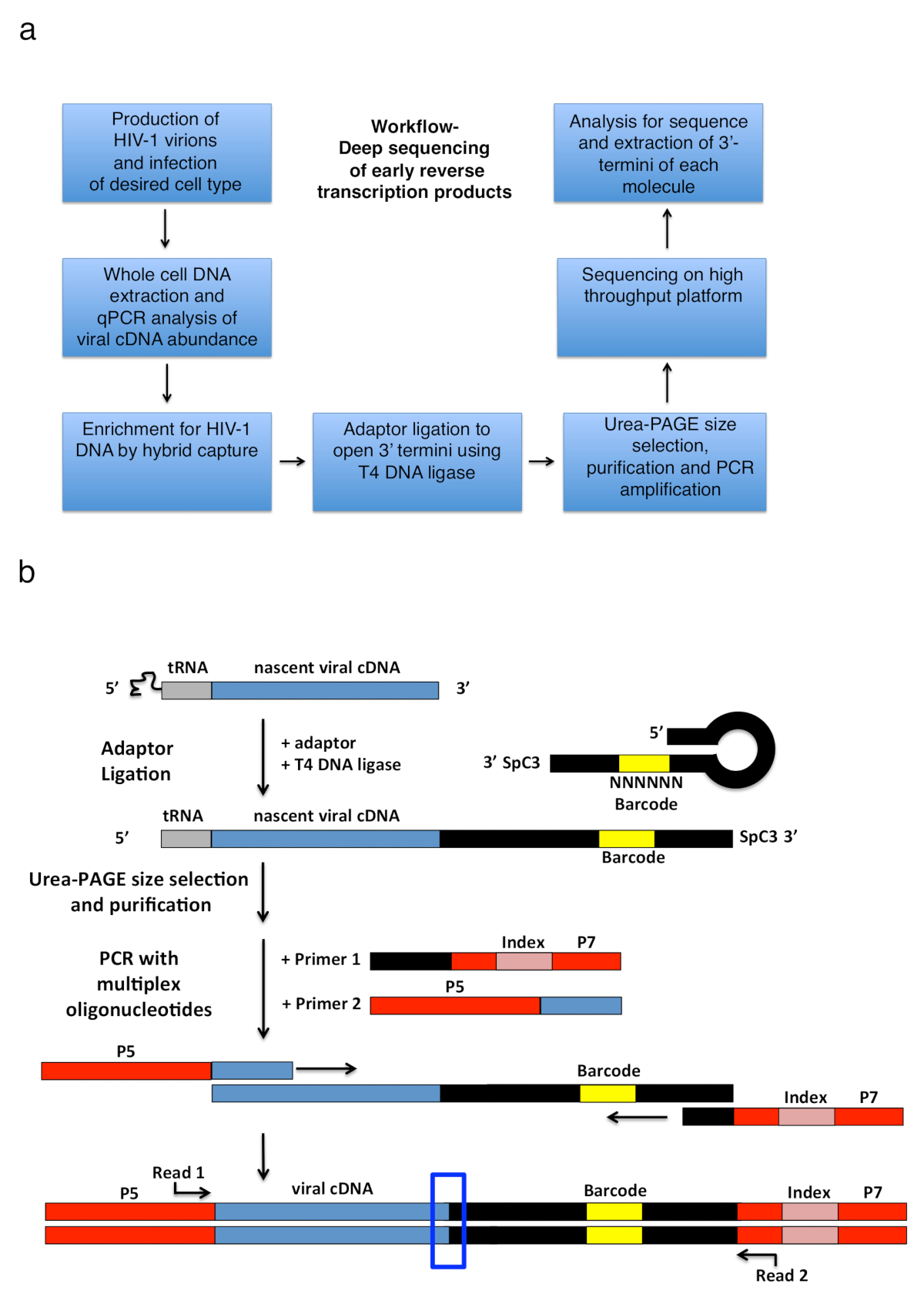
Figura 2 : Struttura di flusso di lavoro e gli schemi della legatura adattatore e strategia di amplificazione PCR. (a) flusso di lavoro che illustra i passi principali per la tecnica descritta per determinare 3'-termini di HIV-1 inversa le trascrizioni in cellule infettate. La figura è adattata da una precedente pubblicazione6. (b) schema della legatura di adattatore e strategia di amplificazione di PCR. Molecole di cDNA nascente di lunghezza variabile che sono stati purificati nei passaggi precedenti sono legati a un adattatore di DNA single-stranded utilizzando ligasi del DNA T4. L'adattatore di tornante (denominato "completo Kwok + MiSeq", Vedi tabella 1) design è stato ispirato da Kwok et al. 11. l'adattatore trasporta un casuale 6 sequenza di codici a barre nt, che consente per appaiamento per facilitare la legatura e contemporaneamente funge da identificatore per letture uniche. Le 3'-estremità dell'adattatore trasporta un distanziatore (SpC3) per evitare auto-legatura. Dei prodotti sono separati da adattatore in eccesso da denaturare elettroforesi del gel di poliacrilamide (pagina). Acidi nucleici nel gel sono macchiati e tagliati in tre pezzi separati, dimensioni uguali il gel nella zona da sopra l'adattatore al pozzo come fatto in25. Dopo eluizione, le precipitazioni e risospensione, i prodotti sono PCR amplificato con iniettori che temprano a sequenza nota dell'adattatore (primer 1, kit del oligonucleotide multiplex, Vedi Tabella materiali) e un primer che trasportano le prime 22 nt del HIV-1 Sequenza 5'-LTR immediatamente dopo il tRNA (primer 2, MP1.0 + 22). La 5'-termini dei primer selezionate portare adattatori per la piattaforma di sequenziamento selezionate (P5 e P7) così come una sequenza di indice per distinguere i singoli campioni eseguiti nella stessa libreria. Punti di partenza della sequenziazione leggere gli iniettori sono indicati. La scatola blu indica la regione di interesse per determinare le originale 3'-estremità della molecola catturata. Questa figura è adattata da una precedente pubblicazione6. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 3 : Risultati rappresentativi. (a) il numero totale di lettura di campioni rappresentativi, trattati con il protocollo descritto. Questo include tutte le sequenze che sono state identificate come uniche letture di molecole di HIV-1 con loro 3'-termini entro il primo 635 nt di meno del cDNA del filo (fino al PPT, Vedi Figura 1). L'infezione con HIV-1 non trasportano A3G produce il maggior numero di letture, mentre A3G inibisce la sintesi del cDNA e quindi riduce il totale leggere totali. Le cellule non infette servito come controllo negativo, mentre un set di oligonucleotidi sintetici fornisce un controllo positivo. b) l'abbondanza relativa di cDNA per ogni lunghezza tra le posizioni di nt 23 e 182 (cDNA full-lenght - sss è 180-182 nt) l' HIV-1NL4.3 sequenza (asse x) è mostrato in blu istogrammi (scala sull'asse y di sinistra). L'abbondanza relativa di cDNA è stato calcolato dal numero assoluto di sequenze di terminazione a un nucleotide specificato all'interno della sequenza di cDNA - sss diviso per la somma di tutte le letture 182nt di misura o meno. Indicato nelle linee rosse tratteggiate sono le percentuali di letture che trasportano le mutazioni di C-a-T/U presso la rispettiva posizione (scala sugli assi y destra–). Figura 3 b viene ripubblicata da una precedente pubblicazione6. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 4 : Risultati rappresentativi dei campioni di controllo. Vengono mostrati due profili per piscine contenenti quantità equimolari di 17 oligonucleotidi sintetici di diversa lunghezza. Questi oligonucleotidi sono sequenze da HIV-1NL4.3 e sono stati selezionati per coprire varie lunghezze e presentare tutte le 4 basi come un nucleotide 3' (cfr. tabella 2). Il grafico superiore mostra il campione di controllo positivo da Figura 3un. Nessuna significativa polarizzazione verso molecola lunghezza o le aperta 3'-estremità viene rilevato. Il grafico inferiore mostra un'altra libreria esegue, che ha prodotto una distorsione minore lunghezza in sequenza. In questo caso, si consiglia di applicare un fattore di normalizzazione, che è derivato dal versante (mostrato in rosa) che rappresenta la differenza di dimensioni. Questa figura è ripubblicata da una precedente pubblicazione6. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}
| Nome di oligo | Lunghezza in nt | Sequenza | Scopo | Produttore (purificazione) | ||||||||||||||
| Full Kwok + MiSeq | 61 | 5'-PHO-tgaagagcctagtcgctgttcannnnnnctgcccatagagagatcggaagagcacacgtct-SpC3-3' | Adattatore | Tecnologie del DNA IDT (HPLC) | ||||||||||||||
| 2xBiotin SS esca | 40 | 5'-biotina-cagtgtggaaaatctctagcagtggcgcccgaacagggac-biotina-3' | Hybrid Capture | MWG Eurofins (HPLC) | ||||||||||||||
| Ss di biotina 1-16 | 22 | 5'-cagtgtggaaaatctctagcag-BiTEG-3' | Hybrid Capture | MWG Eurofins (HPLC | ||||||||||||||
| Biotina tRNA + CTG | 16 | 5'-cagtggcgcccgaaca-BITEG-3' | Hybrid Capture | MWG Eurofins (HPLC) | ||||||||||||||
| MP1.0 + 22 | 82 | 5'-aatgatacggcgaccaccgagatctacactctttccctacacgacgctcttccgatctcactgctagagattttccacactg-3' | Amplificazione di PCR | MWG Eurofins (HPLC | ||||||||||||||
Tabella 1: tabella dei oligonucleotides tra cui lunghezza, sequenze e le modifiche che vengono utilizzate nel protocollo descritto. La tabella è adattata da una precedente pubblicazione6. Per favore clicca qui per scaricare la tabella come file excel.
| Nome di oligo | Lunghezza in nt | Sequenza | Produttore (purificazione) | |||||||||||||
| HTP con lunga C | 120 | 5'-ctgctagagattttccacactgactaaaagggtctgagggatctctagttaccagagtcacacaacagacgggcacacactactttgagcactcaaggcaagctttattgaggcttaagc-3' | MWG Eurofins (HPLC) | |||||||||||||
| HTP con lunga G | 119 | 5'-ctgctagagattttccacactgactaaaagggtctgagggatctctagttaccagagtcacacaacagacgggcacacactactttgagcactcaaggcaagctttattgaggcttaag-3' | MWG Eurofins (HPLC) | |||||||||||||
| HTP con lunghe T | 116 | 5'-ctgctagagattttccacactgactaaaagggtctgagggatctctagttaccagagtcacacaacagacgggcacacactactttgagcactcaaggcaagctttattgaggctt-3' | MWG Eurofins (HPLC) | |||||||||||||
| HTP raggiro A lungo | 118 | 5'-ctgctagagattttccacactgactaaaagggtctgagggatctctagttaccagagtcacacaacagacgggcacacactactttgagcactcaaggcaagctttattgaggcttaa-3' | MWG Eurofins (HPLC) | |||||||||||||
| Con HTP metà C | 76 | 5'-ctgctagagattttccacactgactaaaagggtctgagggatctctagttaccagagtcacacaacagacgggcac-3' | MWG Eurofins (HPLC) | |||||||||||||
| HTP con metà G (a) | 71 | 5'-ctgctagagattttccacactgactaaaagggtctgagggatctctagttaccagagtcacacaacagacg-3' | MWG Eurofins (HPLC) | |||||||||||||
| HTP con metà G (b) | 72 | 5'-ctgctagagattttccacactgactaaaagggtctgagggatctctagttaccagagtcacacaacagacgg-3' | MWG Eurofins (HPLC) | |||||||||||||
| Con HTP metà A | 69 | 5'-ctgctagagattttccacactgactaaaagggtctgagggatctctagttaccagagtcacacaacaga-3' | MWG Eurofins (HPLC) | |||||||||||||
| Con HTP metà T | 85 | 5'-ctgctagagattttccacactgactaaaagggtctgagggatctctagttaccagagtcacacaacagacgggcacacactactt-3' | MWG Eurofins (HPLC) | |||||||||||||
| HTP raggiro A breve | 40 | 5'-ctgctagagattttccacactgactaaaagggtctgaggga-3' | MWG Eurofins (HPLC) | |||||||||||||
| HTP con breve T | 33 | 5'-ctgctagagattttccacactgactaaaagggt-3' | MWG Eurofins (HPLC) | |||||||||||||
| HTP con breve G | 41 | 5'-ctgctagagattttccacactgactaaaagggtctgaggg-3' | MWG Eurofins (HPLC) | |||||||||||||
| HTP con breve C | 34 | 5'-ctgctagagattttccacactgactaaaagggtc-3' | MWG Eurofins (HPLC) | |||||||||||||
| HTP Con 46 (T) | 46 | 5'-ctgctagagattttccacactg actaaaagggtctgagggatctct-3' | MWG Eurofins (HPLC) | |||||||||||||
| HTP Con 83 (C) | 83 | 5'-ctgctagagattttccacactg actaaaagggtctgagggatctctagttaccagagtcacacaacagacgggcacacactac-3' | MWG Eurofins (HPLC) | |||||||||||||
| HTP Con 103 (C) | 103 | 5'-ctgctagagattttccacactg actaaaagggtctgagggatctctagttaccagagtcacacaacagacgggcacacactactttgagcactcaaggcaagc-3' | MWG Eurofins (HPLC) | |||||||||||||
| HTP Con 107 (A) | 107 | 5'-ctgctagagattttccacactg actaaaagggtctgagggatctctagttaccagagtcacacaacagacgggcacacactactttgagcactcaaggcaagcttta-3' | MWG Eurofins (HPLC) | |||||||||||||
Tabella 2: tabella di 17 oligonucleotidi di controllo sintetico usato come un campione di controllo positivo. I oligonucleotides 13 top sono stati scelti sulla base dimensioni [lungo (116-120 nt), mid (69-85 nt), breve (33-41 nt)] così come loro 3'-termini. La tabella è adattata da una precedente pubblicazione6. Per favore clicca qui per scaricare la tabella come file excel.
Discussione
La disponibilità di sequenziamento profondo veloce, affidabile e conveniente ha rivoluzionato molti aspetti nel campo delle scienze della vita, consentendo grande profondità nelle analisi di sequenziamento. Un restante sfida il design innovativo e la creazione del rappresentante librerie di sequenziamento. Qui descriviamo un protocollo per catturare molecole di cDNA virale nascente, in particolare gli intermedi del processo di trascrizione inversa di HIV-1.
La fase più critica in questa strategia è la legatura di un adattatore per le aperte 3'-estremità in maniera quantitativa e imparziale. Efficienze di legature tra due termini di ssDNA, sia inter- e intramolecolari, sono stati studiati e ottimizzati per vari applicazioni11,26,27,28,29. La scelta di utilizzare un adattatore di tornante con T4 DNA ligasi nelle condizioni descritte al punto 3.3 è il risultato dell'ottimizzazione empirica in cui abbiamo valutato diverse ligasi, adattatori e reagenti per la legatura dei oligonucleotides sintetici che rappresentano Sequenze di HIV-1 (tabella 2) (dati non mostrati). In queste reazioni in vitro test, abbiamo confermato che la ligasi del DNA T4 mediato legatura dell'adattatore tornante, come descritto da Kwok et al. 11, ha una distorsione molto bassa e raggiunge vicino a legatura completa di molecole acceptor quando l'adattatore viene utilizzato in eccesso. L'efficienza di legatura era inalterata tramite l'aggiunta di sequenza nucleotidica per eseguire il rendering l'adattatore compatibile per il sistema multiplex primer (Vedi Figura 4). In confronto, abbiamo scoperto che una ligasi del DNA/RNA di termostabile 5' (Vedi "A-ligasi" Tabella materiali per esatte ligasi rispetto qui), che è una ligasi derivati dal RNA che è stata sviluppata in parte per migliorare l'efficienza di legatura con ssDNA come accettore 27, era infatti più efficace a legare due molecole di ssDNA di ligasi di RNA ("ligasi B"), ma aveva una distorsione significativa, con forti differenze di efficienza di legatura anche tra oligonucleotidi con differenze di lunghezza base singola [tabella 2 ; HTP con metà G (a) e (b)]. Inoltre, abbiamo trovato solo una minima distorsione in reazioni con "Ligasi C" combinato con un adattatore che trasportano un randomizzato 5'-termini (una strategia utilizzata per compensare la polarizzazione del nucleotide noto di "Ligasi C"; Vedi ad esempio Ding et al. 30). Tuttavia, la "ligasi C"-mediate intermolecolari legature erano incompleti, rendendo il sistema ligasi del DNA T4 la scelta migliore.
Diversi passaggi di controllo di qualità sopra il corso del protocollo e l'inclusione di controlli positivi e negativi permettono per la rilevazione dei potenziali problemi prima continuazione di dosaggio e forniscono indicazioni per la risoluzione dei sforzi. Le quantificazioni di qPCR ai punti 2.2.2 e 2.3.12 garantiscono che la quantità del materiale in ingresso è sufficiente. Numeri di copia cDNA tipico nella gamma di eluizione (dal punto 2.1) 200 µ l da circa 10.000 a 300.000 per µ l. Il passo di cattura di ibrido può comportare una perdita di HIV-1 complessivo cDNA quantità ma deve risultare in un forte arricchimento del cDNA di HIV-1 specifico su DNA cellulare, che può essere determinato utilizzando appositi primer per quantificare il DNA di genomic prima e dopo arricchimento di qPCR o misurando la concentrazione di DNA totale. Recuperati cDNA di HIV-1 dopo l'ibrido catturare passi dovrebbe essere almeno il 10% dell'input. Basso materiale di partenza può spiegare altrimenti un controllo positivo del oligonucleotide di successo (Vedi punto 3.3.2), ma solo limitato letture raggiunta nei campioni. Bassa legge numeri complessivi potrebbero essere spiegati anche da sovrastima della concentrazione libreria a causa della presenza di specie di DNA irrilevante senza adattatori MiSeq. Questo si tradurrebbe in densità bassa cluster e può essere migliorato determinando la concentrazione delle sequenze di HIV-1 nella libreria di qPCR oltre all'importo totale del DNA di analisi fluorometriche. A causa della natura altamente sensibile del metodo, si dovrebbe prestare particolare attenzione per evitare la contaminazione anche a basso livello, sia da altri campioni (in particolare, provenienti dalle scorte del oligonucleotide controllo alta concentrazione) così come da attrezzature di laboratorio. Lavorare in un UV sterilizzazione workstation PCR è utile a questo proposito. L'elettroforesi automatizzata della biblioteca finale (punto 6.1.2) è un'ulteriore misura di controllo di qualità. La gamma di dimensioni di acido nucleico in genere osservata è tra 150 e 500 nt. primer che può essere rilevato nel controllo facoltativo dopo la PCR, e prima di purificazione (Vedi nota al punto 5.2) dovrebbe essere assente. In un risultato rappresentativo, la curva di intensità di campione ha un picco circa 160-170 nt e un secondo più nitida picco circa 320 a 350 nt. Ciò probabilmente riflette l'abbondanza superiore spesso visto in sia relativamente breve (da 1 a 20 nt inserto) inversione trascritti e forte-fermata Full-Length (lunghezza di 180-182 nt inserto) (Figura 3b).
Mentre il protocollo presentato e selezionato gli iniettori sono specifici per primi costrutti di trascrizione inversa di HIV-1, il metodo è generalmente applicabile a qualsiasi studio con l'obiettivo di determinare aperti 3'-termini di DNA. Le principali modifiche richieste in altri contesti sarà il metodo per la cattura di ibrido e la strategia di disegno di primer. Ad esempio, se la destinazione è essere adattata alla fine trascrizioni di HIV-1, un numero maggiore di oligonucleotidi biotinilati diversi acquisizione ricottura su tutta la lunghezza del cDNA sarebbe consigliabile e probabilmente diminuirà la perdita nel passaggio cattura ibrida. Come accennato nell'introduzione, è importante considerare le limitazioni quando si progetta l'intervallo nel quale devono essere rilevato per evitare diverse fonti di bias 3'-stazione termini. Primo, ci può essere una distorsione nelle reazioni di PCR se i modelli con l'adattatore sono di lunghezza notevolmente variabile. In secondo luogo, la piattaforma di sequenziamento usata qui (per esempio, MiSeq) ha una gamma di lunghezza comodo inserto per il clustering ottimale, e significativamente più brevi e più a lunghi i prodotti non possono essere sequenziati con la stessa efficienza. In parte, questo può essere affrontato dal punto di vista, come è stato fatto calcolando il fattore di correzione per bias lunghezza lineare (Vedi Figura 4, grafico in basso). Tuttavia, se la regione di cui 3'-termini mappatura sono volute è lunga (> 1000 nt), è più consigliabile suddividere le reazioni con le trascrizioni dei e utilizzare più a Monte degli iniettori per valutare 3'-termini a sezioni.
Il programma di analisi è stato scritto in-House per lo scopo specifico di analizzare sia l'ultimo nucleotide della sequenza HIV-1 adiacente alla sequenza fissa adattatore così come la variazione di base di tutte le basi per identificare eventuali mutazioni. I singoli passaggi comprendono quanto segue: in primo luogo, le sequenze di adattatore vengono tagliate usando il toolkit fastx-0.0.13; quindi, vengono rimossi eventuali sequenze che vengono duplicate (cioè sequenze identiche tra cui il codice a barre). Tutte le rimanenti letture uniche quindi sono allineate alla sequenza di HIV-1 con Papillon (http://bowtie-bio.sourceforge.net/index.shtml) con il disallineamento massimo fissato a tre basi. La sequenza di modello è composto dal primo 635 nt del cDNA di HIV-1 (ceppo NL4.3), che comprende la sequenza - sss e il primo prodotto di trasferimento di filo fino alla pista di polipurinico (U5-R-U3-PPT; Vedi Figura 1). In tal modo, il software fornito e modelli sono direttamente adatti solo se il metodo viene utilizzato per la stessa applicazione (rilevazione delle trascrizioni precoce inversione del HIV-1NL4.3). Le regolazioni dovranno prevedere altre sequenze di destinazione. Le posizioni della 3'-termini per ogni lettura sono state determinate dalla posizione nell'allineamento. Chiamate di base per ogni posizione sono registrate e tassi di mutazione sono calcolati dalla copertura totale di ogni base, che varia, come letture sono di diverse lunghezze e inserti lunghi potrebbero non essere interamente coperti mediante il sequenziamento di 125-base in Read2.
Per concludere, riteniamo che il metodo descritto per essere uno strumento prezioso per molti tipi di studi. Ovvie applicazioni includono le indagini dei meccanismi che l'inibizione di trascrizione d'inversione attraverso farmaci antiretrovirali o fattori di restrizione cellulare. Tuttavia, solo relativamente piccoli aggiustamenti dovrebbero essere necessari adattare il sistema a 3'-termini mapping all'interno di altri singoli filamenti intermedi DNA virale, che sono presenti, ad esempio, nella replica di parvovirus. Inoltre, il principio del metodo, in particolare il suo passo di legatura ottimizzato, possa fornire una parte fondamentale della progettazione di preparazione di librerie per la caratterizzazione di eventuali estensioni di 3'-DNA, tra cui allungamenti catalizzati da cellulare DNA double-stranded polimerasi.
Divulgazioni
Gli autori dichiarano che non hanno nulla da rivelare.
Riconoscimenti
Gli autori riconoscono il supporto dai membri del laboratorio Malim, Luis Apolonia, Jernej Ule e Rebecca Oakey. Gli autori ringraziano Matt Arno Centre Genomic del Re College di Londra e Debbie Hughes presso l'University College London (UCL), Istituto di neurologia successivo sequenziamento impianto di generazione, per aiuto con MiSeq l'ordinamento viene eseguito. Il lavoro è stato supportato da UK Medical Research Council (G1000196 e MR/M001199/1 a M.M.), il Wellcome Trust (106223/Z/14/Z a M.M.), settimo programma della Commissione europea quadro (FP7/2007-2013) sotto convenzione di sovvenzione non. PIIF-GA-2012-329679 (per D.P.) e il dipartimento della salute tramite un istituti nazionali per il premio salute ricerca Comprehensive Biomedical Research Center di Guy e St. Thomas' NHS Foundation Trust in collaborazione con College Londra del re e del re College Hospital NHS Foundation Trust.
Materiali
| Name | Company | Catalog Number | Comments |
| 293T cells | ATCC | CRL-3216 | |
| Dulbecco's Modified Eagle's Medium | Gibco | 31966-021 | |
| Penicillin/Streptomycin | Gibco | 15150-122 | |
| Fetal Bovine Serum | Gibco | 10270-106 | |
| HeraCell Vios 250i CO2 Incubator | Thermo Scientific | 51030966 | |
| Laminar flow hood - CAS BioMAT2 | Wolflabs | CAS001-C2R-1800 | |
| 10mm TC-treated culture dish | Corning | 430167 | |
| TrypLE™ Express (1x), Stable Trypsin Replacement Enzyme | Gibco | 12605-010 | |
| OptiMEM® (Minimal Essential Medium) | Gibco | 31985-047 | |
| HIV-1 NL4-3 Infectious Molecular Clone (pNL4-3) | NIH Aids reagent program | 114 | |
| Polyethylenimine (PEI) - MW:25000 | PolySciences Inc | 23966-2 | dissolved at 1mg/ml and adjusted to pH7 |
| RQ1- Rnase free Dnase | Promega | M6101 | |
| Filter 0.22 μm | Triple Red Limited | FPE404025 | |
| 15 mL polypropylene tubes | Corning | CLS430791 | |
| Sucrose | Calbiochem | 573113 | |
| Phosphate Buffered Saline (1x) | Gibco | 14190-094 | |
| Ultracentrifuge tubes | Beckman Coulter | 344060 | |
| Ultracentrifuge | Sorval | WX Ultra Series | Th-641 Rotor |
| Alliance HIV-1 p24 antigen ELISA kit | Perkin Elmer | NEK050001KT | |
| CEM-SS cells | NIH Aids reagent program | 776 | |
| Roswell Park Memorial Institute Medium | Gibco | 31870-025 | |
| CoStar® TC treated multiple well plates | Corning | CLS3513-50EA | |
| Benchtop centrifuge: Heraus™ Multifuge™ X3 FR | Thermo Scientific | 75004536 | |
| TX-1000 Swinging Bucket Rotor | Thermo Scientific | 75003017 | |
| Microcentrifuge: 5424R | Eppendorf | 5404000060 | |
| Total DNA extraction kit (DNeasy Blood and Tissue kit) | Qiagen | 69504 | |
| Nuclease free H2O | Ambion | AM9937 | |
| Cutsmart buffer | New England Biolabs (part of DpnI enzyme) | R0176S | |
| DpnI restriction enzyme | New England Biolabs | R0176S | |
| Oligonucleotides for qPCR | MWG Eurofins | N/A | HPSF purification |
| TaqMan PCR Universal Mastermix | Thermo | 4304437 | |
| LoBind Eppendorf® tubes | Eppendorf | 30108078 | |
| Axygen™ aerosol filter pipette tips, 1000 μL | Fisher Scientific | TF-000-R-S | |
| Axygen™ aerosol filter pipette tips, 200 μL | Fisher Scientific | TF-200-R-S | |
| Axygen™ aerosol filter pipette tips, 20 μL | Fisher Scientific | TF-20-R-S | |
| Axygen™ aerosol filter pipette tips, 10 μL | Fisher Scientific | TF-10-R-S | |
| PCR clean hood | LabCaire | Model PCR-62 | |
| DynaMag™2-magnet | Thermo | 12321D | |
| Streptavidin MagneSphere® paramagnetic particles | Promega | Z5481 | |
| Casein | Thermo Scientific | 37582 | |
| End over end rotator, Revolver™ 360° | Labnet | H5600 | |
| Tris-Base | Fisher Scientific | BP152-5 | |
| Hydrochloric Acid | Sigma | H1758-100ML | |
| EDTA disodium salt dihydrate | Electran (VWR) | 443885J | |
| Sodium Chloride | Sigma | S3014 | |
| Dri-Block® Analog Block Heater | Techne | UY-36620-13 | |
| PCR tubes and domed caps | Thermo Scientific | AB0266 | |
| PCR machine | Eppendorf | Mastercycler® series | |
| T4 DNA ligase | New England Biolabs | M0202M | |
| 40% Polyethylene glycol solution (PEG) in H2O, MW: 8000 | Sigma | P1458-25ML | |
| Betaine solution, 5M | Sigma | B0300-1VL | |
| Gel loading buffer II (formamide buffer) | Thermo Scientific | AM8546G | |
| Precast 6% TBE urea gels | Invitrogen | EC6865BOX | |
| Mini cell electrophoresis system | Invitrogen, Novex | XCell SureLock™ | |
| Tris/Borate/EDTA solution (10x) | Fisher Scientific | 10031223 | |
| Needle 21 G x1 1/2 | VWR | 613-2022 | |
| SYBR Gold nucleic acid stain (10000x) | Life Technologies | S11494 | |
| Dark Reader DR46B transilluminator | Fisher Scientific | NC9800797 | |
| Ammonium acetate | Merck | 101116 | |
| SDS solution 20% (w/v) | Biorad | 161-0418 | |
| Centrifuge tube filter | Appleton Woods | BC591 | |
| Filter Glass Fibre Gf/D 10mm | Whatman (VWR) | 512-0427 | |
| polyadenylic acid (polyA) RNA | Sigma | 10108626001 | |
| Glycogen, molecular biology grade | Thermo Scientific | R0561 | |
| Isopropanol (2-propanol) | Fisher Scientific | 15809665 | |
| Ethanol, molecular biology grade | Fisher Scientific | 10041814 | |
| Accuprime™ Supermix I (DNA polymerase premix) | Life Technologies | 12342-010 | |
| NEBNext® Multiplex Oligo for Illumina (Index Primer Set 1 and 2) | New England Biolabs | E7335S; E7500S | |
| Tapestation D1000 Screentape High sensitivity | Agilent Technologies | 5067- 5584 | |
| Tapestation D1000 Reagents | Agilent Technologies | 5067- 5585 | |
| 2200 Tapestation - automated gel electrophoresis system | Agilent Technologies | G2965AA | |
| Agencourt® AMPure® beads XP | Beckman Coulter | A63880 | |
| Qubit™ dsDNA HS Assay Kit | Invitrogen | Q32851 | |
| Qubit™ 2.0 Fluorometer | Invitrogen | Q32866 | |
| Topo™ TA cloning Kit | Invitrogen | 450071 | |
| Sequencing platform: MiSeq System | Illumina | ||
| Experiment Manager (Sample sheet software) | Illumina | Note: Use TruSeq LT as a template | |
| Miseq™ Reagent kit V3 (150 cycle) | Illumina | MS-102-3001 | |
| Sequencing hub: Basespace | Illumina | https://basespace.illumina.com | |
| Ligase A: Thermostable 5’ App DNA/RNA ligase | NEB | M0319S | Not used in this protocol, but tested in optimization process with results described in the discussion. |
| Ligase B: T4 RNA ligase 1 | NEB | M0204 | Not used in this protocol, but tested in optimization process with results described in the discussion. |
| Ligase C: CircLigase | Epicentre | CL4111K | Not used in this protocol, but tested in optimization process with results described in the discussion. |
Riferimenti
- Herschhorn, A., Hizi, A. Retroviral reverse transcriptases. Cellular and Molecular Life Sciences. 67 (16), 2717-2747 (2010).
- Hu, W. S., Hughes, S. H. HIV-1 reverse transcription. Cold Spring Harbor Perspectives in Medicine. 2 (10), (2012).
- Levin, J. G., Mitra, M., Mascarenhas, A., Musier-Forsyth, K. Role of HIV-1 nucleocapsid protein in HIV-1 reverse transcription. RNA Biology. 7 (6), 754-774 (2010).
- Menendez-Arias, L., Sebastian-Martin, A., Alvarez, M. Viral reverse transcriptases. Virus Research. , (2016).
- Telesnitsky, A., Goff, S. P., Coffin, J. M., Hughes, S. H., Varmus, H. E. . Retroviruses. , (1997).
- Pollpeter, D., et al. Deep sequencing of HIV-1 reverse transcripts reveals the multifaceted antiviral functions of APOBEC3G. Nature Microbiology. 3 (2), 220-233 (2018).
- Frohman, M. A., Dush, M. K., Martin, G. R. Rapid production of full-length cDNAs from rare transcripts: amplification using a single gene-specific oligonucleotide primer. Proceedings of the National Academy of Sciences of the United States of America. 85 (23), 8998-9002 (1988).
- Liu, X., Gorovsky, M. A. Mapping the 5' and 3' ends of Tetrahymena thermophila mRNAs using RNA ligase mediated amplification of cDNA ends (RLM-RACE). Nucleic Acids Research. 21 (21), 4954-4960 (1993).
- Ince, I. A., Ozcan, K., Vlak, J. M., van Oers, M. M. Temporal classification and mapping of non-polyadenylated transcripts of an invertebrate iridovirus. Journal of General Virology. 94, 187-192 (2013).
- Hafner, M., et al. RNA-ligase-dependent biases in miRNA representation in deep-sequenced small RNA cDNA libraries. RNA. 17 (9), 1697-1712 (2011).
- Kwok, C. K., Ding, Y., Sherlock, M. E., Assmann, S. M., Bevilacqua, P. C. A hybridization-based approach for quantitative and low-bias single-stranded DNA ligation. Analytical Biochemistry. 435 (2), 181-186 (2013).
- Abram, M. E., Tsiang, M., White, K. L., Callebaut, C., Miller, M. D. A cell-based strategy to assess intrinsic inhibition efficiencies of HIV-1 reverse transcriptase inhibitors. Antimicrobial Agents and Chemotherapy. 59 (2), 838-848 (2015).
- Bishop, K. N., Verma, M., Kim, E. Y., Wolinsky, S. M., Malim, M. H. APOBEC3G inhibits elongation of HIV-1 reverse transcripts. PLoS Pathogens. 4 (12), 1000231 (2008).
- Zack, J. A., Haislip, A. M., Krogstad, P., Chen, I. S. Incompletely reverse-transcribed human immunodeficiency virus type 1 genomes in quiescent cells can function as intermediates in the retroviral life cycle. Journal of Virology. 66 (3), 1717-1725 (1992).
- Adachi, A., et al. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. Journal of Virology. 59 (2), 284-291 (1986).
- Shah, V. B., Aiken, C. In vitro uncoating of HIV-1 cores. Journal of Visualized Experiments. (57), (2011).
- JoVE Science Education Database. Science Education Database: Basic Methods in Cellular and Molecular Biology: Passaging Cells. Journal of Visualized Experiments. , (2018).
- JoVE Science Education Database. JoVE Science Education Database: Basic Methods in Cellular and Molecular Biology: Using a Hemocytometer to Count Cells. Journal of Visualized Experiments. , (2018).
- Zhou, M. Y., Gomez-Sanchez, C. E. Universal TA cloning. Current Issues in Molecular Biology. 2 (1), 1-7 (2000).
- Zhang, S., Cahalan, M. D. Purifying plasmid DNA from bacterial colonies using the QIAGEN Miniprep Kit. Journal of Visualized Experiments. (6), 247 (2007).
- Mangeat, B., et al. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature. 424 (6944), 99-103 (2003).
- Gillick, K., et al. Suppression of HIV-1 infection by APOBEC3 proteins in primary human CD4(+) T cells is associated with inhibition of processive reverse transcription as well as excessive cytidine deamination. Journal of Virology. 87 (3), 1508-1517 (2013).
- Harris, R. S., et al. DNA deamination mediates innate immunity to retroviral infection. Cell. 113 (6), 803-809 (2003).
- Zhang, H., et al. The cytidine deaminase CEM15 induces hypermutation in newly synthesized HIV-1 DNA. Nature. 424 (6944), 94-98 (2003).
- Konig, J., et al. iCLIP--transcriptome-wide mapping of protein-RNA interactions with individual nucleotide resolution. Journal of Visualized Experiments. (50), (2011).
- Troutt, A. B., McHeyzer-Williams, M. G., Pulendran, B., Nossal, G. J. Ligation-anchored PCR: a simple amplification technique with single-sided specificity. Proceedings of the National Academy of Sciences of the United States of America. 89 (20), 9823-9825 (1992).
- Zhelkovsky, A. M., McReynolds, L. A. Structure-function analysis of Methanobacterium thermoautotrophicum RNA ligase - engineering a thermostable ATP independent enzyme. BMC Molecular Biology. 13 (24), (2012).
- Li, T. W., Weeks, K. M. Structure-independent and quantitative ligation of single-stranded DNA. Analytical Biochemistry. 349 (2), 242-246 (2006).
- Gansauge, M. T., et al. Single-stranded DNA library preparation from highly degraded DNA using T4 DNA ligase. Nucleic Acids Research. 45 (10), 79 (2017).
- Ding, Y., Kwok, C. K., Tang, Y., Bevilacqua, P. C., Assmann, S. M. Genome-wide profiling of in vivo RNA structure at single-nucleotide resolution using structure-seq. Nature Protocols. 10 (7), 1050-1066 (2015).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati