
Method Article
Determinación de las interacciones proteína-ligando Uso diferencial de barrido Fluorimetría
En este artículo
Resumen
Differential scanning fluorimetry is a widely used method for screening libraries of small molecules for interactions with proteins. Here, we present a straightforward method to extend these analyses to provide an estimate of the dissociation constant between a small molecule and its protein partner.
Resumen
Una amplia gama de métodos están disponibles actualmente para la determinación de la constante de disociación entre una proteína y la interacción de moléculas pequeñas. Sin embargo, la mayoría de ellos requieren el acceso a equipos especializados, ya menudo requieren un grado de experiencia para establecer efectivamente experimentos confiables y analizar los datos. Fluorimetry diferencial de barrido (DSF) se está utilizando cada vez más como un método robusto para la selección inicial de las proteínas para interactuar pequeñas moléculas, ya sea para la identificación de socios fisiológicas o para el descubrimiento de golpe. Esta técnica tiene la ventaja de que sólo requiere una máquina de PCR adecuados para la PCR cuantitativa, y así instrumentación adecuada está disponible en la mayoría de las instituciones; una excelente variedad de protocolos ya están disponibles; y hay fuertes antecedentes en la literatura para múltiples usos del método. Un trabajo anterior ha propuesto varios medios de cálculo de constantes de disociación a partir de datos DSF, pero estos son matemáticamente exigente. Aquí, nos demtrar un método para estimar las constantes de disociación de una cantidad moderada de datos experimentales DSF. Estos datos normalmente pueden ser recogidos y analizados en un solo día. Demostramos cómo los diferentes modelos se puede utilizar para ajustar los datos recogidos de eventos de unión simples, y donde los sitios de unión o unión independientes de cooperación presentes. Por último, se presenta un ejemplo de análisis de datos en caso de que no se aplican los modelos estándar. Estos métodos se ilustran con los datos recogidos en las proteínas de control disponibles comercialmente, y dos proteínas de nuestro programa de investigación. En general, nuestro método proporciona una forma sencilla para que los investigadores ganan rápidamente una mayor comprensión de las interacciones ligando-proteína utilizando DSF.
Introducción
Todas las proteínas se unirán, con diferentes afinidades, a una gama diversa de otras moléculas de iones simples a otras macromoléculas grandes. En muchos casos, las proteínas se unen a moléculas pequeñas socios como parte de su función normal (por ejemplo, una quinasa unión a ATP). Otras interacciones pueden no estar relacionadas con la función, pero son útiles como herramientas experimentalmente (por ejemplo, pequeñas moléculas que estabilizan las proteínas para mejorar el éxito de la cristalización, o ayudar en el mantenimiento de las proteínas en solución); mientras que las moléculas pequeñas que se unen a sitios activos y los sitios alostéricos de proteínas pueden actuar como inhibidores, y así modular la actividad de las enzimas.
Hay una amplia gama de técnicas que se pueden utilizar para determinar la afinidad de las proteínas para las moléculas asociadas. Calorimetría de titulación isotérmica 1 es ampliamente visto como un "estándar de oro", ya que proporciona una rica información sobre las reacciones, es libre de la etiqueta, y ha limitado las oportunidades para unrtifacts del experimento. Sin embargo, a pesar de las recientes mejoras en la sensibilidad de la instrumentación y automatización de montaje experimental, todavía es relativamente caro en términos de las necesidades de proteínas, ha en el mejor de un rendimiento bajo a medio, y es el más adecuado a las interacciones con moderada a altas afinidades (10 nm a 100 M K d) 2. Otros métodos libres de etiquetas, tales como resonancia de plasmón superficial o bicapa interferometría 3 oferta rendimientos más altos, y han logrado la sensibilidad para detectar moléculas más pequeñas tan bajas como 100 Da. Sin embargo, los instrumentos de alto rendimiento para estos métodos son relativamente caros, sólo se justifican en el que habrá un caudal continuo de los proyectos pertinentes, y así es probable que sean inaccesibles para muchos laboratorios académicos.
Fluorimetría diferencial de barrido (DSF, o thermofluor) fue descrita por primera vez en 2001 4 como un método para el descubrimiento de fármacos. En este method, las proteínas se incubaron con un colorante fluorescente (se utilizaron colorantes inicialmente naftaleno-sulfónico), que altera su fluorescencia tras la unión a las regiones hidrofóbicas de las proteínas. La muestra de proteína-colorante se calienta a continuación, y monitoriza la fluorescencia a medida que aumenta el calor. El despliegue de la proteína, y la exposición de las partes hidrófobas de la proteína, da lugar a un patrón característico en la fluorescencia como una función de la temperatura (Figura 1A). El experimento se puede realizar en pequeños volúmenes en cualquier instrumento PCR cuantitativa comercial, y por lo tanto en un solo experimento, un gran número de muestras se puede probar de forma simultánea (por lo general 48, 96 o 384 muestras, dependiendo del modelo de instrumento). Los experimentos pueden llevarse a cabo por lo general en torno a una hora, proporcionando la posibilidad de análisis de alto rendimiento de las muestras 5.
Otras mejoras en la metodología han visto la adopción de tintes con mejores propiedades espectrales 6,7 , herramientas genéricas para el análisis de datos, y sugirieron protocolos para la detección inicial de 8,9. La gama de aplicaciones del método se ha extendido, con un enfoque particular en el establecimiento de condiciones óptimas para la preparación y almacenamiento de proteínas de 10, y en la identificación de socios potenciales de unión para ayudar a la cristalización 11. El relativamente alto rendimiento del método, un costo relativamente bajo en proteínas (~ 2 g por reacción), y la aplicabilidad al estudio de moléculas de unión débiles ha hecho DSF una herramienta valiosa para el diseño de fármacos basado fragmento, especialmente en un contexto académico 12-14.
A pesar de la amplia aplicación de DSF a estudiar las interacciones proteína-ligando, pocos estudios han descrito la determinación de las constantes de disociación de estos estudios. Sin embargo, éstos han tendido a producir ecuaciones detalladas que describen el desarrollo de la proteína, con muchos parámetros que deben acoplarse a la escasez de datos o en algunos casos eESTIMADA 7,15-17. Estos métodos son de especial relevancia en los casos difíciles, tales como compuestos fuertemente vinculantes, o proteínas que muestra transiciones inusuales. Sin embargo, para muchos laboratorios, estos análisis detallados son demasiado engorrosos para su uso rutinario. Por tanto, proponemos tratamientos alternativos para diferentes escenarios, y demostrar cómo estos pueden ser utilizados para ajustar los datos resultantes de las diferentes interacciones ligando-proteína. Nuestro método utiliza el instrumento StepOne qPCR, para el cual el software de análisis de datos a medida está disponible; mientras que esto acelera el análisis de datos, los resultados de otros instrumentos pueden ser procesados utilizando métodos previamente publicados 9, y el mismo análisis de aguas abajo se pueden realizar.
Protocolo
1. Determinación de un valor aproximado de la constante de disociación (es decir, dentro de un orden de magnitud)
- Preparar la mezcla se detalla en la Tabla 1.
- Preparar las existencias del ligando de interés en la concentración más alta disponible, y luego a seis de diez diluciones de este. Cuando una K aproximada d es conocido a partir de datos anteriores, el objetivo de tener al menos dos concentraciones por encima y por debajo de la K d.
- Alícuota de 18 l de la mezcla en ocho pocillos en una placa de qPCR. Añadir 2 l de disolvente a la primera también. Añadir 2 l de cada miembro de la serie de dilución ligando (paso 1,2) a un pocillo cada uno de los siete pocillos restantes.
- Coloque un sello qPCR sobre la placa. Para lograr un buen sellado de la placa, colocar un aplicador de mano (véanse los cuadros de reactivos específicos) en el centro de la placa. Suavizar el sello a un lado, y luego repetir en el otro medio dela placa.
- Centrifugar la placa a 500 xg durante dos minutos para eliminar las burbujas de aire.
- Colocar la placa en un instrumento StepOne qPCR. Seleccionar la opción "curva de fusión", los filtros ROX, y elegir la velocidad de rampa rápida (esto proporciona una pausa de 2 min a 25 ° C, seguido por una rampa a 99 ° C durante 40 min, y después una pausa de 2 min). Ejecutar una desnaturalización térmica.
NOTA: Los archivos de comandos para realizar una carrera están disponibles en línea en http: // www.exeter.ac.uk/biosciences/capsular. - Al término de la aplicación del instrumento, haga clic en el botón "Analizar" en la pantalla. Guarde el archivo de resultados.
- Abra la proteína software Shift térmica.
- Crear un nuevo estudio; en la ficha de propiedades, dar a esto un nombre, y en la ficha Condiciones, detalle los ligandos.
- Mover a la pestaña Archivos Experimento, e importar el archivo de resultados de salvado (XXX.eds), y establecer el contenido de cada pocillo (fil plantillaes están disponibles de los autores).
- Mover a la pestaña Análisis, y presione el botón "Analizar".
NOTA: Este analizará los resultados. Es posible exportar los resultados de nuevas investigaciones en Excel utilizando la pestaña Exportar. Los resultados se exportan en un formato de ficha delineado. Lo mejor es abrir el archivo exportado en Excel, e inmediatamente guardar en formato Excel.
- Compruebe que la proteína en la presencia de disolvente solo da un resultado similar al mostrado en la Figura 1A. A continuación, examinar las temperaturas de fusión observados en los resultados en el panel de "réplica". Asegúrese de que este muestra un claro aumento de la temperatura de fusión con el aumento de la concentración de ligando.
NOTA: Idealmente, esto proporcionará una temperatura máxima de fusión claro (suponiendo que la proteína está totalmente ligando unido), y una K aproximada d donde la temperatura de fusión está a medio camino entre la proteína-ligando libre yel máximo.
2. experimental para determinar la constante de disociación
- Preparar la mezcla se detalla en la Tabla 2 como una mezcla maestra.
- Preparar las existencias del ligando a los quince concentraciones diferentes, que se pueden diluir diez veces en el experimento final. Lo ideal es incluir las concentraciones de al menos dos órdenes de magnitud por encima y por debajo del estimado K d, y el centro de las concentraciones en el estimado K d. Centrarse en siete de los puntos dentro de un orden de magnitud del estimado K d, con otros cuatro puntos a cada lado de esta; si hay una opción, incluir más puntos en los valores que están saturado.
NOTA: Si es necesario, es factible para alterar las condiciones experimentales tales que las poblaciones de ligandos son al doble de la concentración experimental, donde la solubilidad del ligando es limitante. - Añadir 120 l de la maestramezclar hasta ocho pocillos en una placa de fondo en U de 96 pocillos, para actuar como un depósito para la dispensación conveniente de la mezcla maestra. Utilice una pipeta de 8 canales para dispensar 18 l en una columna de una placa de PCR. Repita por otros cinco columnas, para dar un total de 48 pozos llenos en un patrón 6 x 8 en la placa.
- Añadir 20 l de las poblaciones de ligando, o el disolvente, para separar pocillos en una placa de fondo en U de 96 pocillos. Usando una pipeta de 8 canales, aspirado de 2 l de ocho acciones diferentes ligandos (o disolvente). Añadir estos a una columna de la placa de PCR que estaba llena de mezcla maestra en el paso 2.3. Repita el procedimiento con los mismos ocho ligando / acciones disolventes para otras dos columnas. Aspirar 2 l de las ocho acciones ligando o disolventes restantes y añadirlos a una cuarta columna en la placa. Repita esto para otras dos columnas. Esto le dará muestras por triplicado para todos 16 ligando y muestras de solventes.
- Coloque un sello qPCR sobre la placa (véase el paso 1.4).
- Centrifugar la placa a 500 xg durantedos min.
- Colocar la placa en el instrumento qPCR. Ejecutar una desnaturalización térmica utilizando los parámetros especificados en el paso 1.6.
- Al término de la aplicación del instrumento, haga clic en el botón "Analizar" en la pantalla. Guarde el archivo de resultados.
- Abra la proteína software Shift térmica. Crear un nuevo estudio; en la ficha de propiedades, dar a esto un nombre, y en la ficha Condiciones, detalle los ligandos.
- Mover a la pestaña Archivos Experimento, e importar el archivo de resultados de salvado (XXX.eds), y establecer el contenido de cada pocillo.
NOTA: los archivos de plantilla están disponibles en línea en http: // www.exeter.ac.uk/biosciences/capsular. - Mover a la pestaña Análisis, y presione el botón "Analizar".
- Elija la ficha "réplicas" en el menú en la parte izquierda de la pantalla para mostrar los resultados como triplicados. Evaluar la fiabilidad de los datos basados en el grado de tensión son los triplicados. Should los triplicados muestran mala reproducibilidad, examinar de cerca los datos en bruto.
- Analizar los datos utilizando tanto el Boltzmann o métodos derivados de evaluar la temperatura de fusión. Seleccione la pestaña "Replicar resultados", y en el "Replicar resultados complot", mueva el "Terreno de:" botón entre "Tm - Boltzmann" y "Tm - Derivado". Seleccione el método que da la mayor reproducibilidad para la muestra. Exportar los resultados de nuevas investigaciones en Excel utilizando la pestaña Exportar.
NOTA: Para ver ejemplos que muestran múltiples transiciones, casi siempre es mejor usar el método derivativo en el modo de fusión múltiple. Los resultados se exportan en un formato de ficha delineado. Lo mejor es abrir el archivo exportado en Excel, e inmediatamente guardar en formato Excel. - Repetir el experimento al menos dos veces, incluyendo una repetición en un día separado, para asegurar la reproducibilidad de los resultados. En caso de análisis de datos (véase el paso 3 a continuación)indican que el valor de K d es significativamente diferente a la estimación original, alterar las concentraciones de ligando en consecuencia (véase el paso 2.2) para garantizar una buena gama de valores en torno a K d.
3. análisis de datos para determinar la constante de disociación bajo desnaturalización térmica
- Crear una tabla en Excel de las concentraciones de ligando y la temperatura de fusión.
- Abra el software GraphPad Prism, y crear una mesa XY. Introduzca los datos, utilizando la columna X de las concentraciones de ligando y la columna de la Y para la fusión de los resultados de temperatura.
NOTA: un ejemplo que se muestra es la figura 1B. Un guión con ecuaciones precargados, y otras instrucciones para el uso del paquete estadístico SPSS, están disponibles en línea en http: // www.exeter.ac.uk/biosciences/capsular. - En la ficha Análisis, seleccione la opción de cambiar los parámetros de análisiss (Ctrl + T). Para entrar en el modelo correcto, seleccionar "Nuevo" y "Crear nueva ecuación". Inserte la ecuación se detalla en la Tabla 3 como "la unión del ligando sitio único".
NOTA: Un ejemplo de estos pasos se muestra en la Figura 1C. Cuando se utiliza la secuencia de comandos con ecuaciones precargados, la ecuación correspondiente se puede dirigir elegido de la lista en lugar de introducir. Derivación de esta ecuación se proporciona en un apéndice. - Seleccione la opción "Reglas para los valores iniciales" caja, y entrar en las reglas para los valores iniciales como se detalla en la Tabla 3.
- Restringir el parámetro P, como "constante igual a" e introducir la concentración final de proteína (en las mismas unidades que el ligando se da en).
- Seleccione Aceptar para realizar el análisis.
NOTA: Un ejemplo de estos pasos se muestra en la Figura 1D. El software de gráficos produce una figura que muestra los datos y el ajuste al modelo. Ejemplos de tanálisis stos se muestran en los datos representativos.
4. Ajustando datos a Modelos de Cooperativas
Para ajustar los datos a un modelo cooperativo, elegir entre cualquiera de un modelo cooperativo simple, o un modelo en el que se definen dos constantes de disociación separadas. Se prefiere el primer enfoque en el caso de cooperatividad negativa, o como una investigación inicial. Sin embargo, en principio es mejor en casos de cooperatividad positiva al modelo de dos diferentes constantes de disociación 18. En este caso, el modelado puede continuar asumiendo ya sea secuencial de unión de ligandos, o de unión de ligandos independiente.
- Siga los mismos pasos iniciales como en protocolo 3. Sin embargo, en la etapa 3.3, inserte una de las ecuaciones en la Tabla 3 que aparece como "modelo cooperativo Simple", "secuencial unión de dos ligandos", o "unión independiente de dos ligandos" 18.
- Seleccione las reglas relevantes para los valores inicialesasociado con cada una de estas ecuaciones en la Tabla 3.
- Examine el ajuste del modelo a los datos. Si los datos se ajustan mal, considerar otro modelo.
NOTA: también es importante examinar cuidadosamente el ajuste de la temperatura de fusión de los datos por el software del Cambio Térmico Proteína (paso 2.9): a veces es necesario modificar los parámetros para obtener los mejores resultados. Otra consideración es si el rango de puntos de datos es ideal, y si hay puntos anómalos: o bien un conjunto limitado de datos a ambos lados de K d, o un punto anómalo (sobre todo en las más altas concentraciones de ligandos), puede afectar significativamente los resultados. - Repita el experimento por lo menos dos veces (vea el paso 2.12) para asegurar la reproducibilidad.
5. Ajustando datos a curvas que muestran Cambios binarios en fusión Temperatura
De vez en cuando, en lugar de una respuesta gradual a ligando, las proteínas han sidoobservado a adoptar una respuesta binaria, en donde la muestra unida se separa claramente de la muestra no unida. Un ejemplo se presenta en los resultados representativos (Figura 4). En este caso, ajuste de las temperaturas de fusión no proporcionar un buen ajuste para K d.
- Exportar la salida de datos en bruto de la Proteína software Shift térmica. Para cada punto de temperatura, calcular la media de fluorescencia para el ligando cero, y las concentraciones más altas de ligandos. Tabular los resultados de cada punto al lado de estos datos.
NOTA: El error creado aquí es menor que el error en las temperaturas de fusión armarios. - Abra el paquete estadístico SPSS. Copie las temperaturas, los dos conjuntos de datos de medios y datos para cada experimento para una ventana de datos en SPSS. En la pestaña de variables, establecer el conjunto de datos media para no ligando como "baja", y el conjunto de datos promedio de la concentración de ligando más alto como "alta".
- Descargue el archivo de sintaxis disponiblesen línea en http: // www.exeter.ac.uk/biosciences/capsular . Seleccione "Ejecutar → Ejecutar todo".
- Copie la proporción resultados unidos a un nuevo libro de Excel, con las concentraciones de ligando pertinentes.
- Abra el software Graphpad, y crear una mesa XY. Introduzca los datos, utilizando la columna X de las concentraciones de ligando y la columna de la Y para la fusión de los resultados de temperatura. En la ficha de análisis, seleccione "Parámetros de análisis de cambio". Para entrar en el modelo correcto, seleccionar "Nuevo" y "Crear nueva ecuación". Introduzca la ecuación dada en la Tabla 3, que aparece como "Análisis de los cambios binarios en temperatura de fusión".
- Seleccione la casilla de "Reglas para los valores iniciales", e introduzca reglas para los valores iniciales que se detallan en la Tabla 3. Restringir el parámetro P, como "constante igual a" y escriba la concentración final de proteína (en las mismas unidades que la ligand se da en).
NOTA: ejemplos de completar estas cajas para el protocolo en la sección 3 se muestran en la Figura 1C, D. - Si hay un buen ajuste, los resultados pueden ser mejorados mediante la extrapolación al resultado esperado en la concentración de ligando infinito. A partir del modelo de la proporción unida a cada concentración de ligando, examinar el valor para el valor más alto de la concentración de ligando. Si esta es 0,99 o mayor, es poco probable que mejorar los resultados de un análisis adicional.
- Si la proporción es inferior a 0,99, se requiere una etapa adicional para corregir los efectos de la proteína no unida en la muestra más alta concentración de ligando. En la etapa 5.2, escribir la proporción de ligando unido en el punto de concentración de ligando más alto (de la etapa 5.7) en la celda R2 (una célula diferente puede ser utilizado, y R2 sustituido apropiadamente en la ecuación en la Tabla 3). Crear una columna adicional después de la media de los resultados más altos de concentración de ligando. En los abetost celular, copia la ecuación que aparece en la Tabla 3, como "La extrapolación a infinito concentración de ligando". Copiar esta fórmula para las células restantes en esta columna.
NOTA: Este cálculo elimina el efecto de la proteína no unida en la más alta concentración de ligando. La diferencia entre la proteína ligando libre y la más alta concentración de ligando se multiplica por el recíproco de la proporción unida a la mayor concentración de ligando para proporcionar la diferencia esperada entre los estados de proteínas totalmente consolidadas y no consolidadas en cada punto de temperatura. Se añade o se resta del estado no ligado para dar el esperado para la proteína de fluorescencia totalmente ligando unido Esta diferencia. - Vuelva a colocar la columna de máxima concentración de ligando en la hoja de datos de SPSS con esta nueva columna, y repita el ajuste de datos.
NOTA: los pasos 5.7 a 5.9 pueden necesitar ser repetidas si el modelo sugiere un cambio significativo de la proporción con destino a los concentrati máximos ligando(si este es el caso, probablemente sería ideal para repetir el experimento con un punto de concentración de ligando superior incluido). - En los casos en que la proteína muestra un comportamiento cooperativo de desplazamiento binario y muestra, la ecuación sugiere en el paso 5.5 debe ser sustituido con los de paso 4.1. Los parámetros de "abajo" "Top" y deben ser sustituidos por 1 y 0, respectivamente.
- Repita el experimento por lo menos dos veces (vea el paso 2.12) para asegurar la reproducibilidad.
Resultados
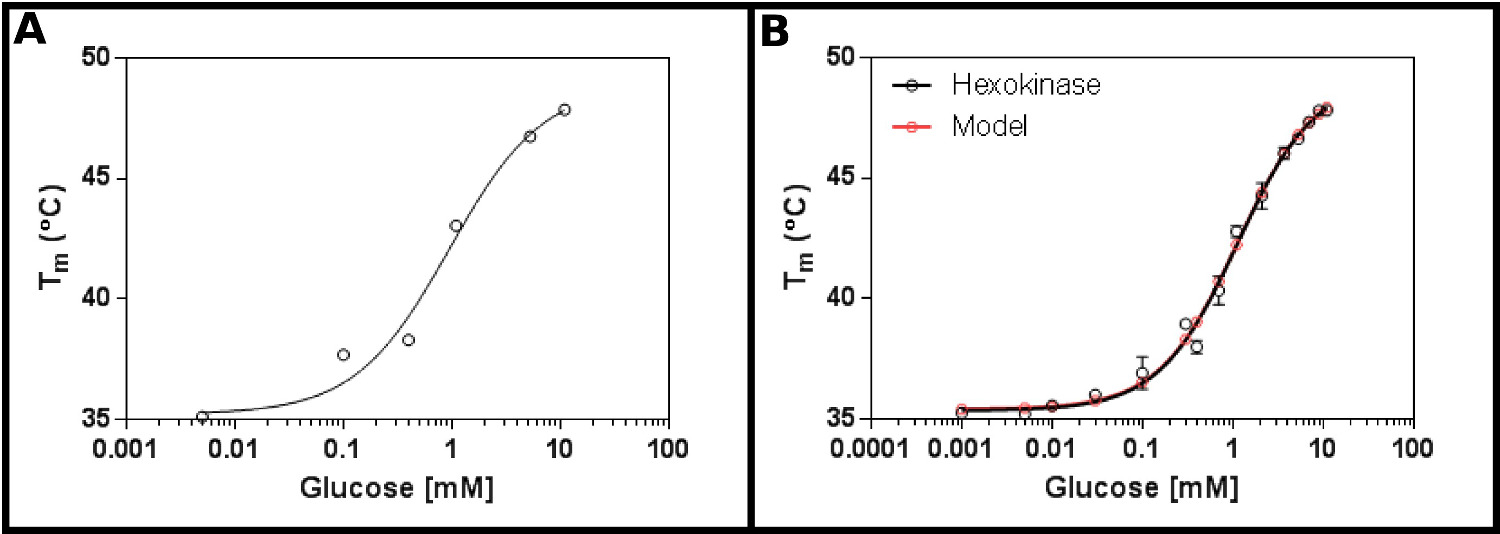
Un sustrato de prueba excelente para este método es la hexoquinasa. Esto tiene las ventajas de ser fácilmente disponibles comercialmente, y que tiene dos sustratos que se encuentran en la mayoría de los laboratorios, y que proporcionan claras y resultados reproducibles en el ensayo. Una pantalla de concentración inicial (Protocolo 1), utilizando la hexoquinasa y glucosa (Figura 2A), sugiere que la probable K d estará en el intervalo de 0,2 a 1,7 mM. Por lo tanto, se realizó una pantalla más grande (Protocolo 2), utilizando las concentraciones mostradas en la Tabla 4 Los resultados (Figura 2B) muestran un buen ajuste a la ecuación de unión al ligando de sitio único (sección Protocolo 3.3) [9], y dio un K d de 1,2 ± 0,1 mM.
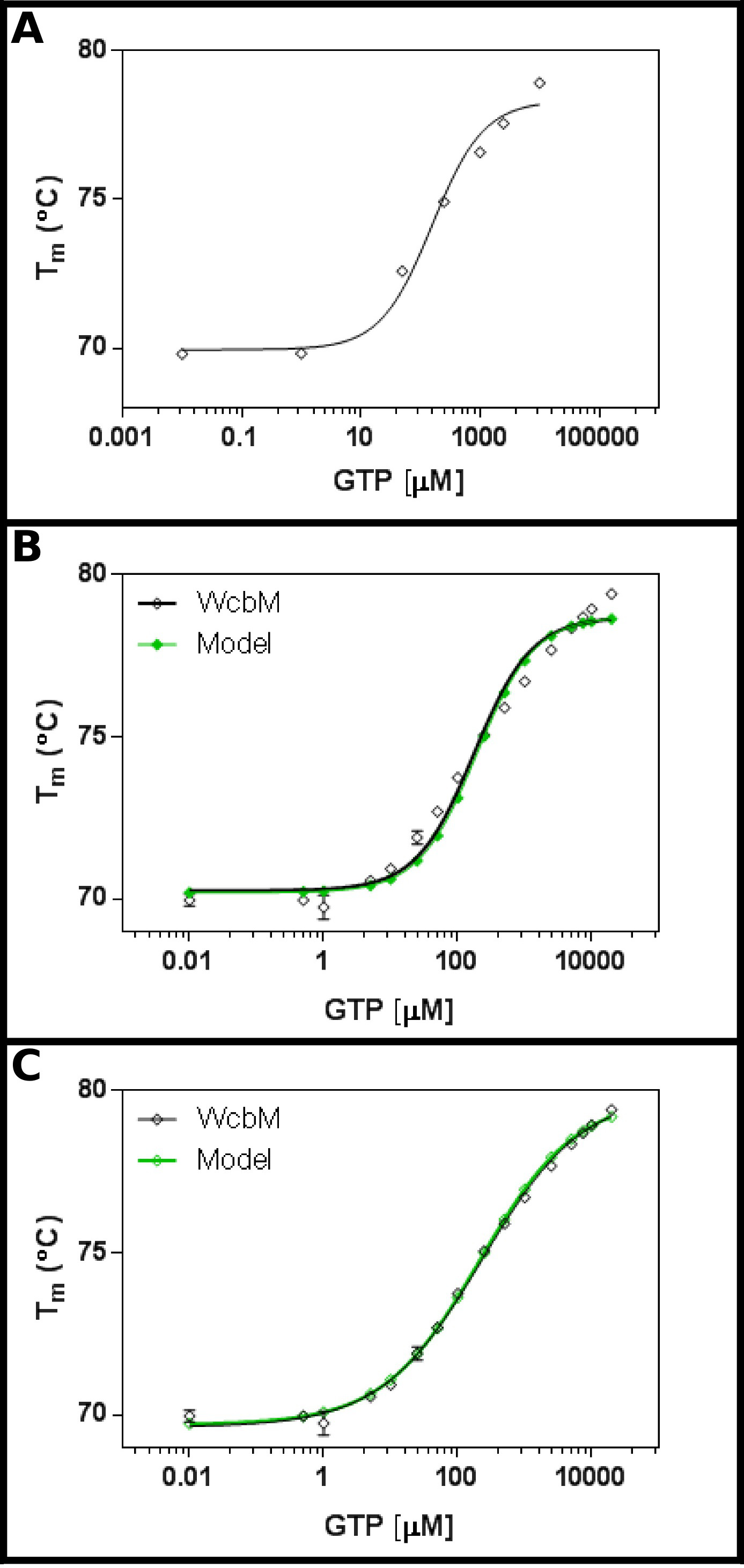
El putativo transferasa heptosa-guanil WCBM 19,20 muestra una fuerte desplazamiento térmico en la unión a GTP (Figura 3A). Una pantalla inicial sugirió que el Kd estaría en el rango de alrededor de 100 mM. Por lo tanto, una pantalla completa se creó, utilizando las concentraciones mostradas en la Tabla 5 de montaje de los resultados a la ecuación 3.3 mostró un ajuste razonable (R 2 de 0,981; Figura 3B). .Sin Embargo, existe una diferencia evidente entre los datos y el modelo, lo que sugiere que se necesita una ecuación diferente. Búsqueda del Banco de Datos de Proteínas 21 con la secuencia WCBM mostró que los homólogos más cercanos para que las estructuras se han determinado de forma dímeros. Los datos fueron analizados utilizando, por tanto, las tres ecuaciones de cooperativa, secuencial, y la unión de dos ligandos (Protocolo 4) independiente. Las estadísticas de ajuste para un modelo cooperativo dio un valor de R 2 de 0,998 y la desviación estándar de los residuos (Sy.x) de 0,215, mientras que ambos modelos de unión secuenciales e independientes dieron un valor de R 2 de 0,992 y un Sy.x de 0,480 y 0,461 respectivamente. Esto sugiere que el modelodando el mejor ajuste a los datos fue el modelo cooperativo: aquí, se observó un K ½ de 230 ± 10 M, con un valor de n de 0,52 ± 0,02 (Figura 3C). Esto indicó una cooperatividad negativa a la unión. Tenga en cuenta que una K ½ se utilizó en este caso en lugar de K d, como las unidades para K d serían bastante insatisfactoria la M 0,52.
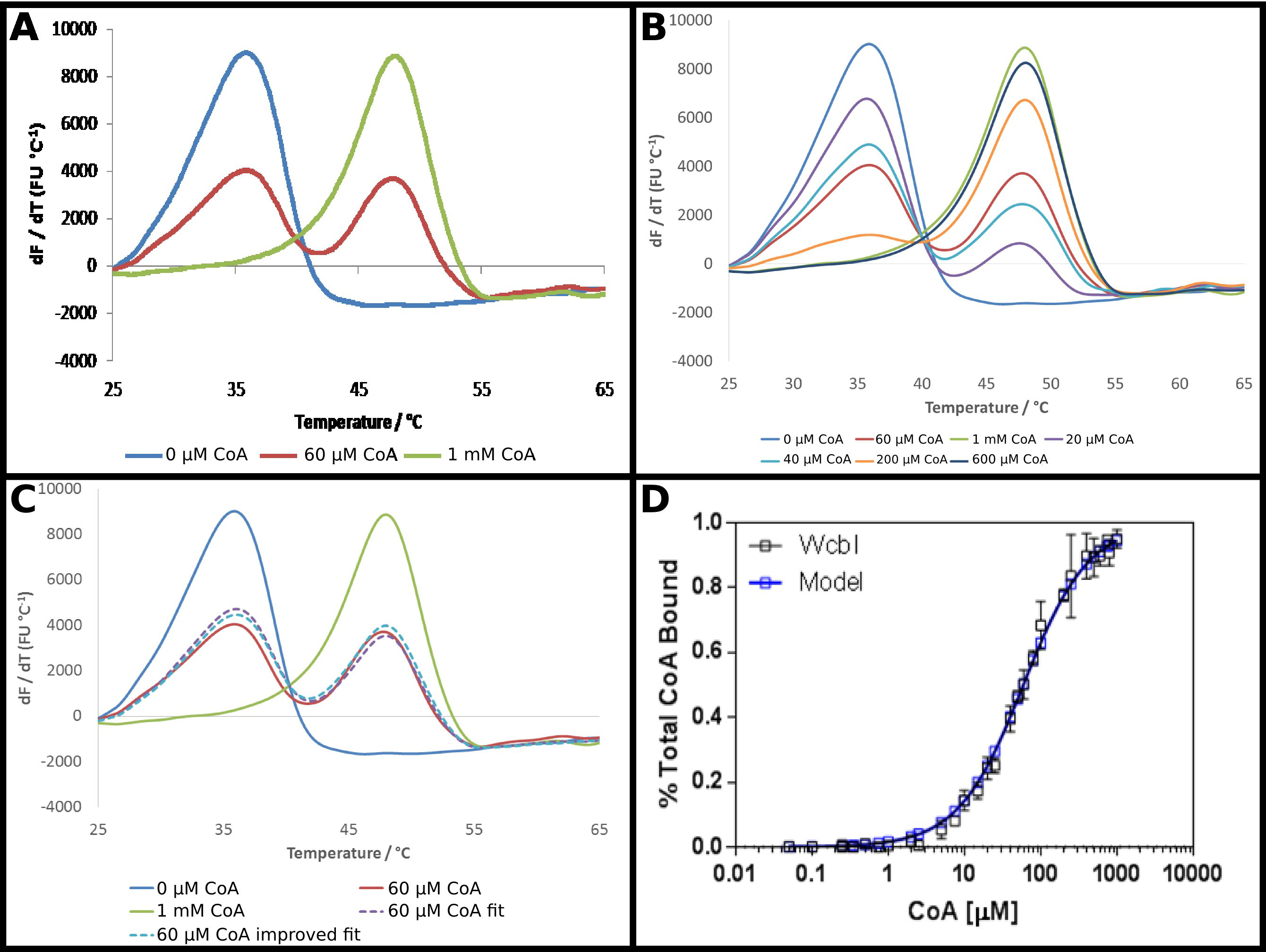
El PIB-6-desoxi-β-D-putativo manno -heptopyranose 2 O -acetylase, WCBI 22, muestra un resultado bastante inusual en fluorimetry diferencial de barrido. En la ausencia de ligandos, que muestra una desnaturalización clara y sencilla (Figura 4A). Coenzima A (CoA) se identificó como un ligando de esta proteína usando DSF, y la afinidad de la proteína para este socio se investigó como se describe en el protocolo. En presencia de altas concentraciones de CoA, un fuerte sse observa hift a una temperatura más alta, con un cambio en la temperatura de fusión de 15 ° C. Sin embargo, a concentraciones intermedias, en lugar de un cambio a una fusión monofásica a una temperatura de fusión intermedio, WCBI mostró un punto de fusión bifásica, con la proteína que aparece a fundirse a la temperatura ya sea-ligando libre, o la temperatura de fusión totalmente unida (Figura 4A) . Las proporciones de las dos especies alteradas de una manera dependiente de la dosis, con el aumento de las concentraciones de sustrato aumento de la proporción que funde a la temperatura más alta (Figura 4B). El análisis directo de estos datos fue un reto: acoplamiento a la ecuación de Boltzmann dio encaja muy pobres, mientras que los métodos derivados de relieve que dos eventos de fusión se producen, pero no ayudan en la demostración de un cambio con el aumento de la concentración de ligando.
Por tanto, un enfoque menos convencional para analizar estos datos se adoptó (Protocolo 5). La fluorescencia dresultados erivative sin ligando y a la mayor concentración de ligando se tomaron como la representación de esencialmente toda la proteína en la temperatura de fusión inferior, o el estado de temperatura de fusión más alto. Los datos derivados restantes se ajustaron en cada punto como la suma de una parte de cada uno de estos dos estados, con la proporción sumada a la unidad (Figura 4C). Los datos obtenidos se ajustan entonces como antes para obtener una K d aparente, usando las mismas ecuaciones como antes. Esto puso de relieve que la "alta" punto ligando es probable que sea sólo el 95% del ligando obligado. Los datos fueron extrapolados a una predicción del resultado para una proteína unida 100%, y los datos de adaptación repiten para dar una K d aparente de 58 ± 2 M. Esto proporciona un excelente ajuste de los resultados experimentales con el modelo de unión (Figura 4D).
fo: contenido-width = "5in" src = "/ files / ftp_upload / 51809 / 51809fig1highres.jpg" width = "500" />
Figura 1. Ejemplos de configuración de la prueba y el análisis. (A) Ejemplo de la forma esperada de un perfil de desnaturalización térmica (tomado de datos para la hexoquinasa de levadura). La forma característica de los datos en bruto muestra un aumento progresivo de la fluorescencia a un máximo, seguido por una disminución de poca profundidad (discutido en más detalle en 9). Esto va acompañado de un único pico en la primera derivada de la fluorescencia. (B) Ejemplo de entrada de datos en Graphpad. La concentración de ligando se da en el eje X, y observó temperaturas de fusión en el eje Y. (C) Ejemplo de definición de ecuación en Graphpad. (D) Ejemplos de establecer correctamente los valores iniciales de las variables, y de la fijación de la concentración de proteína, para permitir una correcta determinación de la constante de disociación.m / files / ftp_upload / 51809 / 51809fig1highres.jpg "target =" _blank "> Haga clic aquí para ver una versión más grande de esta figura.

. Figura 2 Interacción de hexoquinasa con glucosa medida por fluorimetría diferencial de barrido (A) Un experimento inicial probar una amplia gama de concentraciones de glucosa sugiere que la Kd es probable que esté en el intervalo de 0,2 -. 1,7 mM (B) Una detallada experimento, las pruebas de 16 concentraciones de glucosa, permite la determinación de la aparente K D como 1,12 ± 0,05 mM. Los datos se adapta muy bien al modelo de un solo acontecimiento de unión (con la temperatura inferior (T1) y superior (T2) apropiado a 35,4 ± 0,2 º C y 49,3 ± 0,5 º C, respectivamente). Tenga en cuenta queestos datos se recogieron en presencia de 10 mM de MgCl 2. Estas imágenes fueron creadas usando GraphPad. Por favor haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 3 Interacción de WCBM con GTP revela un anti-unión cooperativa. (A) Un experimento inicial probando un amplio intervalo de concentraciones de GTP sugiere que la Kd es probable que esté en el intervalo de 200 -. 500 M (B) Un experimento detallada, las pruebas de 16 concentraciones de GTP, sugiere un valor para la aparente Kd de 120 ± 20 mM. Sin embargo, cuando se utiliza una escala logarítmica para el eje x, hay un significativo discrepancy entre el modelo y los datos. (C) Análisis de los mismos datos con un modelo cooperativo muestra un excelente ajuste a los datos que se utiliza un modelo cooperativo simple. Aquí, un K ½ de 230 ± 20 M se determinó, con el coeficiente de cooperatividad n = 0,52 ± 0,02 (con la parte inferior temperaturas (T1) y superior (T2) de ajuste a 69,63 ± 0,06 ° C y 79,9 ± 0,1 º C respectivamente). Como WCBM parece ser dimérica, esto implica que la enzima es perfectamente anticooperative en su unión a GTP. Estas imágenes fueron creadas usando GraphPad. Por favor haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 4. WCBI muestra un patrón de fusión bifásicaen presencia de su ligando coenzima A (CoA). (A) WCBI, en ausencia de ligando (azul), muestra un patrón de fusión monofásico simple. A altas concentraciones de ligando (1 mM; línea verde), se observa un patrón similar. Sin embargo, a concentraciones de ligando intermedios. (60 M; línea roja), dos picos de fusión distintos, que corresponden a los estados-ligando libre y unido al ligando se observan (B) La transición entre los dos conjuntos de picos es dependiente de la dosis a través de la completa gama de concentraciones. (C) Modelado de la fusión bifásica como una suma de una parte de los resultados de ligando ligando libre y los altos da un buen ajuste a los datos (línea de trazos púrpura, en comparación con la línea roja). Este ajuste se mejora extrapolando el resultado observado para la alta concentración de ligando (donde el modelo sugiere ~ 95% de ocupación) para ocupación completa (línea azul punteada). (D) Los datos obtenidos para la proporción de WCBI unido a CoA sho ws un excelente ajuste a un modelo de unión simple, con una Kd de 58 ± 2 M (estos datos representan datos recogidos en dos días separados, con un poco diferentes concentraciones de ligando elegidos para el segundo día basado en el primer conjunto de resultados). Paneles. (A - C) se prepararon utilizando Excel, y el panel (D) utilizando Graphpad Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Tabla 1 Receta para los experimentos iniciales.
| Reactivo | El volumen en la mezcla de (l) |
| Proteína | Para concentración final de 0,11 mg / ml |
| 0.3 | |
| 0,5 M de HEPES pH 7,0 | 3.7 |
| M NaCl 5 | 5.6 |
| Agua | 180 l |
Esto describe la "mezcla maestra" de proteínas, reactivo de detección y tampón para un experimento inicial de exploración para proporcionar una estimación de K d, como se describe en la sección de protocolo 1. Esta mezcla tampón es apropiado para proteínas genéricos. Cuando los resultados anteriores sugieren otros tampones deben ser utilizados, estos deben ser sustituidos. Si la acción es la proteína a una concentración baja (es decir, menos de 0,3 mg / ml), puede ser necesario reducir la cantidad de tampón adicional añadido para compensar tampón ya está presente en la muestra de proteína.
Cuadro 2 Receta para la determinación de K d.| Reactivo | El volumen en la mezcla de (l) |
| Proteína | Para concentración final de 0,11 mg / ml |
| 5,000x SYPRO Naranja | 1.78 |
| 0,5 M de HEPES pH 7,0 | 22.2 |
| M NaCl 5 | 33.3 |
| Agua | 180 l |
Esto describe la "mezcla maestra" de proteínas, reactivo de detección, y bUffer para una determinación completa de K d para una muestra de proteína, tal como se describe en la sección de protocolo 2. Esta mezcla tampón es apropiado para proteínas genéricos. Cuando los resultados anteriores sugieren otros tampones deben ser utilizados, estos deben ser sustituidos. Si la acción es la proteína a una concentración baja (es decir, menos de 0,3 mg / ml), puede ser necesario reducir la cantidad de tampón adicional añadido para compensar tampón ya está presente en la muestra de proteína.
Cuadro 3 ecuaciones y parámetros para el análisis de datos.
d| Paso en el protocolo experimental | Ecuación requerido | Parámetros requeridos | Descripción de las variables y los parámetros de |
| 3.3 | |||
| Solo ligando sitio de unión | Y = Bottom + ((Top-Bottom) * (1 - ((PK d -X + sqrt (((P + X + K d) ^ 2) - (4 * P * X))) / (2 * P )))) | P: concentración de proteínas. Kd: constante de disociación. P y Kd se dan en las mismas unidades que se utilizaron para las concentraciones de ligando. Superior, Inferior: temperaturas de fusión en la concentración de ligando infinita y no la concentración de ligando, respectivamente. | |
| 3.4 | Bottom = * YMIN | YMIN: Valor mínimo de Y (el más bajo de proteína experimental Tm, en este caso) | |
| Top = * YMAX | YMAX: Máximo valor de Y (el más alto de proteína experimental Tm) | ||
| K d = * X en ymid | Ymid: valor de Y que corresponde a la media de YMIN e Ymax. X es el valor correspondiente X (aquí, la concentración de ligando relevante) | ||
| P = (valor inicial, para estar en forma) | |||
| 4.1 | |||
| Modelo cooperativo simple | Y = Bottom + ((Top-Bottom) * (((X / Kd) ^ n) / (1 + ((x / Kd) ^ n)))) | n: Hcoeficiente enfermo. Esto describe la cooperatividad, u otras propiedades bioquímicas, de la proteína, y no es necesariamente una estimación del número de sitios de unión a ligando en la proteína. Un coeficiente de Hill de uno representa no cooperatividad; valores inferiores a uno indican cooperatividad negativa, y valores mayores que uno cooperatividad positiva. | |
| Bottom = * YMIN | |||
| Top = * YMAX | |||
| K d = * X en ymid | |||
| P = (valor inicial, para estar en forma ) | |||
| n = (valor inicial, para estar en forma) | |||
| La unión de dos ligandos secuencial | Y = Bottom + ((Top-Bottom) * ((X ^ 2) / (K * d K2)) / (1 + (X / K d) + ((x ^ 2) / (K * d K2))) ) | K2: constante de disociación para el segundo evento de unión. | |
| Bottom = * YMIN | |||
| Top = * YMAX | |||
| K2 = * X en ymid | |||
| P = (valor inicial, para estar en forma) | |||
| Unión independiente de dos ligandos | Y = Bottom + ((Top-Bottom) * ((X ^ 2) / (K * d K2)) / (1 + (2 * X / K d) + ((x ^ 2) / (K d K2 *) ))) | ||
| Bottom = * YMIN | |||
| K d = * X en ymid | |||
| K2 = * X en ymid | |||
| P = (valor inicial, para estar en forma) | |||
| 5.5 | |||
| Análisis de los cambios en la temperatura de fusión binarios | Y = 1 - ((PK d -X + sqrt (((P + X + K d) ^ 2) - (4 * P * X))) / (2 * P)) | ||
| Bottom = * YMIN | |||
| Top = * YMAX | |||
| K d = * X en ymid | |||
| P = (valor inicial, para estar en forma) | |||
| 5.8 | |||
| La extrapolación a la concentración de ligando infinita | (C2 - ((1 a $ R $ 2) * B2)) / $ R $ 2 | B2: célula que contiene el resultado sin ligando. C2: célula que contiene el resultado con la máxima ligando. $ R $ 2: celda que contiene la proporción con destino a una concentración máxima del ligando. |
Los pasos 3, 4 y 5 requieren la adición de ecuaciones detalladas en el software de análisis y definición precisa de comenzar parámetros para el análisis de datos. Las ecuaciones para cada paso relevante se muestran, con las selecciones correctas de los parámetros. Se proporciona una explicación del significado de las variables y los parámetros de referencia.
Tabla 4 Concentraciones para la detección de la interacción de la hexoquinasa con glucosa.
| Punto de muestreo | Ligando (Glucose) concentración (mM) |
| 1 | 0 |
| 2 | 0.001 |
| 3 | 0.005 |
| 4 | 0.01 |
| 5 | 0.03 |
| 6 | 0,1 |
| 7 | 0.3 |
| 8 | 0.4 |
| 9 | 0,7 |
| 10 | 1.1 |
| 11 | 2.1 |
| 12 | 3.7 |
| 13 | 5.3 |
| 14 | 7 |
| 15 | 9 |
| 16 | 11 |
Hexoquinasa de la levadura Saccharomyces cerevisiae en ciernes se añadió a la mezcla maestra como se describe en el protocolo, suplementado con 10 mM de MgCl 2 como el magnesio es un cofactor conocido. La estimación inicial de la K d fue de entre 0,5 y 2 mM. Los experimentos se establecieron para proporcionar las concentraciones finales indicadas de glucosa.
Cuadro 5 Concentraciones para la detección de la interacción de WCBM con el PIB.
| Punto de muestreo | Ligando (GTP) de concentración (M) |
| 1 | 0 |
| 2 | 0,5 |
| 3 | 1 |
| 4 | 5 |
| 5 | 10 |
| 6 | 25 |
| 7 | 50 |
| 8 | 100 |
| 9 | 250 |
| 10 | |
| 11 | 1000 |
| 12 | 2500 |
| 13 | 5000 |
| 14 | 7500 |
| 15 | 10000 |
| 16 | 20000 |
WCBM de Burkholderia pseudomallei se añadió a la mezcla maestra según se describe en el protocolo. La estimación inicial de la K d fue de alrededor de 100 mM. Los experimentos se establecieron para proporcionar las concentraciones finales indicadas de GTP, con el objetivo de cubrir al menos dos órdenes de magnitud por encima y por debajo de K d.
Discusión
Fluorimetry diferencial de barrido ha demostrado su poder como un método robusto y versátil para la caracterización de las proteínas, y la identificación de posibles ligandos proteicos. Los éxitos bien documentados en acelerar la estabilización de proteínas, el descubrimiento de fármacos (especialmente en laboratorios tan bien financiados) y cristalización 10,23-25 han convertido en un método atractivo para la selección inicial de los compuestos. Compuestos añadidos a las proteínas muestran un aumento dependiente de la dosis claro en el 7,9 temperatura de fusión aparente. Sin embargo, ha habido pocos intentos de utilizar los resultados de estos experimentos para determinar las constantes de unión aparentes para ayudar en la clasificación de compuestos por su afinidad. A continuación, presentamos un método para determinar sistemáticamente una constante de disociación aparente para las proteínas en presencia de un ligando.
Los resultados presentados aquí demuestran que DSF puede proporcionar rápidamente y con firmeza estimaciones de la constante de disociación parauna combinación de proteína-ligando. Los datos observados se pueden manipular con las herramientas disponibles en el mercado para proporcionar una determinación rápida de K d, sin la necesidad de hacer suposiciones sobre el valor probable de parámetros. El método tiene una ventaja significativa sobre algunos métodos comparables de ser parsimonioso en tanto la proteína y el tiempo requerido. El experimento descrito aquí consumirá 0,13 mg de proteína por experimento (aproximadamente 0,4 mg por repetidos experimentos por triplicado). Esto se compara favorablemente con la calorimetría de titulación isotérmica (ITC), donde un solo experimento con un promedio de proteína de 40 kDa consumirá una cantidad similar. El conjunto completo de los experimentos necesarios para este protocolo consumiría alrededor de 4 horas, incluyendo la preparación, para un único conjunto de experimentos. De nuevo, esto es probable que sea considerablemente más rápido que los métodos como el CCI o la resonancia de plasmón de superficie, mientras que a menudo requieren la optimización de gran alcance considerable para lograr mejores datos.
Nuestros resultados demuestran que sigue habiendo un requisito de examinar cuidadosamente los datos en bruto, el ajuste de estos datos para determinar la temperatura de fusión, y el ajuste de los datos de temperatura de fusión para determinar la constante de disociación. Un primer desafío es la forma de los datos en bruto producidas en la fusión de proteínas. En algunos casos, la forma no puede aproximarse a la observada en la Figura 1A. Problemas comunes incluyen cambios de baja temperatura en la unión del ligando, de alta fluorescencia de fondo, y múltiples transiciones inusuales de temperatura. Cambios de temperatura baja se ven en la unión de un número de ligandos. Para este método, el parámetro más crítico es el error en la medición T m, en comparación con el cambio de temperatura. Los datos por lo general se puede montar razonablemente bien cuando la desviación estándar de las mediciones por triplicado no supere el 10% del cambio de temperatura de fusión entre la proteína no unida y completamente unido. Nuestra experiencia es que cuando dicha temperaturaturnos bibliografía son sólo 2 ° C, esto puede ser suficiente para el montaje de los datos, si los puntos de datos individuales son muy precisos. Una segunda cuestión tiene forma inusualmente curvas. Estos son distintos en las proteínas libres y formas ligando unido, como el ligando de unión afecta los modos de la proteína de despliegue. En estos casos, el usuario debe tener en cuenta si los datos se pueden utilizar con la debida consideración de los modelos a ser utilizado para determinar la temperatura de fusión y la constante de disociación. Otro problema común es que la adición de un cofactor de la proteína (por ejemplo, de MgCl 2 en nuestro ejemplo con hexoquinasa) se requiere para obtener los datos más fiables. Nuestra experiencia ha sido que una cuidadosa consideración de todos los factores que en el experimento en la etapa de toma de lecturas iniciales es esencial para obtener los mejores resultados. Además, los tratamientos teóricos alternativos pueden revelar características de estos datos 15,17. Por último, no es raro que algunas proteínas que contain nativa expuesta regiones hidrófobas para mostrar alta fluorescencia de fondo. Hay un número de soluciones a estos problemas, que han sido ampliamente revisado en otro 6,9.
En particular, el usuario debe considerar la posibilidad de utilizar el Boltzmann o modelos derivados (por ejemplo, Figura 4), y en el caso de uso de derivados, si se funde múltiples deben ser modelados. Los dos métodos de modelado de la desplegamiento térmico se diferencian en que el método de Boltzmann ajusta a los datos experimentales a la ecuación de Boltzmann, asumiendo una forma sigmoidal regular a la curva de desplegamiento. En contraste, el método derivado toma la primera derivada de los datos experimentales en cada punto (panel inferior en la Figura 1A), y considera que la temperatura de fusión para ser el punto más alto de la primera derivada. El método derivado generalmente devuelve una temperatura de fusión más alto en alrededor de 2 - 3 ° C. La mayoría de las proteínas volverán una más consistenteresultado (es decir, el error estándar de la temperatura de fusión para los experimentos por triplicado es menor) para uno de los dos métodos. Esto es generalmente íntimamente relacionada con la forma precisa de la curva de desplegamiento de la proteína, y es necesario para determinar empíricamente el mejor método en cada caso. Cuando se usa el modelo derivado, también es importante tener en cuenta múltiples eventos de fusión. Algunos datos muestran claramente la evidencia para múltiples transiciones, y en estos casos los resultados es probable que sean más fáciles de interpretar si se modelan estas múltiples eventos de fusión. En el contexto de este protocolo, a menudo es el caso que la adición de ligando puede causar una proteína para pasar de tener múltiples transiciones de fusión a una sola transición (por ejemplo, mediante la estabilización de el subdominio térmicamente más frágil), o viceversa. Por lo tanto le abogar por que los datos en bruto se examinan juntos antes de considerar qué enfoque será mejor usar.
Después de la modelling de las temperaturas de fusión individuales, otras cuestiones pueden surgir en la adaptación de éstos a los modelos que se presentan en la sección de protocolo. Es imperativo examinar cuidadosamente el ajuste a la ecuación constante de disociación mediante una escala logarítmica, ya que este análisis pone de manifiesto a menudo discrepancias entre los datos observados y el modelo (e .g., Figura 3). Mientras que los resultados obtenidos son generalmente robusta, cuidado en la interpretación ofrece la oportunidad de extraer mejores resultados, y el más significado, a partir de los datos.
Una cuestión particular planteada por estos datos es la interpretación que se debe colocar en las proteínas que muestran cooperatividad, o múltiples eventos de unión, en DSF. Hemos, hasta la fecha, sólo se observa este fenómeno en las proteínas que se espera que tengan múltiples eventos de unión específicos (por ejemplo, WCBM, una proteína cuya mejor homólogo es un multímetro de 26 años, y que actúa como un multímero en cromatografía de exclusión [de datos nomuestra]). No es del todo claro que la cooperatividad negativa observada en la desnaturalización DSF indica que la enzima en última instancia, mostrar cooperatividad negativa: más bien, esto puede ser una indicación de complejos de unión que debe ser explorado más a fondo el uso de una gama más amplia de métodos. Esto nos sugiere, sin embargo, que es probable que identificar interesantes efectos más amplios estudios de dichas proteínas.
Los valores dados para la constante de disociación utilizando este método son generalmente del mismo orden que los proporcionados por otros métodos, tales como calorimetría de titulación isotérmica y resonancia de plasmón superficial. Sin embargo, los valores absolutos observados son con frecuencia más alta que la observada utilizando estos métodos. Esto es al menos en parte, una consecuencia del hecho de que la constante de disociación se observa a la temperatura de fusión de la proteína con ligando. Este K d es generalmente más alto que a temperaturas fisiológicas. El dissociation constante está relacionado con la temperatura de la reacción por las ecuaciones:
 [1]
[1]
 [2]
[2]
(Donde θ c es la concentración de referencia estándar, Δ R G es el cambio de energía libre de Gibbs de la reacción, R es la constante molar de los gases, Δ H es el cambio de entalpía en la reacción, y Δ S es el cambio de entropía en la reacción .)
Reacciones con constantes de disociación en el rango medible de este método generalmente tendrá un Δ r negativo G, y así el efecto de un aumento de la temperatura en la ecuación [1] serán aumentar la constante de disociación. Tanto los términos Δ Δ H y S que constituyen la energía libre de Gibbs (ecuación [2]) son temperature dependiente 27, y el efecto sobre la constante de disociación se dependen de la magnitud y el signo de estas dependencias de temperatura, y serán necesariamente dependientes de la interacción. En consecuencia, no es inesperado que las constantes de disociación determinadas por este método son a veces mayor que las determinadas por métodos que operan a temperatura ambiente. Dependencia de la temperatura es, por supuesto, también una advertencia de muchos otros métodos, que tienden a proporcionar la constante de disociación a temperaturas inferiores a la temperatura fisiológica.
Otra advertencia del método DSF es que es un método de marcado, a diferencia de ITC. La etiqueta fluorescente utilizado (SYPRO naranja) es hidrófobo, y así en algunos casos puede competir con la unión de ligandos a las proteínas hidrófobas. En consecuencia, es probable que en algunos casos, la constante de disociación obtenida se elevó artificialmente debido a la competencia con la etiqueta. Sin embargo, para la comparación de diversos ligandos, (el uso primario deDSF), las diferencias son poco probable que sea lo suficientemente significativos para afectar el ranking de los compuestos por afinidad.
Un posible inconveniente de este método es el límite de detección que se puede lograr. En principio, no debería ser posible medir con precisión un valor para K d que es menor que 50% de la concentración de proteína, e incluso en este rango de valores es probable que sean de dudosa precisión. Mientras que el límite de detección en este extremo de la gama se puede extender un poco mediante la reducción de las concentraciones de proteína y el tinte, la sensibilidad del instrumento evitar una mayor reducción en la concentración de proteínas. Del mismo modo, el extremo superior de la sensibilidad será determinado por la solubilidad del ligando. Para obtener una estimación robusta matemáticamente para K d, es más importante para obtener datos con un 90% de la proteína presente en la forma unida al ligando, lo que requiere concentraciones de ligando a ser aproximadamente diez veces K d (suponiendo que no cooperatividad). El límite de detección será por lo tanto necesariamente una décima parte de la solubilidad del ligando en el tampón correspondiente. Esto significa que los límites de detección del método variará típicamente entre 1 M y entre 1 y 100 mM, dependiendo de la proteína y el ligando.
En conclusión, fluorimetría diferencial de barrido es una técnica versátil aplicable a una amplia gama de proteínas. Utilizando los métodos presentados aquí, es posible determinar rápida y económicamente la afinidad de una proteína para diferentes ligandos. Esto tiene un gran potencial para su aplicación en la purificación de proteínas y la estabilización, la aclaración de la función o especificidad de las enzimas de metagenomes, y en el descubrimiento de fármacos, sobre todo en los pequeños laboratorios.
Divulgaciones
The authors declare that they have nothing to disclose.
Agradecimientos
This work was funded by grant from the BBSRC (grant number BB/H019685/1 and BB/E527663/1) to the University of Exeter.
Materiales
| Name | Company | Catalog Number | Comments |
| StepOne real time PCR instrument | Life Technologies | 4376357 | DSF can be performed with many other instruments. The StepOne instrument has very convenient software for data analysis. |
| Protein thermal shift software v1.0 | Life Technologies | 4466037 | |
| MicroAmp Fast optical 48-well plates | Life Technologies | 4375816 | |
| Optical sealing tape | Life Technologies | 4375323 | Bio-rad part no. 223-9444 is an alternative supplier |
| U-bottomed 96-well plates | Fisher | 11521943 | |
| SYPRO Orange | Life Technologies | S6650 | For a smaller volume supplier, use Sigma part no. S5692 |
| SPSS statistics version 20 | IBM | Other statistics packages will provide similar functionality | |
| GraphPad Prism 6.02 | GraphPad | Other statistics packages will provide similar functionality | |
| Hand applicator (PA1) | 3M | 75-3454-4264-6 | |
| Hexokinase from Saccharomyces cerevisiae | Sigma-Aldrich | H5000 | |
| Glucose | Fisher scientific | 10141520 |
Referencias
- Freyer, M. W., Lewis, E. A. Isothermal titration calorimetry: experimental design, data analysis, and probing macromolecule/ligand binding and kinetic interactions. Methods Cell Biol. 84, 79-113 (2008).
- Ladbury, J. E. Calorimetry as a tool for understanding biomolecular interactions and an aid to drug design. Biochem Soc Trans. 38, 888-893 (2010).
- Abdiche, Y., Malashock, D., Pinkerton, A., Pons, J. Determining kinetics and affinities of protein interactions using a parallel real-time label-free biosensor, the Octet. Anal Biochem. 377, 209-217 (2008).
- Pantoliano, M. W., et al. High-density miniaturized thermal shift assays as a general strategy for drug discovery. J Biomol Screen. 6, 429-440 (2001).
- Senisterra, G., Chau, I., Vedadi, M. Thermal denaturation assays in chemical biology. Assay Drug Dev Technol. 10, 128-136 (2012).
- Ericsson, U. B., Hallberg, B. M., Detitta, G. T., Dekker, N., Nordlund, P. Thermofluor-based high-throughput stability optimization of proteins for structural studies. Anal Biochem. 357, 289-298 (2006).
- Lo, M. C., et al. Evaluation of fluorescence-based thermal shift assays for hit identification in drug discovery. Anal Biochem. 332, 153-159 (2004).
- Nettleship, J. E., Brown, J., Groves, M. R., Geerlof, A. Methods for protein characterization by mass spectrometry, thermal shift (ThermoFluor) assay, and multiangle or static light scattering. Methods Mol Biol. 426, 299-318 (2008).
- Niesen, F. H., Berglund, H., Vedadi, M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat Protoc. 2, 2212-2221 (2007).
- Geders, T. W., Gustafson, K., Finzel, B. C. Use of differential scanning fluorimetry to optimize the purification and crystallization of PLP-dependent enzymes. Acta Crystallogr Sect F Struct Biol Cryst Commun. 68, 596-600 (2012).
- Vedadi, M., et al. Chemical screening methods to identify ligands that promote protein stability, protein crystallization, and structure determination. Proc Natl Acad Sci USA. 103, 15835-15840 (2006).
- Davis, B. J., Erlanson, D. A. Learning from our mistakes: the 'unknown knowns' in fragment screening. Bioorg Med Chem Lett. 23, 2844-2852 (2013).
- Larsson, A., Jansson, A., Aberg, A., Nordlund, P. Efficiency of hit generation and structural characterization in fragment-based ligand discovery. Curr Opin Chem Biol. 15, 482-488 (2011).
- Scott, D. E., et al. Using a fragment-based approach to target protein-protein interactions. Chembiochem. 14, 332-342 (2013).
- Cimmperman, P., et al. A quantitative model of thermal stabilization and destabilization of proteins by ligands. Biophys. J. 95, 3222-3231 (2008).
- Matulis, D., Kranz, J. K., Salemme, F. R., Todd, M. J. Thermodynamic stability of carbonic anhydrase: measurements of binding affinity and stoichiometry using ThermoFluor. Biochemistry. 44, 5258-5266 (2005).
- Zubriene, A., et al. Measurement of nanomolar dissociation constants by titration calorimetry and thermal shift assay - radicicol binding to Hsp90 and ethoxzolamide binding to CAII. Int J Mol Sci. 10, 2662-2680 (2009).
- Weiss, J. N. The Hill equation revisited: uses and misuses. FASEB J. 11, 835-841 (1997).
- Cuccui, J., et al. Characterization of the Burkholderia pseudomallei K96243 capsular polysaccharide I coding region. Infect Immun. 80, 1209-1221 (2012).
- DeShazer, D., Waag, D. M., Fritz, D. L., Woods, D. E. Identification of a Burkholderia mallei polysaccharide gene cluster by subtractive hybridization and demonstration that the encoded capsule is an essential virulence determinant. Microb Pathog. 30, 253-269 (2001).
- Berman, H., Henrick, K., Nakamura, H. Announcing the worldwide Protein Data Bank. Nat Struct Biol. 10, 980(2003).
- Vivoli, M., Ayres, E., Beaumont, E., Isupov, M., Harmer, N. Structural insights into WcbI, a novel polysaccharide biosynthesis enzyme. IUCr Journal. 1 (1), 28-38 (2014).
- Sorrell, F. J., Greenwood, G. K., Birchall, K., Chen, B. Development of a differential scanning fluorimetry based high throughput screening assay for the discovery of affinity binders against an anthrax protein. J Pharm Biomed Anal. 52, 802-808 (2010).
- Uniewicz, K. A., et al. Differential scanning fluorimetry measurement of protein stability changes upon binding to glycosaminoglycans: a screening test for binding specificity. Anal Chem. 82, 3796-3802 (2010).
- Wan, K. F., et al. Differential scanning fluorimetry as secondary screening platform for small molecule inhibitors of Bcl-XL. Cell Cycle. 8, 3943-3952 (2009).
- Koropatkin, N. M., Holden, H. M. Molecular structure of alpha-D-glucose-1-phosphate cytidylyltransferase from Salmonella typhi. J Biol Chem. 279, 44023-44029 (2004).
- Paleskava, A., Konevega, A. L., Rodnina, M. V. Thermodynamics of the GTP-GDP-operated conformational switch of selenocysteine-specific translation factor SelB. J Biol Chem. 287, 27906-27912 (2012).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados