
Method Article
Détermination des interactions protéine-ligand à l'aide différentielle à balayage Fluorimétrie
Dans cet article
Résumé
Differential scanning fluorimetry is a widely used method for screening libraries of small molecules for interactions with proteins. Here, we present a straightforward method to extend these analyses to provide an estimate of the dissociation constant between a small molecule and its protein partner.
Résumé
Une large gamme de méthodes sont actuellement disponibles pour la détermination de la constante de dissociation entre une protéine et l'interaction des petites molécules. Cependant, la plupart d'entre eux nécessitent l'accès à des équipements spécialisés, et ont souvent besoin d'un degré d'expertise d'établir efficacement des expériences fiables et analyser les données. Fluorimétrie à balayage différentiel (CSD) est de plus en plus utilisée comme une méthode robuste pour le criblage initial de protéines de petites molécules de l'interaction, soit pour l'identification de partenaires de découverte physiologiques ou de toucher. Cette technique présente l'avantage qu'il suffit d'une machine appropriée pour la PCR quantitative PCR, et si approprié instrumentation est disponible dans la plupart des institutions; une excellente gamme de protocoles sont déjà disponibles; et il existe de fortes précédents dans la littérature pour de multiples utilisations du procédé. Les travaux antérieurs a proposé plusieurs moyens de calculer les constantes de dissociation à partir des données de DSF, mais ceux-ci sont mathématiquement exigeant. Ici, nous DEMtrent un procédé d'estimation des constantes de dissociation à partir d'une quantité modérée de DSF des données expérimentales. Ces données peuvent généralement être recueillies et analysées en une seule journée. Nous démontrons comment les différents modèles peuvent être utilisés pour ajuster les données recueillies à partir des événements de liaison simples, et où les sites de liaison ou indépendants de liaison de coopération sommes présents. Enfin, nous présentons un exemple d'analyse de données dans le cas où les modèles standard ne s'appliquent pas. Ces méthodes sont illustrées avec les données recueillies sur les protéines de contrôle disponibles dans le commerce, et deux protéines de notre programme de recherche. Dans l'ensemble, notre méthode fournit un moyen simple pour les chercheurs d'acquérir rapidement d'autres renseignements sur les interactions protéine-ligand à l'aide de DSF.
Introduction
Toutes les protéines se lient avec des affinités variables, à un large éventail d'autres molécules de simples ions à d'autres grandes macromolécules. Dans de nombreux cas, les protéines se lient à des petites molécules partenaires dans le cadre de leur fonction normale (par exemple, une kinase de liaison à l'ATP). Autres interactions peuvent être sans rapport avec la fonction, mais sont expérimentalement utiles comme outils (par exemple, les petites molécules qui stabilisent les protéines pour améliorer la réussite de cristallisation, ou aider à maintenir les protéines en solution); tandis que les petites molécules qui se lient à des sites actifs et les sites allostériques des protéines peuvent agir comme des inhibiteurs, et ainsi de moduler l'activité des enzymes.
Il existe une grande variété de techniques qui peuvent être utilisées pour déterminer l'affinité pour les molécules de protéines partenaires. Calorimétrie de titration isotherme 1 est largement considéré comme un "étalon-or", car il fournit des informations riches sur les réactions, est le marqueur libre, et a limité les possibilités pour unertifacts de l'expérience. Cependant, malgré des améliorations récentes dans la sensibilité de l'instrumentation et de l'automatisation de montage expérimental, il est encore relativement coûteux en termes de besoins en protéines, a au mieux un débit faible à moyen, et est le mieux adapté aux interactions avec modéré à affinités élevées (10 nM à 100 pM Kd) 2. D'autres méthodes étiquettes libres, tels que la résonance des plasmons de surface ou bicouche interférométrie 3 offre des débits plus élevés, et ont réalisé la sensibilité pour détecter des molécules plus petites aussi faibles que 100 Da. Toutefois, les instruments à haut débit pour ces méthodes sont relativement coûteux, sont justifiées que si il y aura un débit continu de projets pertinents, et sont donc susceptibles d'être inaccessibles à de nombreux laboratoires universitaires.
Fluorimétrie différentielle à balayage (DSF ou thermofluor) a été décrite pour la première fois en 2001 en tant que méthode 4 pour la découverte de médicaments. Dans cette méthod, les protéines sont incubées avec un colorant fluorescent (colorants initialement naphtalène-sulfonique ont été utilisés), ce qui modifie sa fluorescence lors de la liaison à des régions hydrophobes des protéines. L'échantillon de protéine-colorant est ensuite chauffé, et la fluorescence contrôlée comme la chaleur monte. Le dépliement de la protéine, et l'exposition des parties hydrophobes de la protéine, donne naissance à un motif caractéristique de la fluorescence en fonction de la température (Figure 1A). L'essai peut être réalisé en de petites quantités dans un instrument PCR quantitative commerciale, et donc en une seule expérience, un grand nombre d'échantillons peuvent être testés en même temps (habituellement 48, 96 ou 384 échantillons, en fonction du modèle de l'instrument). Des expériences peuvent être effectuées habituellement en environ une heure, offrant la possibilité d'une analyse à haut débit de 5 échantillons.
D'autres améliorations de la méthodologie ont vu l'adoption de colorants avec de meilleures propriétés spectrales 6,7 , outils génériques pour l'analyse des données, et suggéré des protocoles de dépistage initial 8,9. La gamme des applications de la méthode a été étendue, avec un accent particulier sur la création de conditions optimales pour la préparation et le stockage des protéines 10, et sur l'identification de partenaires potentiels de liaison pour faciliter la cristallisation 11. Le débit relativement élevé de la méthode, un coût relativement faible en protéines (~ 2 pg par réaction), et l'applicabilité à l'étude de la faiblesse des molécules de liaison a fait DSF un outil précieux pour le fragment basé la conception de médicaments, en particulier dans un contexte académique 12-14.
Malgré la large application de DSF à l'étude des interactions protéine-ligand, peu d'études ont décrit la détermination des constantes de dissociation de ces études. Toutefois, ceux-ci ont tendance à produire des équations détaillées décrivant le déroulement de la protéine, avec de nombreux paramètres qui doivent être montés sur des données éparses ou dans certains cas, estimated 7,15-17. Ces méthodes sont d'une importance particulière dans les cas difficiles, tels que les composés fortement contraignants, ou des protéines présentant des transitions inhabituelles. Cependant, pour de nombreux laboratoires, ces analyses détaillées sont trop lourdes pour une utilisation courante. Nous proposons donc des traitements alternatifs pour les différents scénarios, et montrons comment elles peuvent être utilisées pour ajuster les données résultant de différentes interactions protéine-ligand. Notre méthode utilise l'instrument StepOne qPCR, pour lequel le logiciel d'analyse de données sur mesure est disponible; tout ce qui accélère l'analyse des données, les résultats d'autres instruments peuvent être traitées en utilisant des procédés publiés antérieurement neuf, et en aval à la même analyse peut être effectuée.
Protocole
1 Détermination d'une valeur approximative de la constante de dissociation (c'est à dire, à moins d'un ordre de grandeur)
- Préparer le mélange indiqué dans le tableau 1.
- Préparer les stocks du ligand d'intérêt à la concentration la plus élevée disponible, puis à six dix dilutions de cette. Si un K d approximatif est connu d'après les données antérieures, visent à avoir au moins deux concentrations au-dessus et au-dessous de la valeur de Kd.
- Aliquote de 18 ul du mélange dans huit puits dans une plaque qPCR. Ajouter 2 ul de solvant dans le premier puits. Ajouter 2 ul de chaque membre de la série ligand de dilution (étape 1.2) à un puits de chacune des sept autres puits.
- Placez un joint qPCR sur la plaque. Pour obtenir une bonne étanchéité de la plaque, placer un applicateur à main (voir les tableaux de réactifs spécifiques) dans le milieu de la plaque. Lisser le joint d'un côté, puis répétez de l'autre moitié dela plaque.
- Centrifuger la plaque à 500 g pendant deux minutes pour éliminer les bulles d'air.
- Placer la plaque dans un instrument StepOne qPCR. Sélectionnez l'option "courbe en fusion", les filtres de ROX, et choisir la vitesse de rampe rapide (cela donne une pause de 2 min à 25 ° C, suivie d'une rampe à 99 ° C pendant 40 min, puis 2 min de pause). Exécutez une dénaturation thermique.
REMARQUE: Les fichiers de script pour effectuer une course sont disponibles en ligne à l'adresse http: // www.exeter.ac.uk/biosciences/capsular. - À la fin de la course de l'instrument, cliquez sur le bouton "Analyser" sur l'écran. Enregistrez le fichier de résultat.
- Ouvrez le logiciel Shift thermique des protéines.
- Créer une nouvelle étude; dans l'onglet Propriétés, donner ce nom, et dans l'onglet Conditions, détail des ligands.
- Allez sur l'onglet Fichiers de l'expérience, et importer le fichier enregistré des résultats (des XXX.eds), et définir le contenu de chaque puits (modèle files sont disponibles auprès des auteurs).
- Allez sur l'onglet d'analyse, et appuyez sur le bouton "Analyser".
NOTE: Ce rapport analysera les résultats. Il est possible d'exporter les résultats pour complément d'enquête avec Excel en utilisant l'onglet Exporter. Les résultats sont exportés dans un format délimité onglet. Il est préférable d'ouvrir le fichier exporté dans Excel, et immédiatement enregistrer au format Excel.
- Vérifier que la protéine en présence du solvant seul donne un résultat similaire à celui représenté sur la figure 1A. Ensuite, examinez les températures de fusion observés dans les résultats dans le volet "répliqué". Assurez-vous que cela montre une nette augmentation de la température de fusion avec l'augmentation de la concentration de ligand.
REMARQUE: Idéalement, ce sera de fournir une température maximale de fusion clair (en supposant que la protéine est entièrement ligand lié), et un K approximative d où la température de fusion est à mi-chemin entre la protéine et ligand libre-au maximum.
2. expérimental pour la détermination de la constante de dissociation
- Préparer le mélange détaillées au tableau 2 comme un mélange maître.
- Préparation des stocks de ligand à quinze concentrations différentes, qui sera dilué dix fois dans l'expérience finale. Idéalement, inclure des concentrations d'au moins deux ordres de grandeur au-dessus et au-dessous de la valeur de Kd d'environ, et les concentrations centrer sur la valeur de Kd d'environ. Focus sur sept des points à l'intérieur d'un ordre de grandeur de la valeur de Kd d'environ, avec quatre points de part et d'autre de cette; si il ya un choix, inclure plus de points à des valeurs qui saturent.
REMARQUE: Si nécessaire, il est possible de modifier les conditions expérimentales telles que les stocks de ligands sont au double de la concentration expérimentale, où ligand solubilité est un facteur limitant. - Ajouter 120 ul du maîtremélanger à huit puits dans une plaque de puits à fond en U 96, à agir comme un réservoir pour une distribution commode du mélange maître. Utiliser une pipette à 8 canaux pour distribuer 18 pi dans une colonne d'une plaque de PCR. Répétez l'opération pour une période de cinq colonnes, pour donner un total de 48 puits remplis dans un modèle 6 x 8 sur la plaque.
- Ajouter 20 ul de stocks de ligand, ou le solvant, pour séparer les puits dans une plaque de puits à fond en U 96. En utilisant une pipette à 8 canaux, aspirer 2 pi de huit stocks de ligands différents (ou solvant). Ajouter à une colonne de la plaque de PCR qui a été rempli de mélange maître à l'étape 2.3. Répéter l'opération avec les mêmes huit ligand / stocks de solvants pour deux autres colonnes. Aspirer 2 pi de huit ligands ou de solvants stocks restants et les ajouter à une quatrième colonne de la plaque. Répétez cette opération pour les deux autres colonnes. Cela donnera des échantillons en triple pour les 16 ligand et des échantillons de solvants.
- Placer un joint qPCR sur la plaque (voir l'étape 1.4).
- Centrifuger la plaque à 500 g pourdeux minutes.
- Placer la plaque dans l'instrument qPCR. Exécutez une dénaturation thermique en utilisant les paramètres spécifiés à l'étape 1.6.
- À la fin de la course de l'instrument, cliquez sur le bouton "Analyser" sur l'écran. Enregistrez le fichier de résultat.
- Ouvrez le logiciel Shift thermique des protéines. Créer une nouvelle étude; dans l'onglet Propriétés, donner ce nom, et dans l'onglet Conditions, détail des ligands.
- Allez sur l'onglet Fichiers de l'expérience, et importer le fichier enregistré des résultats (des XXX.eds), et définir le contenu de chaque puits.
REMARQUE: Les fichiers de modèles sont disponibles en ligne à l'adresse http: // www.exeter.ac.uk/biosciences/capsular. - Allez sur l'onglet d'analyse, et appuyez sur le bouton "Analyser".
- Choisissez l'onglet "répliques" dans le menu sur le côté gauche de l'écran pour afficher les résultats sous forme de triple. Évaluer la fiabilité des données sur la base de serrage des trois exemplaires sont. Should triplicats montrent une faible reproductibilité, examiner de près les données brutes.
- Analyser les données en utilisant à la fois la Boltzmann ou méthodes dérivés pour évaluer la température de fusion. Sélectionnez l'onglet "Répliquer les résultats", et dans le "Répliquer résultats complot", basculer le «complot par:" touche entre «Tm - Boltzmann» et «Tm - dérivé". Sélectionnez la méthode qui donne la plus grande reproductibilité de l'échantillon. Exporter les résultats pour complément d'enquête avec Excel en utilisant l'onglet Exporter.
NOTE: Pour les échantillons qui montrent des transitions multiples, il est presque toujours préférable d'utiliser la méthode de la dérivée en mode de fusion multiple. Les résultats sont exportés dans un format délimité onglet. Il est préférable d'ouvrir le fichier exporté dans Excel, et immédiatement enregistrer au format Excel. - Recommencer l'expérience au moins deux fois, y compris un motif sur un autre jour, pour assurer la reproductibilité des résultats. Si l'analyse des données (voir l'étape 3 ci-dessous)indiquent que la valeur de K d est significativement différente de l'estimation initiale, de modifier les concentrations de ligand en conséquence (voir l'étape 2.2) pour assurer une bonne gamme de valeurs autour de K d.
3 Analyse des données pour déterminer la constante de dissociation sous dénaturation thermique
- Insérer un tableau Excel dans des concentrations de ligand et de la température de fusion.
- Ouvrez le logiciel GraphPad Prism, et de créer une table XY. Entrez les données, en utilisant la colonne de X pour les concentrations de ligand et la colonne Y pour la fusion des résultats de température.
REMARQUE: un exemple montré est la figure 1B. Un script avec des équations préchargées, et d'autres instructions pour l'utilisation du logiciel de statistiques SPSS, sont disponibles en ligne à l'adresse http: // www.exeter.ac.uk/biosciences/capsular. - Dans l'onglet Analyse, sélectionnez l'option de modifier le paramètre d'analyses (Ctrl + T). Pour entrer dans le modèle correct, sélectionnez "Nouveau" et "Créer une nouvelle équation". Insérez l'équation détaillé dans le tableau 3 comme «site de liaison ligand unique".
REMARQUE: Un exemple de ces étapes est illustrée à la figure 1C. Lorsque vous utilisez le script avec les équations préchargés, l'équation pertinente peut être dirigé choisi parmi la liste plutôt que saisies. Dérivation de cette équation est fournie en annexe. - Sélectionnez les "Règles de valeurs initiales" boîte, et entrez règles pour les valeurs initiales comme indiqué dans le tableau 3.
- Contraindre le paramètre P, comme "Constant égal à" et entrez la concentration finale de protéines (dans les mêmes unités que le ligand est donnée dans).
- Cliquez sur OK pour effectuer l'analyse.
REMARQUE: Un exemple de ces étapes est illustrée à la figure 1D. Le logiciel graphique produit une figure montrant les données et l'ajustement du modèle. Des exemples de tanalyses es sont présentés dans les données représentatives.
4. adaptation de données à des modèles de coopération
Pour ajuster les données à un modèle coopératif, choisir entre un modèle coopératif simple, ou un modèle où deux constantes de dissociation distincts sont définis. La première approche est préférable dans le cas de coopérativité négative, ou comme une enquête initiale. Toutefois, en principe, il est préférable dans les cas de coopération positive pour modéliser deux dissociation des constantes 18. Dans ce cas, la modélisation peut procéder soit en supposant liaison séquentielle de ligands, ou indépendant de liaison des ligands.
- Suivez les mêmes étapes initiales que dans le protocole 3 Toutefois, à l'étape 3.3, insérer des équations dans le tableau 3 répertorié comme "modèle coopératif simple", "liaison séquentielle de deux ligands", ou "liaison indépendante des deux ligands" 18.
- Sélectionnez les règles pertinentes pour les valeurs initialesassocié à chacune de ces équations dans le tableau 3.
- Étudier l'adaptation du modèle aux données. Si les données s'adapter mal, envisager un autre modèle.
REMARQUE: il est également important d'examiner attentivement la mise en place de la température de fusion de données par le logiciel Shift thermique des protéines (étape 2.9): il est parfois nécessaire de modifier les paramètres ici pour obtenir les meilleurs résultats. Une autre considération est de savoir si la gamme de points de données est idéal, et si il ya des points anormaux: soit un ensemble limité de données de chaque côté de K d, ou un point anormal unique (en particulier à des concentrations les plus élevées de ligand), peut affecter de manière significative les résultats. - Répéter l'expérience au moins deux fois (voir l'étape 2.12) afin d'assurer la reproductibilité.
5. adaptation de données à des courbes montrant Changements binaires dans Température de fusion
De temps en temps, au lieu d'une réponse graduée de ligand, les protéines ont étéobservé à adopter une réponse binaire, où l'échantillon est clairement lié séparé de l'échantillon non lié. Un exemple est fourni dans les résultats représentatifs (figure 4). Dans ce cas, l'ajustement de la température de fusion ne fournira pas un bon ajustement pour K d.
- Exporter la production de données brutes à partir du logiciel de déplacement thermique de la protéine. Pour chaque point de température, calculer la moyenne de fluorescence pour le ligand de zéro, et les concentrations les plus élevées de ligand. Compiler les résultats de chaque point à côté de ces données.
Remarque: l'erreur créée ici est inférieure à l'erreur dans les températures de fusion ajustées. - Ouvrez le logiciel de statistiques SPSS. Copiez les températures, les deux ensembles de données moyennes, et les données pour chaque expérience d'une fenêtre de données dans SPSS. Dans l'onglet variables, définir le jeu de données moyen sans ligand comme «faible», et l'ensemble de données de moyenne pour la plus forte concentration de ligand comme «élevé».
- Télécharger le fichier de syntaxe disponibleen ligne à l'adresse http: // www.exeter.ac.uk/biosciences/capsular . Sélectionnez "Exécuter → Exécuter tous".
- Copiez la proportion des résultats liés à un nouveau classeur Excel, avec les concentrations de ligand pertinents.
- Ouvrez le logiciel Graphpad, et de créer une table XY. Entrez les données, en utilisant la colonne de X pour les concentrations de ligand et la colonne Y pour la fusion des résultats de température. Dans l'onglet d'analyse, sélectionnez "Paramètres d'analyse du changement». Pour entrer dans le modèle correct, sélectionnez "Nouveau" et "Créer une nouvelle équation". Entrez l'équation donnée dans le tableau 3, inscrit comme «Analyse des changements binaires de la température de fusion".
- Cochez la case «Règles pour les valeurs initiales", et entrez règles pour les valeurs initiales détaillées dans le Tableau 3. Contraindre le paramètre P, comme "Constant égal à" et entrez la concentration finale de protéines (dans les mêmes unités que la ligae est donnée dans).
REMARQUE: des exemples de réalisation de ces boîtes pour le protocole de l'article 3 sont présentés à la figure 1C, D. - Si il est un bon ajustement, les résultats peuvent être améliorés en extrapolant le résultat attendu à la concentration de ligand infini. A partir du modèle de la proportion lié à chaque concentration de ligand, examiner la valeur de la valeur la plus élevée de la concentration du ligand. Si cela est 0,99 ou plus, une analyse plus approfondie est peu probable pour améliorer les résultats.
- Si la proportion est inférieure à 0,99, une étape supplémentaire est nécessaire pour corriger les effets de la protéine non liée à la plus forte concentration de l'échantillon de ligand. A l'étape 5.2, écrire la proportion de ligand lié au point de concentration de ligand le plus élevé (de l'étape 5.7) dans R2 cellule (une cellule différente peut être utilisée, de manière appropriée et R2 remplacé dans l'équation dans le tableau 3). Créer une colonne supplémentaire après la moyenne des résultats de concentrations plus élevées de ligand. Dans les sapinst cellule, copier l'équation figurant au tableau 3 que "l'extrapolation à l'infini concentration ligand". Copiez cette formule pour les cellules restantes dans cette colonne.
Note: Ce calcul supprime l'effet de la protéine non liée à la concentration la plus élevée de ligand. La différence entre la protéine de ligand libre et le ligand de concentration plus élevée est multiplié par l'inverse de la proportion lié à la concentration la plus élevée de ligand pour fournir la différence attendue entre les états de protéines complètement liés et non liés à chaque point de température. Cette différence est ajouté ou soustrait de l'état non lié à donner la fluorescence attendu pour la protéine entièrement ligand lié. - Remplacer la colonne de concentration de ligand maximale dans la fiche technique SPSS avec cette nouvelle colonne, et répéter l'ajustement des données.
REMARQUE: les étapes 5.7 à 5.9 peuvent avoir besoin d'être répétée si le modèle suggère un autre changement significatif dans la proportion liée à des concentrations d maximales ligand(si c'est le cas, il serait sans doute idéal pour répéter l'expérience avec un point de concentration de ligand supérieur inclus). - Dans les cas où la protéine présente un comportement coopératif et affiche décalage binaire, l'équation a proposé à l'étape 5.5 doit être substitué avec celles de l'étape 4.1. Le "Top" et les paramètres "bas" devraient être remplacés par 1 et 0 respectivement.
- Répéter l'expérience au moins deux fois (voir l'étape 2.12) afin d'assurer la reproductibilité.
Résultats
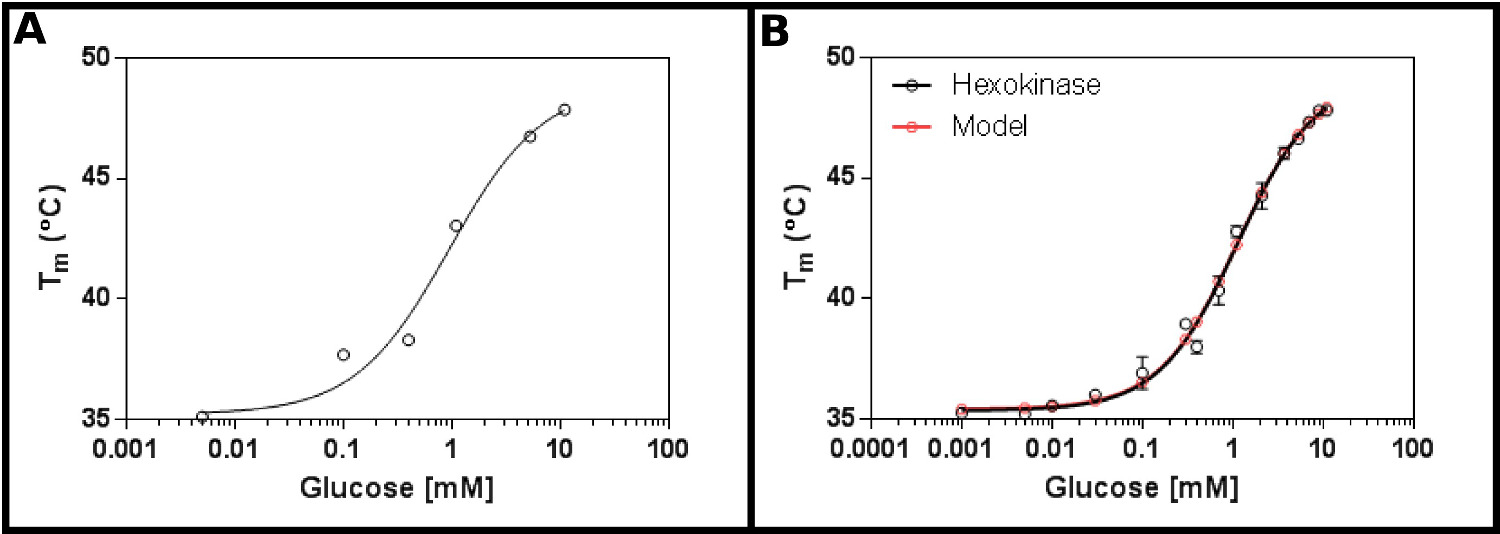
Un excellent substrat de test de cette méthode est l'hexokinase. Ceci présente l'avantage d'être facilement disponibles dans le commerce, et ayant deux substrats que l'on trouve dans la plupart des laboratoires, et qui fournissent des résultats reproductibles, claires dans l'essai. Un écran de concentration initiale (protocole 1), en utilisant l'hexokinase et de glucose (figure 2A), suggère que le K d sera probablement dans la gamme de 0,2 à 1,7 mm. Par conséquent, un plus grand écran (protocole n ° 2) a été réalisée en utilisant les concentrations indiquées dans le tableau 4 Les résultats (figure 2B) montrent un bon ajustement à l'équation de liaison du ligand à un seul site (section Protocole 3.3) [9], et a donné un K d de 1,2 ± 0,1 mM.
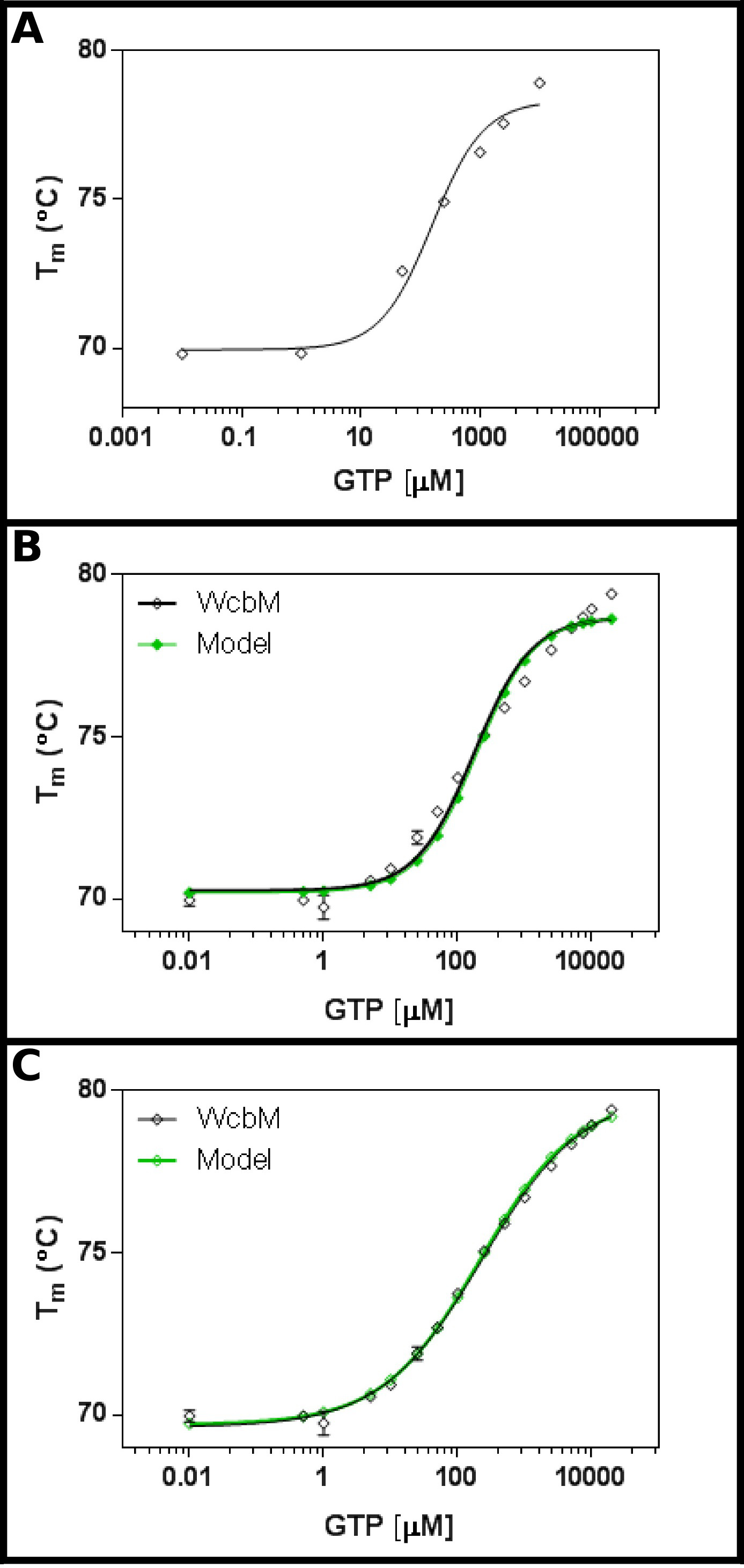
Le putatif heptose guanyl-transférase WCBM 19,20 montre une forte dérive thermique de la liaison de GTP (figure 3A). Un écran initial suggéré que le Kd est dans l'intervalle d'environ 100 pM. Par conséquent, un grand écran a été mis en place, en utilisant les concentrations indiquées dans le tableau 5 Montage des résultats à l'équation 3.3 a montré un ajustement raisonnable (R 2 de 0,981; figure 3B). .Toutefois, Il ya une différence évidente entre les données et la modèle, ce qui suggère que d'une équation différente est nécessaire. La recherche de la banque de données de protéines 21 avec la séquence WCBM montré que les homologues les plus proches pour lesquels les structures ont été déterminées forment des dimères. Les données ont donc été analysées en utilisant les trois équations de coopérative, séquentielle, et indépendant de liaison de deux ligands (protocole n ° 4). Les statistiques de montage pour un modèle coopératif a donné une valeur R 2 de 0,998 et l'écart type des résidus (Sy.x) de 0,215, tandis que les deux modèles de fixations successives et indépendantes ont une valeur R 2 de 0,992 et une Sy.x de 0,480 et 0,461 respectivement. Ceci suggère que le modèledonne le meilleur ajustement aux données a été le modèle coopératif: ici, un K ½ de 230 ± 10 pM a été observée, avec une valeur de n de 0,52 ± 0,02 (figure 3C). Ceci indique une coopérativité négative à la liaison. Notez qu'une ½ K a été utilisé dans ce cas, plutôt que de Kd, comme les unités de K d seraient plutôt insatisfaisante la uM 0,52.
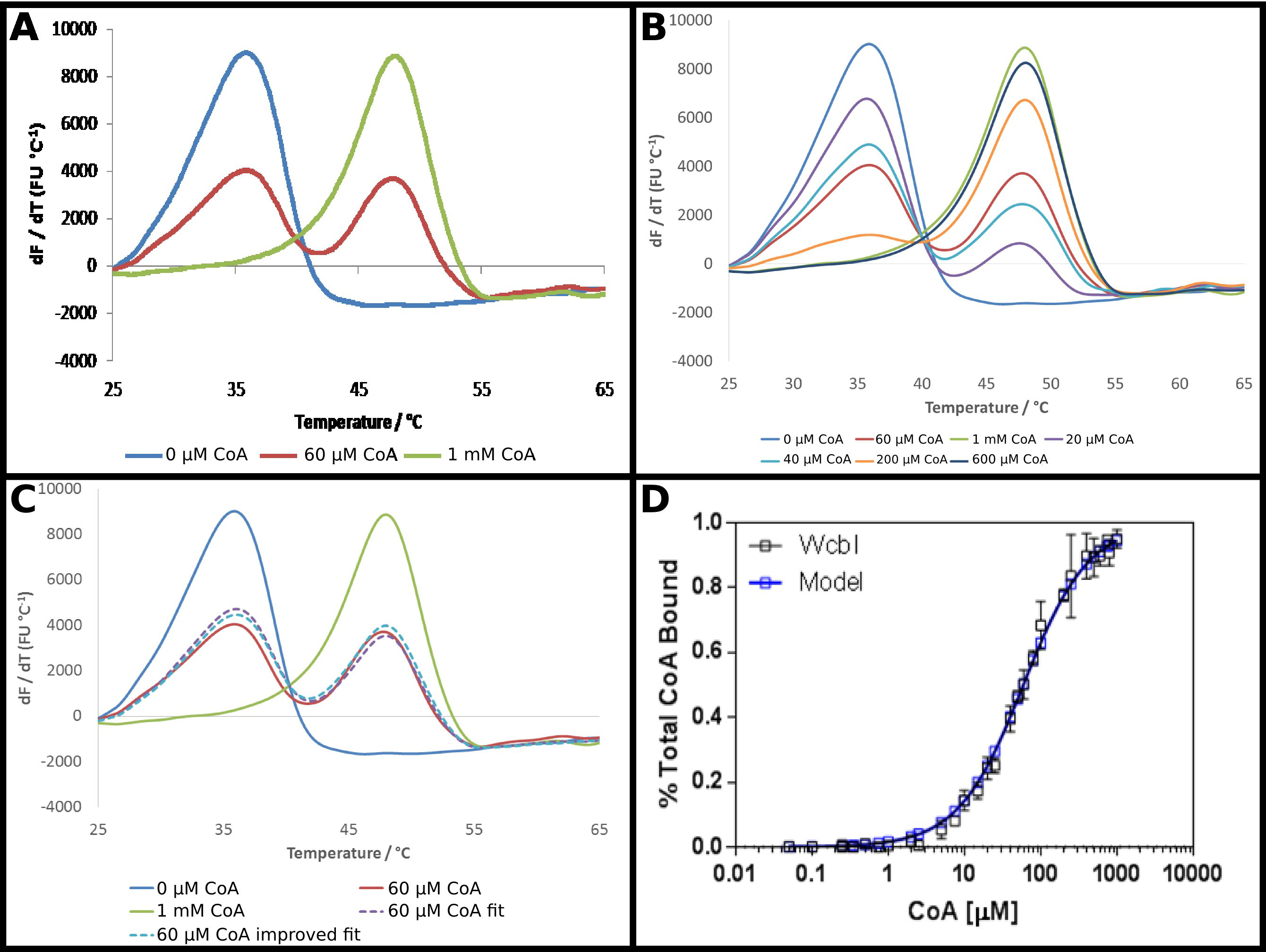
Le PIB-6-désoxy-β-D-manno putatif -heptopyranose 2 O -acetylase, WcbI 22, montre un résultat plutôt inhabituel dans différentielle à balayage fluorimétrie. En l'absence de ligands, il montre une dénaturation clair et simple (figure 4A). Coenzyme A (CoA) a été identifié comme un ligand de cette protéine en utilisant DSF, et l'affinité de la protéine pour ce partenaire a été étudiée de la manière décrite dans le protocole. En présence de concentrations élevées de CoA, une forte shift à une température plus élevée est observée, avec un changement de la température de fusion de 15 ° C. Cependant, à des concentrations intermédiaires, plutôt que d'un passage à une fusion monophasique à une température de fusion intermédiaire, WcbI montré une fusion à deux phases, avec la protéine apparaissant à l'état fondu à une ou l'autre de la température de droits ligand, ou la température de fusion entièrement lié (figure 4A) . Les proportions des deux espèces modifiées d'une manière dépendante de la dose, avec des concentrations croissantes de substrat augmentant la proportion qui fond à une température plus élevée (figure 4B). L'analyse directe de ces données a été difficile: le montage de l'équation de Boltzmann a donné, elle s'adapte très pauvres, tandis que les méthodes dérivés mis en évidence que deux événements de fusion se produisaient, mais n'ont pas aidé à démontrer un changement avec l'augmentation de la concentration de ligand.
Une approche moins conventionnelle pour analyser ces données a donc été adopté (protocole n ° 5). La fluorescence drésultats erivative sans ligand et à la concentration de ligand plus ont été prises comme représentant la quasi-totalité des protéines de la température de fusion plus bas, ou l'état de la température de fusion plus élevé. Les données dérivés restants ont été installés à chaque point comme la somme d'une partie de chacun de ces deux Etats, la proportion a résumé à l'unité (figure 4C). Les données obtenues ont ensuite été montés comme précédemment pour obtenir un Kd apparent, en utilisant les mêmes équations que précédemment. Cela a fait ressortir que le point de ligand "haute" est susceptible d'être seulement 95% ligand lié. Les données ont été ensuite extrapolés à une prédiction du résultat pour une protéine liée de 100%, et les données raccord répétées pour donner un Kd apparent de 58 ± 2 uM. Cela a donné un excellent ajustement des résultats expérimentaux pour le modèle de liaison (figure 4D).
fo: contenu width = "5 pouces" src = "/ files / ftp_upload / 51809 / 51809fig1highres.jpg" width = "500" />
Figure 1: Exemples de configuration du test et de l'analyse. (A) Exemple de la forme attendue d'un profil de dénaturation thermique (pris à partir de données de l'hexokinase de levure). La forme caractéristique des données brutes montre une augmentation progressive de la fluorescence au maximum, suivie d'une baisse peu profond (discuté plus en détail en 9). Ceci est accompagné par un seul pic dans la dérivée première de la fluorescence (B). Exemple de saisie de données dans Graphpad. concentration de ligand est indiquée sur l'axe des X, et observer les températures de fusion sur l'axe des ordonnées (C). Exemple de définition de l'équation dans Graphpad. (D) Exemples de mise correctement les valeurs initiales des variables et de la fixation de la concentration de protéine, afin de permettre une détermination correcte de la constante de dissociation.m / fichiers / ftp_upload / 51809 / 51809fig1highres.jpg "target =" _blank "> S'il vous plaît cliquer ici pour voir une version plus grande de cette figure.

. Figure 2 l'interaction de l'hexokinase de glucose mesuré par analyse différentielle fluorimétrie (A) Une expérience initiale tester une large gamme de concentrations en glucose suggère que le Kd est susceptible d'être comprise dans l'intervalle de 0,2 -. 1,7 mM (B) Un détaillée expérience, tester 16 concentrations de glucose, permet de déterminer le Kd apparent que 1,12 ± 0,05 mM. Les données s'adapte très bien au modèle pour un seul cas de liaison (avec le fond (T1) et supérieure (T2) des températures raccord à 35,4 ± 0,2 ° C et 49,3 ± 0,5 ° C respectivement). Noter queces données ont été recueillies en présence de 10 mM MgCl 2. Ces images ont été préparés en utilisant GraphPad. S'il vous plaît cliquer ici pour voir une version plus grande de cette figure.
{kind=link}

Figure 3 Interaction de WCBM avec GTP révèle une liaison anti-coopérative. (A) Une expérience initiale tester une large gamme de concentrations de GTP suggère que le Kd est susceptible d'être dans la plage de 200 -. 500 uM (B) Une expérience détaillée, en testant 16 les concentrations de GTP, indique une valeur pour le ressort Kd de 120 ± 20 pM. Cependant, quand une échelle logarithmique est utilisée pour l'axe des abscisses, il est important discrétionpation entre le modèle et les données. (C) L'analyse des mêmes données avec un modèle coopératif représente un excellent ajustement aux données où le modèle coopératif simple est utilisé. Ici, un K ½ de 230 ± 20 pM a été déterminée, le coefficient de coopérativité de n = 0,52 ± 0,02 (avec le fond (T1) et supérieure (T2) à des températures de montage 69,63 ± 0,06 ° C et 79,9 ± 0,1 ° C, respectivement). Comme WCBM semble être dimère, ce qui implique que l'enzyme est parfaitement anticooperative dans sa liaison au GTP. Ces images ont été préparés en utilisant GraphPad. S'il vous plaît cliquer ici pour voir une version plus grande de cette figure.
{kind=link}

La figure 4 montre un schéma WcbI de fusion biphasiqueen présence de son ligand coenzyme A (CoA). (A) WcbI, en l'absence de ligand (bleu), représente un motif de fusion monophasique simple. À des concentrations élevées de ligand (1 mM; ligne verte), une tendance similaire est observée. Cependant, à des concentrations de ligand intermédiaire. (60 uM; ligne rouge), deux pics de fusion distincts, correspondant à des états de libre-ligand et un ligand lié sont observés (B) La transition entre les deux séries de pics est proportionnelle à la dose sur toute la gamme de concentrations. (C) Modélisation de la fusion biphasique comme la somme d'une partie des ligands résultats ligand libre et de haute donne un bon ajustement aux données (ligne violette en pointillés, par rapport à la ligne rouge). Cette coupe est améliorée en extrapolant le résultat observé pour une forte concentration de ligand (où le modèle suggère ~ 95% d'occupation) à la pleine occupation (ligne pointillée bleue). (D) Les données obtenues pour la proportion de WcbI lié à CoA sho ws un excellent ajustement à un modèle de liaison simple, avec un Kd de 58 ± 2 pM (les données représentent les données recueillies sur deux jours différents, avec des concentrations de ligand légèrement différentes, choisies pour le deuxième jour sur la base du premier ensemble de résultats). Panneaux. (A - C) ont été préparés en utilisant Excel, et le panneau (D) à l'aide de Graphpad S'il vous plaît cliquer ici pour voir une version plus grande de cette figure.
{kind=link}
Tableau 1 Recette pour les premières expériences.
| Réactif | Volume en mélange (pl) |
| Protéine | Pour une concentration finale de 0,11 mg / ml |
| 0,3 | |
| HEPES 0,5 M pH 7,0 | 3.7 |
| 5 M de NaCl | 5.6 |
| Eau | Pour 180 ul |
Il décrit le "master mix" de protéines, réactif de détection et de tampon pour une expérience de dépistage initial de fournir une estimation de K d, tel que décrit dans la section du protocole 1 Ce mélange tampon est appropriée pour des protéines génériques. Lorsque les résultats précédents suggèrent d'autres tampons doivent être utilisés, ils devraient être remplacés. Si la protéine stock est à une faible concentration (par exemple, moins de 0,3 mg / ml), il peut être nécessaire de réduire la quantité de tampon supplémentaire ajouté au tampon pour compenser une présence dans l'échantillon de protéine.
Tableau 2 Recette pour la détermination de K d.| Réactif | Volume en mélange (pl) |
| Protéine | Pour une concentration finale de 0,11 mg / ml |
| 5,000X SYPRO orange | 1,78 |
| HEPES 0,5 M pH 7,0 | 22.2 |
| 5 M de NaCl | 33,3 |
| Eau | Pour 180 ul |
Il décrit le "master mix" de protéines, réactif de détection, et buffer pour une détermination complète de K d pour un échantillon de protéine, tel que décrit dans la section 2 du protocole Ce mélange tampon est appropriée pour des protéines génériques. Lorsque les résultats précédents suggèrent d'autres tampons doivent être utilisés, ils devraient être remplacés. Si la protéine stock est à une faible concentration (par exemple, moins de 0,3 mg / ml), il peut être nécessaire de réduire la quantité de tampon supplémentaire ajouté au tampon pour compenser une présence dans l'échantillon de protéine.
Tableau 3 équations et des paramètres d'analyse des données.
d| Étape dans le protocole expérimental | Équation nécessaire | Paramètres nécessaires | Description des variables et des paramètres |
| 3.3 | |||
| Seul site de liaison du ligand | Y = Bas + ((Haut-Bas) * (1 - ((PK-X d + sqrt (((P + X + K d) ^ 2) - (4 * P * X))) / (2 * P )))) | P: concentration en protéine. Kd: constante de dissociation. P et Kd sont donnés dans les mêmes unités qui ont été utilisées pour les concentrations de ligand. Haut, Bas: des températures de fusion à une concentration de ligand infinie et aucune concentration de ligand respectivement. | |
| 3.4 | Bas = * YMIN | YMIN: valeur minimum de Y (plus bas protéine expérimentale Tm, dans ce cas) | |
| Haut = * YMAX | YMAX: valeur maximale de Y (le plus élevé de protéines expérimental Tm) | ||
| K d = * X à ymid | Ymid: valeur de Y qui correspond à la moyenne de YMIN et YMAX. X est la valeur correspondant à X (ici, la concentration de ligand pertinent) | ||
| P = (valeur initiale, pour être en forme) | |||
| 4.1 | |||
| Modèle coopératif simple | Y = Bas + ((Haut-Bas) * (((X / Kd) ^ n) / (1 + ((X / Kd) ^ n)))) | n: Hmauvais coefficient. Ceci décrit la coopérativité, ou d'autres propriétés biochimiques de la protéine, et n'est pas nécessairement une estimation du nombre de sites de liaison de ligand dans la protéine. Un coefficient de Hill de l'un représente pas coopérativité; des valeurs inférieures à un indiquent coopérativité négative, et les valeurs supérieures à un coopérativité positive. | |
| Bas = * YMIN | |||
| Haut = * YMAX | |||
| K d = * X à ymid | |||
| P = (valeur initiale, pour être en forme ) | |||
| n = (valeur initiale, pour être en forme) | |||
| Séquentiel de liaison de deux ligands | Y = Bas + ((Haut-Bas) * ((X ^ 2) / (K d * K2)) / (1 + (X / K d) + ((X ^ 2) / (K d * K2))) ) | K2: constante pour la deuxième événement de liaison dissociation. | |
| Bas = * YMIN | |||
| Haut = * YMAX | |||
| K2 = * X à ymid | |||
| P = (valeur initiale, pour être en forme) | |||
| Indépendant et contraignant de deux ligands | Y = Bas + ((Haut-Bas) * ((X ^ 2) / (K d * K2)) / (1 + (2 * X / K d) + ((X ^ 2) / (K d * K2) ))) | ||
| Bas = * YMIN | |||
| K d = * X à ymid | |||
| K2 = * X à ymid | |||
| P = (valeur initiale, pour être en forme) | |||
| 5.5 | |||
| Analyse des changements binaires de la température de fusion | Y = 1 - ((PK-X d + sqrt (((P + X + K d) ^ 2) - (4 * P * X))) / (2 * P)) | ||
| Bas = * YMIN | |||
| Haut = * YMAX | |||
| K d = * X à ymid | |||
| P = (valeur initiale, pour être en forme) | |||
| 5.8 | |||
| L'extrapolation à la concentration de ligand infini | (C2 - ((1 $ R $ 2) * B2)) / $ R $ 2 | B2: cellule contenant le résultat sans ligand. C2: cellule contenant le résultat avec le ligand maximale. $ R $ 2: cellule contenant la proportion liée à la concentration de ligand maximale. |
Les étapes 3, 4 et 5 nécessitent l'ajout d'équations détaillées dans le logiciel d'analyse et de définition précise des paramètres de départ pour l'analyse des données. Les équations pour chaque étape pertinente sont présentés, avec les sélections correctes des paramètres. Une explication de la signification des variables et des paramètres est fourni à titre de référence.
Tableau 4: Concentrations pour le dépistage de l'interaction de l'hexokinase avec du glucose.
| point de l'échantillon | Ligand (glucose) la concentration (mM) |
| 1 | 0 |
| 2 | 0,001 |
| 3 | 0,005 |
| 4 | 0,01 |
| 5 | 0,03 |
| 6 | 0,1 |
| 7 | 0,3 |
| 8 | 0,4 |
| 9 | 0,7 |
| 10 | 1.1 |
| 11 | 2.1 |
| 12 | 3.7 |
| 13 | 5.3 |
| 14 | 7 |
| 15 | 9 |
| 16 | 11 |
L'hexokinase de la bourgeonnante Saccharomyces cerevisiae de la levure a été ajoutée au mélange maître tel que décrit dans le protocole, additionné de 10 mM de MgCl2 en tant que magnésium est un cofacteur connu. L'estimation initiale de la valeur de Kd était entre 0,5 et 2 mM. Les expériences ont été mises en place pour fournir des concentrations finales indiquées de glucose.
Tableau 5: Concentrations pour le dépistage de l'interaction de WCBM PIB.
| point de l'échantillon | Ligand (GTP) concentration (M) |
| 1 | 0 |
| 2 | 0,5 |
| 3 | 1 |
| 4 | 5 |
| 5 | 10 |
| 6 | 25 |
| 7 | 50 |
| 8 | 100 |
| 9 | 250 |
| 10 | |
| 11 | 1000 |
| 12 | 2500 |
| 13 | 5000 |
| 14 | 7500 |
| 15 | 10000 |
| 16 | 20000 |
WCBM de Burkholderia pseudomallei a été ajouté au mélange maître tel que décrit dans le protocole. L'estimation initiale de la valeur de Kd est d'environ 100 pM. Des expériences ont été mis en place pour obtenir les concentrations finales indiquées de GTP, visant à couvrir au moins deux ordres de grandeur ci-dessus et ci-dessous K d.
Discussion
Différentielle à balayage fluorimétrie a démontré sa puissance comme une méthode robuste et polyvalent pour la caractérisation des protéines, et l'identification des ligands protéiques potentiels. Les succès bien documentés dans d'accélérer la stabilisation des protéines, la découverte de médicaments (en particulier dans les laboratoires moins bien financés) et la cristallisation 10,23-25 ont fait une méthode intéressante pour le dépistage initial de composés. Les composés ajoutés aux protéines montrent une augmentation dépendante de la dose dans le ressort clairement de 7,9 température de fusion. Cependant, il ya eu quelques tentatives d'utiliser les résultats de ces expériences pour déterminer les constantes de liaison apparentes pour aider dans le classement des composés pour leur affinité. Ici, nous présentons une méthode systématique permettant de déterminer une constante de dissociation apparente pour des protéines en présence d'un ligand.
Les résultats présentés ici démontrent que DSF peut rapidement et vigoureusement fournir des estimations de la constante de dissociation pourune combinaison de la protéine-ligand. Les données observées peuvent être manipulés avec des outils disponibles dans le commerce pour fournir une détermination rapide de K d, sans la nécessité d'émettre des hypothèses sur la valeur probable de paramètres. La méthode a un avantage considérable sur certaines des méthodes comparables de parcimonie en protéines et en temps requis. L'expérience décrite ici va consommer 0,13 mg de protéine par essai (environ 0,4 mg pour des expériences répétées en triple exemplaire). Cela se compare favorablement avec calorimétrie de titration isotherme (ITC), où une expérience unique avec une protéine de 40 kDa moyenne va consommer un montant similaire. L'ensemble des expériences requises pour ce protocole consommer environ 4 heures, y compris la préparation, pour une seule série d'expériences. Encore une fois, cela est susceptible d'être beaucoup plus rapide que les procédés tels que le CCI ou résonance de plasmon de surface, qui ont souvent besoin tout puissant optimisation considérable pour atteindre les meilleures données.
Nos résultats démontrent qu'il subsiste un besoin d'examiner soigneusement les données brutes, l'ajustement de ces données afin de déterminer la température de fusion, et l'ajustement des données de température de fusion pour déterminer la constante de dissociation. Une première difficulté est la forme de données brutes produites dans la fusion de la protéine. Dans certains cas, la forme peut ne pas se rapprocher de celle observée sur la figure 1A. Les problèmes courants sont faibles changements de température sur la liaison de ligand, une forte fluorescence de fond, et de multiples transitions inhabituelles de température. Déplacements à basse température sont visibles sur la liaison d'un certain nombre de ligands. Pour cette méthode, le paramètre le plus critique est l'erreur sur la mesure de la T m, par rapport au changement de température. Les données peuvent généralement être monté normalement lorsque l'écart type de mesures triples ne dépasse pas 10% de la variation de température de fusion entre la protéine non liée et entièrement lié. Notre expérience est que lorsque cette températuredéplacements rature ne sont de 2 ° C, cela peut être suffisant pour ajuster les données, si les points de données individuels sont très précis. Une deuxième question est particulièrement en forme des courbes. Celles-ci diffèrent souvent entre les protéines libres et les formulaires de ligand lié, comme la liaison ligand affecte les modes de la protéine déroulement. Dans ces cas, l'utilisateur doit déterminer si les données peuvent être utilisés en tenant compte appropriée des modèles à utiliser pour la détermination de la température de fusion et la constante de dissociation. Un autre problème commun est que l'addition d'un cofacteur de la protéine (par exemple, MgCl 2 dans notre exemple avec l'hexokinase) est nécessaire pour obtenir les données les plus fiables. Notre expérience a été que l'examen minutieux de tous les facteurs susceptibles de l'expérience au stade de la prise de mesures initiales est essentielle pour obtenir les meilleurs résultats. En outre, les traitements théoriques alternatifs peuvent révéler des caractéristiques de ces données 15,17. Enfin, il n'est pas rare que certaines protéines qui contain nativement exposée régions hydrophobes à montrer une forte fluorescence de fond. Il ya un certain nombre de solutions à ces problèmes, qui ont été largement décrites ailleurs 6,9.
En particulier, l'utilisateur doit se demander si l'utilisation de l'Boltzmann ou modèles dérivés (figure 4), et dans le cas de l'utilisation d'instruments dérivés, si plusieurs fond doivent être modélisés. Les deux méthodes de modélisation du dépliement thermique diffèrent en ce que la méthode de Boltzmann correspond aux données expérimentales à l'équation de Boltzmann, en supposant une forme sigmoïdale régulière de la courbe de dépliement. En revanche, la méthode de la dérivée prend la première dérivée des données expérimentales à chaque point (en bas sur la figure 1A), et considère que la température de fusion pour être le point de la plus haute dérivée première. Procédé dérivé renvoie généralement une température de fusion supérieure d'environ 2-3 ° C. La plupart des protéines reviendront un plus cohérenterésultat (à savoir, l'erreur-type de la température de fusion pour les expériences en triple est plus faible) pour une des deux méthodes. Il s'agit généralement intimement liée à la forme précise de la courbe de dépliement des protéines, et il est nécessaire de déterminer la meilleure méthode empiriquement dans chaque cas. Lorsque le modèle dérivé est utilisé, il est également important de tenir compte de multiples événements de fusion. Certaines données montrent clairement preuve de transitions multiples, et dans ces cas, les résultats sont susceptibles d'être plus facile à interpréter si ces événements de fusion multiples sont modélisés. Dans le cadre de ce protocole, il est souvent le cas que l'addition de ligand peut provoquer une protéine de passer d'avoir plusieurs transitions de fusion à une seule transition (par exemple, en stabilisant le sous-domaine le plus thermiquement fragiles), ou vice versa. Nous serions donc plaider pour que les données brutes sont examinés ensemble avant d'envisager l'approche sera préférable d'utiliser.
À la suite de la modélisation des températures de fusion différentes, d'autres problèmes peuvent survenir dans le montage de ces modèles présentés dans la section de protocole. Il est impératif d'examiner attentivement la forme de l'équation constante de dissociation à l'aide d'une échelle logarithmique, comme cette analyse met en évidence souvent des écarts entre les données observées et le modèle (par ex., Figure 3). Alors que les résultats obtenus sont généralement robustes, les soins d'interprétation offre la possibilité d'extraire de meilleurs résultats, et le plus de sens, à partir des données.
Un problème particulier soulevé par ces données est l'interprétation qui doit être placé sur les protéines qui montrent coopérativité, ou des événements multiples de liaison, DSF. Nous avons, à ce jour, seulement observé ce phénomène dans les protéines qui sont censés avoir de multiples événements de liaison spécifiques (par exemple, WCBM, une protéine dont le meilleur homologue est un multimère 26, et qui agit comme un multimère sur la chromatographie d'exclusion stérique [données nonreprésenté]). Il n'est pas du tout évident que la coopérativité négative observée dans DSF dénaturation indique que l'enzyme sera finalement montrer coopérativité négative: non, cela peut être une indication de la liaison complexe qui doit être étudié de manière plus approfondie en utilisant un large éventail de méthodes. Cela ne nous suggèrent, cependant, que plus des études approfondies de ces protéines sont susceptibles d'identifier des effets intéressants.
Les valeurs indiquées pour la constante de dissociation à l'aide de ce procédé sont généralement du même ordre que ceux fournis par d'autres méthodes, telles que la calorimétrie de titration isotherme et la résonance de plasmon de surface. Toutefois, les valeurs absolues sont observées fréquemment supérieur à celui observé en utilisant ces méthodes. Ceci est au moins en partie une conséquence du fait que la constante de dissociation est observée à la température de fusion de la protéine avec un ligand. Cette valeur de Kd est généralement plus élevée que celle à des températures physiologiques. Le dissociation constant est lié à la température de la réaction par les équations:
 [1]
[1]
 [2]
[2]
(Où c θ est la concentration de référence standard, Δ R G est la variation de la réaction de Gibbs d'énergie libre, R est la constante molaire des gaz, Δ H est la variation d'enthalpie de la réaction, et Δ S est la variation d'entropie dans la réaction .)
Les réactions avec des constantes de dissociation dans la plage de mesure de ce procédé ont généralement un négatif Δ r G, et ainsi l'effet d'une augmentation de la température de l'équation [1] sera d'augmenter la constante de dissociation. Les deux termes Δ H et Δ S qui constituent l'énergie libre de Gibbs (équation [2]) sont température dépendante 27, et l'effet sur la constante de dissociation dépendront de l'ampleur et le signe de ces dépendances de température, et ne seront pas nécessairement interaction dépend. Par conséquent, il n'est pas surprenant que les constantes de dissociation calculées par cette méthode sont parfois supérieures à celles qui sont déterminées par des procédés qui fonctionnent à température ambiante. Influence de la température est, bien entendu, également une mise en garde de nombreuses autres méthodes, qui ont tendance à fournir la constante de dissociation à une température inférieure à la température physiologique.
Une autre mise en garde de la méthode de DSF est qu'il s'agit d'une méthode marquée, à la différence de l'ITC. Le marqueur fluorescent utilisé (SYPRO Orange) est hydrophobe, et donc dans certains cas, ne peut rivaliser avec la liaison des ligands hydrophobes des protéines. Par conséquent, il est probable que dans certains cas, la constante de dissociation obtenu sera augmenté artificiellement raison de la compétition avec l'étiquette. Cependant, pour la comparaison de divers ligands, (la principale utilisation deDSF), les différences sont peu susceptibles d'être suffisamment importante pour influer sur le classement des composés par affinité.
Un inconvénient potentiel de cette méthode est la limite de détection que l'on peut obtenir. En principe, il ne devrait pas être possible de mesurer avec précision une valeur de Kd qui est inférieure à 50% de la concentration de protéine, et même des valeurs de cette gamme sont susceptibles d'être d'une précision douteuse. Bien que la limite de détection à cette extrémité de l'intervalle peut être prolongée par une petite réduction de la concentration de protéine et de colorant, la sensibilité de l'instrument permet d'éviter en outre la réduction de la concentration en protéine. De même, l'extrémité supérieure de la sensibilité est déterminée par la solubilité du ligand. Pour obtenir une estimation mathématique robuste K d, il est très important d'obtenir des données de 90% de la protéine présente dans le formulaire lié au ligand, ce qui nécessite des concentrations de ligands à être approximatifly dix fois Kd (en supposant l'absence coopérativité). La limite de détection sera donc nécessairement un dixième de la solubilité du ligand dans le tampon approprié. Cela signifie que les limites de détection de la méthode sera généralement comprise entre 1 pM et entre 1 et 100 mM, en fonction de la protéine et le ligand.
En conclusion, la fluorimétrie différentielle à balayage est une technique polyvalent applicable à une large gamme de protéines. En utilisant les procédés présentés ici, il est possible de déterminer rapidement et à moindre coût l'affinité d'une protéine pour différents ligands. Cela a un grand potentiel pour des applications dans la purification de protéines et de stabilisation, élucider la fonction ou la spécificité des enzymes de métagénomes, et dans la découverte de médicaments, en particulier dans les petits laboratoires.
Déclarations de divulgation
The authors declare that they have nothing to disclose.
Remerciements
This work was funded by grant from the BBSRC (grant number BB/H019685/1 and BB/E527663/1) to the University of Exeter.
matériels
| Name | Company | Catalog Number | Comments |
| StepOne real time PCR instrument | Life Technologies | 4376357 | DSF can be performed with many other instruments. The StepOne instrument has very convenient software for data analysis. |
| Protein thermal shift software v1.0 | Life Technologies | 4466037 | |
| MicroAmp Fast optical 48-well plates | Life Technologies | 4375816 | |
| Optical sealing tape | Life Technologies | 4375323 | Bio-rad part no. 223-9444 is an alternative supplier |
| U-bottomed 96-well plates | Fisher | 11521943 | |
| SYPRO Orange | Life Technologies | S6650 | For a smaller volume supplier, use Sigma part no. S5692 |
| SPSS statistics version 20 | IBM | Other statistics packages will provide similar functionality | |
| GraphPad Prism 6.02 | GraphPad | Other statistics packages will provide similar functionality | |
| Hand applicator (PA1) | 3M | 75-3454-4264-6 | |
| Hexokinase from Saccharomyces cerevisiae | Sigma-Aldrich | H5000 | |
| Glucose | Fisher scientific | 10141520 |
Références
- Freyer, M. W., Lewis, E. A. Isothermal titration calorimetry: experimental design, data analysis, and probing macromolecule/ligand binding and kinetic interactions. Methods Cell Biol. 84, 79-113 (2008).
- Ladbury, J. E. Calorimetry as a tool for understanding biomolecular interactions and an aid to drug design. Biochem Soc Trans. 38, 888-893 (2010).
- Abdiche, Y., Malashock, D., Pinkerton, A., Pons, J. Determining kinetics and affinities of protein interactions using a parallel real-time label-free biosensor, the Octet. Anal Biochem. 377, 209-217 (2008).
- Pantoliano, M. W., et al. High-density miniaturized thermal shift assays as a general strategy for drug discovery. J Biomol Screen. 6, 429-440 (2001).
- Senisterra, G., Chau, I., Vedadi, M. Thermal denaturation assays in chemical biology. Assay Drug Dev Technol. 10, 128-136 (2012).
- Ericsson, U. B., Hallberg, B. M., Detitta, G. T., Dekker, N., Nordlund, P. Thermofluor-based high-throughput stability optimization of proteins for structural studies. Anal Biochem. 357, 289-298 (2006).
- Lo, M. C., et al. Evaluation of fluorescence-based thermal shift assays for hit identification in drug discovery. Anal Biochem. 332, 153-159 (2004).
- Nettleship, J. E., Brown, J., Groves, M. R., Geerlof, A. Methods for protein characterization by mass spectrometry, thermal shift (ThermoFluor) assay, and multiangle or static light scattering. Methods Mol Biol. 426, 299-318 (2008).
- Niesen, F. H., Berglund, H., Vedadi, M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat Protoc. 2, 2212-2221 (2007).
- Geders, T. W., Gustafson, K., Finzel, B. C. Use of differential scanning fluorimetry to optimize the purification and crystallization of PLP-dependent enzymes. Acta Crystallogr Sect F Struct Biol Cryst Commun. 68, 596-600 (2012).
- Vedadi, M., et al. Chemical screening methods to identify ligands that promote protein stability, protein crystallization, and structure determination. Proc Natl Acad Sci USA. 103, 15835-15840 (2006).
- Davis, B. J., Erlanson, D. A. Learning from our mistakes: the 'unknown knowns' in fragment screening. Bioorg Med Chem Lett. 23, 2844-2852 (2013).
- Larsson, A., Jansson, A., Aberg, A., Nordlund, P. Efficiency of hit generation and structural characterization in fragment-based ligand discovery. Curr Opin Chem Biol. 15, 482-488 (2011).
- Scott, D. E., et al. Using a fragment-based approach to target protein-protein interactions. Chembiochem. 14, 332-342 (2013).
- Cimmperman, P., et al. A quantitative model of thermal stabilization and destabilization of proteins by ligands. Biophys. J. 95, 3222-3231 (2008).
- Matulis, D., Kranz, J. K., Salemme, F. R., Todd, M. J. Thermodynamic stability of carbonic anhydrase: measurements of binding affinity and stoichiometry using ThermoFluor. Biochemistry. 44, 5258-5266 (2005).
- Zubriene, A., et al. Measurement of nanomolar dissociation constants by titration calorimetry and thermal shift assay - radicicol binding to Hsp90 and ethoxzolamide binding to CAII. Int J Mol Sci. 10, 2662-2680 (2009).
- Weiss, J. N. The Hill equation revisited: uses and misuses. FASEB J. 11, 835-841 (1997).
- Cuccui, J., et al. Characterization of the Burkholderia pseudomallei K96243 capsular polysaccharide I coding region. Infect Immun. 80, 1209-1221 (2012).
- DeShazer, D., Waag, D. M., Fritz, D. L., Woods, D. E. Identification of a Burkholderia mallei polysaccharide gene cluster by subtractive hybridization and demonstration that the encoded capsule is an essential virulence determinant. Microb Pathog. 30, 253-269 (2001).
- Berman, H., Henrick, K., Nakamura, H. Announcing the worldwide Protein Data Bank. Nat Struct Biol. 10, 980(2003).
- Vivoli, M., Ayres, E., Beaumont, E., Isupov, M., Harmer, N. Structural insights into WcbI, a novel polysaccharide biosynthesis enzyme. IUCr Journal. 1 (1), 28-38 (2014).
- Sorrell, F. J., Greenwood, G. K., Birchall, K., Chen, B. Development of a differential scanning fluorimetry based high throughput screening assay for the discovery of affinity binders against an anthrax protein. J Pharm Biomed Anal. 52, 802-808 (2010).
- Uniewicz, K. A., et al. Differential scanning fluorimetry measurement of protein stability changes upon binding to glycosaminoglycans: a screening test for binding specificity. Anal Chem. 82, 3796-3802 (2010).
- Wan, K. F., et al. Differential scanning fluorimetry as secondary screening platform for small molecule inhibitors of Bcl-XL. Cell Cycle. 8, 3943-3952 (2009).
- Koropatkin, N. M., Holden, H. M. Molecular structure of alpha-D-glucose-1-phosphate cytidylyltransferase from Salmonella typhi. J Biol Chem. 279, 44023-44029 (2004).
- Paleskava, A., Konevega, A. L., Rodnina, M. V. Thermodynamics of the GTP-GDP-operated conformational switch of selenocysteine-specific translation factor SelB. J Biol Chem. 287, 27906-27912 (2012).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.