Method Article
Generación de plásmidos vectores que expresan proteínas de la bandera de etiquetado bajo la regulación del promotor factor de elongación humano-1α Usando Asamblea Gibson
En este artículo
Resumen
Synthesis of custom plasmids is labor and time consuming. This protocol describes the use of Gibson assembly cloning to reduce the work and duration of custom DNA cloning procedure. The protocol described also produces reliable tagged protein constructs for mammalian expression at similar cost to the traditional cut-and-paste DNA cloning.
Resumen
Asamblea Gibson (GA) clonación ofrece una alternativa rápida, fiable y flexible para métodos de clonación de ADN convencionales. Utilizamos GA para crear plásmidos a medida para la expresión de genes exógenos en células madre embrionarias de ratón (mESCs). La expresión de genes exógenos bajo el control del SV40 o promotores de citomegalovirus humano disminuye rápidamente después de la transfección en mESCs. Un remedio para esta expresión disminuida es utilizar el promotor de factor de elongación humano 1-alfa (hEF1α) para conducir la expresión génica. Los vectores plásmidos que contienen hEF1α no son tan ampliamente disponible como SV40 o plásmidos conteniendo CMV, especialmente aquellos que también contienen N-terminal 3xFLAG-tags. El protocolo descrito aquí es un método rápido para crear plásmidos que expresan etiquetada con FLAG CSTF-64 y CSTF-64 mutante bajo la regulación del promotor expressional hEF1α. GA utiliza una mezcla de exonucleasa del ADN, ADN polimerasa y ligasa de ADN para hacer clonación de extremos de fragmentos de ADN posible superposición.Con base en los ADN de plantilla que teníamos disponible, hemos diseñado nuestros constructos para ser montados en una sola secuencia. Nuestro diseño utiliza cuatro fragmentos de ADN: pcDNA 3.1 vector columna vertebral, hEF1α promotor parte 1, parte 2 hEF1α promotor (que contenía 3xFLAG-tag comprar de un fragmento de ADN sintético de doble cadena), y, o bien CSTF-64 o específica CSTF-64 mutante. Las secuencias de estos fragmentos se cargan en una herramienta de generación de imprimación para diseñar cebadores de PCR adecuados para la generación de los fragmentos de ADN. Después de la PCR, los fragmentos de ADN se mezclaron con el vector que contiene el marcador selectivo y la reacción de clonación GA fue montado. Se aislaron plásmidos a partir de colonias bacterianas transformadas individuales. Pantalla inicial de los plásmidos se realizó por digestión de restricción, seguido por secuenciación. En conclusión, GA nos permitió crear plásmidos a medida para la expresión de genes en 5 días, incluyendo la construcción de pantallas y verificación.
Introducción
Procedimientos de clonación de ADN convencionales se basan en el uso de enzimas de restricción para escindir el ADN y ADN ligasa para unir los fragmentos de ADN juntos. Generación de construcciones de expresión personalizados que contienen diferentes fragmentos de ADN es un procedimiento secuencial que incluye la escisión del ADN con una y / o múltiples endonucleasas de restricción y la posterior inserción de fragmentos de ADN a través de la ligadura. El principal inconveniente de este procedimiento es que las enzimas de restricción adecuadas para uno de los fragmentos de ADN pueden ser difíciles de identificar (es decir, podría tener varios sitios de escisión) de representación de clonación de ADN con éxito de la proteína de longitud completa de interés imposible. Por lo tanto, la generación de expresión personalizada construye bajo la regulación transcripcional de promotores específicos del tipo de células eficiente con proteína-etiquetas personalizadas requiere diseño muy cuidado. También es una técnica de tiempo y mano de obra que consume. Recientemente, varios informes describen metodologías para armar multíparaLe diferentes fragmentos de ADN sintético en una secuencia continua, al mismo tiempo, ya sea en reacciones de uno o de dos pasos sin el uso de enzimas de restricción 1-3. La reacción de la clonación de un solo paso (con exclusión de todos los pasos preparatorios), depende de la utilización de una mezcla de exonucleasa del ADN, ADN polimerasa, ADN ligasa 2,3 y los extremos solapados de fragmentos de ADN (Figura 1). Puesto que no hay uso de enzimas de restricción, fragmentos de ADN de cualquier tamaño y composición de la secuencia (con exclusión de secuencias altamente repetitivas) se pueden fusionar juntos en una construcción sin costuras. Recientemente, un kit comercial (montaje Gibson; GA) para las reacciones de clonación de un solo paso se hizo disponible. Este kit permite un montaje rápido y eficiente y costo de los fragmentos de ADN en un solo vector con los promotores de la medida y etiquetas de proteínas.
Los vectores de expresión de plásmidos disponibles ampliamente usados para expresar proteínas exógenas en los modelos de cultivo de células de mamíferos son a menudo bajo la reg transcripcionalulación del citomegalovirus viral (CMV) o el virus 40 (SV40) promotores Simian. Estos promotores virales proporcionan la expresión transitoria robusta de las proteínas exógenos en la mayoría de los modelos basados en cultivos celulares de mamíferos. Sin embargo, la generación de líneas celulares que expresan de manera estable proteínas exógenas es a menudo éxito debido a silenciamiento transcripcional del CMV o promotores de SV40 durante el proceso de establecimiento de 4,5. Además, los promotores de SV40 y CMV viral no suficientemente promover la expresión de proteínas exógenas en células del linaje linfoide o células madre embrionarias 6,7. La solución a la limitación inherente de los promotores virales es el uso de promotores fuertes no virales constitutivos 8-10. Un promotor fuerte no viral constitutiva bien caracterizado de origen humano es el promotor del factor de elongación 1α (hEF1α) (hEF1α está implicado en la catálisis de la asociación dependiente de GTP de aminoacil-tRNA a los ribosomas 11). Sin embargo,vectores de expresión que contienen el promotor hEF1α no son tan ampliamente disponibles como el promotor viral que contiene plásmidos, especialmente los que también contiene 3 × FLAG en el extremo terminal amino de la proteína de interés.
La proteína del factor de estimulación de escisión de 64.000 MW (CSTF-64) está implicado en el procesamiento del extremo 3 'de la mayoría de los mRNAs 12,13, incluyendo mRNAs histonas replicación dependiente de 14,15. CSTF-64 se expresa en todos los tejidos somáticos 12. Su motivo de reconocimiento de ARN se une a GU-ricos secuencias de ARN en naciente transcripciones aguas abajo de la escisión y poliadenilación sitio 16. Esta unión de CSTF-64 a la pre-mRNA promueve la escisión endonucleolítica eficiente de la transcripción naciente.
Aquí, un protocolo se describe que utiliza amplificación por PCR de los fragmentos de ADN, un kit de clonación de montaje Gibson (que incluye células bacterianas químicamente competentes) para producir vectores personalizados de m-etiquetados 3xFLAGouse CSTF-64 o CSTF-64 mutante a su extremo amino terminal bajo la expresión de hEF1α promotor 1.
Protocolo
1. En Silico Diseño del plásmido y la generación de los cebadores superpuestos
NOTA: El objetivo de este paso es ensamblar secuencia de nucleótidos completa de la construcción y el diseño de los cebadores que se utilizan para generar fragmentos con extremos solapados para GA clonación.
- Diseñar una secuencia de nucleótidos continua para representar el plásmido final.
- Obtener o una lista de los plásmidos reales y los fragmentos de ADN que se utilizarán como plantillas en PCR.
NOTA: los fragmentos de ADN que no son fácilmente disponibles - como diferentes combinaciones de etiquetas y promotores - se pueden pedir como fragmentos individuales o múltiples sintéticos de ADN bicatenario (sADN). Estos fragmentos están comercialmente disponibles a costos relativamente bajos y pueden ser de hasta 2 kb de longitud (véase la Tabla de Materiales). - Divida la secuencia de nucleótidos continua de la construcción ensamblada en el paso 1.1 en fragmentos de ADN adecuados para la PCR. Confirmado dem que los fragmentos coinciden plásmidos disponibles y fragmentos sADN. Evite fragmentos de ADN más pequeño que 200 nt.
- Acceda a la herramienta de generación de imprimación (ver Tabla de Equipo). Seleccione el menú "Configuración de Preferencias". Elegir parámetros haciendo clic en la pestaña "PREFS cambio" en la ventana emergente "Cambiar la configuración de la Asamblea Gibson".
NOTA: Diseño de cebadores también se puede realizar sin el uso de la herramienta de generación de imprimación. Sin embargo, el uso de la herramienta de generación de imprimación simplifica el proceso. - Empezar a construir el constructo seleccionando el menú "Construir Construir". Inserte los fragmentos de ADN de división en la forma secuencial herramienta de generación de imprimación de 5 'a 3'.
NOTA: El fragmento de ADN utilizado como esqueleto del plásmido puede ser convenientemente dividida en dos piezas. En el producto final de la PCR 'final de este primer fragmento y el extremo 3' 5 final de la última fragmento están unidos entre sí en una DN continuoUn fragmento que representa el esqueleto del vector. - Pegue el primer fragmento de ADN que representa el extremo 5 'del ADN del vector en formato FASTA (una representación basada en texto de secuencia de nucleótidos) en la ventana emergente "Enter Vector o Fragment". Nombre del fragmento de ADN. Elegir la forma adecuada para obtener el fragmento de ADN, ya sea como PCR, RE Digesto o de síntesis. Haga clic en la pestaña "CONTINUAR".
- Si hay una necesidad de añadir nucleótidos extra / sitios de restricción en el cruce de la construcción final utilizar los espacios "Fwd o cebador Rev espaciadores" previstas en la opción "Agregar un fragmento de inserción a la asamblea" ventana. Añadir nucleótidos extra / sitios de restricción a sólo uno de los fragmentos de ADN y no a ambos fragmentos. Haga clic en la pestaña "DONE".
- Repita este procedimiento para todos los fragmentos hasta que el constructo es completa. Seleccione el menú "Ver Primers" y revisar las secuencias de cebadores.
- Repita para todas las construcciones (vector backbones e inserciones), o de fragmentos de ADN que son diferentes.
2. La amplificación de los fragmentos de ADN por PCR utilizando Hot Start Corrección ADN polimerasa (2x Master Mix)
NOTA: El objetivo de este paso es obtener suficiente ADN para la reacción de ensamblaje usando PCR.
- Compra los cebadores de PCR diseñados en el paso anterior como productos y desaladas en la escala más pequeña posible. Diluir a 10 mM en agua o TE (Tris-HCl 10, pH 7,9, EDTA 1 mM).
- Compra fragmentos de ADN que no son fácilmente disponibles como fragmentos sADN.
- Diluir todos los fragmentos de ADN, incluyendo los fragmentos de sADN que serán utilizados como plantillas para las PCRs a 1 ng / l en agua.
- Montar las reacciones de PCR a temperatura ambiente. Brevemente, utilizar 2,5 l (de 10 mM solución madre) de la respectiva cada cebador de la pareja de cebadores, 1 l (de 1 ng solución / l de stock) del fragmento de ADN plantilla, 25 μl de ADN polimerasa corrección de pruebas de arranque en caliente (ADN pol; mezcla maestra 2x; véase la Tabla de Materiales) y 19 l de agua. Mezclar el tubo mediante parpadeo suave y recoger las gotas de líquido por centrifugación breve.
- Amplificar simultáneamente en tubos separados los fragmentos de ADN de tamaño similar de acuerdo con las recomendaciones para el ADN pol. Realizar 25 a 28 ciclos de PCR o determinar el número de ciclos que producen suficiente rendimiento de ADN.
- Ejecutar 10% del volumen de reacción PCR (5 l) en una electroforesis en gel de agarosa estándar teñido con bromuro de etidio (/ ml de concentración final 0,2 g). Asegúrese de que una única banda de ADN que representa el producto de PCR es visible. Determinar el tamaño y la cantidad relativa de los fragmentos de ADN utilizando ADN estándares de peso molecular. Si es necesario, repita el PCR para obtener suficiente cantidad de fragmentos de ADN.
3. DpnI La digestión de los productos de PCR
NOTA: El objetivo de este paso es Digest plantilla de ADN plásmido residual de los PCRs en la Sección 2. DpnI digerirá ADN de plásmido sólo si es metilado, tal como ocurre al plásmido de ADN crecido en la presa + cepas bacterianas. Por lo tanto, no tratan con DpnI si plásmido de ADN se utiliza como un fragmento de ADN en la reacción de GA.
- Añadir 2 l de enzima de restricción DpnI a los 45 l de los productos de PCR producidos en la Sección 2. Incubar a 37 ° C durante 1 hr. Continúe con la Sección 4 o congelación a -20 ° C hasta que se necesite.
4. purificación y concentración de los fragmentos de ADN sobre purificación de ADN de los granos magnéticos
NOTA: El objetivo de este paso es purificar y concentrar los productos de PCR obtenidos en las secciones 2 y 3. Otros métodos de purificación de PCR se pueden utilizar también.
- Equilibrar las perlas magnéticas de purificación de ADN a temperatura ambiente. Vuelva a suspender las cuentas por breve vórtex.
- Transferir las PCR wi digerido pre-º DpnI a un tubos de 1,5 ml y añadir 81 l de perlas magnéticas de purificación de ADN a cada tubo. Incubar la mezcla a temperatura ambiente durante 10 min.
- Colocar los tubos en el colector magnética durante aproximadamente 2 min. Deseche el líquido transparente con una pipeta. Lavar dos veces con 200 l de etanol al 80% durante 30 segundos.
- Permitir que los pellets se sequen. Mantenga la tapa de los tubos abierta, con los tubos colocados en el colector magnético.
- Vuelva a suspender las perlas secas en 10 l de 10 mM Tris-HCl, pH 8,0. Incubar a temperatura ambiente durante 2 min. Centrifuga brevemente para recoger el líquido en la parte inferior de los tubos.
- Colocar los tubos en el colector magnético durante 2 min. Retire 8,5-10 l de la solución clara y colocarlo en un nuevo tubo previamente etiquetado. Determinar la concentración de los fragmentos de ADN mediante espectroscopia UV (véase la Tabla de Equipo).
5. Asamblea Clonación de reacción y transformación de los productos en E. coli
NOTA: El objetivo de este paso es calcular 3: 1 de inserción: vector y llevar a cabo la reacción de ensamblaje.
- Utilice al menos 100 ng del fragmento de ADN que representa el esqueleto del vector o fragmento de ADN que lleva el marcador selectivo. Calcular el exceso molar de 3 veces para los fragmentos de ADN que se utilizarán como insertos.
- Convertir el molar exceso en ng necesaria de cada inserción particular.
NOTA: Una forma cómoda de hacer los cálculos es el uso de una aplicación basada en la web (véase la Tabla de Equipo). - Mezcle cantidades calculadas de fragmentos de ADN en un tubo de PCR, ajustar el volumen de 10 l. Añadir 10 l de la mezcla maestra GA (2x, consulte la Tabla de Materiales). Incubar la reacción a 50 ° C durante 1 hora para el montaje de 4 - 6 o fragmentos de 15 min para el montaje de fragmentos de 2-3 en un ciclador térmico PCR.
- Proceda con la transformación del producto de ensamblaje en E. competente coli o congele los productos a -20° C, hasta que se necesite.
- Siga el procedimiento de transformación que acompaña a la células electro competentes química o. Por lo general, utilizar 2 l de la reacción de ensamblaje por reacción de transformación.
- Después de la transformación es completa, difundir las células transformadas en placas de agar suplementado con el antibiótico selectivo apropiado.
6. El plásmido Aislamiento, Restricción Digestión enzimática y secuenciación
NOTA: El objetivo de este paso es aislar ADN plasmídico a partir de E. coli, a continuación, para verificar el constructo mediante digestión de restricción y secuenciación.
- Propagar varias colonias individuales para mini preparaciones y aislamiento de plásmidos. Durante la noche de cultivo líquido LB Uso 2-5 ml complementa con antibiótico adecuado. Aislar los plásmidos correspondientes siguiendo el procedimiento que se describe en el kit de mini-prep que se utiliza.
- Determinar la cantidad y la concentración de los plásmidos obtenidos utilizando Spectrofotómetro.
- Realizar la digestión de restricción con una o varias endonucleasas de restricción a ~ 0,5 g de los plásmidos purificados. Use enzimas de restricción que proporcionan patrón distinto de característica de digestión para las construcciones de ADN.
- Confirmar el éxito de clonación de ADN mediante secuenciación de ADN utilizando cebadores específicos o estándar. Analizar los datos de secuenciación para la exactitud.
Resultados
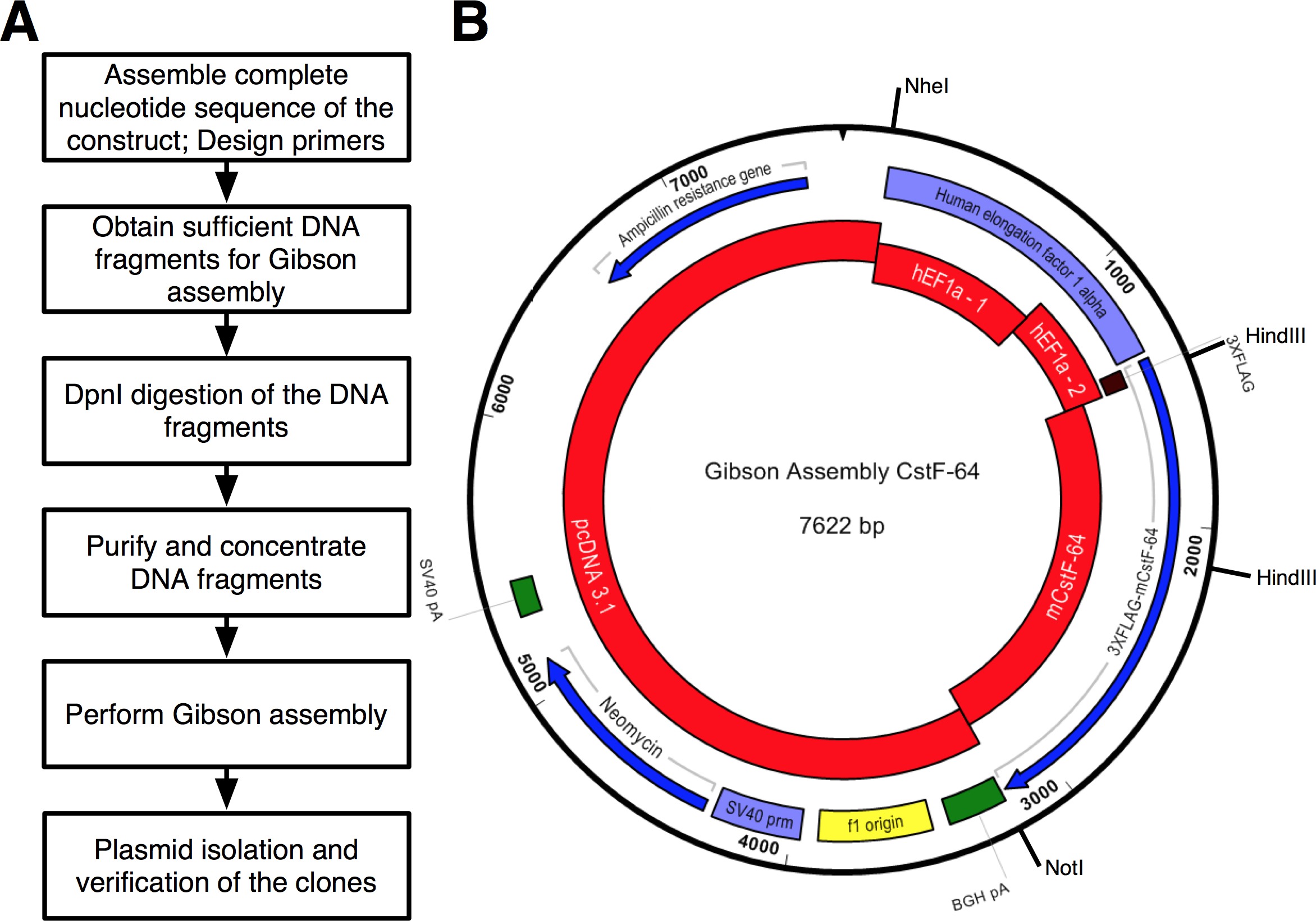
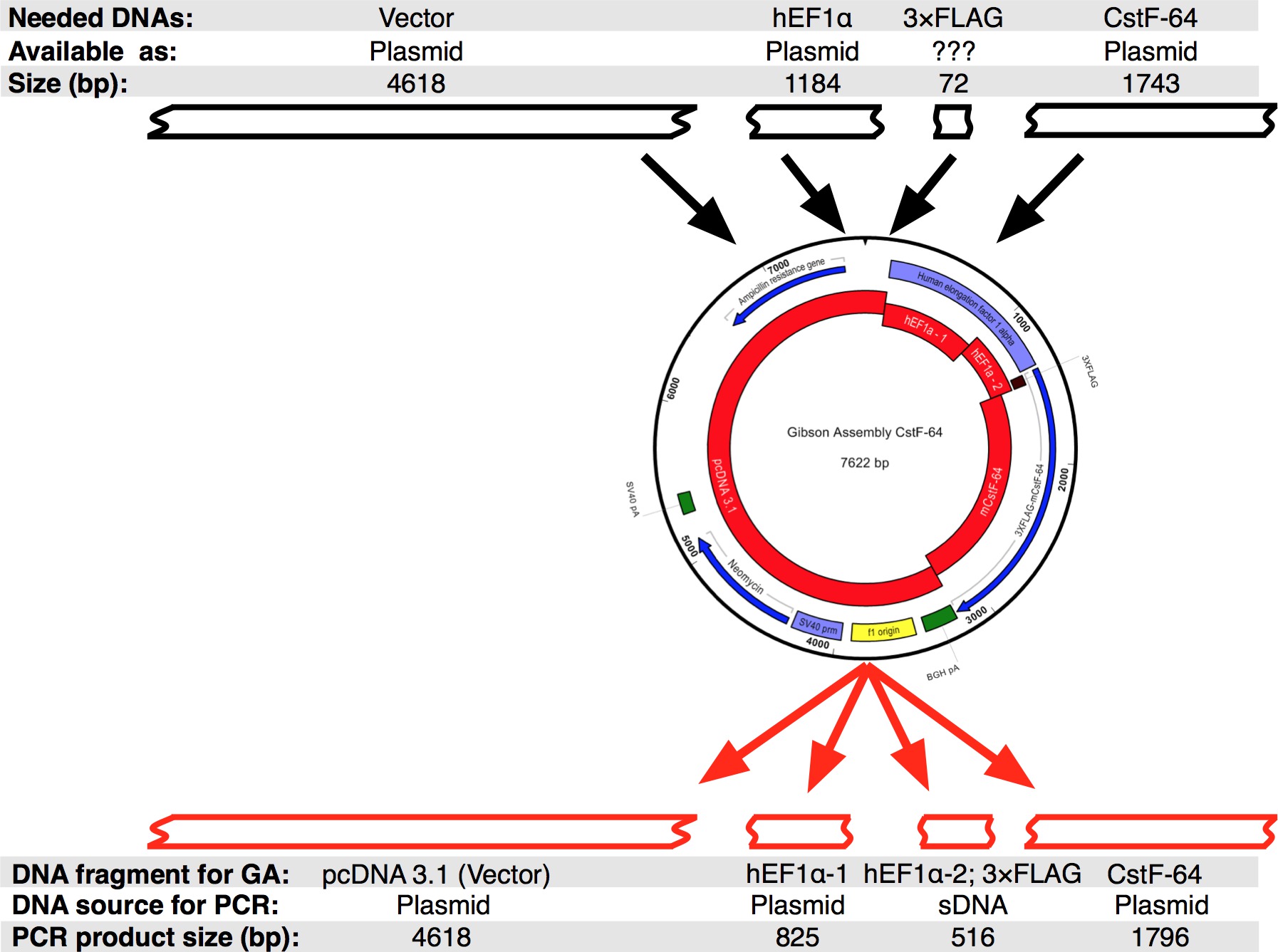
Un flujo de trabajo del protocolo que se siguió se muestra en la Figura 2A. Queríamos clonar CSTF-64 y mutantes CSTF-64 proteínas fusionadas a 3xFLAG etiqueta bajo la regulación del promotor expressional hEF1α (Figura 2B y Figura 3). Un hEF1α plásmido que contiene seguido 3xFLAG-tag no estaba disponible para nosotros. Sin embargo, los siguientes plásmidos estaban disponibles: pcDNA 3.1 myc-Su (A; un generoso regalo de Michaela Jansen), hEF1α plásmido que contiene (un generoso regalo de Mladen Yovchev) y ratón CSTF-64 plásmidos 12 (Figura 3). La secuencia completa para la construcción (s) se ensambla utilizando las aplicaciones de edición de nucleótidos y de texto (Figura 3; véase la tabla de Equipo). Posteriormente, la secuencia (s) se dividió en cuatro piezas convenientes (Figura 2B, bloques de color rojo y la Figura 3) correspondientes a los ADN plasmídicos disponibles. Amplificación primers fueron diseñados utilizando la herramienta de generación de imprimación (véase la Tabla de Equipo) con limitaciones de 4-6 fragmentos con mínima superposición de 25 nt, creada en la ventana emergente "Cambiar la configuración de la Asamblea Gibson". Sitios de restricción NheI y NotI se incluyeron en el diseño del cebador con el propósito de identificar los plásmidos correctamente reunidos. NheI sitio se encuentra en la secuencia del cebador entre pcDNA 3.1 y el extremo 5 'del promotor hEF1α. Noti sitio está ubicado después del codón de parada (UGA) de CSTF-64 y pcDNA 3.1 vector columna vertebral. Durante la digestión simultánea con el fragmento de ADN ambas enzimas que consiste en el promotor hEF1α, 3xFLAG y CSTF-64 o mutante CSTF-64 se dará a conocer (véase más adelante y en la Figura 2B). Los cebadores se ordenan en la escala más pequeña posible y desalados. La segunda parte del promotor hEF1α fragmento de ADN que contiene 3xFLAG-tag (490 pb, la Figura 2B, Figura 3) se adquirió como un solo fragmento sDNA (see Tabla de Materiales). Los fragmentos de ADN utilizados en la reacción de ensamblaje fueron amplificados utilizando ADN pol (véase la Tabla de Materiales). Fragmentos de ADN de hEF1α promotor parte 1, parte 2 hEF1α promotor, de cuerpo entero y CSTF-64 mutante se amplificaron de forma simultánea en un tubos separados durante 28 ciclos (Figura 4A), siguiendo las recomendaciones del proveedor del ADN pol (véase la Tabla de Materiales , para cada ciclo de desnaturalización fue de 7 segundos a 98 ° C, recocido 45 segundos a 55 ° C, el alargamiento de 90 segundos a 72 ° C). Inicialmente, la columna vertebral pcDNA 3.1 se amplificó durante 22 ciclos (utilizando las mismas condiciones que el anterior con la excepción del tiempo de elongación, que fue ajustado a 3 min a 72 ° C). Sin embargo, el rendimiento de ADN resultante no era suficiente para ser utilizado en una reacción de ensamblaje (Figura 4A). Por lo tanto, se realizó una amplificación adicional para obtener suficiente ADN.
Productos de PCR obtienened partir de una plantilla de plásmido debe ser digerido con la enzima de restricción DpnI para eliminar el ADN de plásmido, que de otro modo contaminar los productos de reacción resultantes de montaje y producirá colonias bacterianas falsos positivos resistentes a los medicamentos. Por lo tanto, los productos de PCR fueron digeridos con la enzima de restricción DpnI, que escinde ADN de plásmido aislado metilado y hemi-metilado de la presa + E. cepas de E. coli. Productos de PCR obtenidos utilizando fragmentos de ADN sintéticos, como plantillas no necesitan ser digerido con DpnI ya que el ADN sintetizado químicamente no contiene bases metiladas o hemi-metilado.
Los fragmentos de ADN se purificaron y se concentraron sobre perlas magnéticas de purificación de ADN (véase la Tabla de Materiales) como se describe en el paso de protocolo 4. Las PCRs para el pcDNA 3.1 vector columna vertebral se combinaron juntos y la cantidad de perlas magnéticas de purificación de ADN utilizada se ajustó en consecuencia. El rendimiento de ADN se determinóutilizando un espectrofotómetro (Tabla 1 y la Tabla de Equipo). Reacciones de montaje para CSTF-64 y mutantes CSTF-64 construcciones se ensamblaron en hielo (Tabla 1). Un exceso molar de 3 veces de los fragmentos de ADN consideradas como "insertos" fue utilizado (Tabla 1, Figura 3). El volumen final de los fragmentos de ADN mixtas se ajustó a 10 l con agua y 10 l de mezcla maestra de montaje (2x) se añadió. Las reacciones se mezclaron y se incubaron a 50 ° C durante 1 hr. Reacción de control positivo también se montó de acuerdo a la recomendación del manual del kit GA y se incubaron simultáneamente con los CSTF-64 y mutantes CSTF-64 reacciones. Como se recomienda en el protocolo, 2 l de la cada una de las reacciones de ensamblaje se transformaron en el E. químicamente competentes coli suministra con el kit de clonación de montaje (véase la Tabla de Materiales). La transformación se llevó a cabo como se describe en el kitmanual. Los clones positivos se seleccionaron en placas de agar con ampicilina / LB. 6 colonias por cada reacción de ensamblaje fueron seleccionados al azar para ser reproducido. Los ADN de plásmido se aisló utilizando un mini kit de aislamiento de plásmido (véase la Tabla de Materiales). En la digestión in silico de las construcciones con las enzimas de restricción NheI y NotI resultaron en dos fragmentos con tamaños de 4.590 pb, 3032 pb para CSTF-64 y 4.590 pb, 2711 pb para mutante CSTF-64 (Figura 2B y Figura 4B). La digestión con las enzimas de restricción HindIII y NotI resultaron en tres fragmentos con los siguientes tamaños: 5.872 pb, 1005 pb y 745 pb (CSTF-64) y 5872 pb, 1005 pb y 424 pb (CSTF-64 mutante, la Figura 2B y Figura 4C). De hecho, la digestión de los plásmidos aislados muestra los patrones característicos esperados (Figura 4B, C). Tenga en cuenta que el fragmento de ADN 424 pb producido por la digestión con HindIII y NotI de la CSTF-64 mutanteplásmidos en la figura 4C se tiñen débilmente debido a su pequeño tamaño. 2 de los 6 plásmidos aislados fueron enviados para la secuenciación. Hemos secuenciado el promotor hEF1α y CSTF-64 o mutante CSTF-64 partes de las construcciones para verificar que no hay deleciones, inserciones o sustituciones. Recomendamos encarecidamente la secuenciación de las construcciones de ADN que resultan de este o de cualquier protocolo basado en PCR. La secuenciación mostró que uno de cada plásmido secuenciado contenía la secuencia esperada en la región de hEF1α, 3xFLAG-tag y CSTF-64 o CSTF-64 mutante. Cada uno de los otros plásmidos tenían un punto de mutación introducida durante la amplificación del fragmento de ADN correspondiente. Expresión del plásmido que contiene CSTF-64 en células madre embrionarias de ratón, produce abundante cantidad de proteína exógena comparable a la de tipo salvaje expresión 17.

Figura 1. Representación esquemática del mecanismo de Asamblea Gibson. Fragmentos de ADN con la superposición de los extremos se ensamblaron isotérmicamente en una sola secuencia continua. La superposición Final primera se mastica de nuevo por 5 'exonucleasa, que es calentar gradualmente inactivado. En consecuencia, los diferentes fragmentos de ADN con extremos superpuestos se recocer isotérmicamente. ADN polimerasa llenará los vacíos y la ADN ligasa termoestable productos ligados los nicks. Por favor, haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 2. (A) Diagrama de flujo del protocolo descrito para la clonación de montaje. (B) Representación del plásmido, PGA-CSTF-64 generado usando el kit de GA rojos - fragmentos de ADN utilizados en la reacción de ensamblaje:. PcDNA3,1; hEF1α promotor parte 1 (hEF1a - 1); hEF1α promotor parte 2 (hEF1a - 2) ordenó como un ADN sintético; ratón CSTF-64 (mCstF-64). Marcos de lectura abierta - azul. Violet - promotores virales y no virales. Verdes -. Regiones de escisión y poliadenilación Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 3. Diseño de la asamblea Gibson CSTF-64 plásmido. Negro cajas representan los fragmentos de ADN que estaban disponibles para diseñar un único conjunto Gibson CSTF-64 en secuencia silico. Posteriormente, la secuencia se divide en cuatro piezas de ADN, que fueron amplificados por PCR. Tenga en cuenta que debido al pequeño tamaño de la 3xFLAG-etiqueta de la secuencia fue diseñado como un sDNA junto con la pr hEF1α parte omoter 2. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 4. PCR de los fragmentos de ADN utilizados en las reacciones de clonación y representante de la digestión con enzimas de restricción de los plásmidos obtenidos (A) PCR usando representativos de arranque en caliente de alta fidelidad mezcla maestra 2x para los fragmentos de ADN utilizados en las reacciones de montaje:. (B) Representante plásmidos de hEF1α, de cuerpo entero CSTF-64, pcDNA 3.1 constructo (PGA-CSTF-64) y hEF1α, mutante CSTF-64, pcDNA 3.1 constructo (PGA-mutCstF-64) digerido con NheI y NotI. (C) los mismos plásmidos como en B digeridos con las enzimas HindIII y NotI."_blank"> Haga clic aquí para ver una versión más grande de esta figura.
| Nombre de fragmentos de ADN | El tamaño esperado (bp) | Concn. (Ng / l) | Diluido a (ng / l) | l utiliza en CSTF GA-64 de diluido | l utiliza en GA mutCstF-64, desde diluido | Relación molar (ins: vec) |
| pcDNA 3.1 (vector) | 4618 | 158 | sin diluir | 1 | 1 | |
| hEF1_ promotor parte 1 | 825 | 213 | 75 | 1 | 1 | 3: 1 |
| hEF1_ promotor parte 2 para CSTF-64 | 516 | 229 | 50 | 1 | 3: 1 | |
| CSTF-64 | 1796 | 161 | sin diluir | 1 | 3: 1 | |
| hEF1_ promotor parte 2 para mutante CSTF-64 | 516 | 199 | 50 | 1 | 3: 1 | |
| CSTF-64 mutante | 1448 | 201 | 171 | 1 | 3: 1 |
Tabla 1. Rendimiento de fragmentos de ADN después de la concentración de perlas magnéticas, dilución y puesta en marcha de las reacciones de ensamblaje.
Discusión
El uso exitoso de GA clonación debe ser siempre precedido por un diseño cuidadoso de la construcción completa (Figura 2 y Figura 3).
También es muy recomendable la verificación cuidadosa de las secuencias de cebadores diseñados por la herramienta de generación de imprimación. Los cebadores para GA se pueden generar sin el uso de la herramienta de generación de imprimación. Sin embargo, el uso de la herramienta es muy recomendable, ya que simplifica el proceso. Generalmente, el cebador para la clonación GA debe tener dos secuencias funcionalmente diferentes. La primera secuencia es fragmento específico de ADN, y permite la amplificación del fragmento de PCR utilizando. La segunda secuencia se solapa con el fragmento adyacente, que es necesario para el montaje GA. Una secuencia típica-DNA-fragmento específico sería 18-22 nt de longitud. Secuencias específicas de ADN fragmento utilizados para amplificar el mismo ADN deben tener temperaturas de fusión similares y contenido de GC. Secuencia de superposición debe ser al menos de 15nt de longitud con una temperatura de fusión de al menos 48 ° C. El conjunto de más de 4 fragmentos de ADN requerirá la secuencia de superposición que ser al menos 20 nt. Superposiciones más largos permitirán un mayor especificidad de la hibridación que resulta en fragmentos de ADN más correctamente ensamblados. Se recomienda evitar secuencias que están sesgadas en su GC o AT contenido en el desarrollo de secuencias superpuestas, porque las secuencias sesgadas podrían comprometer montaje adecuada del ADN.
También sugerimos usar como un control negativo, que no debe producir ningún colonias de bacterias resistentes a los fármacos, sólo el fragmento de ADN correspondiente a la cadena principal del vector en una reacción de GA. Por otra parte, uno de los fragmentos de ADN que comprenden las "inserciones" puede omitirse de la reacción GA, que también debería dar lugar a colonias bacterianas resistentes a los medicamentos. La razón no hay colonias crecerán será la falta de extremos superpuestos de los fragmentos de ADN adyacentes, lo que hará que el montaje de un complplásmido ete.
En el protocolo descrito, la superposición de secuencias de 25 nt para generar Se utilizaron los conjuntos de cebadores, debido al número de fragmentos de ADN utilizados (Figura 3). La recomendación de la página web de la herramienta de generación de imprimación (véase la Tabla de Equipo) es el uso de al menos 20 nt secuencias superpuestas de montar 4-6 fragmentos de ADN. Además, la superposición de secuencia más larga asegure la complementación adecuada de las cadenas de ADN (véase la Figura 1) aumentar el número de productos ensamblados con precisión.
Actualmente, varios sistemas para la clonación sin fisuras están disponibles. Sin embargo, algunos de estos sistemas todavía utilizan enzimas de restricción (es decir, la puerta de oro de clonación 18). Otros utilizan mezclas de propiedad de enzimas basados en virus vaccinia ADN polimerasa y la proteína de unión a ADN de una sola hebra de la misma fuente biológica 19. Ambos sistemas son limitados en comparación con GA por el shorter longitud de secuencias que se solapan. Debido a la superposición de secuencias más cortas podrían no proporcionar la suficiente especificidad para la etapa de hibridación de fragmentos de ADN adyacentes, haciendo que el montaje correcto de más de 23 fragmentos de ADN problemáticos. Estas deficiencias no están presentes en el sistema GA.
El tamaño de los fragmentos de ADN a amplificar se debe también considerarse a fin de no exceder el tamaño fiable amplificable por PCR (es decir, menos de 8 kpb de longitud). Incluso con las mejoras en la función de las polimerasas de ADN en los últimos 10 años, grandes fragmentos de ADN se amplificaron con menos eficiencia y precisión. Si es necesario, los fragmentos de ADN más grandes podrían ser obtenidos de otras fuentes alternativas de PCR, por ejemplo, mediante el aislamiento del plásmido y digestión del ADN con enzimas de restricción apropiadas. En concreto, para el protocolo descrito en el manuscrito actual, nuestro racional para utilizar PCR se basó en el tamaño de los fragmentos de ADN disponibles, que eran todos menos de5 kpb. Si hay más de un producto PCR identificado por electroforesis en gel de agarosa, se recomienda la purificación en gel del fragmento de tamaño conveniente utilizando cualquiera de las técnicas de biología molecular disponibles o kits apropiados. En el protocolo actual se utiliza una ADN polimerasa termoestable (véase la Tabla de Materiales). Sin embargo, cualquier ADN polimerasa proporcionando alta fidelidad y rendimiento será adecuado para ser utilizado con este protocolo. Si ADN polimerasas de alta fidelidad diferentes de lo descrito en el protocolo están disponibles, utilice las condiciones establecidas como se describe en los manuales respectivos. En el protocolo actual, E. químicamente competentes coli células se utilizan que se suministra con el kit de GA. E. Alternativamente, química o electro competente cepas de E. coli tales como DH5a o DH10B pueden ser utilizados.
El montaje de la clonación de mezcla maestra 2x es fácil de usar con manos en un tiempo mínimo. Sin embargo, se requiere de pipeteado preciso debido a los pequeños volúmenes neEDED a mezclar juntos. Una buena técnica de biología molecular debe ser ejercido en todo momento así.
La clonación GA ofrece posibilidades ilimitadas para una construcción de fragmentos de ADN, plásmidos y vectores, que son más de 3 kpb de tamaño. Además tiene un impacto más amplio en el campo de la biología sintética, ya que permite la síntesis y ensamblaje de, por ejemplo, un (Mycoplasma mycoides) genoma entero bacteriana o una levadura (Saccharomyces cerevisiae) cromosoma 20,21. La técnica también es aplicable a la clonación convencional necesita para generar construcciones sin costura.
En conclusión, GA clonación ofrece alternativa rápida, fiable y flexible para el procedimiento de clonación de ADN convencional.
Divulgaciones
Open Access publication and production fees were supplied by New England BioLabs Inc.
Agradecimientos
Nos gustaría dar las gracias a Michaela Jansen en el Centro de Ciencias de la Salud de Texas Tech University, Lubbock, TX y Mladen Yovchev en la Universidad de Pittsburgh Medical Center, Pittsburgh, PA para el pcDNA proporcionar generosamente 3.1 myc-Su (A) y hEF1α promotor que contiene plásmidos . Las investigaciones realizadas en esta publicación fue apoyada por el Instituto Nacional Eunice Kennedy Shriver de Salud Infantil y Desarrollo Humano de los Institutos Nacionales de Salud con el número premio R01HD037109 (a CCM). El apoyo adicional fue desde el Instituto Bush W. Laura para la Salud de la Mujer (a CCM y PNG). El contenido es responsabilidad exclusiva de los autores y no representa necesariamente las opiniones oficiales de los Institutos Nacionales de Salud.
Los autores declaran que no tienen intereses financieros en competencia.
Materiales
| Name | Company | Catalog Number | Comments |
| Name of Material/ Equipment | Company | Catalog Number | Comments/Description |
| Synthetic double-stranded DNA fragment (sDNA) | Integrated DNA Technologies | We used gBlocks, which can be up to 2 kb in length. However, there are many commercial sources of synthetic DNA available. | |
| DNA purification magnetic beads | Beckman Coulter | A63880 | Agencourt AMPure XP - PCR Purification system |
| Plasmid Isolation Mini Kit | Omega Bio-Tek Inc | D6942-01 | E.Z.N.A. Plasmid Mini Kit I |
| Gibson Assembly Cloning Kit | New England BioLabs | E5510S | |
| DNA polymerase | New England BioLabs | M0494S | Q5 Hot Start High-Fidelity 2X Master Mix |
| DpnI | New England BioLabs | R0176S | |
| NheI | New England BioLabs | R3131S | |
| NotI | New England BioLabs | R3189S | |
| HindIII | New England BioLabs | R3104S | |
| Name of Material/ Equipment | Company | Catalog Number | Comments/Description |
| Spectrophotometer | Thermo Scientific | NanoDrop device | |
| EditSeq | DNASTAR | Part of Lasergene Core Suite | |
| SeqBuilder | DNASTAR | Part of Lasergene Core Suite | |
| Word | Microsoft | Part of Microsoft Office | |
| NEBuilder | New England BioLabs | primer generation tool: http://nebuilder.neb.com | |
| NEBioCalculator | New England BioLabs | ligation calculator: http://nebiocalculator.neb.com/#!/ligation |
Referencias
- Gibson, D. G. Enzymatic assembly of overlapping DNA fragments. Methods in enzymology. 498, 349-361 (2011).
- Gibson, D. G., Smith, H. O., Hutchison, C. A., Venter, J. C., Merryman, C. Chemical synthesis of the mouse mitochondrial genome. Nature methods. 7, 901-903 (2010).
- Gibson, D. G., et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nature methods. 6, 343-345 (2009).
- Bowtell, D. D., Johnson, G. R., Kelso, A., Cory, S. Expression of genes transferred to haemopoietic stem cells by recombinant retroviruses. Molecular biology & medicine. 4, 229-250 (1987).
- Challita, P. M., Kohn, D. B. Lack of expression from a retroviral vector after transduction of murine hematopoietic stem cells is associated with methylation in vivo. Proceedings of the National Academy of Sciences of the United States of America. 91, 2567-2571 (1994).
- Lutzko, C., Senadheera, D., Skelton, D., Petersen, D., Kohn, D. B. Lentivirus vectors incorporating the immunoglobulin heavy chain enhancer and matrix attachment regions provide position-independent expression in B lymphocytes. Journal of Virology. 77, 7341-7351 (2003).
- Meilinger, D., et al. Np95 interacts with de novo DNA methyltransferases, Dnmt3a and Dnmt3b, and mediates epigenetic silencing of the viral CMV promoter in embryonic stem cells. EMBO reports. 10, 1259-1264 (2009).
- Chan, K. K., Wu, S. M., Nissom, P. M., Oh, S. K., Choo, A. B. Generation of high-level stable transgene expressing human embryonic stem cell lines using Chinese hamster elongation factor-1 alpha promoter system. Stem cells and development. 17, 825-836 (2008).
- Chung, S., et al. Analysis of different promoter systems for efficient transgene expression in mouse embryonic stem cell lines. Stem cells. 20, 139-145 (2002).
- Qin, J. Y., et al. Systematic comparison of constitutive promoters and the doxycycline-inducible promoter. PloS one. 5, e10611 (2010).
- Lund, A., Knudsen, S. M., Vissing, H., Clark, B., Tommerup, N. Assignment of human elongation factor 1alpha genes: EEF1A maps to chromosome 6q14 and EEF1A2 to 20q13.3. Genomics. 36, 359-361 (1996).
- Wallace, A. M., et al. Two distinct forms of the 64,000 Mr protein of the cleavage stimulation factor are expressed in mouse male germ cells. Proceedings of the National Academy of Sciences of the United States of America. 96, 6763-6768 (1999).
- MacDonald, C. C., McMahon, K. W. Tissue-specific mechanisms of alternative polyadenylation: testis, brain, and beyond. Wiley interdisciplinary reviews. RNA. 1, 494-501 (2010).
- Sabath, I., et al. 3'-End processing of histone pre-mRNAs in Drosophila: U7 snRNP is associated with FLASH and polyadenylation factors. Rna. 19, 1726-1744 (2013).
- Yang, X. C., et al. A complex containing the CPSF73 endonuclease and other polyadenylation factors associates with U7 snRNP and is recruited to histone pre-mRNA for 3'-end processing. Molecular and cellular biology. 33, 28-37 (2013).
- Grozdanov, P. N., Macdonald, C. C. High-Throughput Sequencing of RNA Isolated by Cross-Linking and Immunoprecipitation (HITS-CLIP) to Determine Sites of Binding of CstF-64 on Nascent RNAs. Methods in molecular biology. 1125, 187-208 (2014).
- Youngblood, B. A., Grozdanov, P. N., MacDonald, C. C. CstF-64 supports pluripotency and regulates cell cycle progression in embryonic stem cells through histone 3' end processing. Nucleic acids research. , (2014).
- Engler, C., Kandzia, R., Marillonnet, S. A one pot, one step, precision cloning method with high throughput capability. PloS one. 3, e3647 (2008).
- Irwin, C. R., Farmer, A., Willer, D. O., Evans, D. H. In-fusion(R) cloning with vaccinia virus DNA polymerase. Methods in molecular biology. 890, 23-35 (2012).
- Annaluru, N., et al. Total synthesis of a functional designer eukaryotic chromosome. Science. 344, 55-58 (2014).
- Gibson, D. G., et al. Creation of a bacterial cell controlled by a chemically synthesized genome. Science. 329, 52-56 (2010).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados