
Method Article
Erzeugung von Plasmid-Vektoren, die FLAG-markierten Proteine Nach der Verordnung der Menschen Elongation Factor-1α Promoter Mit Gibson Assembly
In diesem Artikel
Zusammenfassung
Synthesis of custom plasmids is labor and time consuming. This protocol describes the use of Gibson assembly cloning to reduce the work and duration of custom DNA cloning procedure. The protocol described also produces reliable tagged protein constructs for mammalian expression at similar cost to the traditional cut-and-paste DNA cloning.
Zusammenfassung
Gibson Anordnung (GA) Klonierung bietet eine schnelle, zuverlässige und flexible Alternative zu herkömmlichen DNA-Klonierungsverfahren. Wir verwendeten GA angepasste Plasmide für die Expression von exogenen Genen in mausembryonalen Stammzellen (mESCs) zu erstellen. Expression exogener Gene unter der Kontrolle des SV40 oder menschliche Cytomegalovirus-Promotoren vermindert schnell nach Transfektion in mESCs. Eine Abhilfe für diese verminderte Expression ist es, die menschliche Elongationsfaktor-1 alpha (hEF1α) Promoter zu verwenden, um die Genexpression zu fahren. Plasmidvektoren, die hEF1α sind nicht so weit verbreitet wie SV40 oder CMV-haltigen Plasmide, vor allem jenen, die auch N-terminale 3xFLAG-Tags zur Verfügung. Das hier beschriebene Protokoll ist eine schnelle Methode, um die Plasmide FLAG-markierten CstF-64 und CstF-64-Mutante unter der Ausdrucks Regulierung des hEF1α Promotors erstellen. GA verwendet eine Mischung aus DNA-Exonuclease, DNA-Polymerase und DNA-Ligase, um die Klonierung von überlappenden Enden der DNA-Fragmente zu ermöglichen.Auf der Grundlage der Vorlage DNAs wir zur Verfügung hatten, haben wir unsere Konstrukte in einer einzigen Sequenz zusammengesetzt werden. Unser Design verwendet vier DNA-Fragmente: pcDNA 3.1 Vektor-Rückgrat, hEF1α Promoter Teil 1, Teil 2 hEF1α Promoter entweder CstF-64 oder spezifische CstF-64-Mutante (die 3xFLAG-Tag als doppelsträngige synthetische DNA-Fragment enthielt gekauft), und. Die Sequenzen dieser Fragmente wurden an einen Primer Generierungswerkzeug geeignete PCR-Primer für die Erzeugung der DNA-Fragmente zu entwerfen hochgeladen. Nach der PCR wurden DNA-Fragmente mit dem Vektor, der den Selektionsmarker und die GA Klonierung Reaktion wurde zusammengemischt. Plasmide von einzelnen transformierten bakteriellen Kolonien wurden isoliert. Einstieg der Plasmide wurde durch Restriktionsverdau durchgeführt, gefolgt von einer Sequenzierung. Abschließend erlaubt GA uns, maßgeschneiderte Plasmide für die Genexpression in 5 Tagen zu erstellen, einschließlich konstruieren Bildschirme und Verifikation.
Einleitung
Herkömmliche DNA-Klonierungsverfahren beruhen auf der Verwendung von Restriktionsenzymen, um die DNA und die DNA-Ligase zu spalten, um die DNA-Fragmente miteinander zu verbinden. Erzeugung von benutzerdefinierten Expressionskonstrukte, die verschiedene DNA-Fragmente ist ein sequentielles Verfahren, das die Spaltung der DNA mit einem und / oder mehreren Restriktionsendonukleasen und die anschließende Insertion von DNA-Fragmenten durch Ligation umfasst. Der Hauptnachteil dieses Verfahrens ist, dass geeignete Restriktionsenzyme für eines der DNA-Fragmente können schwer zu identifizieren (dh kann mehrere Spaltstellen) Rendern erfolgreichen DNA-Klonierung der Volllängen-Protein von Interesse möglich. Daher Generation von kundenspezifischen Expressionskonstrukte unter der Transkriptionsregulation der effizienten zelltypspezifische Promotoren mit kundenspezifischen Protein-Tags erfordert eine sehr sorgfältige Gestaltung. Es ist auch ein zeit- und arbeitsaufwendige Technik. Vor kurzem beschrieben Methoden zur multip montieren mehrere Berichtele verschiedenen synthetischen DNA-Fragmenten in einer kontinuierlichen Sequenz gleichzeitig in entweder ein- oder zweistufigen Reaktionen ohne die Verwendung von Restriktionsenzymen 1-3. Das einstufige Klonierung Reaktion (unter Ausschluss aller vorbereitenden Schritte), hängt von der Verwendung einer Mischung von DNA-Exonuclease, DNA-Polymerase, DNA-Ligase 2,3 und die überlappenden Enden der DNA-Fragmente (1). Da es keine Verwendung von Restriktionsenzymen, DNA-Fragmente können von beliebiger Größe und Sequenzzusammensetzung (ohne hoch repetitive Sequenzen) zusammen in einer nahtlosen Konstruktion fusioniert werden. Vor kurzem wurde ein im Handel erhältlichen Kits (Gibson Anordnung; GA) für die Ein-Schritt-Klonierung Reaktionen verfügbar wurde. Dieses Kit ermöglicht eine schnelle und kostengünstige Montage von beliebigen DNA-Fragmente in einem einzigen Vektor mit kundenspezifischen Promotoren und Protein-Tags.
Die verwendet werden, um exogene Proteine in Säugetier-Zellkulturmodellen ausdrücken weithin erhältlichen Plasmid-Expressionsvektoren sind häufig unter der Transkriptions regordnung des viralen Cytomegalovirus (CMV) oder Simian Virus 40 (SV40) Promotoren. Diese viralen Promotoren stellen robuste transiente Expression der exogenen Proteine in den meisten Säugerzellkultur-Modellen. Jedoch ist von Zelllinien, die stabil exogene Proteine oft wegen Transkriptions Silencing der CMV- oder SV40-Promotoren während des Herstellungsvorgangs 4,5 erfolglos. Darüber hinaus sind die SV40 und CMV virale Promo nicht ausreichend die Expression von exogenen Proteinen in Zellen aus der lymphoiden Linie oder embryonale Stammzellen 6,7 fördern. Die Lösung der inhärenten Beschränkungen der viralen Promotoren ist es, starke konstitutive nicht-viralen Promotoren 8-10 zu verwenden. Ein gut charakterisiertes starken konstitutiven nichtviralen Promotor von menschlichem Ursprung ist die Elongationsfaktor 1α (hEF1α) Promotor (hEF1α ist bei der Katalyse des GTP-abhängige Assoziation von Aminoacyl-tRNA an Ribosomen 11 beteiligt sind). JedochExpressionsvektoren, die das hEF1α Promotor nicht so stark wie der virale Promotor-enthaltenden Plasmiden verfügbar, insbesondere solche, ebenfalls enthaltend 3 × FLAG am Amino-terminalen Ende des Proteins von Interesse.
Das 64.000 MW Spaltung Stimulationsfaktor-Protein (CstF-64) wird in das 3'-End-Verarbeitung der meisten mRNAs 12,13 einschließlich replikationsabhängigen Histon-mRNAs 14,15 beteiligt. CstF-64 ist in allen Körpergeweben 12 ausgedrückt. Seine RNA-Erkennungsmotiv bindet an GU-reiche RNA-Sequenzen auf im Entstehen begriffene Transkripte hinter der Spaltung und Polyadenylierung Seite 16. Diese Bindung CstF-64 auf die prä-mRNA fördert die effiziente endonukleolytische Spaltung des entstehenden Niederschrift.
Hier wird ein Protokoll beschrieben, das die PCR-Amplifikation der DNA-Fragmente verwendet, um eine Gibson Anordnung Klonierungskit (das chemisch kompetenten Bakterienzellen enthält) individuelle Vektoren 3xFLAG markierten m herzustellenouse CstF-64 oder eine Mutante CstF-64 ihre aminoterminalen Ende unter dem Ausdruck hEF1α Promotor 1.
Protokoll
1. In Silico Gestaltung der Plasmide und Erzeugung des Überlappungs Grundierungen
HINWEIS: Das Ziel dieses Schrittes ist die vollständige Nucleotidsequenz des Konstrukts zusammen und entwerfen die Primer verwendet werden, um Fragmente mit überlappenden Enden für GA Klonen erzeugen.
- Entwerfen Sie eine kontinuierliche Nukleotidsequenz, die endgültige Plasmid darstellen.
- Zu erhalten, oder eine Liste der tatsächlichen Plasmide und DNA-Fragmente, die als Matrizen bei der PCR verwendet werden.
HINWEIS: DNA-Fragmente, die nicht leicht verfügbar sind - wie andere Kombination von Tags und Promotoren - kann als eine einzelne oder mehrere synthetische Doppelstrang-DNA-Fragmente (sDNA) bestellt werden. Diese Fragmente sind zu relativ geringen Kosten im Handel erhältlich und kann bis zu 2 kb Länge (siehe Tabelle der Materialien) sein. - Unterteilen den kontinuierlichen Nucleotidsequenz des Konstrukts in Schritt 1.1 in DNA-Fragmente für PCR zusammengebaut. Confirm, die die Fragmente entsprechen erhältlichen Plasmiden und sDNA Fragmente. Vermeiden DNA-Fragmente kleiner als 200 nt.
- Zugriff auf die Grundierung Werkzeuggeneration (siehe Tabelle der Ausrüstung). Wählen Sie das Menü "Set Preferences". Wählen Sie die entsprechenden Einstellungen, indem Sie auf der Registerkarte "CHANGE PREFS" im "Change Gibson Assembly-Einstellungen" Popup-Fenster.
HINWEIS: Primer Design kann auch ohne die Verwendung des Primers Werkzeug Generation durchgeführt werden. Die Verwendung des Primers Werkzeugerzeugung vereinfacht jedoch das Verfahren. - Starten Sie den Aufbau des Konstrukts, indem Sie das Menü "Build-Construct". Stecken Sie das gespaltene DNA-Fragmente in der Grundierung Generierungstool sequentiell von 5 'nach 3' Ende.
ANMERKUNG: Das DNS-Fragment als Plasmidrückgrat eingesetzt bequem in zwei Teile aufgeteilt werden. In der PCR-Endprodukt das 5'-Ende dieser ersten Fragment und das 3'-Ende des letzten Fragments zusammen in einer kontinuierlichen DN verbundenEin Fragment, das die Vektor-Hauptkette. - Fügen Sie den ersten DNA-Fragment das 5'-Ende der Vektor-DNA in FASTA-Format (eine textbasierte Darstellung der Nukleotid-Sequenz) in die "Enter Vektor oder Insert Fragment" Popup-Fenster darstellt. Benennen Sie die DNA-Fragment. Wählen Sie die passende Art und Weise, um das DNA-Fragment zu erhalten, entweder als PCR, RE Digest oder Synthese. Klicken Sie auf die Registerkarte "WEITER".
- Wenn es notwendig ist, zusätzliche Nukleotide / Restriktionsstellen an der Kreuzung der endgültige Konstrukt hinzuzufügen benutzen Sie die "FWD oder REV Primer Spacer" Bereiche im Fenster "Add einen Einsatz Fragment der Montage" zur Verfügung gestellt. Fügen Sie zusätzliche Nukleotide / Restriktionsstellen, um nur eine der DNA-Fragmente und nicht an beiden Fragmenten. Klicken Sie auf "DONE" Registerkarte.
- Wiederholung für alle Fragmente, bis das Konstrukt abgeschlossen ist. Wählen Sie das Menü "Ansicht Primer" und überprüfen Sie die Primer-Sequenzen.
- Wiederholen Sie dies für alle Konstrukte (Vektor backbones und Einsätze), oder für die DNA-Fragmente, die unterschiedlich sind.
2. Verstärkung der DNA-Fragmente durch PCR mit Hot Start Korrektur DNA Polymerase (2x Master Mix)
ANMERKUNG: Das Ziel dieses Schrittes ist es, ausreichend DNA für die Montage mit Hilfe der PCR-Reaktion erhalten.
- Erwerben der PCR-Primer in dem vorhergehenden Schritt als entsalzt Produkte und in möglichst geringem Umfang gestaltet. Verdünnen auf 10 & mgr; M in Wasser oder TE (10 mM Tris-HCl, pH 7,9, 1 mM EDTA).
- Erwerben DNA-Fragmente, die nicht ohne weiteres als sDNA Fragmente vorhanden sind.
- Verdünne alle DNA-Fragmente, einschließlich der sDNA Fragmente, die als Vorlagen für die PCRs bis 1 ng / & mgr; l in Wasser verwendet wird.
- Montieren Sie die PCR-Reaktionen bei Raumtemperatur. Kurz gesagt, verwenden 2,5 ul (10 & mgr; M Stammlösung) der jedem jeweiligen Primer des Primerpaares, 1 & mgr; l (von 1 ng / & mgr; l Vorratslösung) des Matrizen-DNA-Fragment, 25 μl Warmstart Korrekturlesen-DNA-Polymerase (DNA pol; 2x Mastermix, siehe Tabelle der Materialien) und 19 & mgr; l Wasser. Mischen Sie den Schlauch durch leichtes Schnippen und sammeln Sie die Flüssigkeitströpfchen durch kurze Zentrifugation.
- Gleichzeitig verstärken in getrennten Röhren DNA-Fragmente von ähnlicher Größe entsprechend den Empfehlungen für die DNA-pol. Führen 25-28 PCR-Zyklen oder bestimmen die Anzahl der Zyklen, die eine ausreichende DNA-Ausbeute zu produzieren.
- Laufen 10% des PCR-Reaktionsvolumen (5 ul) auf einem Standard-Agarosegel-Elektrophorese und Ethidiumbromid (0,2 ug / ml Endkonzentration) gefärbt. Stellen Sie sicher, dass eine einzelne DNA-Bande, die den PCR-Produkt zu sehen ist. Bestimmung der Größe und der relativen Menge der DNA-Fragmente unter Verwendung von DNA-Molekulargewichtsstandards. Wiederholen Sie gegebenenfalls die PCR, um eine ausreichende Menge an DNA-Fragmente zu erhalten.
3. DpnI Verdau der PCR-Produkte
HINWEIS: Das Ziel dieses Schrittes ist es DIGEst Restplasmid Templat-DNA aus den PCRs in Abschnitt 2. DpnI wird Plasmid-DNA zu spalten, wenn es methyliert, wie er bei den in dam + Bakterienstämme gezüchtet Plasmid-DNA. Daher nicht mit DpnI behandelt, wenn die Plasmid-DNA wird als DNA-Fragment in der GA-Reaktion verwendet werden.
- Hinzufügen 2 ul Restriktionsenzym DpnI an der 45 ul der in Abschnitt 2. Inkubieren hergestellt PCR-Produkte bei 37 ° C für 1 Stunde. Fahren Sie mit Abschnitt 4 oder Einfrieren bei -20 ° C, bis sie benötigt.
4. Reinigung und Konzentration der DNA-Fragmente in DNA Purification Magnetic Beads
ANMERKUNG: Das Ziel dieses Schrittes ist es, zu reinigen und zu konzentrieren, die PCR-Produkte in den Abschnitten 2 und 3. Andere PCR-Reinigungsverfahren erhalten werden, können ebenso verwendet werden.
- Äquilibrieren die DNA-Aufreinigung magnetischen Kügelchen auf Raumtemperatur. Resuspendieren die Perlen durch kurzes Vortexen.
- Übertragen Sie die PCRs vorverdaut with DpnI auf ein 1,5-ml-Röhrchen und fügen Sie 81 ul der DNA-Aufreinigung magnetischen Kügelchen in jedes Röhrchen. Inkubieren des Gemisches bei Raumtemperatur für 10 min.
- Die Röhrchen auf dem Magnetsammler für etwa 2 min. Entsorgen Sie die klare Flüssigkeit mit einer Pipette. Zweimaliges Waschen mit 200 ul 80% Ethanol für 30 sec.
- Ermöglichen, dass die Pellets zu trocknen. Halten den Deckel der Rohre offen ist, wobei die Rohre auf dem Magnetsammler positioniert ist.
- Re-suspendieren die getrockneten Perlen in 10 ul 10 mM Tris-HCl, pH 8,0. Inkubiere bei Raumtemperatur für 2 min. Spin kurz, um die Flüssigkeit am Boden der Röhrchen zu sammeln.
- Positionieren Sie die Rohre auf dem Magnetsammler für 2 min. Entfernen von 8,5 bis 10 & mgr; l der klaren Lösung und legen Sie sie in eine neue Pre-markierten Röhrchen. Man bestimmt die Konzentration der DNA-Fragmente, die durch UV-Spektroskopie (siehe Tabelle Equipment).
5. Montage Klonen Reaktion und Transformation der Produkte in E. coli
Hinweis: Das Ziel dieses Schrittes ist die Berechnung 3: 1-Verhältnis von Insert: Vektor und führen Sie die Montage Reaktion.
- Verwenden mindestens 100 ng DNA-Fragment, das das Vektorgerüst und DNA Fragment, das die selektiven Marker. Berechnen der 3-fachen molaren Überschuß für die DNA-Fragmente, die als Einlage verwendet wird.
- Konvertieren Sie die Mol-Überschuß in ng benötigt jedes einzelnen Einsatz.
HINWEIS: Ein bequemer Weg, um die Berechnungen zu tun ist, um eine Web-basierte Anwendung verwenden (siehe Tabelle der Ausrüstung). - Mischen Sie berechneten Mengen von DNA-Fragmenten in einem PCR-Röhrchen, die Lautstärke auf 10 ul. Fügen Sie 10 ul der GA-Mastermix (2x siehe Tabelle der Materialien). Inkubieren der Reaktion bei 50 ° C für 1 Stunde zur Montage von 4 - 6 Fragmente oder 15 min für die Montage von 2-3 Fragmente in einem PCR-Thermocycler.
- Fahren Sie mit der Transformation der Montage Produkt in kompetente E. coli oder Einfrieren der Produkte bei -20° C, bis es gebraucht wird.
- Folgen Sie dem Transformationsverfahren, die die chemische oder elektrokompetenten Zellen begleitet. Normalerweise benutzen 2 ul der Versammlung Reaktion pro Transformationsansatz.
- Nachdem die Umwandlung abgeschlossen ist, verteilt die transformierten Zellen auf Agarplatten mit den entsprechenden selektiven Antibiotikum ergänzt.
6. Plasmidisolierung, Restriktionsenzymverdau und Sequenzierung
ANMERKUNG: Das Ziel dieses Schrittes ist die Plasmid-DNA aus E. Isolieren coli, anschließend, um das Konstrukt durch Restriktionsverdau und Sequenzierung verifizieren.
- Propagieren mehrere Einzelkolonien für Minipreps und Plasmidisolierung. Verwenden Sie 2-5 ml LB Flüssigkultur über Nacht mit einer geeigneten Antibiotika ergänzt. Isolieren der entsprechenden Plasmide nach dem Verfahren, das in dem Mini prep Kits, die verwendet wird, beschrieben wird.
- Bestimmung der Menge und Konzentration der Plasmide mit spektro erhaltenenPhotometer.
- Führen Restriktionsverdau mit einer oder mehreren Restriktionsendonukleasen, um ~ 0,5 & mgr; g der gereinigten Plasmide. Verwenden Restriktionsenzyme, die unterschiedliche Muster der Verdauung charakteristisch für die DNA-Konstrukte bereitzustellen.
- Bestätigen DNA-Klonierung Erfolg durch DNA-Sequenzierung unter Verwendung spezifischer Primer oder Standard. Analysieren Sie die Sequenzdaten für die Richtigkeit.
Ergebnisse
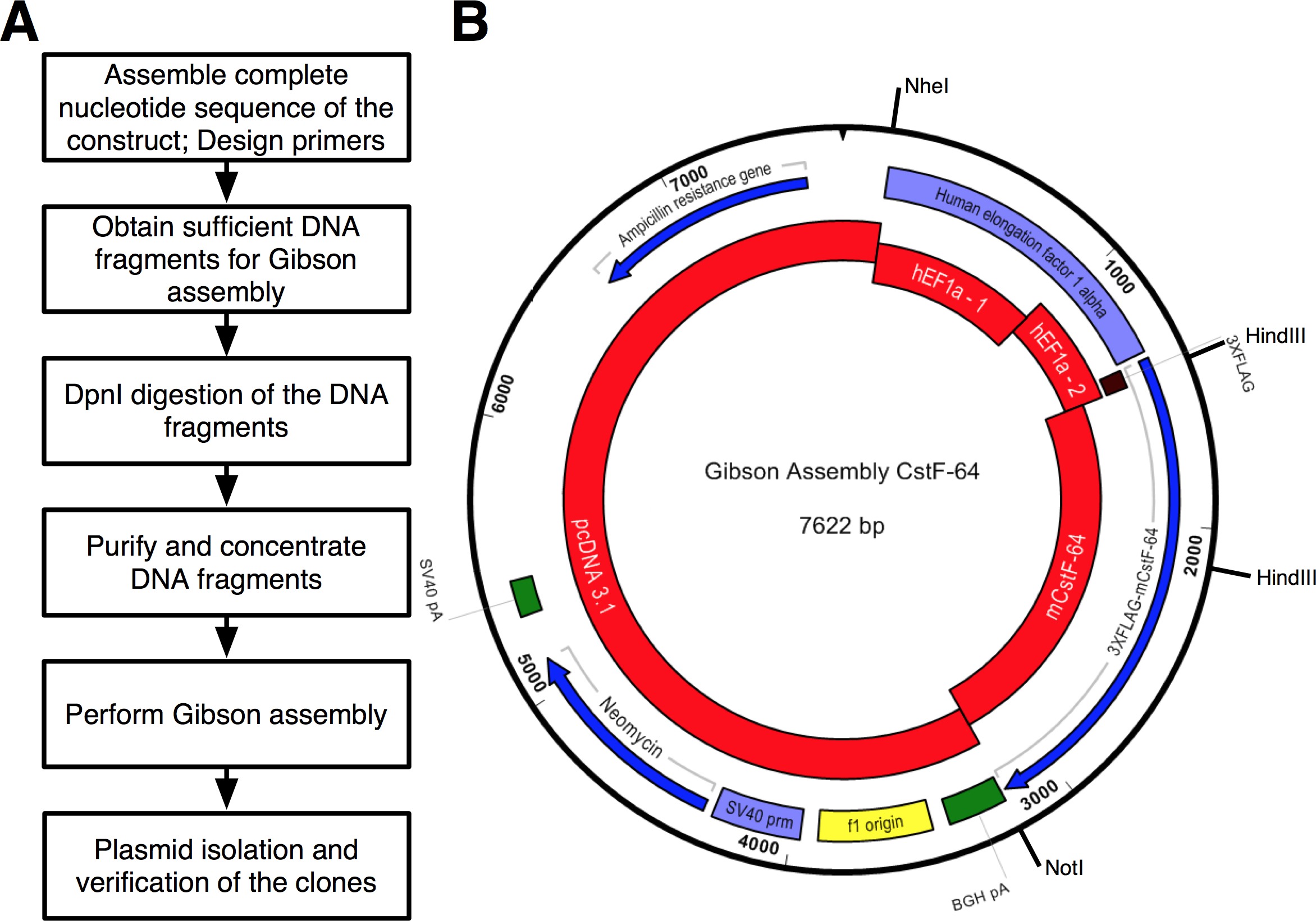
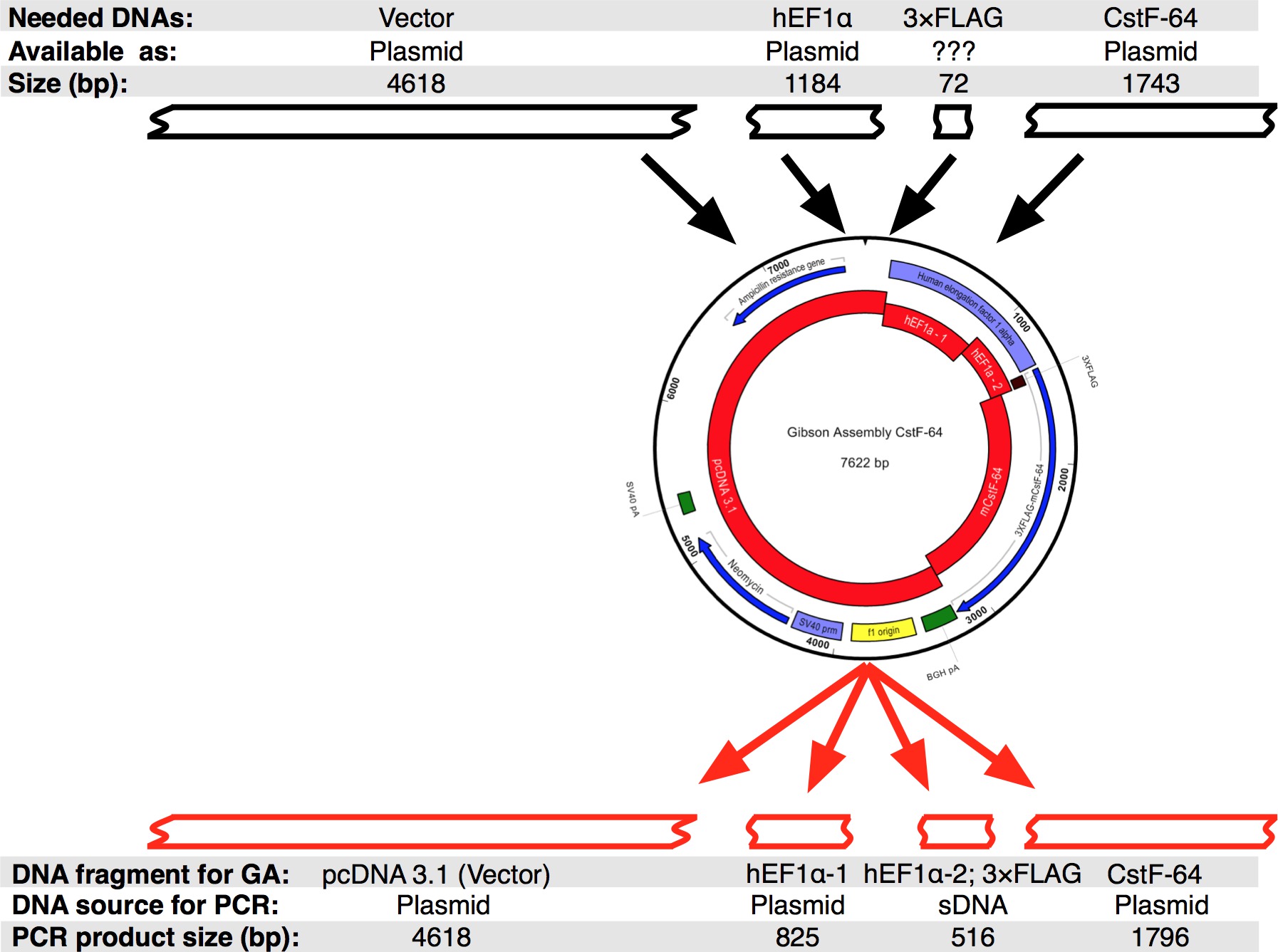
Ein Arbeitsablauf des Protokolls, gefolgt wurde, ist in 2A gezeigt. Wir wollten CstF-64 und mutierten CstF-64 Proteine 3xFLAG-Tag unter dem Ausdrucks Regulierung hEF1α Promotor (2B und 3) fusioniert zu klonen. Ein Plasmid, das hEF1α gefolgt von 3xFLAG-Tag nicht zur Verfügung zu uns. Allerdings waren die folgenden Plasmide verfügbar: pcDNA 3.1 myc-His (A, ein großzügiges Geschenk von Michaela Jansen), hEF1α das Plasmid (ein großzügiges Geschenk von Mladen Yovchev) und Maus CstF-64 Plasmide 12 (Abbildung 3). Die gesamte Sequenz für das Konstrukt (e) wurde mit den Nukleotid-und Textbearbeitungsprogramme (siehe Tabelle der Anlagen Abbildung 3) montiert. Anschließend wurde die Sequenz (en) in vier praktischen Stücke (2B roten Blöcke und Figur 3), die den erhältlichen Plasmid DNAs spalten. Verstärkung primers wurden mit Primer Generierungstool (siehe Tabelle der Anlagen) mit Einschränkungen von 4 entwickelt - 6 Fragmente mit minimaler Überlappung von 25 nt, in der "Change Gibson Assembly-Einstellungen" Popup-Fenster festlegen. NheI und NotI-Restriktionsstellen wurden in die Primer-Design für den Zweck der Identifizierung korrekt montiert Plasmide enthalten. NheI-Stelle in dem Primer-Sequenz zwischen pcDNA 3.1 und 5 'Ende des hEF1α Promotor. NotI-Stelle nach dem Stopp-Codon (UGA) des CstF-64 und pcDNA 3.1 Vektorgerüst entfernt. Bei gleichzeitiger Spaltung mit beide Enzyme DNA-Fragment, bestehend aus hEF1α Promotor 3xFLAG und CstF-64 oder Mutanten CstF-64 wird freigegeben (siehe unten und 2B). Primer wurden auf kleinstem Maßstab bestellt und entsalzt. Der zweite Teil des hEF1α Promotor enthaltenden 3xFLAG-tag-DNA-Fragment (490 bp, 2B, 3) als ein einzelnes sDNA-Fragment (s gekauftee Tabelle der Materialien). DNA-Fragmente in der Versammlung Reaktion verwendet wurden mit DNA pol (siehe Tabelle der Materialien) amplifiziert. DNA-Fragmente von hEF1α Promoter Teil 1, hEF1α Promoter Teil 2, in voller Länge und mutierten CstF-64 wurden gleichzeitig in einem separaten Röhrchen für 28 Zyklen (4A) verstärkt wird, im Anschluss an die Empfehlungen des Lieferanten, der das DNA-pol (siehe Tabelle der Materialien für jeden Zyklus Denaturierung dauerte 7 sec bei 98 ° C, Annealing 45 sec bei 55 ° C, Dehnung von 90 sec bei 72 ° C). Anfänglich wurde die pcDNA 3.1 Rückgrat 22 Zyklen (mit den gleichen Bedingungen wie oben mit der Ausnahme der Dehnung Zeit, die 3 Minuten bei 72 ° C eingestellt war) amplifiziert. Jedoch war das resultierende DNA-Ausbeute nicht ausreichend, um in einem Assoziationsreaktion (4A) verwendet werden. Daher wurde eine zusätzliche Amplifizierung durchgeführt, um ausreichende DNA zu erhalten.
PCR-Produkte zu erhalten,ed aus einem Plasmid Vorlage muss mit DpnI Restriktionsenzym verdaut werden, um die Plasmid-DNA, die sonst die sich ergebende Anordnung Reaktionsprodukte kontaminieren und falsch-positiven Drogen-resistenten Bakterienkolonien produzieren zu entfernen. Daher wurden die PCR-Produkte mit dem Restriktionsenzym DpnI, spaltet, methyliert und Hemi-methylierte Plasmid-DNA aus dam + E. getrennt verdaut coli-Stämmen. Erhaltenen PCR-Produkte unter Verwendung von synthetischen DNA-Fragmente als Matrizen müssen nicht mit DpnI da chemisch synthetisierte DNA verdaut enthält nicht methyliert oder hemi-methylierte Basen.
DNA-Fragmente wurden gereinigt und über DNA-Aufreinigung magnetischen Kügelchen konzentriert (siehe Tabelle der Materialien), wie im Protokoll Schritt 4. Die PCRs für den pcDNA 3.1 Vektor-Rückgrat wurden vereinigt beschrieben, und die Menge der DNA-Reinigung magnetische Kügelchen verwendet wurde entsprechend angepasst. Die DNA-Ausbeute bestimmtVerwendung eines Spektrophotometers (Tabelle 1 und Tabelle Equipment). Aufbaureaktionen zum CstF-64 und mutante CstF-64-Konstrukte wurden auf Eis (Tabelle 1) zusammengesetzt. A 3-fachen molaren Überschuß von den DNA-Fragmenten als "Einsätze" betrachtet wurde (Tabelle 1, Abbildung 3). Das endgültige Volumen der gemischten DNA-Fragmente wurden zu 10 & mgr; l mit Wasser und 10 ul Anordnung Mastermix (2x) wurde zugegeben. Die Reaktionen wurden gemischt und bei 50 ° C für 1 h inkubiert. Positive Kontrollreaktion wurde nach der Empfehlung des Kit manuelle GA montiert und gleichzeitig mit den CstF-64 und mutierten CstF-64 Reaktionen inkubiert. Wie im Protokoll empfohlen wurden 2 ul der jeder der Montage Reaktionen in chemisch kompetente E. transformierten coli geliefert mit der Montage Klonierungskit (siehe Tabelle der Materialien). Die Transformation wurde durchgeführt wie in dem Kit beschriebenHandbuch. Positive Klone wurden auf Ampicillin-Agar / LB-Platten selektiert. 6 Kolonien pro jeder Baugruppe Reaktion wurden zufällig ausgewählt, um vermehrt werden. Plasmid-DNAs wurden unter Verwendung eines Plasmid-Isolation Mini Kit (siehe Tabelle der Materialien) isoliert. In silico Verdauung der Konstrukte mit den Restriktionsenzymen NheI und NotI ergab zwei Fragmente mit Größen von 4590 bp, 3032 bp für CstF-64 und 4.590 bp, 2.711 bp auf mutante CstF-64 (2B und 4B). Verdauung mit den Restriktionsenzymen HindIII und NotI ergab drei Fragmente mit den Größen: 5.872 bp, 1005 bp und 745 bp (CstF-64) und 5.872 bp, 1005 bp und 424 bp (Mutante CstF-64, Figur 2B und 4C). Tatsächlich Verdau der isolierten Plasmide zeigten die erwarteten charakteristischen Mustern (4B, C). Man beachte, dass die durch Spaltung mit HindIII und NotI des CstF-64-Mutante produziert 424 bp DNA-Fragment,Plasmide auf 4C schwach aufgrund seiner geringen Größe gefärbt. 2 der 6 isolierten Plasmide wurden für die Sequenzierung gesandt. Wir sequenzierten das hEF1α Promotor und CstF-64 oder eine Mutante CstF-64 Teile des Konstrukts, um zu überprüfen, dass es keine Deletionen, Insertionen oder Substitutionen. Wir empfehlen die Sequenzierung der DNA-Konstrukte hieraus resultierenden oder einer PCR-basiertes Protokoll. Die Sequenzierung zeigte, dass eines von jedem Plasmid sequenziert die erwartete Sequenz im Bereich von hEF1α, 3xFLAG-tag und CstF-64 oder eine Mutante CstF-64 enthielt. Jede der anderen Plasmide hatten eine Punktmutation in der Verstärkung des entsprechenden DNA-Fragment eingeführt wird. Expression des Plasmids, welches CstF-64 in embryonale Stammzellen der Maus, hergestellt reichliche Menge an exogenem Protein vergleichbar mit Wildtyp-Expressions 17.

Abbildung 1. Schematische Darstellung der Gibson Assembly-Mechanismus. DNA-Fragmente mit überlappenden Enden wurden isotherm in einem einzigen kontinuierlichen Abfolge zusammengesetzt. Die überlappenden Enden zuerst zurück durch 5'-Exonuclease, die allmählich durch Hitze inaktiviert gekaut. Folglich werden verschiedene DNA-Fragmente mit überlappenden Enden annealen isotherm. DNA-Polymerase wird in die Lücken und thermostabilen DNA-Ligase Ligaten der Kerben zu füllen. Bitte klicken Sie hier, um eine größere Version dieses Bild anzuzeigen.
{kind=link}

Abbildung 2. (A) Flussdiagramm des Protokolls beschriebenen Montage Klonen. (B) Darstellung der Plasmid pGA-CstF-64 erzeugt unter Verwendung von GA-Kit Red - DNA-Fragmente in der Bindungsreaktion verwendet. PcDNA3.1; hEF1α Promoter Teil 1 (hEF1a - 1); hEF1α Promoter Teil 2 (hEF1a - 2) bestellt als synthetische DNA; Maus CstF-64 (mCstF-64). Blue - offene Leserahmen. Violet - virale und nicht-virale Promotoren. Grün -. Spaltung und Polyadenylierung Regionen Bitte klicken Sie hier, um eine größere Version dieses Bild anzuzeigen.
{kind=link}

Abbildung 3: Aufbau des Gibson Montage CstF-64-Plasmid. Schwarz Kästchen stellen die DNA-Fragmente, die für einen einzelnen Gibson Montage CstF-64 in silico Sequenz entwerfen waren. Anschließend wurde die Sequenz in vier DNA-Stücken, die durch PCR amplifiziert wurden aufgeteilt. Beachten Sie, dass aufgrund der geringen Größe des 3xFLAG-Tag die Sequenz als sDNA zusammen mit dem hEF1α pr konzipiert omoter Teil 2. Bitte klicken Sie hier, um eine größere Version dieses Bild anzuzeigen.
{kind=link}

Abbildung 4. PCR der DNA-Fragmente in den Klonen Reaktionen und Vertreter Restriktionsenzymverdau der erhaltenen Plasmide (A) Vertreter PCRs mit heißem starten High-Fidelity-2x Mastermix für die DNA-Fragmente in der Montage Reaktionen eingesetzt:. (B) Vertreter Plasmide hEF1α, in voller Länge CstF-64, pcDNA 3.1-Konstrukt (PGA-CstF-64) und hEF1α, mutierte CstF-64, pcDNA 3.1-Konstrukt (PGA-mutCstF-64) mit NheI und NotI verdaut. (C) die gleichen Plasmide wie in B, gespalten mit HindIII und NotI Enzyme."_blank"> Bitte klicken Sie hier, um eine größere Version dieses Bild anzuzeigen.
| Namen von DNA-Fragmenten | Erwartete Größe (bp) | Konz. (Ng / ul) | Verdünnt (ng / ul) | ul in GA CstF-64 verwendet, von verwässerte | ul verwendet in GA mutCstF-64, aus verdünnten | Molverhältnis (Ins: vec) |
| pcDNA 3.1 (Vektor) | 4618 | 158 | unverdünnt | 1 | 1 | |
| hEF1_ Promoter Teil 1 | 825 | 213 | 75 | 1 | 1 | 3: 1 |
| hEF1_ Promoter Teil 2 für CstF-64 | 516 | 229 | 50 | 1 | 3: 1 | |
| CstF-64 | 1796 | 161 | unverdünnt | 1 | 3: 1 | |
| hEF1_ Promoter Teil 2 für die Mutante CstF-64 | 516 | 199 | 50 | 1 | 3: 1 | |
| Mutante CstF-64 | 1448 | 201 | 171 | 1 | 3: 1 |
Tabelle 1. Ausbeute von DNA-Fragmenten nach der Konzentration auf magnetische Kügelchen, Verdünnung und aus der Montage Reaktionen einstellen.
Diskussion
Der erfolgreiche Einsatz der GA Klonierung stets durch eine sorgfältige Konstruktion der gesamten Konstrukt (Abbildung 2 und Abbildung 3) vorausgehen.
Eine sorgfältige Überprüfung der Primersequenzen durch den Primer Werkzeuggeneration konzipiert ist auch sehr zu empfehlen. Primer für GA kann ohne die Verwendung des Primers Werkzeuggeneration erzeugt werden. Jedoch ist die Verwendung des Werkzeugs sehr empfehlen, da es den Prozess vereinfacht. Im Allgemeinen muss die Grundierung für GA Klonen zwei funktionell verschiedene Sequenzen. Die erste Sequenz-DNA-Fragment-spezifisch, und erlaubt die Amplifikation des Fragments unter Verwendung von PCR. Die zweite Sequenz überlappt mit der angrenzenden Fragment, das für GA Montage notwendig ist. Eine typische DNA-Fragment-spezifische Sequenz würde 18-22 nt lang. DNA-Fragment-spezifischen Sequenzen verwendet werden, um die gleiche DNA amplifizieren müssen ähnliche Schmelztemperaturen und dem GC-Gehalt. Überlappende Sequenz sollte mindestens 15nt in der Länge mit einer Schmelztemperatur von mindestens 48 ° C. Die Anordnung von mehr als 4 DNA-Fragmente wird die überlappende Sequenz erfordern mindestens 20 nt betragen. Mehr Überschneidungen wird eine erhöhte Spezifität des Glühen was zu mehr richtig zusammengebaut DNA-Fragmente zu ermöglichen. Es wird empfohlen, um Sequenzen, die in ihrer GC- oder AT-Gehalt in den Entwicklungs überlappende Sequenzen verzerrt werden, da schräg Sequenzen möglicherweise richtige DNA Anordnung beeinträchtigen vermeiden.
Wir empfehlen die Verwendung als negative Kontrolle, die keine Arzneimittel-resistente Bakterienkolonien, nur die DNA-Fragment, das dem Vektor-Rückgrat in einem GA Reaktion nicht produzieren sollten. Alternativ kann eines der DNA-Fragmente, umfassend die "Einsätze" kann aus der GA-Reaktion, die auch in nicht Arzneimittel-resistente Bakterienkolonien führen soll weggelassen werden. Der Grund, keine Kolonien wachsen wird das Fehlen von überlappenden Enden der benachbarten DNA-Fragmente sein, welche den Zusammenbau eines kompl rendertete Plasmid.
Beschrieben in der Protokoll überlappende Sequenzen von 25 nt zur Erzeugung der Primersätze verwendet wurden, da die Anzahl der DNA-Fragmente verwendet (Abbildung 3). Die Empfehlung des Werkzeugs Website Primer Generation (siehe Tabelle der Ausrüstung) ist es, mindestens 20 nt überlappende Sequenzen zu verwenden, um 4 zu montieren - 6 DNA-Fragmente. Außerdem mehr überlappende Sequenz die richtige Komplementation der DNA-Stränge zu gewährleisten (siehe Abbildung 1) Erhöhung der Anzahl der genau zusammengesetzten Produkte.
Derzeit mehrere Systeme für die nahtlose Klonen zur Verfügung. Jedoch verwenden einige dieser Systeme noch Restriktionsenzymen (dh Golden Gate Klonen 18). Andere verwenden eigene Mischungen der Enzyme auf Vaccinia-Virus-DNA-Polymerase und DNA-Einzelstrangbindeprotein aus derselben biologischen Quelle 19. Beide Systeme sind im Vergleich zu GA vom Shor begrenztter Länge der überlappenden Sequenzen. Weil kürzere überlappende Sequenzen stellt möglicherweise ausreichende Spezifität zu dem Glühschritt benachbarter DNA-Fragmente, wodurch die korrekte Montage von mehr als 23-DNA-Fragmente problematisch. Diese Mängel sind nicht in der GA-System vorhanden ist.
Die Grße der DNA-Fragmente, die amplifiziert werden soll auch berücksichtigt, um nicht die Größe von PCR zuverlässig amplifizierbare (dh weniger als 8 kbp Länge) überschritten wird. Selbst mit den Verbesserungen in der Funktion der DNA-Polymerasen in den letzten 10 Jahren werden große DNA-Fragmente mit weniger Effizienz und Genauigkeit verstärkt werden. Falls erforderlich, könnten größere DNA-Fragmente aus anderen Quellen Alternative zur PCR, beispielsweise erhalten werden, indem Plasmid-Isolierung und geeignete DNA-Verdauung mit Restriktionsenzymen. Insbesondere für die im aktuellen Manuskript beschriebenen Protokoll unserer rational PCR verwendet wurde auf die Größe der verfügbaren DNA-Fragmente, die alle kleiner sind basierend5 kbp. Wenn es mehr als ein PCR-Produkt durch Agarosegelelektrophorese identifiziert wird Gelreinigung des gewünschten Fragmentgröße unter Verwendung von einer der verfügbaren Techniken der Molekularbiologie oder entsprechende Kits empfohlen. In der aktuellen Protokoll eine thermostabile DNA-Polymerase verwendet (siehe Tabelle der Materialien). Jedoch kann jede DNA-Polymerase, hohe Genauigkeit und Ausbeute wird geeignet mit diesem Protokoll verwendet werden kann. Wenn High-Fidelity DNA-Polymerasen, anders als im Protokoll beschrieben sind, verwenden Sie die Einrichtung Bedingungen wie in den entsprechenden Handbüchern beschrieben. In der aktuellen Protokoll, chemisch kompetente E. coli-Zellen verwendet werden, die mit dem GA-Kit geliefert werden. Alternativ chemisch oder elektro kompetente E. coli-Stämme, wie DH5a oder DH10B verwendet werden.
Die Montage Klonen 2x Mastermix ist einfach, mit minimalen hands-on Zeit verwenden. Jedoch ist eine genaue Pipettieren wegen der kleinen Volumina ne erforderlicheEDED miteinander vermischt werden. Geeignet Molekularbiologie Technik benötigt, um jederzeit als auch ausgeübt werden.
Der GA Klonen bietet unbegrenzte Möglichkeiten für eine Konstruktion von DNA-Fragmenten, Plasmide und Vektoren, die länger als 3 kbp groß sind. Zusätzlich hat es eine breitere Wirkung auf dem Gebiet der synthetischen Biologie, weil es die Synthese und Anordnung von zum Beispiel eine ganze Bakterien (Mycoplasma mycoides) Genom oder eine Hefe (Saccharomyces cerevisiae) Chromosom 20,21. Die Technik ist auch anwendbar auf konventionelle Klonen muss nahtlose Konstrukte zu erzeugen.
Abschließend bietet GA Klonierung schnelle, zuverlässige und flexible Alternative zu herkömmlichen DNA Klonierungsverfahren.
Offenlegungen
Open Access publication and production fees were supplied by New England BioLabs Inc.
Danksagungen
Wir möchten Michaela Jansen an der Texas Tech University Health Sciences Center, Lubbock, TX und Mladen Yovchev an der University of Pittsburgh Medical Center, Pittsburgh für die großzügige Bereitstellung pcDNA 3.1 myc-His (A) und hEF1α Promoter die Plasmide danken, PA . Forschung in dieser Veröffentlichung berichtet, wurde von der Eunice Kennedy Shriver National Institute of Child Health und Human Development der National Institutes of Health unter Verleihungsnummer R01HD037109 (CCM) unterstützt. Zusätzliche Unterstützung war von Laura W. Bush Institut für Frauengesundheit (CCM und PNG). Der Inhalt ist allein in der Verantwortung der Autoren und nicht unbedingt die offizielle Meinung der National Institutes of Health.
Die Autoren erklären, dass sie keine konkurrierenden finanziellen Interessen.
Materialien
| Name | Company | Catalog Number | Comments |
| Synthetic double-stranded DNA fragment (sDNA) | Integrated DNA Technologies | We used gBlocks, which can be up to 2 kb in length. However, there are many commercial sources of synthetic DNA available. | |
| DNA purification magnetic beads | Beckman Coulter | A63880 | Agencourt AMPure XP - PCR Purification system |
| Plasmid Isolation Mini Kit | Omega Bio-Tek Inc | D6942-01 | E.Z.N.A. Plasmid Mini Kit I |
| Gibson Assembly Cloning Kit | New England BioLabs | E5510S | |
| DNA polymerase | New England BioLabs | M0494S | Q5 Hot Start High Fidelity 2x Master Mix |
| DpnI | New England BioLabs | R0176S | |
| NheI | New England BioLabs | R3131S | |
| NotI | New England BioLabs | R3189S | |
| HindIII | New England BioLabs | R3104S | |
| Spectrophotometer | Thermo Scientific | NanoDrop device | |
| EditSeq | DNASTAR | Part of Lasergene Core Suite | |
| SeqBuilder | DNASTAR | Part of Lasergene Core Suite | |
| Word | Microsoft | Part of Microsoft Office | |
| NEBuilder | New England BioLabs | primer generation tool: http://nebuilder.neb.com | |
| NEBioCalculator | New England BioLabs | ligation calculator: http://nebiocalculator.neb.com/#!/ligation |
Referenzen
- Gibson, D. G. Enzymatic assembly of overlapping DNA fragments. Methods in enzymology. 498, 349-361 (2011).
- Gibson, D. G., Smith, H. O., Hutchison, C. A., Venter, J. C., Merryman, C. Chemical synthesis of the mouse mitochondrial genome. Nature methods. 7, 901-903 (2010).
- Gibson, D. G., et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nature methods. 6, 343-345 (2009).
- Bowtell, D. D., Johnson, G. R., Kelso, A., Cory, S. Expression of genes transferred to haemopoietic stem cells by recombinant retroviruses. Molecular biology & medicine. 4, 229-250 (1987).
- Challita, P. M., Kohn, D. B. Lack of expression from a retroviral vector after transduction of murine hematopoietic stem cells is associated with methylation in vivo. Proceedings of the National Academy of Sciences of the United States of America. 91, 2567-2571 (1994).
- Lutzko, C., Senadheera, D., Skelton, D., Petersen, D., Kohn, D. B. Lentivirus vectors incorporating the immunoglobulin heavy chain enhancer and matrix attachment regions provide position-independent expression in B lymphocytes. Journal of Virology. 77, 7341-7351 (2003).
- Meilinger, D., et al. Np95 interacts with de novo DNA methyltransferases, Dnmt3a and Dnmt3b, and mediates epigenetic silencing of the viral CMV promoter in embryonic stem cells. EMBO reports. 10, 1259-1264 (2009).
- Chan, K. K., Wu, S. M., Nissom, P. M., Oh, S. K., Choo, A. B. Generation of high-level stable transgene expressing human embryonic stem cell lines using Chinese hamster elongation factor-1 alpha promoter system. Stem cells and development. 17, 825-836 (2008).
- Chung, S., et al. Analysis of different promoter systems for efficient transgene expression in mouse embryonic stem cell lines. Stem cells. 20, 139-145 (2002).
- Qin, J. Y., et al. Systematic comparison of constitutive promoters and the doxycycline-inducible promoter. PloS one. 5, e10611 (2010).
- Lund, A., Knudsen, S. M., Vissing, H., Clark, B., Tommerup, N. Assignment of human elongation factor 1alpha genes: EEF1A maps to chromosome 6q14 and EEF1A2 to 20q13.3. Genomics. 36, 359-361 (1996).
- Wallace, A. M., et al. Two distinct forms of the 64,000 Mr protein of the cleavage stimulation factor are expressed in mouse male germ cells. Proceedings of the National Academy of Sciences of the United States of America. 96, 6763-6768 (1999).
- MacDonald, C. C., McMahon, K. W. Tissue-specific mechanisms of alternative polyadenylation: testis, brain, and beyond. Wiley interdisciplinary reviews. RNA. 1, 494-501 (2010).
- Sabath, I., et al. 3'-End processing of histone pre-mRNAs in Drosophila: U7 snRNP is associated with FLASH and polyadenylation factors. Rna. 19, 1726-1744 (2013).
- Yang, X. C., et al. A complex containing the CPSF73 endonuclease and other polyadenylation factors associates with U7 snRNP and is recruited to histone pre-mRNA for 3'-end processing. Molecular and cellular biology. 33, 28-37 (2013).
- Grozdanov, P. N., Macdonald, C. C. High-Throughput Sequencing of RNA Isolated by Cross-Linking and Immunoprecipitation (HITS-CLIP) to Determine Sites of Binding of CstF-64 on Nascent RNAs. Methods in molecular biology. 1125, 187-208 (2014).
- Youngblood, B. A., Grozdanov, P. N., MacDonald, C. C. CstF-64 supports pluripotency and regulates cell cycle progression in embryonic stem cells through histone 3' end processing. Nucleic acids research. , (2014).
- Engler, C., Kandzia, R., Marillonnet, S. A one pot, one step, precision cloning method with high throughput capability. PloS one. 3, e3647 (2008).
- Irwin, C. R., Farmer, A., Willer, D. O., Evans, D. H. In-fusion(R) cloning with vaccinia virus DNA polymerase. Methods in molecular biology. 890, 23-35 (2012).
- Annaluru, N., et al. Total synthesis of a functional designer eukaryotic chromosome. Science. 344, 55-58 (2014).
- Gibson, D. G., et al. Creation of a bacterial cell controlled by a chemically synthesized genome. Science. 329, 52-56 (2010).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten