
Method Article
Generazione di plasmidi vettori che esprimono proteine FLAG-tagged Ai sensi del regolamento di Human Factor-allungamento 1α Promotore Uso Gibson Assembly
In questo articolo
Riepilogo
Synthesis of custom plasmids is labor and time consuming. This protocol describes the use of Gibson assembly cloning to reduce the work and duration of custom DNA cloning procedure. The protocol described also produces reliable tagged protein constructs for mammalian expression at similar cost to the traditional cut-and-paste DNA cloning.
Abstract
Assemblaggio Gibson (GA) clonazione offre un rapido, affidabile e flessibile alternativa ai tradizionali metodi di clonazione del DNA. Abbiamo usato GA per creare plasmidi personalizzati per l'espressione di geni esogeni nelle cellule staminali embrionali di topo (mESCs). L'espressione di geni esogeni sotto il controllo del SV40 o promotori citomegalovirus umano diminuisce rapidamente dopo trasfezione in mESCs. Un rimedio per questa espressione diminuita è utilizzare il promotore umano allungamento factor-1 alfa (hEF1α) per guidare l'espressione genica. Vettori plasmide contenente hEF1α non sono così ampiamente disponibili come SV40- o plasmidi-CMV contenente, in particolare quelli che contengono anche N-terminale 3xFLAG-tags. Il protocollo qui descritto è un metodo rapido per creare plasmidi esprimenti FLAG-tagged CSTF-64 e CSTF-64 mutante ai sensi del regolamento espressivo del promotore hEF1α. GA utilizza una miscela di esonucleasi DNA, DNA polimerasi e DNA ligasi per fare la clonazione di fini di frammenti di DNA possibili sovrapposizioni.Sulla base dei modelli di DNA che avevamo a disposizione, abbiamo progettato i nostri costrutti da assemblare in una singola sequenza. Il nostro design utilizzato quattro frammenti di DNA: pcDNA 3.1 vettore backbone, hEF1α promoter parte 1, parte 2 hEF1α promotore (che conteneva 3xFLAG-tag acquistato come un frammento a doppia elica del DNA sintetico), e sia CSTF-64 o specifici CSTF-64 mutante. Le sequenze di questi frammenti sono stati caricati su un fondo strumento di generazione di progettare adeguati primer PCR per generare i frammenti di DNA. Dopo la PCR, frammenti di DNA sono stati mescolati con il vettore contenente il marcatore selettivo e la reazione clonazione GA è stato assemblato. Plasmidi da singole colonie batteriche trasformate sono stati isolati. Schermata iniziale dei plasmidi è stato fatto per restrizione digestione, seguito dal sequenziamento. In conclusione, GA ha permesso di creare plasmidi personalizzate per l'espressione genica in 5 giorni, tra cui la costruzione di schermi e di verifica.
Introduzione
Convenzionali procedure di clonazione DNA basano sull'uso di enzimi di restrizione di fendere il DNA e DNA ligasi per unire i frammenti di DNA insieme. Generazione di costrutti di espressione personalizzate contenenti diversi frammenti di DNA è una procedura sequenziale che include scissione del DNA con uno e / o più enzimi di restrizione e il successivo inserimento di frammenti di DNA mediante legatura. Il principale svantaggio di questa procedura è che opportuni enzimi di restrizione per uno dei frammenti di DNA possono essere difficili da identificare (cioè, potrebbe avere diversi siti di taglio) rendendo successo clonazione DNA della proteina intera lunghezza di interesse impossibile. Pertanto, la generazione di espressione personalizzata costruisce sotto la regolazione trascrizionale di cellule efficienti promotori specifici del tipo di proteina-tag personalizzati richiede molto attenta progettazione. E 'anche una tecnica di tempo e di manodopera consumano. Recentemente, diverse relazioni descritte le metodologie per assemblare multipLe differenti frammenti di DNA sintetico in una sequenza continua allo stesso tempo sia in reazioni una o due fasi, senza l'impiego di enzimi di restrizione 1-3. La reazione clonazione one-step (escludendo tutti i passi di preparazione), dipende dall'uso di una miscela di esonucleasi DNA, DNA polimerasi, DNA ligasi 2,3 e le estremità sovrapposte di frammenti di DNA (Figura 1). Poiché non vi è alcun uso di enzimi di restrizione, frammenti di DNA di qualsiasi dimensione e la composizione di sequenza (escluso sequenze altamente ripetitive) possono essere fusi insieme in una costruzione senza soluzione di continuità. Recentemente, un kit commerciale (montaggio Gibson, GA) per le reazioni clonazione di uno stadio è diventato disponibile. Questo kit permette una rapida ed efficace dei costi di assemblaggio di frammenti di DNA in un unico vettore con i promotori personalizzati e tag proteiche.
I vettori di espressione plasmide ampiamente disponibili utilizzati per esprimere le proteine esogene in modelli di coltura cellulare di mammifero sono spesso sotto il reg trascrizionalelamento del citomegalovirus virale (CMV) o Simian virus 40 (SV40) promotori. Questi promotori virali forniscono robusta espressione transiente delle proteine esogene nella maggior parte dei modelli basati colture cellulari di mammifero. Tuttavia, la generazione di linee cellulari stabilmente che esprimono proteine esogene è spesso senza successo a causa del silenziamento trascrizionale del CMV o promotori SV40 durante il processo di creazione di 4,5. Inoltre, i promotori SV40 e CMV virale non promuovono sufficientemente l'espressione di proteine esogene in cellule della linea linfoide o cellule staminali embrionali 6,7. La soluzione dei limiti dei promotori virali è usare forti promotori non virali costitutivi 8-10. Una forte promoter non virale costitutiva ben caratterizzato di origine umana è il promotore fattore di allungamento 1α (hEF1α) (hEF1α è coinvolto nella catalisi dell'associazione GTP-dipendente-tRNA ai ribosomi 11). Tuttavia,vettori di espressione contenenti il promotore hEF1α non sono ampiamente disponibili come promotore virale contenente plasmidi, specialmente quelli anche contenente 3 × FLAG all'estremità terminale amminico della proteina di interesse.
Il fattore di stimolazione scissione delle proteine 64.000 MW (CSTF-64) è coinvolto nella lavorazione all'estremità 3 'della maggior parte dei mRNA 12,13, tra cui replica-dipendente istoni mRNA 14,15. Tale task force-64 è espressa in tutti i tessuti somatici 12. Il suo motivo riconoscimento RNA si lega alla GU-ricche sequenze di RNA su trascrizioni nascenti a valle del sito di scissione e poliadenilazione 16. Questo legame di CSTF-64 al pre-mRNA promuove efficiente taglio endonucleolitico del trascritto nascente.
Qui, un protocollo è descritto che utilizza l'amplificazione PCR dei frammenti di DNA, un kit di clonazione gruppo Gibson (che comprende cellule batteriche chimicamente competenti) per produrre vettori personalizzati di m 3xFLAG-tagouse CSTF-64 o mutante CSTF-64 per il loro terminale amino sotto l'espressione di hEF1α promoter 1.
Protocollo
1. In Silico Progettazione della plasmidi e Generazione dei primer sovrapposti
NOTA: L'obiettivo di questa fase è quello di assemblare completa sequenza nucleotidica del costrutto e progettare i primer da utilizzare per generare frammenti con estremità sovrapposte per la clonazione GA.
- Progettare una sequenza nucleotidica continua a rappresentare il plasmide finale.
- Ottenere o elencare i plasmidi reali e frammenti di DNA che saranno utilizzati come modelli di PCR.
NOTA: frammenti di DNA che non sono prontamente disponibili - come la diversa combinazione di tag e promotori - possono essere ordinati come un unico o più frammenti sintetici DNA a doppia elica (SDNA). Questi frammenti sono disponibili in commercio a costi relativamente bassi e possono essere fino a 2 kb di lunghezza (vedi Tabella dei Materiali). - Dividere la sequenza nucleotidica continuo del costrutto assemblato al passo 1.1 in frammenti di DNA adatti per PCR. Confirm che i frammenti corrispondono plasmidi disponibili e frammenti SDNA. Evitare DNA frammenti più piccoli di 200 nt.
- Accedere allo strumento fondo generazione (vedi Tabella delle attrezzature). Selezionare il menu "Preferenze". Scegliere impostazioni appropriate facendo clic sulla scheda "PREFS CHANGE" nella finestra pop-up "Modifica impostazioni Gibson montaggio".
NOTA: Primer disegno può essere eseguita senza l'utilizzo dello strumento di primer generazione. Tuttavia, l'utilizzo dello strumento di primer generazione semplifica il processo. - Iniziare a costruire il costrutto selezionando il menu "Build Construct". Inserire i frammenti di DNA di divisione nello strumento di generazione in sequenza primer 5 'a 3'.
NOTA: Il frammento di DNA utilizzato come dorsale plasmide può essere convenientemente diviso in due parti. Nel prodotto finale PCR 'fine di questo primo frammento e 3' del 5 fine dell'ultimo frammento sono legati insieme in una continua DNUn frammento che rappresenta la spina dorsale vettoriale. - Incollare il primo frammento di DNA che rappresentano 5 'del DNA vettoriali in formato FASTA (una rappresentazione testuale della sequenza nucleotidica) nella finestra di pop-up "Enter vettore o Inserisci Fragment". Nome del frammento di DNA. Scegli il modo appropriato per ottenere il frammento di DNA, sia come PCR, RE Digest o sintesi. Fare clic sulla scheda "CONTINUA".
- Se vi è la necessità di aggiungere nucleotidi supplementari / siti di restrizione al bivio del costrutto finale utilizzare gli spazi "FWD o REV fondo distanziatori" fornite nella finestra "Aggiungi un frammento inserto all'assemblea". Aggiungere nucleotidi supplementare / siti di restrizione per uno solo dei frammenti di DNA e non di entrambi i frammenti. Clicca sulla scheda "DONE".
- Ripetere per tutti i frammenti fino a quando il costrutto è completa. Selezionare il menu "View Primer" e rivedere le sequenze di primer.
- Ripetere l'operazione per tutti i costrutti (vettore backbones e inserti), o per i frammenti di DNA che sono diversi.
2. Amplificazione dei frammenti di DNA mediante PCR utilizzando Hot Start Riletture DNA polimerasi (2x Master Mix)
NOTA: L'obiettivo di questa fase è quello di ottenere DNA sufficiente per la reazione assieme utilizzando PCR.
- L'acquisto del primer PCR disegnati nella fase precedente come prodotti dissalate e alla scala più piccola possibile. Li Diluire a 10 micron in acqua o TE (10 mM Tris-HCl, pH 7,9, 1 mM EDTA).
- Acquisto frammenti di DNA che non sono facilmente disponibili, come frammenti SDNA.
- Diluire tutti i frammenti di DNA, tra cui i frammenti SDNA che saranno utilizzati come modelli per la PCR a 1 ng / ml in acqua.
- Montare le reazioni PCR a temperatura ambiente. In breve, utilizzare 2,5 ml (da 10 micron soluzione madre) del ciascuna rispettiva innesco della coppia di primer, 1 ml (da 1 ng / ml soluzione madre) del frammento DNA stampo, 25 μl di polimerasi caldo revisione inizio DNA (pol DNA; 2x master mix, vedi Tabella dei Materiali) e 19 ml di acqua. Mescolare il tubo delicatamente sfogliando e raccogliere le gocce di liquido da una breve centrifugazione.
- Amplifica simultaneamente in tubi separati di DNA frammenti di dimensioni simili in base alle raccomandazioni per la pol DNA. Eseguire 25 - 28 cicli di PCR o di determinare il numero di cicli che producono rendimenti DNA sufficiente.
- Eseguire il 10% del volume di reazione PCR (5 mL) su un gel di agarosio standard di colorato con bromuro di etidio (0,2 mcg / ml concentrazione finale). Assicurarsi che una banda di DNA a singolo che rappresenta il prodotto della PCR è visibile. Determinare la dimensione e la quantità relativa di frammenti di DNA utilizzando DNA standard di peso molecolare. Se necessario, ripetere la PCR per ottenere quantità sufficienti di frammenti di DNA.
3. DpnI digestione dei prodotti di PCR
NOTA: L'obiettivo di questa fase è quello di Digev plasmide residua DNA stampo da PCR nella sezione 2. DpnI digerire il DNA plasmidico solo se è metilato, come avviene per DNA plasmidico coltivato in diga + ceppi batterici. Pertanto, non trattare con DpnI se sarà usato DNA plasmidico come un frammento di DNA nella reazione GA.
- Aggiungere 2 ml di enzima di restrizione DpnI ai 45 ml di prodotti di PCR ottenuti nella sezione 2. Incubare a 37 ° C per 1 ora. Procedere con la sezione 4 o congelare a -20 ° C fino al momento dell'uso.
4. Purificazione e concentrazione dei frammenti di DNA nel corso di purificazione del DNA Beads Magnetic
NOTA: L'obiettivo di questa fase è quello di purificare e concentrare i prodotti di PCR ottenuti nelle sezioni 2 e 3. Altri metodi di purificazione PCR possono essere utilizzati come bene.
- Equilibrare la purificazione del DNA sfere magnetiche a temperatura ambiente. Re-sospensione le microsfere da brevi vortex.
- Trasferire i PCR pre-digeriti with DpnI ad un provette da 1,5 ml e aggiungere 81 ml di purificazione del DNA sfere magnetiche ad ogni provetta. Incubare la miscela a temperatura ambiente per 10 min.
- Posizionare i tubi sul collettore magnetica per circa 2 min. Eliminare il liquido chiaro con una pipetta. Lavare due volte con 200 ml di etanolo all'80% per 30 sec.
- Lasciare i pellet asciugare. Mantenere il coperchio dei tubi aperta, con i tubi posizionati sul collettore magnetica.
- Risospendere le microsfere essiccate in 10 ml di 10 mM Tris-HCl, pH 8,0. Incubare a temperatura ambiente per 2 min. Spin brevemente per raccogliere il liquido sul fondo delle provette.
- Posizionare i tubi del collettore magnetica per 2 min. Rimuovere 8,5-10 ml di soluzione limpida e metterlo in un nuovo tubo pre-etichettati. Determinare la concentrazione di frammenti di DNA mediante spettroscopia UV (vedi Tabella delle attrezzature).
5. Assemblea clonazione di reazione e trasformazione dei prodotti in E. coli
NOTA: L'obiettivo di questa fase è quello di calcolare rapporto 3: 1 dell'inserto: vettoriale e eseguire la reazione di assemblaggio.
- Utilizzare almeno 100 ng del frammento di DNA che rappresentano la spina dorsale vettoriale o frammento di DNA che trasporta il marcatore selettivo. Calcolare l'eccesso molare di 3 volte per i frammenti di DNA che verranno utilizzati come inserti.
- Convertire il molare-eccesso in ng necessaria di ogni particolare inserto.
NOTA: Un modo conveniente per fare i calcoli è quello di utilizzare una applicazione web-based (vedi Tabella delle attrezzature). - Mescolare gli importi calcolati di frammenti di DNA in un tubo di PCR, regolare il volume a 10 ml. Aggiungere 10 ml di master mix GA (2x, vedi Tabella dei Materiali). Incubare la reazione a 50 ° C per 1 ora per il montaggio di 4 - 6 frammenti o 15 min per il montaggio di 2-3 frammenti in un ciclatore termico PCR.
- Procedere con la trasformazione del prodotto in assemblea competente E. coli o congelare i prodotti a -20° C, fino al momento dell'uso.
- Seguire la procedura di trasformazione che accompagna il cellule elettro competenti chimico o. Solitamente, utilizzare 2 ml di reazione assieme a reazione di trasformazione.
- Dopo la trasformazione è completa, diffondere le cellule trasformate su piastre di agar integrate con l'antibiotico selettivo appropriata.
6. Isolamento plasmidi, enzimi di restrizione e Sequencing
NOTA: L'obiettivo di questa fase è quello di isolare DNA plasmidico da E. coli, poi verificare il costrutto di digestione per restrizione e sequenziamento.
- Propagazione diverse colonie singole per mini preparazioni e isolamento plasmide. Pernottamento LB coltura liquida Usa 2-5 ml integrato con antibiotico appropriato. Isolare i plasmidi corrispondenti seguendo la procedura descritta nel mini kit di preparazione che viene utilizzato.
- Determinare la quantità e la concentrazione dei plasmidi ottenuti usando spectrofotometro.
- Eseguire la digestione di restrizione con uno o più enzimi di restrizione a ~ 0,5 ug di plasmidi purificati. Utilizzare enzimi di restrizione che forniscono modello distinto di digestione caratteristico per i costrutti di DNA.
- Confermare DNA clonazione successo dal sequenziamento del DNA utilizzando primer specifici o standard. Analizzare i dati di sequenziamento per la precisione.
Risultati
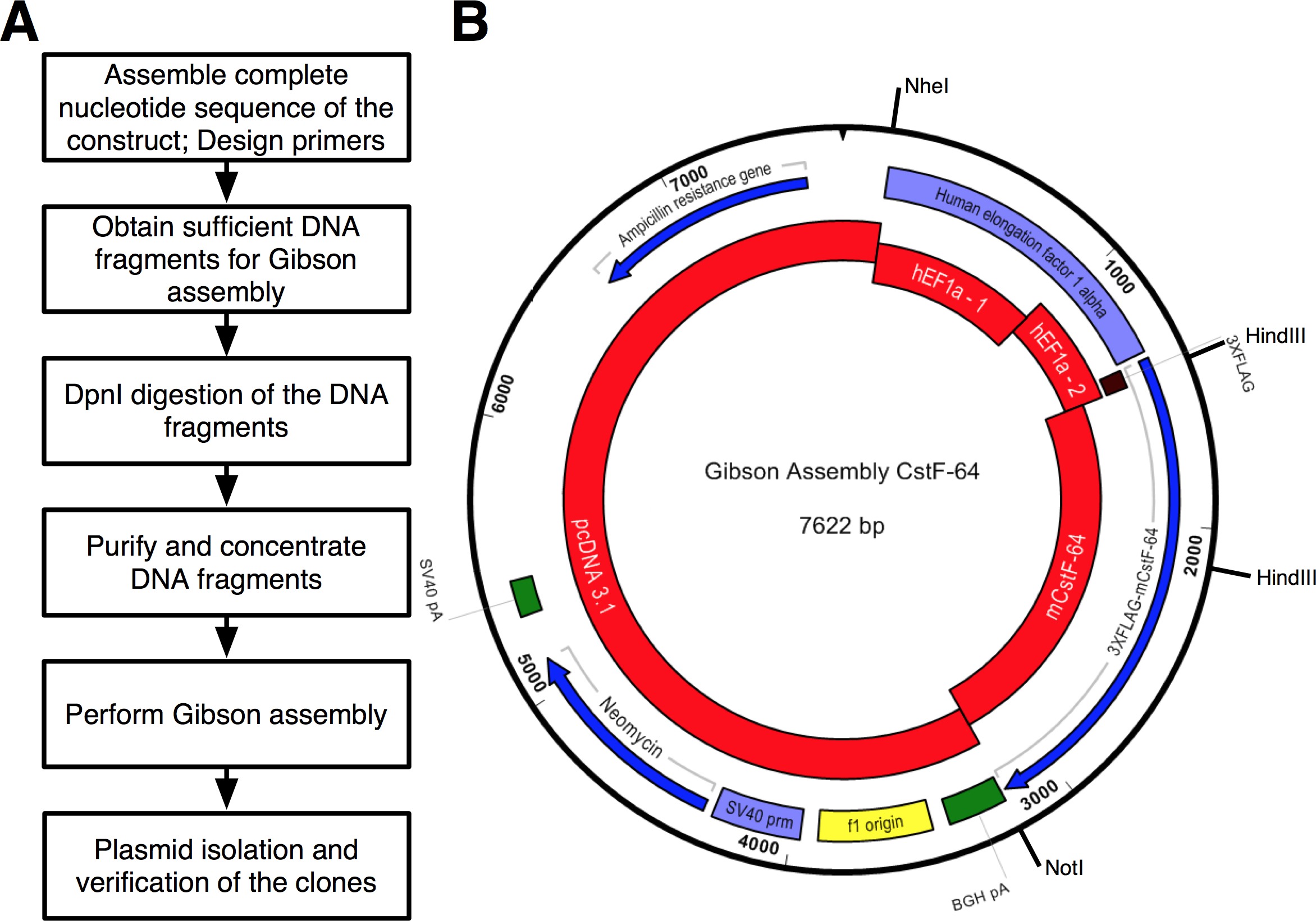
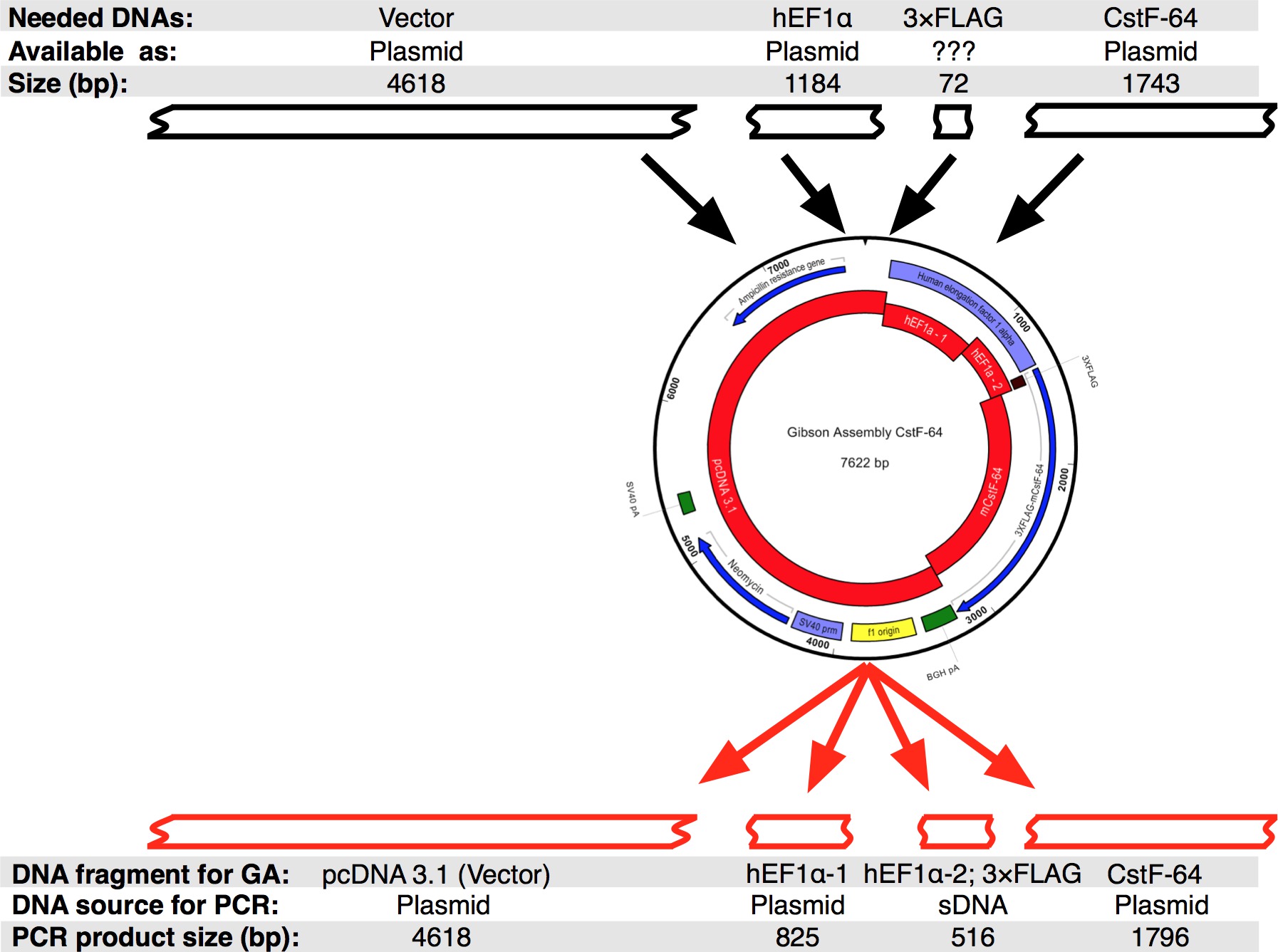
Un flusso di protocollo che è stato seguito è mostrata nella Figura 2A. Abbiamo voluto clonare CSTF-64 e mutanti CSTF-64 proteine fuse a 3xFLAG-tag ai sensi del regolamento espressivo di hEF1α promotore (Figura 2B e Figura 3). Un hEF1α plasmide contenente seguita da 3xFLAG-tag non era disponibile a noi. Tuttavia, i seguenti plasmidi erano disponibili: pcDNA 3.1 myc-His (A; un dono generoso da Michaela Jansen), hEF1α contenente plasmide (un dono generoso da Mladen Yovchev) e mouse CSTF-64 plasmidi 12 (Figura 3). L'intera sequenza per il costrutto (s) è stato assemblato utilizzando le applicazioni nucleotide e il testo di modifica (Figura 3, vedere Tabella delle attrezzature). Successivamente, la sequenza (s) è stato suddiviso in quattro pezzi convenienti (Figura 2B, blocchi rossi e Figura 3) corrispondente al DNA plasmide disponibili. Amplificazione primers sono stati progettati utilizzando lo strumento di generazione di fondo (vedi tabella di macchinari) con vincoli di 4-6 frammenti con sovrapposizione minima di 25 nt, istituito nella finestra pop-up "Modifica impostazioni Gibson montaggio". NheI e NotI siti di restrizione sono stati inclusi nel progetto di fondo al fine di identificare plasmidi correttamente montati. Sito NheI è situata nella sequenza innesco tra pcDNA 3.1 e 5 'del promotore hEF1α. Sito NotI si trova dopo il codone di stop (UGA) di CSTF-64 e pcDNA 3.1 vettore backbone. Dopo la digestione simultanea con entrambi gli enzimi frammento di DNA costituito da hEF1α promoter, 3xFLAG e CSTF-64 o mutante CSTF-64 saranno rilasciati (vedi sotto e Figura 2B). I primer sono stati ordinati in scala più piccola possibile e dissalati. La seconda parte del promotore hEF1α frammento di DNA contenente 3xFLAG-tag (490 bp, Figura 2B, Figura 3) è stato acquistato come un singolo frammento SDNA (see Tabella dei Materiali). Frammenti di DNA utilizzati nella reazione di montaggio sono stati amplificati usando pol DNA (vedi Tabella dei Materiali). Frammenti di DNA di hEF1α promoter parte 1, parte 2 hEF1α promoter, a figura intera e mutante CSTF-64 sono stati amplificati simultaneamente in tubi separati per 28 cicli (Figura 4A), seguendo le raccomandazioni del fornitore della pol DNA (vedi Tabella dei Materiali , per ogni ciclo di denaturazione era 7 sec a 98 ° C, annealing a 45 sec a 55 ° C, allungamento 90 sec a 72 ° C). Inizialmente, la spina dorsale pcDNA 3.1 è stato amplificato per 22 cicli (utilizzando le stesse condizioni come sopra con l'eccezione del tempo di allungamento, che è stato impostato a 3 min a 72 ° C). Tuttavia, la resa DNA risultante non era sufficiente per essere utilizzato in una reazione di montaggio (Figura 4A). Pertanto, un ulteriore amplificazione è stata eseguita per ottenere DNA sufficiente.
Prodotti di PCR ottengonoed da un modello plasmide deve essere digerito con enzima di restrizione DpnI per rimuovere il DNA plasmidico, che altrimenti contaminare i risultanti prodotti di reazione di assemblaggio e produrrà colonie batteriche farmacoresistenti falsi-positivi. Pertanto, i prodotti PCR sono stati digeriti con DpnI enzima di restrizione, che scinde metilato e emi-metilato DNA plasmide isolato dalla diga + E. ceppi coli. I prodotti di PCR ottenuti utilizzando frammenti di DNA sintetico, come modelli non hanno bisogno di essere digerito con DpnI dal DNA sintetizzato chimicamente non contiene basi metilate o emi-metilato.
Frammenti di DNA sono stati purificati e concentrati su di purificazione del DNA sfere magnetiche (vedi Tabella dei Materiali), come descritto nel passaggio protocollo 4. Le PCR per la pcDNA 3.1 vettore backbone sono stati combinati insieme e la quantità di purificazione del DNA sfere magnetiche utilizzato è stato modificato di conseguenza. La resa del DNA è stata determinatautilizzando uno spettrofotometro (Tabella 1 e Tabella delle attrezzature). Le reazioni di montaggio per CSTF-64 e mutanti CSTF-64 costrutti sono stati assemblati su ghiaccio (Tabella 1). Un eccesso molare di 3 volte dei frammenti di DNA considerati come "inserti" è stata utilizzata (Tabella 1, Figura 3). Il volume finale dei frammenti di DNA misti è stato regolato a 10 ml con acqua e 10 ml di master mix assemblaggio (2x) è stato aggiunto. Le reazioni sono state mescolate ed incubate a 50 ° C per 1 ora. Reazione di controllo positivo è stato anche montato secondo le raccomandazioni del manuale del kit GA e incubato simultaneamente Tale task force-64 reazioni CSTF-64 e mutanti. Come raccomandato nel protocollo, 2 ml di ciascuna delle reazioni di montaggio sono stati trasformati in chimicamente competente E. coli fornito con il kit di clonazione di montaggio (vedi Tabella dei Materiali). La trasformazione è stata effettuata come descritto nel kitmanuale. Cloni positivi sono stati selezionati sulla ampicillina agar piastre / LB. 6 colonie per ogni reazione di assemblaggio sono stati scelti a caso per essere propagate. DNA plasmidici sono stati isolati con un mini kit di isolamento plasmide (vedi Tabella dei Materiali). In silico digestione dei costrutti con gli enzimi di restrizione NheI e NotI portato in due frammenti con dimensioni di 4.590 bp, 3.032 bp per CSTF-64 e 4.590 bp, 2.711 bp per mutante CSTF-64 (Figura 2B e Figura 4B). Digestione con gli enzimi di restrizione HindIII e NotI portato in tre frammenti con le seguenti dimensioni: 5.872 bp, 1005 bp e 745 bp (CSTF-64) e 5872 bp, 1005 bp e 424 bp (mutante CSTF-64, figura 2B e Figura 4C). Infatti, la digestione dei plasmidi isolati visualizzati i modelli caratteristici attesi (Figura 4B, C). Si noti che il frammento di DNA 424 bp prodotto mediante digestione con HindIII e NotI del CSTF-64 mutanteplasmidi sulla figura 4C è debolmente macchiato grazie alle sue piccole dimensioni. 2 dei 6 plasmidi isolati sono stati inviati per il sequenziamento. Abbiamo sequenziato il promotore hEF1α, e CSTF-64 o mutante CSTF-64 parti dei costrutti per verificare che non vi siano delezioni, inserzioni o sostituzioni. Consigliamo vivamente sequenziamento dei costrutti di DNA derivati da questo o da qualsiasi protocollo basato-PCR. Il sequenziamento ha dimostrato che uno di ciascun plasmide sequenziato conteneva la sequenza prevista nella regione di hEF1α, 3xFLAG-tag e CSTF-64 o mutante CSTF-64. Ciascuno degli altri plasmidi aveva una mutazione puntiforme introdotta durante l'amplificazione del frammento di DNA corrispondente. Espressione del plasmide contenente CSTF-64 in cellule staminali embrionali di topo, ha prodotto abbondanti quantità di proteina esogena paragonabile a wild type espressione 17.

Figura 1. Rappresentazione schematica del meccanismo Gibson Assembly. Frammenti di DNA con sovrapposizione estremità erano isotermicamente assemblati in una singola sequenza continua. La sovrapposizione finisce prima vengono masticati indietro di 5 'esonucleasi, che sta gradualmente calore inattivato. Di conseguenza, diversi frammenti di DNA con estremità sovrapposte saranno temprare isotermicamente. DNA polimerasi riempirà i vuoti e termostabile DNA ligasi ligates i nick. Clicca qui per vedere una versione più grande di questa figura.
{kind=link}

Figura 2. (A) Diagramma di flusso del protocollo descritto per clonazione assemblaggio. (B) Rappresentazione del plasmide, PGA-Tale task force-64 generato utilizzando kit GA Red - frammenti di DNA utilizzati nella reazione di montaggio:. PcDNA3.1; hEF1α promoter parte 1 (hEF1a - 1); hEF1α promoter parte 2 (hEF1a - 2) ordinato da DNA sintetico; Mouse CSTF-64 (mCstF-64). Blu - quadri di lettura aperti. Violet - promotori virali e non virali. Verde -. Le regioni scissione e poliadenilazione Cliccate qui per vedere una versione più grande di questa figura.
{kind=link}

Figura 3. Progettazione del gruppo Gibson CSTF-64 plasmide. Nero scatole rappresentano i frammenti di DNA che erano disponibili per la progettazione di un unico gruppo Gibson CSTF-64 in sequenza silico. Successivamente, la sequenza è stata divisa in quattro pezzi di DNA, che sono state amplificate mediante PCR. Si noti che a causa delle piccole dimensioni del 3xFLAG-tag della sequenza è stato progettato come un SDNA insieme al pr hEF1α parte omoter 2. Clicca qui per vedere una versione più grande di questa figura.
{kind=link}

Figura 4. PCR dei frammenti di DNA utilizzati nelle reazioni di clonazione e rappresentante enzimi di restrizione dei plasmidi ottenuti (A) PCR utilizzando rappresentativi hot iniziare alta fedeltà mix 2x master per i frammenti di DNA utilizzati nelle reazioni di montaggio:. (B) rappresentativa plasmidi di hEF1α, full length Tale task force-64, pcDNA 3.1 costrutto (PGA-CSTF-64) e hEF1α, mutante Tale task force-64, pcDNA 3.1 costrutto (PGA-mutCstF-64) digerito con NheI e NotI. (C) gli stessi plasmidi come in B digeriti con HindIII e NotI enzimi."_blank"> Clicca qui per vedere una versione più grande di questa figura.
| Nome di frammenti di DNA | Dimensioni previste (bp) | Concn. (Ng / ml) | Diluito a (ng / ml) | microlitri utilizzato in GA CSTF-64 da diluito | microlitri utilizzato in GA mutCstF-64, dal diluito | Rapporto molare (ins: vec) |
| pcDNA 3.1 (vettore) | 4.618 | 158 | diluito | 1 | 1 | |
| hEF1_ promoter parte 1 | 825 | 213 | 75 | 1 | 1 | 3: 1 |
| hEF1_ promoter parte 2 per CSTF-64 | 516 | 229 | 50 | 1 | 3: 1 | |
| Tale task force-64 | 1.796 | 161 | diluito | 1 | 3: 1 | |
| hEF1_ promoter parte 2 per mutante CSTF-64 | 516 | 199 | 50 | 1 | 3: 1 | |
| mutante CSTF-64 | 1.448 | 201 | 171 | 1 | 3: 1 |
Tabella 1. Resa di frammenti di DNA dopo la concentrazione su sfere magnetiche, la diluizione e impostare delle reazioni di montaggio.
Discussione
Uso di successo della clonazione GA deve essere sempre preceduta da una attenta progettazione del costrutto completo (Figura 2 e Figura 3).
Verifica attenta delle sequenze di primer disegnati dallo strumento fondo di generazione è anche altamente raccomandato. Primer per GA possono essere generati senza l'utilizzo dello strumento di primer generazione. Tuttavia, l'uso dello strumento è altamente suggerisce, perché semplifica il processo. Generalmente, il primer per clonazione GA deve avere due sequenze funzionalmente differenti. La prima sequenza è frammento di DNA-specifica, e consente l'amplificazione del frammento mediante PCR. La seconda sequenza si sovrappone con il frammento adiacente, che è necessaria per il montaggio GA. Una tipica sequenza di DNA-frammento specifico sarebbe 18-22 nt lunghezza. Sequenze specifiche di DNA-frammento usato per amplificare lo stesso DNA deve avere temperature di fusione simili e contenuti GC. Sequenza sovrapposizione dovrebbe essere almeno di 15nt in lunghezza, con una temperatura di fusione di almeno 48 ° C. L'assemblaggio di più di 4 frammenti di DNA richiederà la sequenza sovrapposizione essere almeno 20 nt. Sovrapposizioni più lunghi permettono una maggiore specificità della ricottura conseguente frammenti di DNA più propriamente assemblati. Si raccomanda di evitare le sequenze che sono sbilanciato a loro GC o AT contenuti in via di sviluppo di sequenze sovrapposte, perché le sequenze inclinate potrebbero compromettere il montaggio DNA corretta.
Si suggerisce anche utilizzando come controllo negativo, che non dovrebbe produrre alcun colonie batteriche farmacoresistenti, solo il frammento di DNA corrispondente al vettore backbone in una reazione GA. In alternativa, uno dei frammenti di DNA che compongono i "inserti" può essere omesso dalla reazione GA, che dovrebbe anche tradursi in nessun colonie batteriche resistenti ai farmaci. Il motivo non colonie cresceranno sarà la mancanza di estremità sovrapposte di frammenti di DNA adiacenti, il che renderebbe l'assemblaggio di un complplasmide ete.
Nel protocollo descritto, sequenze di sovrapposizione 25 nt a generare stati usati i set di primer, a causa del numero di frammenti di DNA utilizzato (Figura 3). La raccomandazione del sito web strumento di generazione di fondo (vedi tabella di macchinari) è quello di utilizzare almeno 20 sequenze nt sovrapposti da montare 4 - 6 frammenti di DNA. Inoltre, sovrapponendo più sequenza a garantire il buon complementazione dei filamenti di DNA (vedi Figura 1) aumentare il numero di prodotti assemblati con precisione.
Attualmente, diversi sistemi per la clonazione senza soluzione di continuità sono disponibili. Tuttavia, alcuni di questi sistemi usano ancora gli enzimi di restrizione (cioè, Golden Gate clonazione 18). Altri usano miscele proprietarie di enzimi a base di virus del vaccino DNA polimerasi e legame al DNA delle proteine a singolo filamento dalla stessa fonte biologica 19. Entrambi i sistemi sono limitate rispetto alla GA dalla shorLunghezza ter delle sequenze sovrapposte. Poiché sequenze sovrapposte brevi potrebbero non fornire sufficiente specificità alla fase di ricottura di frammenti di DNA adiacenti, rendendo il corretto montaggio di più di 23 frammenti di DNA problematici. Queste carenze non sono presenti nel sistema GA.
La dimensione dei frammenti di DNA da amplificare dovrebbe essere considerato anche in modo da non superare la dimensione affidabile amplificabile mediante PCR (cioè, meno di 8 kbp di lunghezza). Anche con i miglioramenti nella funzione della DNA polimerasi negli ultimi 10 anni, grandi frammenti di DNA saranno amplificati con meno efficienza e precisione. Se necessario, frammenti di DNA più grandi possono essere ottenuti da altre fonti alternative di PCR, ad esempio, per l'isolamento plasmide e appropriata la digestione del DNA con enzimi di restrizione. In particolare, per il protocollo descritto nel manoscritto attuale, il nostro razionale utilizzare PCR è basata sulla dimensione dei frammenti di DNA disponibili, che erano tutti inferiori5 kbp. Se vi è più di un prodotto PCR identificato mediante elettroforesi su gel di agarosio, la purificazione gel della dimensione del frammento desiderabile raccomanda utilizzando una qualsiasi delle tecniche di biologia molecolare o kit disponibili appropriati. Nel protocollo corrente viene utilizzata una DNA polimerasi termostabile (vedi Tabella dei Materiali). Tuttavia qualsiasi DNA polimerasi fornire alta fedeltà e rendimento sarà adatto per essere utilizzato con questo protocollo. Se ad alta fedeltà DNA polimerasi diversi da quelli indicati nel protocollo sono disponibili, utilizzare le condizioni come descritto nei rispettivi manuali. Nel protocollo attuale, chimicamente competente E. coli sono usati che vengono forniti con il kit GA. In alternativa, chimicamente o elettro competente E. ceppi coli come DH5α o DH10B possono essere usati.
L'assemblea clonazione master mix 2x è facile da usare con hands-on minimo tempo. Tuttavia, pipettaggio accurato è necessario a causa dei piccoli volumi neEDED da miscelare insieme. Buona tecnica di biologia molecolare deve essere esercitato in ogni momento pure.
La clonazione GA offre possibilità illimitate per una costruzione di frammenti di DNA, plasmidi e vettori, che sono più di 3 kbp dimensioni. Inoltre ha un impatto più ampio nel campo della biologia sintetica, perché permette la sintesi e l'assemblaggio di, ad esempio, un intero batterica (Mycoplasma mycoides) genoma o un lievito (Saccharomyces cerevisiae) cromosoma 20,21. La tecnica è applicabile anche alla clonazione convenzionale deve generare costrutti senza soluzione di continuità.
In conclusione, la clonazione GA offre rapida, affidabile e flessibile alternativa alla procedura di clonazione DNA convenzionali.
Divulgazioni
Open Access publication and production fees were supplied by New England BioLabs Inc.
Riconoscimenti
Vorremmo ringraziare Michaela Jansen alla Texas Tech University Health Sciences Center, Lubbock, TX e Mladen Yovchev presso l'Università di Pittsburgh Medical Center, Pittsburgh, PA per la fornitura generosamente pcDNA 3.1 myc-His (A) e hEF1α promotore contenenti plasmidi . La ricerca riportata in questa pubblicazione è stato sostenuto dal Eunice Kennedy Shriver National Institute of Child Health and Human Development dei National Institutes of Health con il numero premio R01HD037109 (a CCM). Ulteriore supporto è stato dal Laura W. Bush Istituto per la salute della donna (a CCM e PNG). Il contenuto è di esclusiva responsabilità degli autori e non rappresentano necessariamente il punto di vista ufficiale del National Institutes of Health.
Gli autori dichiarano di non avere interessi finanziari in competizione.
Materiali
| Name | Company | Catalog Number | Comments |
| Synthetic double-stranded DNA fragment (sDNA) | Integrated DNA Technologies | We used gBlocks, which can be up to 2 kb in length. However, there are many commercial sources of synthetic DNA available. | |
| DNA purification magnetic beads | Beckman Coulter | A63880 | Agencourt AMPure XP - PCR Purification system |
| Plasmid Isolation Mini Kit | Omega Bio-Tek Inc | D6942-01 | E.Z.N.A. Plasmid Mini Kit I |
| Gibson Assembly Cloning Kit | New England BioLabs | E5510S | |
| DNA polymerase | New England BioLabs | M0494S | Q5 Hot Start High Fidelity 2x Master Mix |
| DpnI | New England BioLabs | R0176S | |
| NheI | New England BioLabs | R3131S | |
| NotI | New England BioLabs | R3189S | |
| HindIII | New England BioLabs | R3104S | |
| Spectrophotometer | Thermo Scientific | NanoDrop device | |
| EditSeq | DNASTAR | Part of Lasergene Core Suite | |
| SeqBuilder | DNASTAR | Part of Lasergene Core Suite | |
| Word | Microsoft | Part of Microsoft Office | |
| NEBuilder | New England BioLabs | primer generation tool: http://nebuilder.neb.com | |
| NEBioCalculator | New England BioLabs | ligation calculator: http://nebiocalculator.neb.com/#!/ligation |
Riferimenti
- Gibson, D. G. Enzymatic assembly of overlapping DNA fragments. Methods in enzymology. 498, 349-361 (2011).
- Gibson, D. G., Smith, H. O., Hutchison, C. A. 3rd, Venter, J. C., Merryman, C. Chemical synthesis of the mouse mitochondrial genome. Nature methods. 7, 901-903 (2010).
- Gibson, D. G., et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nature methods. 6, 343-345 (2009).
- Bowtell, D. D., Johnson, G. R., Kelso, A., Cory, S. Expression of genes transferred to haemopoietic stem cells by recombinant retroviruses. Molecular biology & medicine. 4, 229-250 (1987).
- Challita, P. M., Kohn, D. B. Lack of expression from a retroviral vector after transduction of murine hematopoietic stem cells is associated with methylation in vivo. Proceedings of the National Academy of Sciences of the United States of America. 91, 2567-2571 (1994).
- Lutzko, C., Senadheera, D., Skelton, D., Petersen, D., Kohn, D. B. Lentivirus vectors incorporating the immunoglobulin heavy chain enhancer and matrix attachment regions provide position-independent expression in B lymphocytes. Journal of Virology. 77, 7341-7351 (2003).
- Meilinger, D., et al. Np95 interacts with de novo DNA methyltransferases, Dnmt3a and Dnmt3b, and mediates epigenetic silencing of the viral CMV promoter in embryonic stem cells. EMBO reports. 10, 1259-1264 (2009).
- Chan, K. K., Wu, S. M., Nissom, P. M., Oh, S. K., Choo, A. B. Generation of high-level stable transgene expressing human embryonic stem cell lines using Chinese hamster elongation factor-1 alpha promoter system. Stem cells and development. 17, 825-836 (2008).
- Chung, S., et al. Analysis of different promoter systems for efficient transgene expression in mouse embryonic stem cell lines. Stem cells. 20, 139-145 (2002).
- Qin, J. Y., et al. Systematic comparison of constitutive promoters and the doxycycline-inducible promoter. PloS one. 5, e10611(2010).
- Lund, A., Knudsen, S. M., Vissing, H., Clark, B., Tommerup, N. Assignment of human elongation factor 1alpha genes: EEF1A maps to chromosome 6q14 and EEF1A2 to 20q13.3. Genomics. 36, 359-361 (1996).
- Wallace, A. M., et al. Two distinct forms of the 64,000 Mr protein of the cleavage stimulation factor are expressed in mouse male germ cells. Proceedings of the National Academy of Sciences of the United States of America. 96, 6763-6768 (1999).
- MacDonald, C. C., McMahon, K. W. Tissue-specific mechanisms of alternative polyadenylation: testis, brain, and beyond. Wiley interdisciplinary reviews. RNA. 1, 494-501 (2010).
- Sabath, I., et al. 3'-End processing of histone pre-mRNAs in Drosophila: U7 snRNP is associated with FLASH and polyadenylation factors. Rna. 19, 1726-1744 (2013).
- Yang, X. C., et al. A complex containing the CPSF73 endonuclease and other polyadenylation factors associates with U7 snRNP and is recruited to histone pre-mRNA for 3'-end processing. Molecular and cellular biology. 33, 28-37 (2013).
- Grozdanov, P. N., Macdonald, C. C. High-Throughput Sequencing of RNA Isolated by Cross-Linking and Immunoprecipitation (HITS-CLIP) to Determine Sites of Binding of CstF-64 on Nascent RNAs. Methods in molecular biology. 1125, 187-208 (2014).
- Youngblood, B. A., Grozdanov, P. N., MacDonald, C. C. CstF-64 supports pluripotency and regulates cell cycle progression in embryonic stem cells through histone 3' end processing. Nucleic acids research. , (2014).
- Engler, C., Kandzia, R., Marillonnet, S. A one pot, one step, precision cloning method with high throughput capability. PloS one. 3, e3647(2008).
- Irwin, C. R., Farmer, A., Willer, D. O., Evans, D. H. In-fusion(R) cloning with vaccinia virus DNA polymerase. Methods in molecular biology. 890, 23-35 (2012).
- Annaluru, N., et al. Total synthesis of a functional designer eukaryotic chromosome. Science. 344, 55-58 (2014).
- Gibson, D. G., et al. Creation of a bacterial cell controlled by a chemically synthesized genome. Science. 329, 52-56 (2010).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati