Method Article
Geração de plasmídeo vectores que expressam proteínas marcadas-bandeira nos termos do regulamento de Alongamento Human Fator-1α Promotor Usando Assembleia Gibson
Neste Artigo
Resumo
Synthesis of custom plasmids is labor and time consuming. This protocol describes the use of Gibson assembly cloning to reduce the work and duration of custom DNA cloning procedure. The protocol described also produces reliable tagged protein constructs for mammalian expression at similar cost to the traditional cut-and-paste DNA cloning.
Resumo
Montagem Gibson (GA) clonagem oferece uma alternativa rápida, confiável e flexível para métodos de clonagem de DNA convencionais. Usamos GA para criar plasmídeos personalizadas para expressão de genes exógenos em células-tronco embrionárias (mESCs). A expressão de genes exógenos, sob o controlo do SV40 ou promotores de citomegalovírus humanos diminui rapidamente após a transfecção em mESCs. Um remédio para esta expressão diminuída é usar o promotor humano fator de alongamento-1 alfa (hEF1α) para conduzir a expressão do gene. Vetores plasmídeo contendo hEF1α não são tão amplamente disponível como SV40 ou plasmídeos contendo CMV, especialmente aqueles que contêm também N-terminal 3xFLAG-tags. O protocolo descrito aqui é um método rápido para criar plasmídeos que expressam FLAG-marcado CstF-64 e CstF-64 mutante sob a regulação expressional do promotor hEF1α. GA utiliza uma mistura de exonuclease de ADN, polimerase de ADN e ligase de ADN para fazer a clonagem de extremidades sobrepostas de fragmentos de ADN possíveis.Com base nos DNAs de modelo que tínhamos disponível, nós projetamos nossas construções a ser montado em uma única seqüência. Nosso projeto usou quatro fragmentos de DNA: pcDNA backbone 3.1 vetor, hEF1α promotor parte 1, parte 2 hEF1α promotor (que continha 3xFLAG-tag comprado como um fragmento de DNA de cadeia dupla sintético), e quer CstF-64 ou específica CstF-64 mutante. As sequências destes fragmentos foram carregados para uma ferramenta de geração de iniciador para conceber iniciadores de PCR adequados para gerar os fragmentos de DNA. Após PCR, os fragmentos de DNA foram misturados com o vector contendo o marcador selectivo e a reacção clonagem GA foi montado. Plasmídeos de colônias bacterianas transformadas individuais foram isolados. Ecrã inicial dos plasmídeos foi realizada por digestão de restrição, seguido de sequenciação. Em conclusão, GA nos permitiu criar plasmídeos personalizadas para a expressão do gene em 5 dias, incluindo construção de telas e verificação.
Introdução
Procedimentos de clonagem de ADN convencionais contam com a utilização de enzimas de restrição para clivar o ADN e ligase de ADN para juntar os fragmentos de DNA em conjunto. Geração de construções de expressão que contenham diferentes mercadorias de fragmentos de ADN é um processo sequencial que inclui a clivagem do ADN com uma e / ou várias endonucleases de restrição e a subsequente inserção de fragmentos de ADN através da ligadura. O inconveniente principal deste procedimento é que as enzimas de restrição adequadas para um dos fragmentos de DNA pode ser difícil de identificar (ou seja, pode ter vários locais de clivagem) tornando a clonagem bem sucedida de ADN da proteína de comprimento completo de interesse impossível. Portanto, a geração de expressão personalizada constrói sob a regulação da transcrição de promotores específicos para o tipo de célula eficiente com personalizados proteína-tags requer design muito cuidado. É também uma técnica que consome tempo e trabalho. Recentemente, vários estudos descrevem metodologias para montar multipdiferentes fragmentos de ADN sintético le numa sequência contínua, ao mesmo tempo, quer em reacções de um ou dois passos, sem a utilização de enzimas de restrição 1-3. A reacção de clonagem de um passo (excluindo todos os passos de preparação), depende da utilização de uma mistura de exonuclease de ADN, ADN-polimerase, ligase de ADN de 2,3 e as extremidades sobrepostas de fragmentos de ADN (Figura 1). Uma vez que não há nenhuma utilização de enzimas de restrição, os fragmentos de ADN de qualquer tamanho e composição da sequência (excluindo as sequências altamente repetitivas) podem ser fundidos em conjunto num constructo sem costura. Recentemente, um kit comercial (montagem Gibson; GA) para as reações de clonagem de uma etapa tornou-se disponível. Este kit permite uma montagem rápida e eficiente custo de quaisquer fragmentos de DNA em um único vector com os promotores personalizadas e tags de proteína.
Os vectores de expressão de plasmídeos disponíveis amplamente utilizados para expressar proteínas exógenas em modelos de cultura de células de mamíferos são muitas vezes sob a reg transcricionalulação do citomegalovírus viral (CMV) ou vírus SV40 (40) os promotores de Simian. Estes promotores virais proporcionam a expressão transiente robusta das proteínas exógenas na maioria dos modelos baseados em cultura de células de mamíferos. No entanto, a geração de linhas celulares que expressam estavelmente proteínas exógenas é frequentemente mal sucedida por causa de silenciamento transcricional de CMV ou promotores de SV40, durante o processo de estabelecimento de 4,5. Além disso, os promotores de SV40, CMV viral e não serão suficientes para promover a expressão de proteínas exógenas em células da linhagem linfóide ou células estaminais embrionárias 6,7. A solução para a limitação inerente de promotores virais é a utilização de promotores fortes constitutivos não virais 8-10. Um promotor constitutivo forte bem caracterizados não-viral de origem humana é o promotor do factor de alongamento 1α (hEF1α) (hEF1α está envolvido na catálise da associação dependente de GTP de aminoacil-ARNt de ribossomas 11). No entanto,Os vectores de expressão contendo o promotor hEF1α não são tão amplamente quanto a-promotor viral contendo plasmídeos, especialmente aqueles que contenham também 3 × FLAG na extremidade do terminal amino da proteína de interesse.
A proteína de factor de estimulação de clivagem de 64.000 MW (CstF-64) é envolvido na direcção 3 'de processamento final da maioria dos mRNAs de histonas 12,13, incluindo ARNms dependentes de replicação 14,15. CstF-64 é expresso em todos os tecidos somáticos 12. Seu motivo de reconhecimento RNA se liga a seqüências de RNA GU-ricos em transcrições nascentes a jusante da clivagem e poliadenilação local 16. Esta ligação de CstF-64 para o pré-mRNA promove a clivagem endonucleolítica eficiente do transcrito nascente.
Aqui, é descrito um protocolo que utiliza amplificação por PCR dos fragmentos de ADN, um kit de clonagem de montagem Gibson (que inclui células bacterianas quimicamente competentes) para produzir vectores de mercadorias m 3xFLAG-tagouse CstF-64 ou mutante CstF-64 para a sua extremidade terminal amino sob a expressão de hEF1α promotor 1.
Protocolo
In Silico 1. Concepção do plasmídeo e Geração dos iniciadores sobrepostos
NOTA: O objectivo deste passo é o de montar sequência de nucleótidos completa da construção e conceber os iniciadores para ser utilizada para gerar fragmentos com extremidades sobrepostas para GA clonagem.
- Projete uma sequência de nucleotídeos contínua para representar o plasmídeo final.
- Obter ou listar os plasmídeos reais e os fragmentos de ADN que vão ser utilizadas como moldes em PCR.
NOTA: fragmentos de DNA que não estão prontamente disponíveis - como a combinação de diferentes marcas e promotores - pode ser encomendado como um único ou vários fragmentos sintéticos de cadeia dupla de DNA (DNAs). Estes fragmentos estão comercialmente disponíveis a custo relativamente baixo e pode ser de até 2 kb de comprimento (ver Tabela de Materiais). - Dividir a sequência de nucleótidos da construção contínua montado na etapa 1.1 em fragmentos de DNA adequados para a PCR. Confirm que os fragmentos corresponder plasmídeos disponíveis e fragmentos DNAs. Evitar os fragmentos de DNA menores do que 200 nt.
- Acesse a ferramenta de geração de primer (ver Tabela de Equipamentos). Selecione o menu "Definir preferências". Escolha as configurações apropriadas, clicando na aba "MUDANÇA PREFS" na janela pop-up "Alterar configurações de montagem Gibson".
NOTA: Primer desenho também pode ser realizada sem a utilização da ferramenta de geração de iniciador. No entanto, a utilização da ferramenta de geração de iniciador simplifica o processo. - Comece a construir a construção, selecionando o menu "Build Construct". Insira os fragmentos de DNA de divisão no sequencialmente ferramenta de geração de iniciador de 5 'para 3'.
NOTA: O fragmento de ADN utilizado como uma espinha dorsal de plasmídeo pode ser convenientemente dividido em duas partes. No produto final de PCR 'final deste primeiro fragmento e a 3' da extremidade 5 do último fragmento são ligados entre si num DN contínuaUm fragmento representando o esqueleto do vector. - Cole o primeiro fragmento de DNA que representa a extremidade 5 'do DNA vetor no formato FASTA (uma representação baseada em texto de sequência de nucleotídeos) na janela pop-up "Enter Vector ou Insert Fragment". Nomeie o fragmento de DNA. Escolha a forma adequada para obter o fragmento de DNA, quer como PCR, RE Digest ou Synthesis. Clique na aba "Continuar".
- Se houver a necessidade de acrescentar nucleotídeos extra / sítios de restrição na junção da construção final utilizar os espaços "espaçador Fwd ou Rev cartilha", previsto no "Adicionar um fragmento de inserção para a montagem" janela. Adicionar nucleótidos extra / locais de restrição para apenas um dos fragmentos de DNA e não para ambos os fragmentos. Clique na aba "DONE".
- Repetir para todos os fragmentos até que a construção esteja completa. Selecione o menu "Ver Primers" e rever as seqüências de primers.
- Repita o procedimento para todas as construções (vector backbones e pastilhas), ou para fragmentos de ADN que são diferentes.
2. Amplificação dos fragmentos de ADN por PCR Utilizando Hot Start Revisão ADN Polimerase (2x Master Mix)
NOTA: O objectivo deste passo é obter ADN suficiente para a reacção de PCR usando a montagem.
- Compre os iniciadores de PCR concebidos no passo anterior como produtos dessalinizada e na menor escala possível. Dilui-se-lhes a 10 uM em água ou TE (10 mM Tris-HCl, pH 7,9, EDTA 1 mM).
- Comprar fragmentos de DNA que não estão prontamente disponíveis como fragmentos de DNAs.
- Dilui-se todos os fragmentos de ADN, incluindo os fragmentos de DNAs que serão usados como moldes para as PCRs para 1 ng / mL em água.
- Montar as reacções de PCR à temperatura ambiente. Resumidamente, usar 2,5 ul (10 uM de solução de reserva) da respectiva cada iniciador do par de iniciadores, 1 ul (solução a partir de 1 ng / ul de estoque) do fragmento de ADN molde, 25 μl de polimerase quente revisão início DNA (DNA pol; 2x mix mestre; consulte a Tabela de Materiais) e 19 mL de água. Misture o tubo através de um toque suave e recolher as gotículas de líquido por centrifugação breve.
- Amplificar simultaneamente em tubos separados fragmentos de DNA de tamanho similar de acordo com as recomendações para a pol DNA. Realizar 25-28 ciclos de PCR ou determinar o número de ciclos que produzem um rendimento de ADN suficiente.
- Executar 10% do volume de reacção de PCR (5 ul) numa electroforese em gel de agarose padrão manchado com brometo de etídio (0,2 ug / ml de concentração final). Assegure-se que uma única banda de ADN que representa o produto de PCR é visível. Determinar o tamanho e quantidade relativa dos fragmentos de ADN utilizando ADN padrões de peso molecular. Se necessário, repetir a PCR para se obter uma quantidade suficiente de fragmentos de ADN.
3. Dpnl digestão dos produtos de PCR
NOTA: O objetivo deste passo é Diger plasmídeo residual molde de ADN a partir dos PCRs na Secção 2. Dpnl irá digerir o ADN plasmídeo somente se é metilado, tal como ocorre ao plasmídeo de ADN cultivadas em barragem + estirpes bacterianas. Portanto, não se tratar com Dpnl ADN plasmídeo será utilizado como um fragmento de ADN na reacção GA.
- Adicionar 2 ul da enzima de restrição Dpnl para os 45 ul dos produtos de PCR produzidos na Secção 2. Incubar a 37 ° C durante 1 h. Prossiga com o ponto 4 ou congelar a -20 ° C até ser necessário.
4. purificação e concentração dos fragmentos de DNA sobre DNA Purificação esferas magnéticas
NOTA: O objectivo deste passo é o de purificar e concentrar os produtos de PCR obtidos nos pontos 2 e 3. Outros métodos de purificação de PCR podem ser usados também.
- Equilibrar a purificação DNA esferas magnéticas à temperatura ambiente. Re-suspender as contas por breve vortex.
- Transfira os PCRs wi pré-digeridoth Dpnl para a tubos de 1,5 ml e adicionar 81 uL de pérolas magnéticas de purificação do ADN para cada tubo. Incubar a mistura à temperatura ambiente durante 10 min.
- Colocar os tubos no colector magnético durante cerca de 2 min. Descartar o líquido limpo usando uma pipeta. Lavar duas vezes com 200 ul de etanol a 80% durante 30 segundos.
- Permitir que as pelotas para secar. Manter a tampa aberta dos tubos, com os tubos posicionados no colector magnético.
- Re-suspender as pérolas secas em 10 ul de 10 mM Tris-HCl, pH 8,0. Incubar à temperatura ambiente durante 2 min. Girar rapidamente para recolher o líquido, na parte inferior dos tubos.
- Posicionar os tubos sobre o colector magnético durante 2 min. Remover 8,5-10 ul da solução límpida e introduzi-la num tubo novo pré-rotulados. Determinar a concentração dos fragmentos de DNA por espectroscopia de UV (ver Tabela de equipamento).
Reação 5. Assembléia Clonagem e Transformação dos produtos em E. coli
NOTA: O objetivo desta etapa é calcular 3: 1 de inserção: vector e realizar a reação de montagem.
- Use pelo menos 100 ng de fragmento de ADN representando o esqueleto do vector ou fragmento de DNA contendo o marcador selectivo. Calcula-se o excesso molar de 3 vezes para os fragmentos de ADN que vão ser utilizadas como inserções.
- Converter o excesso molar em ng necessária de cada inserto em particular.
NOTA: Uma maneira conveniente de fazer os cálculos é usar um aplicativo baseado na web (ver Tabela de Equipamentos). - Misture quantidades calculadas de fragmentos de ADN em um tubo de PCR, ajustar o volume para 10 mL. Adicionar 10 ml da mistura principal GA (2x, consulte a Tabela de Materiais). Incubar a reacção a 50 ° C durante 1 hora para o conjunto de 4 - 6 ou fragmentos de 15 min para a montagem de fragmentos de 2-3 num termociclador de PCR.
- Prossiga com a transformação do produto em conjunto E. competente coli ou congelar os produtos a -20° C, até ser necessário.
- Siga o processo de transformação que acompanha a células electro competentes ou químicas. Normalmente, utilizar 2 uL da reacção de montagem por reacção de transformação.
- Após a transformação está completa, a propagação das células transformadas em placas de agar suplementado com o antibiótico selectivo apropriado.
6. O plasmídeo isolamento, restrição de digestão enzimática e Sequencing
NOTA: O objectivo deste passo é isolar o DNA de plasmídeo a partir de E. coli, em seguida, para verificar a construção, por digestão de restrição e sequenciação.
- Propagar várias colónias individuais para mini-preps e isolamento de plasmídeos. Durante a noite cultura LB líquido Uso 2-5 ml suplementado com antibiótico apropriado. Isolar os plasmídeos correspondentes, seguindo o procedimento descrito no mini-kit de preparação que é usado.
- Determinar a quantidade e concentração dos plasmídeos obtidos usando espectrofotometriafotômetro.
- Executar a digestão de restrição com uma ou várias endonucleases de restrição para ~ 0,5 ug dos plasmídeos purificados. Utilização de enzimas de restrição que proporcionam padrão distinto de digestão característicos para as construções de DNA.
- Confirme DNA sucesso clonagem por sequenciação de ADN usando primers específicos ou padrão. Analisar os dados de sequenciamento de precisão.
Resultados
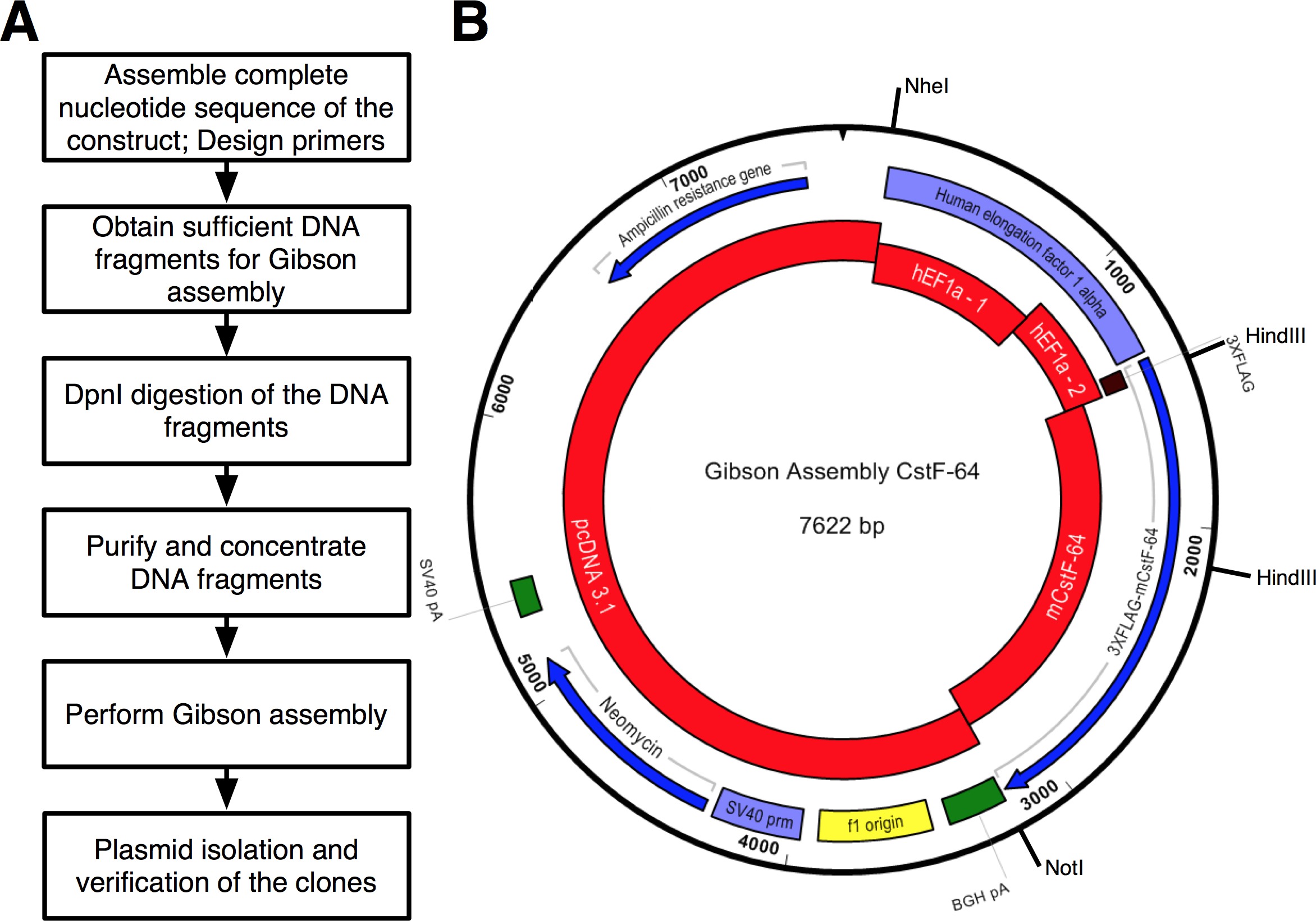
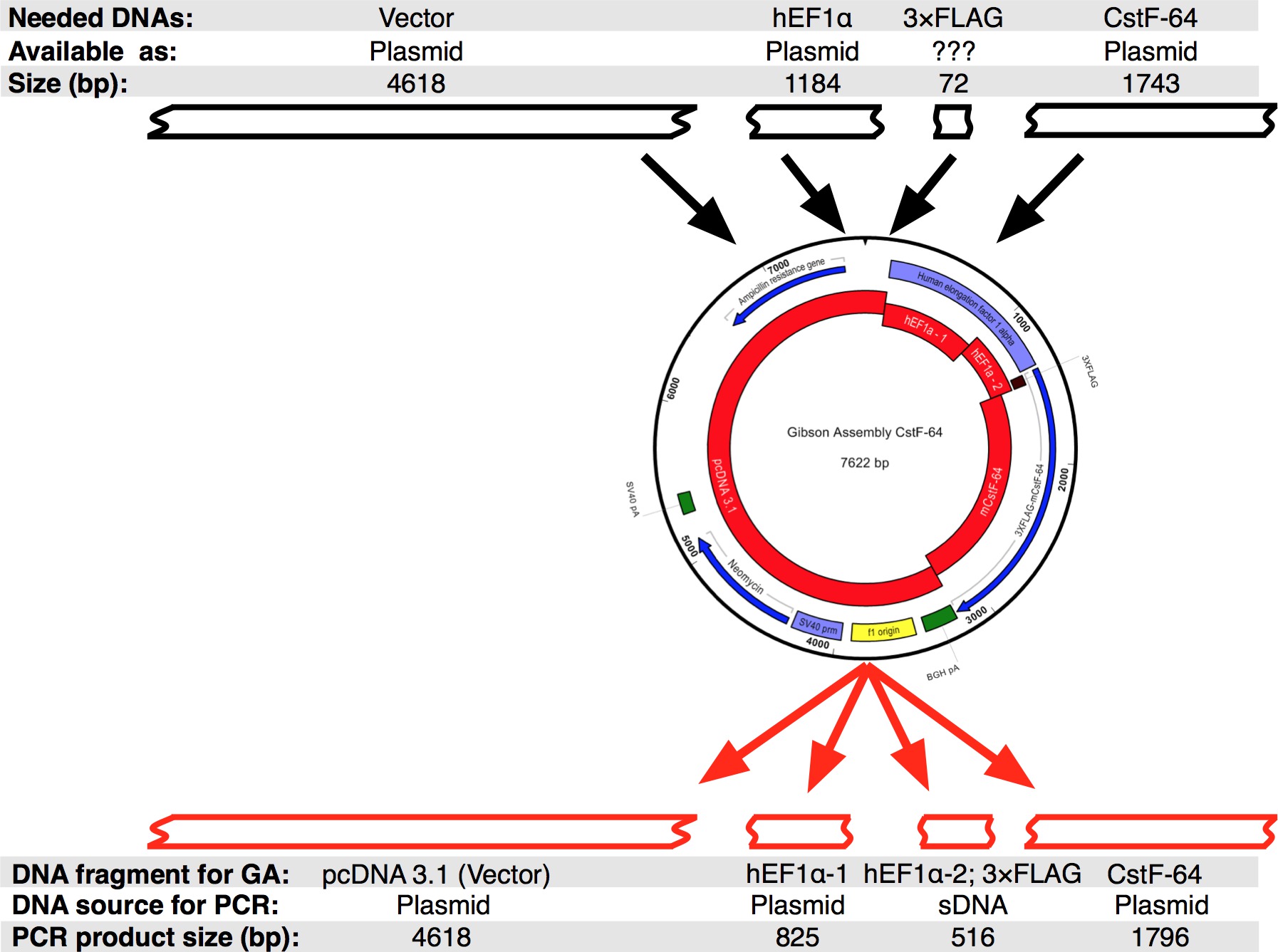
Um fluxo de trabalho do protocolo que foi seguido é mostrado na Figura 2A. Queríamos clonar CstF-64 e mutantes CstF-64 proteínas fundidas para 3xFLAG-tag ao abrigo do regulamento expressional de hEF1α promotor (Figura 2B e Figura 3). A hEF1α plasmídeo contendo seguido por 3xFLAG-tag não estava disponível para nós. No entanto, os seguintes plasmídeos estavam disponíveis: pcDNA 3.1 myc-His (A; uma oferta generosa de Michaela Jansen), hEF1α contendo plasmídeo (uma oferta generosa de Mladen Yovchev) e mouse CstF-64 plasmídeos 12 (Figura 3). Toda a sequência para a construção (s) foi montado utilizando os aplicativos de edição de texto e de nucleotídeos (Figura 3; veja tabela de equipamento). Subsequentemente, a sequência (s) foi dividida em quatro pedaços convenientes (Figura 2B, blocos vermelho e Figura 3) correspondentes aos ADN plasmídicos disponíveis. Amplification primers foram projetados usando a ferramenta de geração de primer (ver Tabela de Equipamentos) com restrições de 4-6 fragmentos com sobreposição mínima de 25 nt, criada na janela pop-up "Alterar configurações de montagem Gibson". Sítios de restrição NheI e NotI foram incluídas na concepção de iniciadores para a finalidade de identificar os plasmídeos correctamente montados. NheI está localizado na sequência de iniciador entre pcDNA 3.1 e na extremidade 5 'do promotor hEF1α. Local Notl situa-se após o codão de paragem (UGA) de CstF-64 e pcDNA 3.1 vector de espinha dorsal. Após a digestão simultânea com ambos os enzimas fragmento de ADN que consiste em hEF1α promotor, 3xFLAG e CstF-64 mutante ou CstF-64 será libertado (ver abaixo e na Figura 2B). Primers foram encomendados em menor escala possível e dessalinizada. A segunda parte do promotor hEF1α contendo fragmento de DNA 3xFLAG-tag (490 bp, figura 2B, Figura 3) foi comprado como um único fragmento DNAs (see Table of Materials). Os fragmentos de DNA usados na reacção de reunião foram amplificados utilizando pol do ADN de (ver Tabela de Materiais). Fragmentos de DNA de hEF1α promotor parte 1, parte 2 hEF1α promotor, de corpo inteiro e mutante CstF-64 foram amplificados simultaneamente em um tubos separados por 28 ciclos (Figura 4A), seguindo as recomendações do fornecedor da pol DNA (ver Tabela de Materiais , para cada ciclo de desnaturação foi de 7 segundos a 98 ° C, 45 s de recozimento a 55 ° C, alongamento de 90 segundos a 72 ° C). Inicialmente, a espinha dorsal pcDNA 3.1 foi amplificado durante 22 ciclos (utilizando as mesmas condições como acima com a excepção de o tempo de alongamento, o qual foi ajustado para 3 min a 72 ° C). No entanto, o rendimento de ADN resultante não foi suficiente para ser usado numa reacção de montagem (Figura 4A). Portanto, uma amplificação adicional foi realizado para obter suficiente ADN.
Os produtos de PCR obtered partir de um molde de plasmídeo deve ser digerido com a enzima de restrição Dpnl para remover o ADN de plasmídeo, que de outra forma contaminar os produtos resultantes da reacção de montagem e irá produzir colónias bacterianas resistentes a drogas falso-positivos. Por conseguinte, os produtos de PCR foram digeridos com a enzima de restrição Dpnl, que cliva o DNA de plasmídeo desnaturado e hemi-metilado isolado a partir de barragem + E. estirpes de E. coli. Os produtos de PCR obtidos a partir de fragmentos de ADN sintéticos, como moldes não precisam de ser digerido com Dpnl já que o ADN sintetizado quimicamente não contém bases metiladas ou hemi-metilado.
Os fragmentos de ADN foram purificados e concentrados através de purificação do ADN pérolas magnéticas (ver Tabela de Materiais) como descrito no passo protocolo 4. Os PCRs para o vector pcDNA 3.1 espinha dorsal foram combinados em conjunto e a quantidade de purificação de ADN esferas magnéticas utilizado foi ajustado em conformidade. O rendimento de ADN foi determinadautilizando um espectrofotômetro (Tabela 1 e Tabela de Equipamentos). Reacções de Montagem de CstF-64 e mutantes CstF-64 construções foram montadas em gelo (Tabela 1). Um excesso molar de 3 vezes de fragmentos de ADN consideradas como "pastilhas" foi utilizada (Tabela 1, Figura 3). O volume final dos fragmentos de ADN mistos foi ajustado a 10 ul com água e 10 ul de mistura de base de montagem (2x) foi adicionado. As reacções foram misturadas e incubadas a 50 ° C durante 1 h. Reacção de controlo positiva, também foi montada de acordo com a recomendação do manual do kit GA e incubadas simultaneamente com os CstF-64 e mutantes CstF-64 reações. Tal como recomendado no protocolo, 2 ul da cada uma das reacções foram transformadas de montagem na E. quimicamente competente coli fornecido com o kit de clonagem de montagem (ver Tabela de Materiais). A transformação foi realizada como descrito no kitmanual. Os clones positivos foram seleccionados em placas de agar / LB ampicilina. 6 colónias por cada reação montagem foram selecionados aleatoriamente para ser propagada. Os DNAs de plasmídeo foram isolados utilizando uma mini-kit de isolamento de plasmídeo (ver Tabela de Materiais). Na digestão in silico das construções com as enzimas de restrição Nhel e NotI resultou em dois fragmentos com tamanhos de 4590 pb, 3032 pb para o CstF-64 e 4590 pb, 2711 pb para o mutante CstF-64 (Figura 2B e Figura 4B). A digestão com as enzimas de restrição HindIII e NotI resultou em três fragmentos com os seguintes tamanhos: 5872 pb, 1005 pb e 745 pb (CstF-64) e 5872 pb, 1005 pb e 424 pb (mutante CstF-64, Figura 2B e Figura 4C). De facto, a digestão dos plasmídeos isolados apresentado os padrões característicos esperados (Figura 4B, C). Note-se que o fragmento de ADN de 424 pb produziu por digestão com HindIII e Notl do mutante de CstF-64plasmídeos na Figura 4C é fracamente manchado devido ao seu pequeno tamanho. 2 dos 6 plasmídeos isolados foram enviados para o seqüenciamento. Nós sequenciado o promotor hEF1α, e CstF-64 mutante ou CstF-64 partes das construções para verificar que não existem deleções, inserções ou substituições. Recomendamos seqüenciamento das construções de DNA resultantes deste ou de qualquer protocolo baseado em PCR. A sequenciação revelou que uma de cada plasmídeo sequenciado continha a sequência esperada na região de hEF1α, 3xFLAG-tag e CstF-64 mutante ou CstF-64. Cada um dos outros plasmídeos tinham um ponto de mutação introduzidas durante a amplificação do fragmento de ADN correspondente. A expressão do plasmídeo contendo CstF-64 de ratinho em células estaminais embrionárias, produziu uma quantidade abundante de proteínas exógenas comparável à expressão do tipo selvagem 17.

Figura 1. Representação esquemática do mecanismo de montagem Gibson. Os fragmentos de ADN com extremidades sobrepostas isotermicamente foram montados numa única sequência contínua. A sobreposição termina primeiro são mastigados de volta por 5 'exonuclease, que é gradualmente inativados pelo calor. Por conseguinte, fragmentos de ADN com diferentes extremidades que se podem sobrepor recozer isotermicamente. DNA polimerase para preencher as lacunas e ligase DNA termoestável ligates os nicks. Por favor, clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 2. (A) Diagrama de fluxo do protocolo descrito para a clonagem de montagem. (B) Representação do plasmídeo, pGA-CstF-64 gerado utilizando o kit de GA vermelhos - fragmentos de DNA usados na reacção de montagem:. PcDNA3,1; hEF1α promotor part 1 (hEF1a - 1); hEF1α promotor part 2 (hEF1a - 2) pedido como um DNA sintético; rato CstF-64 (mCstF-64). Azuis - grelhas de leitura abertas. Violeta - promotores virais e não virais. Verde -. Regiões de clivagem e poliadenilação Por favor, clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 3. projeto do conjunto Gibson CstF-64 plasmídeo. Preto caixas representam os fragmentos de DNA que estavam disponíveis para projetar um conjunto único Gibson CstF-64 em sequência silico. Subsequentemente, a sequência foi dividido em quatro partes de ADN, os quais foram amplificados por PCR. Note-se que, devido à pequena dimensão do-tag 3xFLAG a sequência foi concebido como um DNAs juntamente com o pr hEF1α omoter parte 2. Por favor, clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 4. A PCR dos fragmentos de ADN utilizados nas reacções de clonagem e a digestão com enzimas de restrição representante dos plasmídeos obtidos (A) PCRs usando representativos quente iniciar de alta fidelidade master mix 2x para os fragmentos de ADN utilizados em reacções de montagem:. (B) representativas plasmídeos de hEF1α, comprimento completo CstF-64, pcDNA 3.1 construto (pGA-CstF-64) e hEF1α, mutante CstF-64, pcDNA 3.1 construto (pGA-mutCstF-64), digerido com NheI e NotI. (C) os mesmos plasmídeos como em B digerido com as enzimas HindIII e NotI."_blank"> Clique aqui para ver uma versão maior desta figura.
| Nome de fragmentos de DNA | O tamanho esperado (pb) | Concn. (Ng / mL) | Diluída a (ng / mL) | ul utilizado em GA CstF-64 a partir diluído | ul utilizado em mutCstF GA-64, a partir diluído | Razão molar (ins: VEC) |
| pcDNA 3.1 (vector) | 4.618 | 158 | undiluted | 1 | 1 | |
| hEF1_ parte promotor 1 | 825 | 213 | 75 | 1 | 1 | 3: 1 |
| hEF1_ promotor parte 2 para CstF-64 | 516 | 229 | 50 | 1 | 3: 1 | |
| CstF-64 | 1.796 | 161 | undiluted | 1 | 3: 1 | |
| hEF1_ promotor parte 2 para o mutante CstF-64 | 516 | 199 | 50 | 1 | 3: 1 | |
| mutante CstF-64 | 1.448 | 201 | 171 | 1 | 3: 1 |
Tabela 1. Produção de fragmentos de ADN após concentração em esferas magnéticas, diluição e definir-se as reacções de montagem.
Discussão
O uso bem sucedido de clonagem GA deve ser sempre precedida por uma concepção cuidadosa da construção completa (Figura 2 e Figura 3).
Verificação cuidadosa das seqüências dos iniciadores desenhados pela ferramenta de geração de cartilha também é altamente recomendável. Os primers para a GA podem ser gerados, sem a utilização da ferramenta de geração de iniciador. No entanto, a utilização da ferramenta é altamente recomendável, porque simplifica o processo. Geralmente, o iniciador para a clonagem GA deve ter duas sequências funcionalmente diferentes. A primeira sequência é um fragmento de ADN específico, e permite a amplificação do fragmento utilizando PCR. A segunda sequência coincide com o fragmento adjacente, o que é necessário para a montagem GA. Uma sequência típica de ADN específica para o fragmento seria 18-22 nt de comprimento. Sequências específicas de ADN-fragmento utilizados para amplificar o ADN mesma deve ter temperaturas de fusão semelhantes e conteúdo em GC. Sequência de sobreposição deve ser pelo menos de 15nt de comprimento com uma temperatura de fusão de pelo menos 48 ° C. O conjunto de mais de 4 fragmentos de DNA exigirá a sequência de sobreposição de modo a ser pelo menos 20 nt. Sobreposições mais longos permitirá o aumento da especificidade do recozimento resultando em fragmentos de DNA mais devidamente montado. Recomenda-se evitar seqüências que estão tortos em seu GC ou AT conteúdo no desenvolvimento de sequências sobrepostas, porque sequências distorcidas podem comprometer montagem DNA adequado.
Também sugerem utilizar como controlo negativo, o qual não deve produzir colónias de bactérias resistentes a drogas, apenas o fragmento de ADN correspondente à estrutura do vector numa reacção de GA. Alternativamente, um dos fragmentos de DNA compreendendo as "pastilhas" pode ser omitido da reacção de GA, a qual também deverá resultar em nenhum colónias bacterianas resistentes a drogas. A razão pela qual não há colónias irão crescer vai ser a falta de sobreposição extremidades dos fragmentos de ADN adjacentes, o que irá tornar o conjunto de um complplasmídeo ete.
No protocolo descrito, sequências sobrepostas de 25 nt foram usadas para gerar os conjuntos de iniciadores, porque o número de fragmentos de DNA usadas (Figura 3). A recomendação do website ferramenta de geração de primer (ver Tabela de Equipamentos) é a utilização de pelo menos 20 sequências de nt sobrepor para montar 4-6 fragmentos de DNA. Além disso, a sobreposição de sequência mais longa vai garantir a complementação adequada das cadeias de ADN (ver Figura 1) aumentar o número de produtos montados com precisão.
Actualmente, vários sistemas para clonagem estão disponíveis sem costura. No entanto, alguns destes sistemas ainda utilizar enzimas de restrição (por exemplo, Golden Gate clonagem 18). Outros usam misturas patenteadas de enzimas baseados em vírus vaccinia polimerase de ADN e da proteína de ligação ao ADN de cadeia simples a partir da mesma fonte biológica 19. Ambos os sistemas são limitados em comparação com GA por o shcomprimento ter de sobreposição de sequências. Porque sequências sobrepostas mais curtos podem não fornecer especificidade suficiente para a etapa de recozimento de fragmentos de DNA adjacentes, tornando a montagem correta de mais de 23 fragmentos de DNA problemáticos. Estes defeitos não estão presentes no sistema de GA.
O tamanho dos fragmentos de ADN a ser amplificado deve ser também considerado, de modo a não exceder o tamanho de maneira confiável amplificável por PCR (isto é, menos de 8 kpb em comprimento). Mesmo com as melhorias na função de polimerases de ADN dos últimos 10 anos, grandes fragmentos de ADN serão amplificados com menos eficiência e precisão. Se necessário, os fragmentos de ADN maiores podem ser obtidas a partir de outras fontes alternativas para PCR, por exemplo, através do isolamento de plasmídeo e de ADN adequada digestão com enzimas de restrição. Especificamente, para o protocolo descrito no manuscrito actual, o nosso racional para utilizar PCR foi baseada no tamanho dos fragmentos de ADN disponíveis, que foram todos inferiores a5 kbp. Se houver mais do que um produto de PCR identificado por electroforese em gel de agarose, purificação em gel do fragmento de tamanho desejável é recomendada a utilização de qualquer das técnicas de biologia molecular ou kits disponíveis adequados. No protocolo atual de um DNA polimerase termoestável é utilizado (ver Tabela de Materiais). No entanto, qualquer ADN-polimerase de alta fidelidade e proporcionando rendimento será adequado para ser utilizado com este protocolo. Se polimerases de DNA de alta fidelidade diferentes do que o descrito no protocolo estão disponíveis, use as condições estabelecidas, tal como descrito nos respectivos manuais. No protocolo atual, E. quimicamente competente coli são usados que são fornecidos com o kit GA. E. Alternativamente, química ou electro competente estirpes de E. coli DH5a tal como DH10B ou pode ser usado.
O conjunto de clonagem mix mestre 2x é fácil de usar com as mãos sobre o mínimo de tempo. No entanto, pipetagem precisa é necessária por causa dos pequenos volumes neEDED a ser misturados em conjunto. Boa técnica de biologia molecular deve ser exercido em todos os momentos assim.
A clonagem GA oferece possibilidades ilimitadas para a construção dos fragmentos de DNA, plasmídeos e vectores que são mais longos do que 3 kpb de tamanho. Além disso, tem um impacto mais amplo no campo da biologia sintética, porque permite a síntese e montagem de, por exemplo, um (Mycoplasma mycoides) genoma bacteriano inteira ou uma levedura (Saccharomyces cerevisiae) cromossoma 20,21. A técnica é também aplicável a clonagem convencional necessita para gerar as construções sem costura.
Em conclusão, GA clonagem oferece alternativa rápida, confiável e flexível para o procedimento de clonagem de DNA convencional.
Divulgações
Open Access publication and production fees were supplied by New England BioLabs Inc.
Agradecimentos
Gostaríamos de agradecer a Michaela Jansen no Centro de Ciências da Saúde da Universidade Texas Tech, Lubbock, TX e Mladen Yovchev na Universidade de Pittsburgh Medical Center, Pittsburgh, PA para o generosamente fornecer pcDNA 3.1 myc-His (A) e hEF1α promotor contendo plasmídeos . Research informou nesta publicação foi apoiada pelo Kennedy Shriver Instituto Nacional de Saúde Infantil e Desenvolvimento Humano dos Institutos Nacionais de Saúde Eunice sob o número prêmio R01HD037109 (a CCM). Suporte adicional foi do Instituto Bush, Laura W. de Saúde da Mulher (para CCM e PNG). O conteúdo é da exclusiva responsabilidade dos autores e não representam, necessariamente, a posição oficial do National Institutes of Health.
Os autores declaram que não têm interesses financeiros concorrentes.
Materiais
| Name | Company | Catalog Number | Comments |
| Name of Material/ Equipment | Company | Catalog Number | Comments/Description |
| Synthetic double-stranded DNA fragment (sDNA) | Integrated DNA Technologies | We used gBlocks, which can be up to 2 kb in length. However, there are many commercial sources of synthetic DNA available. | |
| DNA purification magnetic beads | Beckman Coulter | A63880 | Agencourt AMPure XP - PCR Purification system |
| Plasmid Isolation Mini Kit | Omega Bio-Tek Inc | D6942-01 | E.Z.N.A. Plasmid Mini Kit I |
| Gibson Assembly Cloning Kit | New England BioLabs | E5510S | |
| DNA polymerase | New England BioLabs | M0494S | Q5 Hot Start High-Fidelity 2X Master Mix |
| DpnI | New England BioLabs | R0176S | |
| NheI | New England BioLabs | R3131S | |
| NotI | New England BioLabs | R3189S | |
| HindIII | New England BioLabs | R3104S | |
| Name of Material/ Equipment | Company | Catalog Number | Comments/Description |
| Spectrophotometer | Thermo Scientific | NanoDrop device | |
| EditSeq | DNASTAR | Part of Lasergene Core Suite | |
| SeqBuilder | DNASTAR | Part of Lasergene Core Suite | |
| Word | Microsoft | Part of Microsoft Office | |
| NEBuilder | New England BioLabs | primer generation tool: http://nebuilder.neb.com | |
| NEBioCalculator | New England BioLabs | ligation calculator: http://nebiocalculator.neb.com/#!/ligation |
Referências
- Gibson, D. G. Enzymatic assembly of overlapping DNA fragments. Methods in enzymology. 498, 349-361 (2011).
- Gibson, D. G., Smith, H. O., Hutchison, C. A., Venter, J. C., Merryman, C. Chemical synthesis of the mouse mitochondrial genome. Nature methods. 7, 901-903 (2010).
- Gibson, D. G., et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nature methods. 6, 343-345 (2009).
- Bowtell, D. D., Johnson, G. R., Kelso, A., Cory, S. Expression of genes transferred to haemopoietic stem cells by recombinant retroviruses. Molecular biology & medicine. 4, 229-250 (1987).
- Challita, P. M., Kohn, D. B. Lack of expression from a retroviral vector after transduction of murine hematopoietic stem cells is associated with methylation in vivo. Proceedings of the National Academy of Sciences of the United States of America. 91, 2567-2571 (1994).
- Lutzko, C., Senadheera, D., Skelton, D., Petersen, D., Kohn, D. B. Lentivirus vectors incorporating the immunoglobulin heavy chain enhancer and matrix attachment regions provide position-independent expression in B lymphocytes. Journal of Virology. 77, 7341-7351 (2003).
- Meilinger, D., et al. Np95 interacts with de novo DNA methyltransferases, Dnmt3a and Dnmt3b, and mediates epigenetic silencing of the viral CMV promoter in embryonic stem cells. EMBO reports. 10, 1259-1264 (2009).
- Chan, K. K., Wu, S. M., Nissom, P. M., Oh, S. K., Choo, A. B. Generation of high-level stable transgene expressing human embryonic stem cell lines using Chinese hamster elongation factor-1 alpha promoter system. Stem cells and development. 17, 825-836 (2008).
- Chung, S., et al. Analysis of different promoter systems for efficient transgene expression in mouse embryonic stem cell lines. Stem cells. 20, 139-145 (2002).
- Qin, J. Y., et al. Systematic comparison of constitutive promoters and the doxycycline-inducible promoter. PloS one. 5, e10611 (2010).
- Lund, A., Knudsen, S. M., Vissing, H., Clark, B., Tommerup, N. Assignment of human elongation factor 1alpha genes: EEF1A maps to chromosome 6q14 and EEF1A2 to 20q13.3. Genomics. 36, 359-361 (1996).
- Wallace, A. M., et al. Two distinct forms of the 64,000 Mr protein of the cleavage stimulation factor are expressed in mouse male germ cells. Proceedings of the National Academy of Sciences of the United States of America. 96, 6763-6768 (1999).
- MacDonald, C. C., McMahon, K. W. Tissue-specific mechanisms of alternative polyadenylation: testis, brain, and beyond. Wiley interdisciplinary reviews. RNA. 1, 494-501 (2010).
- Sabath, I., et al. 3'-End processing of histone pre-mRNAs in Drosophila: U7 snRNP is associated with FLASH and polyadenylation factors. Rna. 19, 1726-1744 (2013).
- Yang, X. C., et al. A complex containing the CPSF73 endonuclease and other polyadenylation factors associates with U7 snRNP and is recruited to histone pre-mRNA for 3'-end processing. Molecular and cellular biology. 33, 28-37 (2013).
- Grozdanov, P. N., Macdonald, C. C. High-Throughput Sequencing of RNA Isolated by Cross-Linking and Immunoprecipitation (HITS-CLIP) to Determine Sites of Binding of CstF-64 on Nascent RNAs. Methods in molecular biology. 1125, 187-208 (2014).
- Youngblood, B. A., Grozdanov, P. N., MacDonald, C. C. CstF-64 supports pluripotency and regulates cell cycle progression in embryonic stem cells through histone 3' end processing. Nucleic acids research. , (2014).
- Engler, C., Kandzia, R., Marillonnet, S. A one pot, one step, precision cloning method with high throughput capability. PloS one. 3, e3647 (2008).
- Irwin, C. R., Farmer, A., Willer, D. O., Evans, D. H. In-fusion(R) cloning with vaccinia virus DNA polymerase. Methods in molecular biology. 890, 23-35 (2012).
- Annaluru, N., et al. Total synthesis of a functional designer eukaryotic chromosome. Science. 344, 55-58 (2014).
- Gibson, D. G., et al. Creation of a bacterial cell controlled by a chemically synthesized genome. Science. 329, 52-56 (2010).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados