
Method Article
ギブソンアセンブリを使用して、ヒト伸長因子1αプロモーターの制御下でFLAGタグ化タンパク質を発現するプラスミドベクターの作製
要約
Synthesis of custom plasmids is labor and time consuming. This protocol describes the use of Gibson assembly cloning to reduce the work and duration of custom DNA cloning procedure. The protocol described also produces reliable tagged protein constructs for mammalian expression at similar cost to the traditional cut-and-paste DNA cloning.
要約
ギブソン·アセンブリ(GA)クローニングは、従来のDNAクローニング方法に、迅速で信頼性が高く、柔軟な代替手段を提供しています。我々は、マウス胚性幹細胞(たmESC)における外来遺伝子の発現のためのカスタマイズされたプラスミドを作成するためにGAを用いた。 SV40またはヒトサイトメガロウイルスプロモーターの制御下にある外来遺伝子の発現は、たmESCへのトランスフェクションの後にすぐに減少する。この減少した発現のための治療薬は、遺伝子発現を駆動するために、ヒト伸長因子1α(HEF1α)プロモーターを使用することである。 HEF1αを含むプラスミドベクターは、SV40-またはCMV-含むプラスミド、特にも含むN末端3xFLAGタグなどとして広く利用可能ではありません。ここで説明するプロトコルは、HEF1αプロモーターの表情調節下FLAGタグCstF-64とCstF-64変異体を発現するプラスミドを作成するための迅速な方法である。 GAは、可能なDNA断片の末端の重複クローニングを行うためにDNAエキソヌクレアーゼ、DNAポリメラーゼ及びDNAリガーゼのブレンドを使用する。私たちが利用できる持っていた鋳型DNAに基づいて、我々は我々の構築物は、単一のシーケンスに組み立てられるように設計されています。をpcDNA 3.1ベクター骨格、HEF1αプロモーターパート1、(二本鎖合成DNA断片として購入3xFLAGタグを含まれている)に従えば容易プロモーター部2、及びCstF-64または特定CstF-64のどちらかの変異体:私たちのデザインは、4つのDNA断片を使用していました。これらのフラグメントの配列は、DNA断片を生成するための適切なPCRプライマーを設計するためのプライマー生成ツールにアップロードされた。 PCR後、DNA断片は、選択マーカーを含むベクターと混合し、GAクローニング反応を組み立てた。個々の形質転換された細菌コロニーからプラスミドを単離した。プラスミドの初期画面は、配列決定に続いて、制限酵素消化によって行った。結論として、GAは、私たちは画面と検証を構築するなど、5日間での遺伝子発現のためにカスタマイズされたプラスミドを作成することができました。
概要
従来のDNAクローニング方法は、一緒にDNA断片を結合するためにDNAリガーゼとDNAを切断する制限酵素の使用に依存している。異なるDNA断片を含むカスタム発現構築物の生成は、一または複数の制限エンドヌクレアーゼおよびライゲーションを介してDNA断片の挿入に続くDNAの切断を含むシーケンシャルな手順である。この方法の主な欠点は、DNA断片のいずれかの適当な制限酵素は不可能関心の全長タンパク質の正常なDNAのクローニングをレンダリング( すなわち、いくつかの切断部位を有し得る)を識別することは困難かもしれないということである。そのため、カスタム式の生成は、カスタマイズタンパク質タグを用いる効率的な細胞型特異的プロモーターの転写調節下の構築物は、非常に慎重な設計が必要です。また、時間と労力のかかる手法である。最近では、いくつかの報告がmultipを組み立てるための方法論を説明した連続したシーケンスでル異なる合成DNA断片のいずれか一次元または二段反応で同時に制限を使用することなく1-3酵素 。ワンステップクローニング反応(すべての準備ステップを除く)は、DNAエキソヌクレアーゼ、DNAポリメラーゼ、DNAリガーゼ2,3及びDNA断片の重複端部( 図1)の混合物の使用に依存する。制限酵素の無駄がないので、(高度に反復配列を除く)任意のサイズおよび配列組成のDNA断片は、シームレス構造物で一緒に融合することができる。最近では、市販のキット(ギブソンアセンブリと、GA)ワンステップクローニング反応のために利用できるようになった。このキットは、カスタマイズされたプロモーターおよびタンパク質タグを有する単一のベクター内の任意のDNA断片の迅速かつ費用効率的な組み立てを可能にする。
哺乳動物細胞培養モデルにおいて外因性タンパク質を発現させるために使用広く入手可能なプラスミド発現ベクターは、転写regの下にあることが多いウイルス、サイトメガロウイルス(CMV)またはシミアンウイルス40(SV40)プロモーターのulation。これらのウイルスプロモーターは、哺乳動物細胞培養ベースのモデルの大部分において外因性タンパク質の強固な一過性発現を提供する。しかし、安定して外因性タンパク質を発現する細胞株の生成が原因確立プロセス4,5の間にCMVまたはSV40プロモーターの転写サイレンシングがしばしば成功しない。また、SV40およびCMVウイルスプロモーターが十分6,7リンパ系統または胚性幹細胞由来の細胞における外因性タンパク質の発現を促進しない。ウイルスプロモーターの固有の制限を解決するには、強力な構成非ウイルスプロモーター8-10を使用することです。ヒト由来の1つのよく特徴付け強力な構成の非ウイルスプロモーターは、伸長因子1α(HEF1α)プロモーター(HEF1αがリボソーム11にアミノアシルtRNAのGTP依存関連の触媒作用に関与している)である。しかし、HEF1αプロモーターを含む発現ベクターは、特にものはまた、目的のタンパク質のアミノ末端に3×FLAGを含むプラスミドを含むウイルスプロモーターとして広く利用できない。
64000 MW切断刺激因子タンパク質(CstF-64)は、複製依存ヒストンmRNAを14,15を含むほとんどのmRNA 12,13の3 '末端プロセシングに関与している。 CstF-64は、すべての体細胞組織12で表されます。そのRNA認識モチーフは、切断およびポリアデニルサイト16の下流新生転写上のGUリッチRNA配列に結合する。 CstF-64 mRNA前駆体への結合はこれが新生転写物の効率的なエンドヌクレアーゼ的切断を促進する。
ここでは、プロトコルは、3xFLAGタグ化メートルのカスタムベクトルを生成するためにDNAフラグメントのPCR増幅、(化学的コンピテント細菌細胞を含む)ギブソン·アセンブリクローニングキットを使用して、記載されているウーズCstF-64または変異CstF-64HEF1αプロモーター1の発現下でのアミノ末端まで。
プロトコル
プラスミドと重複プライマーの世代のシリコデザイン1.
注:このステップの目的は、構築物の完全なヌクレオチド配列をアセンブルし、GAのクローニングのための端部を重複するフラグメントを生成するために使用するプライマーを設計することである。
- 最終プラスミドを表現するための継続的な塩基配列を設計します。
- PCRでテンプレートとして使用される実際のプラスミドおよびDNA断片を取得またはリスト。
注:容易に入手できないDNA断片 - そのようなタグやプロモーターの異なる組み合わせとしては、 - 単一または複数の合成二本鎖DNA断片(SDNA)として注文することができます。これらの断片は、比較的低コストで商業的に入手可能であり、長さは2キロバイト( 材料の表を参照)までとすることができる。 - PCRに適したDNAフラグメントにステップ1.1で組み立てコンストラクトの連続ヌクレオチド配列を分割します。 Confirフラグメントが利用可能なプラスミドとSDNAフラグメントを比較メートル。 DNAは、NT 200より小さな断片は避けてください。
- プライマー生成ツールにアクセスします( 機器の表を参照)。 「セットの設定」メニューを選択します。 「変更ギブソンアセンブリ設定」のポップアップウィンドウ内の「変更PREFS」タブをクリックすることにより、適切な設定を選択してください。
注:プライマーの設計は、プライマーの生成ツールを使用せずに行うことができる。しかし、プライマー生成ツールを使用すると、プロセスを簡素化します。 - 「構築物の構築」メニューを選択することにより、構造物の構築を開始。最後の5 'から3'のプライマー生成ツールを順次に分割DNA断片を挿入する。
NOTE:プラスミドバックボーンとして使用されたDNA断片は、好都合に2分割することができる。最終的なPCR産物の最後の断片の5 '末端、この最初のフラグメントと3'末端は、連続した1つのDNに連結されているベクター骨格を表すフラグメント。 - 「ベクトルまたは挿入断片を入力して「ポップアップウィンドウにFASTA形式のベクターDNA(ヌクレオチド配列のテキストベースの表現)の5 '末端を表す第DNA断片を貼り付けます。 DNA断片に名前を付けます。どちらPCR、REダイジェストや合成など、DNA断片を得るための適切な方法を選択してください。 「CONTINUE」タブをクリックします。
- 「アセンブリに挿入断片を追加」ウィンドウに表示される「FWDまたは改訂プライマースペーサー」はスペースを使用し、最終的な構造物の接合部で余分なヌクレオチド/制限部位を付加する必要がある場合。両方のフラグメントにのみ1 DNA断片のとしないように余分なヌクレオチド/制限部位を追加します。 「DONE」タブをクリックします。
- 構築物が完了するまで、すべてのフラグメントのために繰り返します。 「表示プライマー」メニューを選択し、プライマー配列を確認します。
- (すべての構築物についてベクトルbを繰り返しますackbonesおよび挿入)、または異なるDNA断片である。
ホットスタート校正DNAポリメラーゼを用いたPCRによりDNA断片の2増幅(2Xマスターミックス)
注:このステップの目的は、PCRを使用して、集合反応のための十分なDNAを得ることである。
- 脱塩した製品として、前のステップでかつ可能な限り最小のスケールで設計されたPCRプライマーを購入します。水中10μMまたはTE(10mMのトリス-HCl、pHが7.9、1mMのEDTA)にそれらを希釈する。
- SDNA断片として容易に入手できないDNA断片を購入する。
- 水中での1ng /μlのへのPCRのテンプレートとして使用されるSDNAフラグメントを含む全てのDNAフラグメントを、希釈する。
- 室温でのPCR反応を組み立てます。簡単に説明すると、プライマー対の各プライマー(10μMストック溶液から)を2.5μl、鋳型DNA断片(の1ng /μlのストック溶液から)を1μl、25μを使用ホットスタートプルーフリーディングDNAポリメラーゼのLと19μlの水(; 2XマスターミックスDNA POLはマテリアルの表を参照のこと)。穏やかなフリックでチューブを混合し、短時間の遠心分離によって液滴を集める。
- DNAのPOL用の推奨に従って同じようなサイズの別々のチューブのDNA断片を同時に増幅する。 28 PCRサイクルまたは十分なDNAの収量を生産するサイクル数を決定する - 25を実行します。
- エチジウムブロマイド(0.2 / mlの最終濃度)で染色し、標準的なアガロースゲル電気泳動でPCR反応容量(5μl)を、10%を実行する。 PCR産物を表す単一のDNAバンドが表示されていることを確認してください。 DNA分子量標準を用いたDNA断片の大きさと相対量を決定します。必要に応じて、DNAフラグメントの十分な量を得るために、PCRを繰り返す。
PCR産物の3のDpnI消化
注:このステップの目的は、DIGEにあるDpnIを第2節でのPCRからのSTの残留プラスミド鋳型DNAは、 ダム+細菌の菌株で成長させプラスミドDNAに生じるような、それがメチル化されている場合にのみ、プラスミドDNAを消化し ます。プラスミドDNAは、GA反応におけるDNA断片として使用される場合、したがって、DpnIで治療しない。
- 1時間37℃でインキュベート第2節で生産されたPCR産物の45μlに制限酵素DpnIの2を添加する。必要になるまで-20℃で第4節又は凍結を続行します。
4.精製およびDNA精製磁気ビーズ上のDNA断片の濃度
注:このステップの目的は、セクション2および3の他のPCR精製法で得られたPCR産物を同様に使用することができる、精製·濃縮することである。
- 室温にDNAを精製した磁気ビーズを平衡化する。短時間のボルテックスでビーズを再懸濁。
- のPCRを予め消化のWiを転送1.5ミリリットルチューブにDpnIを目と各チューブにDNA精製磁気ビーズの81μlを添加する。 10分間室温で混合物をインキュベートする。
- 約2分間の磁気コレクタ上にチューブを置きます。ピペットを用いて透明な液体を捨てる。 30秒間の80%エタノール200μlで二回洗浄する。
- ペレットが乾燥することができます。磁気コレクタ上に配置チューブを、オープンチューブの蓋をしてください。
- 再懸濁10mMトリス-HCl、pH8.0の10μlの乾燥したビーズ。 2分間室温でインキュベートする。チューブの底に液体を収集するために簡単にスピン。
- 2分間磁気コレクターにチューブを置きます。澄んだ溶液10μlと新予め標識チューブに入れ - 8.5を削除します。 UV分光法によるDNA断片の濃度を測定( 機器の表を参照)。
E.における製品の5.組み立てクローニング反応と変容大腸菌の
注:挿入物比1:ベクトル集合反応を実行するこのステップの目的は、3を計算することである。- ベクターバックボーンまたは選択マーカーを有するDNA断片を表すDNAフラグメントの少なくとも100ngのを使う。インサートとして使用されるDNA断片のための3倍モル過剰量を計算する。
- それぞれの特定の挿入物の必要なNGで、モル過剰に変換します。
注:計算を行うための便利な方法は、Webベースのアプリケーションを使用することです( 機器の表を参照)。 - 10μlに音量を調整する、PCRチューブにDNAフラグメントの計算された量を混合する。 GAマスターミックス10μlのを追加します(2X、 マテリアルの表を参照のこと)。 6断片またはPCRサーマルサイクラーで2-3の断片の組み立てのために15分 - 4の組み立てのために1時間50℃で反応をインキュベートする。
- コンピテントで組立製品の転換を進めます大腸菌や-20℃で製品を凍結する°Cは、必要になるまで。
- 化学薬品やエレクトロコンピテント細胞に付随する形質転換手順に従ってください。通常、形質転換反応あたりの集合反応2μlのを使用しています。
- 変換が完了した後、適切な選択抗生物質を補充した寒天プレート上で形質転換細胞を広げた。
6.プラスミド単離、制限酵素消化およびシーケンシング
注:このステップの目的は、EからプラスミドDNAを単離することである大腸菌は 、その後制限消化および配列決定によって構成物を確認します。
- ミニプレップとプラスミド単離するためのいくつかの単一コロニーを伝播します。適切な抗生物質を補充した2〜5ミリリットル一晩のLB液体培地を使用してください。使用されているミニプレップキットに記載されている手順に従うことにより、対応するプラスミドを分離します。
- 分光を用いて得られた量とプラスミドの濃度を決定する光度計。
- 1または精製プラスミドのいくつかの制限エンドヌクレアーゼと〜0.5μgの制限消化を実行します。 DNA構築のための消化特性の異なるパターンを提供する制限酵素を使用してください。
- 特定のまたは標準的なプライマーを用いてDNA配列決定によりDNAクローニングの成功を確認する。精度のための配列決定データを分析します。
結果
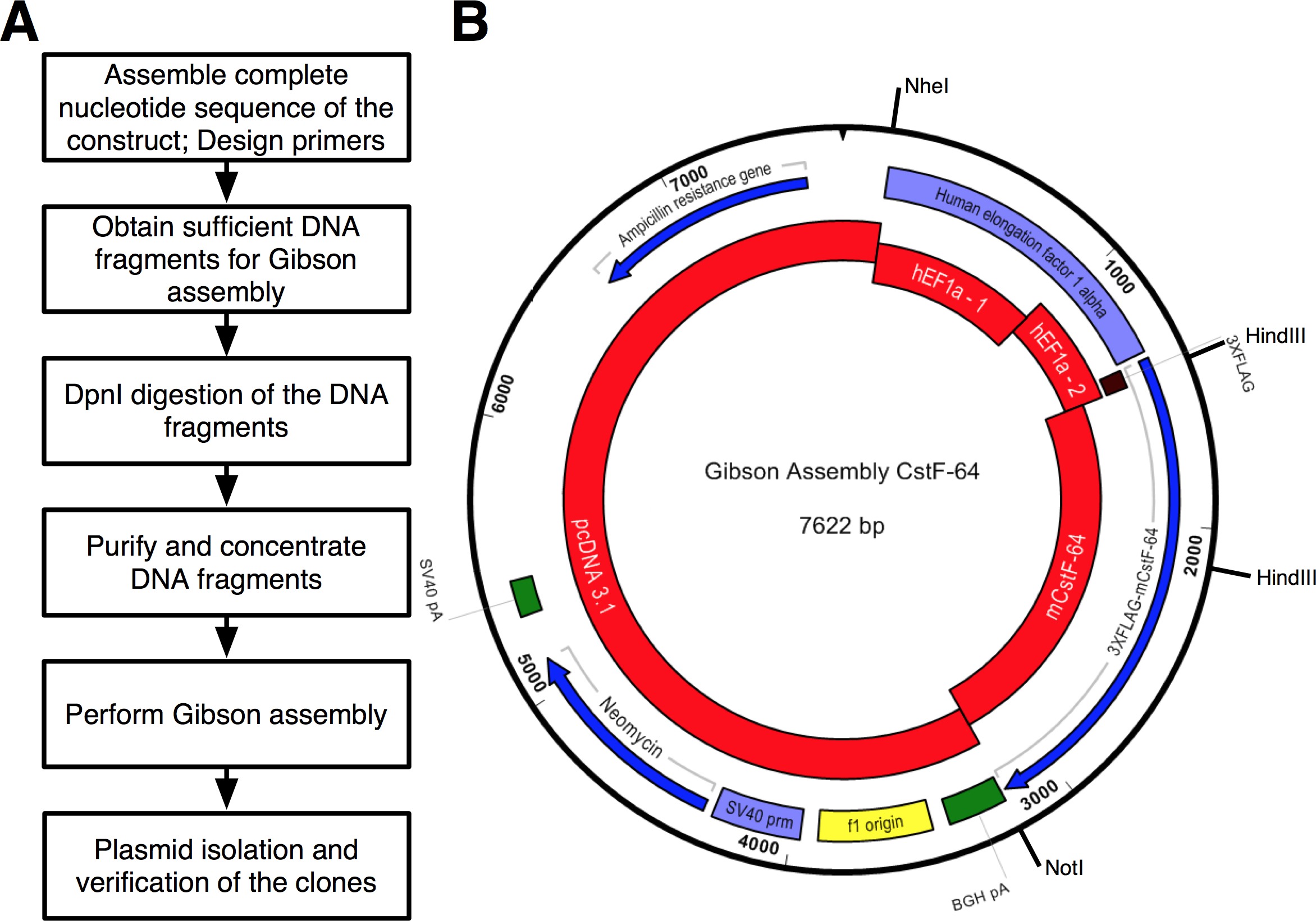
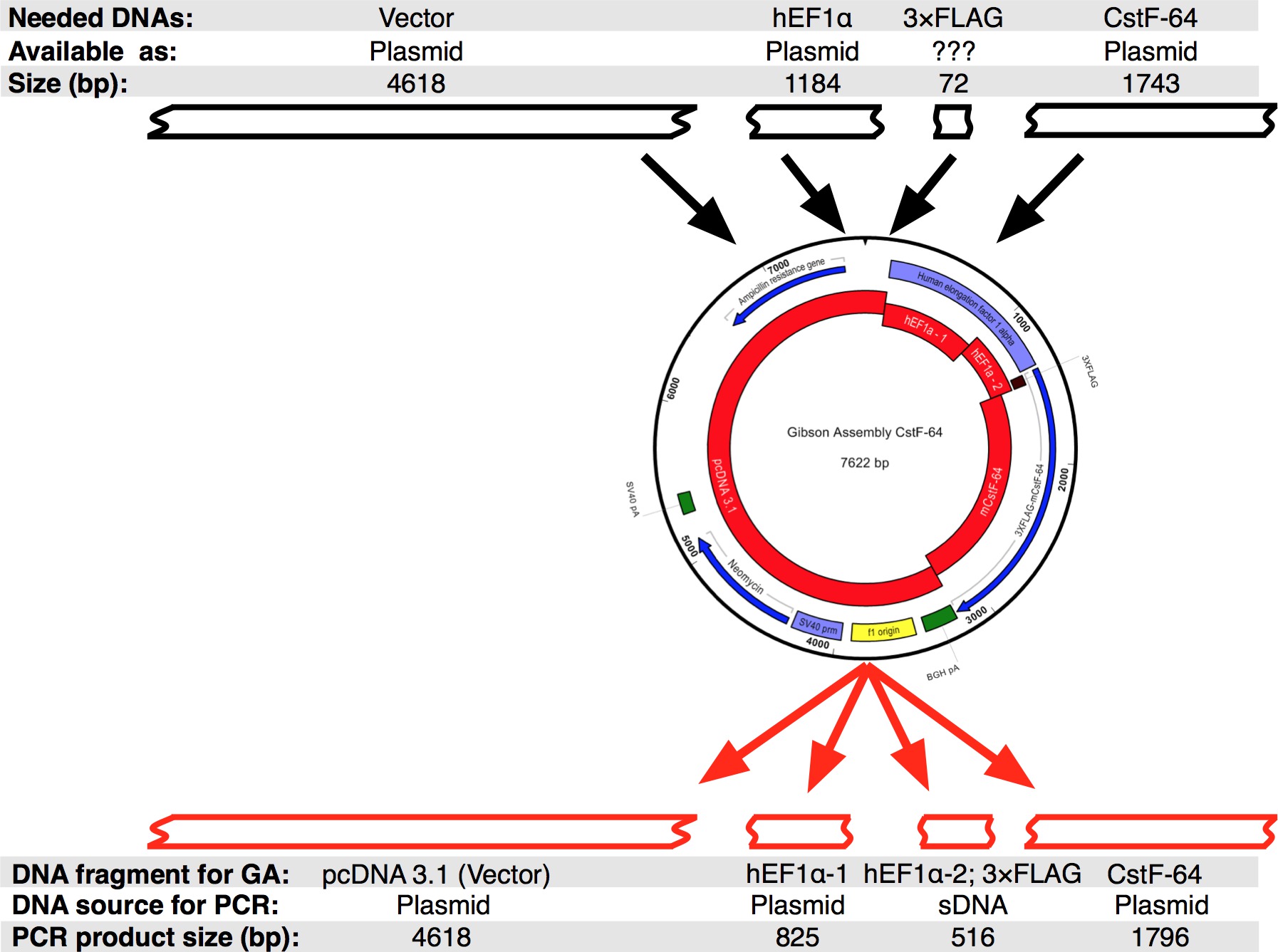
続いたプロトコルのワークフローは、 図2Aに示されている。私たちは、HEF1αプロモーターの表情レギュレーション( 図2Bおよび図3)の下で3xFLAGタグに融合CstF-64及び変異CstF-64タンパク質のクローンを作成したかった。 HEF1αを含むプラスミドは、私たちには利用できなかった3xFLAGタグが続く。ただし、以下のプラスミドが利用可能であった:をpcDNA 3.1 MYC-彼の(A;ミカエラヤンセンからの寛大な贈り物)、HEF1α含むプラスミド(ムラデンYovchevからの寛大な贈り物)およびマウスCstF-64プラスミド12( 図3)。構築物(単数または複数)のための全体の配列は、ヌクレオチドおよびテキスト編集アプリケーション(; 機器の表を図3を参照) を使用して組み立てた。その後、配列(単数または複数)は、利用可能なプラスミドDNAに対応する4つの便利なピース( 図2B、赤ブロックおよび図3)に分割した。増幅Primers 4の制約( 機器の表を参照)プライマー生成ツールを使用して設計された- 25の最小限のオーバーラップで6断片NT、「変更ギブソンアセンブリ設定」ポップアップウィンドウで設定します。 NheI及びNotI制限部位は、適切に組み立てられたプラスミドを同定する目的のためのプライマーの設計に含めた。 NheI部位にサブクローニングさとHEF1αプロモーターの5 '末端との間のプライマー配列に位置している。のNotIサイトはCstF-64とのpcDNA 3.1ベクター骨格の終止コドン(UGA)の後に位置しています。 HEF1αプロモーターからなる酵素DNA断片の両方で同時消化により、3xFLAGとCstF-64または変異CstF-64が発売されます(下記参照および図2B)。プライマーは、可能な限り最小の規模で注文して脱塩した。 3xFLAGタグDNA断片(490 bpの、 図2B、図3)を含むHEF1αプロモーターの第2の部分は、単一のSDNAフラグメント(単数として購入した。材料の ee 表 )。アセンブリ反応に使用されるDNA断片は、DNA polの( 材料の表を参照)を用いて増幅した。 HEF1αプロモーターパート1、HEF1αプロモーターパート2、全長及び変異CstF-64のDNA断片が、DNA POLの供給業者の推奨に従って、28サイクル( 図4A)のために別々のチューブで同時に増幅した( 材料の表を参照してください。各サイクルの変性、98℃、アニーリング55℃で45秒、伸長72℃90秒)で7秒であった。最初に、をpcDNA 3.1骨格は、(72℃で3分とした伸長時間を除いて、上記と同じ条件を用いて)、22サイクル増幅した。しかし、得られたDNAの収量は、アセンブリ反応( 図4A)で使用するには十分ではなかった。したがって、追加の増幅は十分なDNAを得た。
PCR産物を得るプラスミド鋳型からedはそうでなければ結果のアセンブリ反応生成物を汚染すると、偽陽性の薬剤耐性細菌コロニーを生成したプラスミドDNAを除去するためのDpnI制限酵素で消化されなければならない。従って、PCR産物は、切断するメチル化および半メチル化プラスミドDNAは、 ダム+ E.から単離された制限酵素DpnIで消化し た。 coli株 。化学的に合成されたDNAはメチル化またはヘミ - メチル化塩基を含んでいないので、テンプレートはDpnIで消化する必要がないように、PCR産物は、合成DNA断片を用いて得られた。
プロトコルのステップ4に記載のようにDNA断片ををpcDNA 3.1ベクター骨格のためのPCRとを組み合わせた使用DNA精製磁気ビーズの量に応じて調整した( 材料の表を参照)DNA精製磁気ビーズ上で精製し、濃縮した。 DNA収量を決定した分光光度計( 機器の表1および表 ) を使用して。 CstF-64及び変異CstF-64コンストラクトのアセンブリ反応は、氷( 表1)上で組み立てた。 「インサート」と考えDNA断片の3倍モル過剰( 表1、図3)を用いた。混合DNA断片の最終容量を水で10μlに調整し、組立マスターミックス(2×)10μlを添加した。反応物を混合し、1時間50℃でインキュベートした。ポジティブコントロール反応もGAキットのマニュアルの勧告に従って組み立ててCstF-64及び変異CstF-64の反応と同時にインキュベートした。プロトコールで推奨されているように、組立反応のそれぞれの2μlを化学的にコンピテントに転換した大腸菌は、アセンブリクローニングキット( 材料の表を参照)が供給。キットに記載のように形質転換を行ったマニュアル。陽性クローンは、アンピシリン寒天/ LBプレート上で選択した。各アセンブリ反応あたり6個のコロニーを無作為に伝播されるように選択された。プラスミドDNAは、( 材料の表を参照) をプラスミド単離ミニキットを用いて単離した。制限を有する構築物のインシリコ消化はNheIおよびNotIでは4590 BP、CstF-64と4590 bpのための3032塩基対のサイズの二つの断片が得られた酵素で 、変異体CstF-64( 図2Bおよび図4B)のための2711 bpの。制限での消化をHindIIIおよびNotIは、以下のサイズを持つ3つの断片が得られた酵素:(5872塩基対、1005塩基対、および745塩基対(CstF-64)と5872塩基対、1005塩基対、および424塩基対の変異体CstF-64、 図2B及び図4C)。確かに、単離したプラスミドの消化は、予想される特徴的なパターン( 図4B、C)を表示さ。 CstF-64変異体のHindIIIおよびNotIでの消化によって生成さ424 bpのDNA断片ことに注意してください図4Cにプラスミドが弱く、その小さいサイズのために染色される。 6単離したプラスミドのうち、図2に示すように、配列決定のために送った。我々は欠失、挿入または置換がないことを確認するために従えば容易にプロモーターを配列決定し、CstF-64または変異CstF-64コンストラクトの部品。我々は非常にこのまたはPCRベースのプロトコルから得られたDNA構築物のシークエンシングをお勧めします。配列決定は、各配列決定されたプラスミドの一つがHEF1α、3xFLAGタグとCstF-64または変異CstF-64の領域で期待されるシーケンスを含んでいることを示した。他のプラスミドのそれぞれは、対応するDNA断片の増幅中に導入された点突然変異を有していた。マウス胚性幹細胞にCstF-64を含有するプラスミドの発現は、野生型の表現17と同等の外因性タンパク質の豊富な量を産生した。

ギブソン組立機構の1模式図。重複末端を有するDNA断片を等温的に単一の連続した順序で組み立てられた。重複は最初徐々に熱不活性化されている5 'エキソヌクレアーゼによってバック噛むされ終了します。このため、重複する端部と異なるDNA断片が等温的にアニーリングする。 DNAポリメラーゼは、ギャップを埋めるし、耐熱性DNAリガーゼがニックを結紮する。 この図の拡大版をご覧になるにはこちらをクリックしてください。
{kind=link}

図2のアセンブリクローニングに記載したプロトコルの(A)のフローチャート。 (B)プラスミドの表現、てpGA CstF-64 GAキットを使用して生成された赤-集合反応で使用されるDNA断片:をpcDNA3.1。 HEF1αプロモーターパート1(hEF1a - 1); HEF1αプロモーターパート2(hEF1a - 2)合成DNAとして注文。マウスCstF-64(mCstF-64)。ブルー - オープンリーディングフレーム。バイオレット - ウイルスおよび非ウイルスプロモーター。グリーン- 。切断およびポリアデニル化領域は、 この図の拡大版をご覧になるにはこちらをクリックしてください。
{kind=link}

ギブソンアセンブリCstF-64プラスミドの図3.デザイン。ブラックボックスは、インシリコシーケンスでシングルギブソン組立CstF-64を設計することが可能であったDNA断片を表す。続いて、配列をPCRによって増幅した4つのDNA断片、に分けた。原因3xFLAGタグのサイズが小さい配列が一緒に従えば容易にPRしてSDNAとして設計されたことに注意してください omoterパート2。 この図の拡大版をご覧になるにはこちらをクリックしてください。
{kind=link}

クローニング反応および得られたプラスミドの代表的な制限酵素消化に使用されるDNA断片の図4. PCR(A)代表のPCRホットアセンブリ反応に使用されるDNA断片のための高忠実度の2×マスターミックスを開始使用して:(B)代表HEF1α、完全長CstF-64、のpcDNA 3.1構成体(PGA-CstF-64)とHEF1α、変異体CstF-64、のpcDNA 3.1構成体(PGA-mutCstF-64)NheIおよびNotIで消化のプラスミド。 (C)Bと同様のプラスミドをHindIIIおよびNotI酵素で消化し た。"_blank">この図の拡大版をご覧になるにはこちらをクリックしてください。
| DNAフラグメントの名前 | 予想されるサイズ(bp)の | Concn。 (ng /μLで) | に希釈した(ng /μLで) | 希薄化後からGA CstF-64で使用μL | μlの希釈から、GA mutCstF-64で使用される | モル比(INS:VEC) |
| をpcDNA 3.1(ベクトル) | 4618 | 158 | 希釈されていない | 1 | 1 | |
| hEF1_プロモーターパート1 | 825 | 213 | 75 | 1 | 1 | 3:1 |
| CstF-64用のhEF1_プロモーターパート2 | 516 | 229 | 50 | 1 | 3:1 | |
| Cstf-64 | 1,796 | 161 | 希釈されていない | 1 | 3:1 | |
| 変異体CstF-64用のhEF1_プロモーター部2 | 516 | 199 | 50 | 1 | 3:1 | |
| 変異体CstF-64 | 1,448 | 201 | 171 | 1 | 3:1 |
表1.磁気ビーズ上に濃縮した後のDNA断片の収量、希釈およびアセンブリ反応のセットアップ。
ディスカッション
GAクローニングの成功した使用は、常に完全な構築物( 図2と図3)の注意深い設計が先行されるべきである。
プライマー生成ツールによって設計されたプライマー配列の慎重な検証も強くお勧めします。 GAのためのプライマーは、プライマー生成ツールを使用せずに生成されてもよい。しかし、ツールの使用は、プロセスを簡素化しているため非常にお勧めです。一般的に、GAクローニングのためのプライマーは、2つの機能的に異なる配列を持っている必要があります。第一の配列は、DNA断片特異的であり、PCRを用いて、断片の増幅を可能にする。第二の配列は、GAアセンブリのために必要である隣接フラグメント、と重なっている。典型的なDNA断片特異的配列の長さは18〜22 ntのだろう。同じDNAを増幅するために使用したDNA断片の特定の配列は、類似の融解温度及びGC含量を持っている必要があります。重複配列は、15の少なくともあるべきである塩基、少なくとも48℃の融解温度を有する、長さ。 4つ以上のDNAフラグメントのアセンブリは、少なくとも20塩基であることが重複配列を必要とする。長い重複がより適切に組み立てられたDNA断片をもたらしアニーリングの特異性の増加が可能になります。それは、歪んだ配列は、適切なDNAアセンブリを損なう可能性があるため、彼らのGCまたは重複配列を開発する上でコンテンツATスキューされる配列を避けることをお勧めします。
また、単にDNA断片はGA反応においてベクター骨格に対応する、任意の薬剤耐性細菌コロニーを生成するべきでない、ネガティブコントロールとして使用して示唆している。あるいは、「挿入」を含むDNA断片のいずれもない薬剤耐性細菌コロニーを生じるはずであるGA反応から省略され得る。はコロニーが成長しません理由はCOMPLの組み立てをレンダリングします隣接するDNA断片の重複両端の欠如、となりますETEプラスミド。
プロトコルにおいてプライマーセットを生成するためのnt 25の重複配列、記載があるため( 図3)を使用したDNA断片の数、使用された。 6 DNAフラグメント-プライマー生成ツールウェブサイトの推薦4を組み立てるためには、少なくとも20塩基の重複配列を使用することである( 機器の表を参照)。また、長いDNA鎖の適切な相補性を確保する重複配列を正確に組み立てられた製品の数を増加させる( 図1参照)。
現在、シームレスクローニングするためのいくつかのシステムが利用可能です。しかしながら、これらのシステムのいくつかは、まだ( すなわち、ゴールデンゲート18をクローニング)制限酵素を使用。他のものは、同一の生物学的供給源19からのワクシニアウイルスDNAポリメラーゼおよび一本鎖DNA結合タンパク質に基づく酵素の独自のブレンドを使用する。両方のシステムは、ショアによりGAに比べて限定されている重複配列のTERの長さ。短い重複配列が問題のある23以上のDNA断片の適切なアセンブリをレンダリングする、隣接するDNA断片のアニーリング工程に十分な特異性を提供しない可能性があるため。これらの欠点は、GAシステムには存在しない。
(長さ、すなわち、8未満kbpの)PCRにより確実に増幅可能な大きさを超えないように、増幅されるDNA断片の大きさも考慮されるべきである。でも、過去10年間におけるDNAポリメラーゼの機能の改善で、大きなDNA断片は、以下の効率および精度で増幅される。必要に応じて、より大きなDNAフラグメントは、プラスミド単離および制限酵素による適切なDNA消化により、例えば PCRに代わる他のソースから得られるであろう。具体的には、PCRを使用して私たちの合理的な現在の原稿に記載されているプロトコル、すべての未満であった利用可能なDNA断片の大きさに基づいていた5 kbpの。アガロースゲル電気泳動によって同定し、複数のPCR産物が存在する場合、望ましい断片サイズのゲル精製は、利用可能な分子生物学の技法または適切なキットのいずれかを使用して推奨される。現在のプロトコルでは、熱安定性DNAポリメラーゼは、( 材料の表を参照)が使用される。しかし、高い忠実度および収率を提供する任意のDNAポリメラーゼは、このプロトコルで使用するのに適しているであろう。プロトコールに記載さとは異なる高忠実度DNAポリメラーゼが利用可能な場合は、それぞれのマニュアルに記載されているように、セットアップ条件を使用しています。現在のプロトコルでは、化学的にコンピテントE. coli細胞を、GAキットに付属しているが使用される。また、化学的または電気コンピテントE.そのようなDH5α又はDH10Bなどの大腸菌株を使用することができる。
2Xマスターミックスをクローニングアセンブリは、最小限のハンズオンタイムで使いやすいです。しかし、正確なピペッティングは、NEが小さいため、ボリュームが要求される一緒に混合するeded。良好な分子生物学技術は、同様に、常に注意が必要である。
GAのクローニングは、サイズが3 kbpのより長いDNA断片、プラスミドおよびベクターの構築のための無限の可能性を提供しています。それは例えば、全体の細菌( マイコプラズマ·ミコイデス )ゲノムまたは酵母(サッカロミセス·セレビシエ)染色体20,21の合成およびアセンブリを可能にするため、また、それは、合成生物学の分野において広範な影響を有する。技術はまた、シームレスな構築物を生成する従来のクローニングニーズに適用可能である。
結論として、GAのクローニングは、従来のDNAクローニング手順に、迅速な信頼性と柔軟な代替手段を提供しています。
開示事項
Open Access publication and production fees were supplied by New England BioLabs Inc.
謝辞
我々は気前よく提供するpcDNA 3.1 MYC-彼の(A)とHEF1αプロモーターを含むプラスミドについて、テキサス工科大学健康科学センター、ラボック、テキサス州とムラデンYovchevピッツバーグ大学医療センターでは、ピッツバーグでのPAをミカエラヤンセンに感謝したいと思います。この出版物で報告された研究は、受賞番号(CCM)はR01HD037109の下で成育医療国立衛生研究所の人間開発のユーニス·ケネディ·シュライバー国立研究所によってサポートされていました。追加のサポートは、女性の健康のためにローラ·W·ブッシュ研究所(CCMおよびPNG)であった。内容はもっぱら著者の責任であり、必ずしも国立衛生研究所の公式見解を示すものではありません。
著者は、彼らが競合する経済的利益を持っていないことを宣言。
資料
| Name | Company | Catalog Number | Comments |
| Synthetic double-stranded DNA fragment (sDNA) | Integrated DNA Technologies | We used gBlocks, which can be up to 2 kb in length. However, there are many commercial sources of synthetic DNA available. | |
| DNA purification magnetic beads | Beckman Coulter | A63880 | Agencourt AMPure XP - PCR Purification system |
| Plasmid Isolation Mini Kit | Omega Bio-Tek Inc | D6942-01 | E.Z.N.A. Plasmid Mini Kit I |
| Gibson Assembly Cloning Kit | New England BioLabs | E5510S | |
| DNA polymerase | New England BioLabs | M0494S | Q5 Hot Start High Fidelity 2x Master Mix |
| DpnI | New England BioLabs | R0176S | |
| NheI | New England BioLabs | R3131S | |
| NotI | New England BioLabs | R3189S | |
| HindIII | New England BioLabs | R3104S | |
| Spectrophotometer | Thermo Scientific | NanoDrop device | |
| EditSeq | DNASTAR | Part of Lasergene Core Suite | |
| SeqBuilder | DNASTAR | Part of Lasergene Core Suite | |
| Word | Microsoft | Part of Microsoft Office | |
| NEBuilder | New England BioLabs | primer generation tool: http://nebuilder.neb.com | |
| NEBioCalculator | New England BioLabs | ligation calculator: http://nebiocalculator.neb.com/#!/ligation |
参考文献
- Gibson, D. G. Enzymatic assembly of overlapping DNA fragments. Methods in enzymology. 498, 349-361 (2011).
- Gibson, D. G., Smith, H. O., Hutchison, C. A., Venter, J. C., Merryman, C. Chemical synthesis of the mouse mitochondrial genome. Nature methods. 7, 901-903 (2010).
- Gibson, D. G., et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nature methods. 6, 343-345 (2009).
- Bowtell, D. D., Johnson, G. R., Kelso, A., Cory, S. Expression of genes transferred to haemopoietic stem cells by recombinant retroviruses. Molecular biology & medicine. 4, 229-250 (1987).
- Challita, P. M., Kohn, D. B. Lack of expression from a retroviral vector after transduction of murine hematopoietic stem cells is associated with methylation in vivo. Proceedings of the National Academy of Sciences of the United States of America. 91, 2567-2571 (1994).
- Lutzko, C., Senadheera, D., Skelton, D., Petersen, D., Kohn, D. B. Lentivirus vectors incorporating the immunoglobulin heavy chain enhancer and matrix attachment regions provide position-independent expression in B lymphocytes. Journal of Virology. 77, 7341-7351 (2003).
- Meilinger, D., et al. Np95 interacts with de novo DNA methyltransferases, Dnmt3a and Dnmt3b, and mediates epigenetic silencing of the viral CMV promoter in embryonic stem cells. EMBO reports. 10, 1259-1264 (2009).
- Chan, K. K., Wu, S. M., Nissom, P. M., Oh, S. K., Choo, A. B. Generation of high-level stable transgene expressing human embryonic stem cell lines using Chinese hamster elongation factor-1 alpha promoter system. Stem cells and development. 17, 825-836 (2008).
- Chung, S., et al. Analysis of different promoter systems for efficient transgene expression in mouse embryonic stem cell lines. Stem cells. 20, 139-145 (2002).
- Qin, J. Y., et al. Systematic comparison of constitutive promoters and the doxycycline-inducible promoter. PloS one. 5, e10611 (2010).
- Lund, A., Knudsen, S. M., Vissing, H., Clark, B., Tommerup, N. Assignment of human elongation factor 1alpha genes: EEF1A maps to chromosome 6q14 and EEF1A2 to 20q13.3. Genomics. 36, 359-361 (1996).
- Wallace, A. M., et al. Two distinct forms of the 64,000 Mr protein of the cleavage stimulation factor are expressed in mouse male germ cells. Proceedings of the National Academy of Sciences of the United States of America. 96, 6763-6768 (1999).
- MacDonald, C. C., McMahon, K. W. Tissue-specific mechanisms of alternative polyadenylation: testis, brain, and beyond. Wiley interdisciplinary reviews. RNA. 1, 494-501 (2010).
- Sabath, I., et al. 3'-End processing of histone pre-mRNAs in Drosophila: U7 snRNP is associated with FLASH and polyadenylation factors. Rna. 19, 1726-1744 (2013).
- Yang, X. C., et al. A complex containing the CPSF73 endonuclease and other polyadenylation factors associates with U7 snRNP and is recruited to histone pre-mRNA for 3'-end processing. Molecular and cellular biology. 33, 28-37 (2013).
- Grozdanov, P. N., Macdonald, C. C. High-Throughput Sequencing of RNA Isolated by Cross-Linking and Immunoprecipitation (HITS-CLIP) to Determine Sites of Binding of CstF-64 on Nascent RNAs. Methods in molecular biology. 1125, 187-208 (2014).
- Youngblood, B. A., Grozdanov, P. N., MacDonald, C. C. CstF-64 supports pluripotency and regulates cell cycle progression in embryonic stem cells through histone 3' end processing. Nucleic acids research. , (2014).
- Engler, C., Kandzia, R., Marillonnet, S. A one pot, one step, precision cloning method with high throughput capability. PloS one. 3, e3647 (2008).
- Irwin, C. R., Farmer, A., Willer, D. O., Evans, D. H. In-fusion(R) cloning with vaccinia virus DNA polymerase. Methods in molecular biology. 890, 23-35 (2012).
- Annaluru, N., et al. Total synthesis of a functional designer eukaryotic chromosome. Science. 344, 55-58 (2014).
- Gibson, D. G., et al. Creation of a bacterial cell controlled by a chemically synthesized genome. Science. 329, 52-56 (2010).
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請さらに記事を探す
This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved