
Method Article
Una plataforma de metil-Seq de rata para identificar cambios epigenéticos asociados a la exposición de estrés
En este artículo
Resumen
Aquí, describimos el protocolo y la implementación de metil-Seq, una plataforma de epigenómica, utilizando un modelo de rata para identificar cambios epigenéticos asociados a la exposición de estrés crónico. Los resultados demuestran que la plataforma de metil-Seq de la rata es capaz de detectar diferencias de metilación que surgen de la exposición del estrés en ratas.
Resumen
Medida genomas de una amplia variedad de animales, hay una creciente necesidad de herramientas que pueden capturar dinámicos cambios epigenéticos en estos modelos animales. La rata es un animal modelo en particular donde una herramienta epigenética puede complementar muchos estudios farmacológicos y de comportamiento para proveer valiosa información mecanicista. Para ello, adaptamos el SureSelect sistema de captura de objetivo (denominado metil-Seq) de la rata, que puede evaluar los niveles de metilación de ADN en el genoma de la rata. El diseño de la rata había dirigida promotores, las islas de CpG, costas de la isla y regiones ricas en GC de todos los genes RefSeq.
Para implementar la plataforma en un experimento de rata, ratas Sprague Dawley machos fueron expuestas a estrés crónico variable durante 3 semanas, después de lo cual se recolectaron muestras de sangre para la extracción de ADN genómica. Metil-Seq bibliotecas fueron construidas de las muestras de ADN de rata por corte, ligadura de adaptador, enriquecimiento objetivo, conversión de bisulfito y multiplexación. Las bibliotecas fueron ordenadas en una plataforma de secuenciación de próxima generación y las lecturas secuenciadas fueron analizadas para identificar DMRs entre ADN de ratas tensionadas y unstressed. DMRs candidato superior fueron validados independientemente por pirosecuenciación de bisulfito para confirmar la robustez de la plataforma.
Los resultados demuestran que la plataforma de metil-Seq de rata es una útil herramienta epigenética que puede captar cambios de metilación inducidos por la exposición al estrés.
Introducción
Avances en la secuenciación de alto rendimiento han llevado a una gran cantidad de secuencias genómicas para el modelo y organismos no-modelo. La disponibilidad de tales secuencias ha facilitado la investigación en genética, genómica comparativa y transcriptómica. Por ejemplo, secuencias genómicas disponibles son altamente útiles para alinear los datos de la secuencia de experimentos de ChIP-Seq que enriquecen de ADN basado en su asociación con histona modificaciones1o secuenciación de bisulfito, que mide la metilación del ADN por detección de uracilo forma de conversión de bisulfito de unmethylated cytosines2. Sin embargo, ha habido retrasos en la implementación de plataformas epigenómicas que incorporan datos de secuenciación disponibles en su diseño debido a la falta de datos comentadas de secuencias reguladoras específicas de especies que pueden influir en funciones de los genes.
En particular, la metilación del ADN es uno de las más ampliamente estudiadas modificaciones epigenéticas en el ADN que pueden aprovechar los datos genómicos disponibles para la construcción de una plataforma de methylomic. Un ejemplo es una plataforma basada en arreglos de discos para el metiloma humano3, que ha sido ampliamente utilizado en diversas disciplinas de la oncología a psiquiatría4,5. Por desgracia, plataformas similares para los modelos animales no humanos son escasas, ya que hay prácticamente no hay plataformas ampliamente utilizado que han aprovechado la secuencia genomic en su diseño inicial.
Un método común para evaluar el paisaje de methylomic de modelos animales no humanos es reducida representación bisulfito de secuenciación (RRBS)6. Este enfoque supera el coste de la secuenciación de genoma completo bisulfito que, ofreciendo un paisaje de methylomic integral, ofrece cobertura a profundidad de lectura inferior debido al costo y limitada información funcional en grandes áreas pobres en genes del genoma2 . RRBS implica restricción recopilación y selección del tamaño de ADN genómico para enriquecer para altamente rico en GC secuencias como las islas de CpG que son comúnmente encontradas cerca de promotores de genes y se cree que desempeñan un papel en la regulación de genes7. Mientras que el método RRBS se ha utilizado en un número de importantes estudios, su dependencia de las enzimas de restricción no es sin limitaciones y desafíos notables. Por ejemplo, enriquecimiento de secuencias ricas en GC en RRBS es enteramente dependiente de la presencia de secuencias específicas reconocidas por la enzima de restricción y posterior preselección por electroforesis. Esto significa que se excluyen las áreas genómicas que contienen estos sitios de restricción durante la selección de tamaño. Además, las comparaciones entre especies están desafiando a menos que los mismos sitios de restricción están presentes en el mismo loci entre las diferentes especies.
Un enfoque para superar las limitaciones de RRBS es utilizar un método de enriquecimiento que se aprovecha de la secuencia genómica Publicada en el diseño de la plataforma. La plataforma humana basada en la matriz utiliza sondas de cartilla diseñadas contra GPC específica de alelo específico (CG vs TG después de la conversión de bisulfito) blanco recocido y cartilla extensión. Su diseño refleja no sólo la secuencia genómica humana disponible, sino regiones reguladoras verificadas experimentalmente de varias líneas de investigación, tales como codificar y ENSEMBL8. A pesar de su amplio uso en las investigaciones de la methylomic humana, una plataforma similar no existe para los animales modelo. Además, el formato basado en el arreglo pone limitación significativa en el área de superficie disponible para la colocación de la sonda. En los últimos años, se hicieron esfuerzos para combinar la especificidad objetivo de diseño de la sonda de captura y la característica de alto rendimiento de secuenciación de próxima generación. Tal empeño ha resultado en el sistema enriquecimiento objetivo basado en la secuenciación del genoma del ratón (ratón metil-Seq), que fue utilizado para identificar las diferencias específicas del cerebro o inducida por glucocorticoides en metilación9,10. Plataformas similares para otros modelo y modelo no animales son necesarios para facilitar la investigación epigenómicas en estos animales.
Aquí, demostramos la aplicación de esta novedosa plataforma para llevar a cabo análisis de methylomic en la rata. La rata ha servido como un modelo animal importante en farmacología, metabolismo, Neuroendocrinología y comportamiento. Por ejemplo, hay una creciente necesidad de entender los mecanismos subyacentes que dan lugar a toxicidad de la droga, obesidad, respuesta de estrés o adicción a las drogas. Una plataforma de alto rendimiento capaz de captar cambios de methylomic asociados con estas condiciones aumentaría nuestra comprensión de los mecanismos. Puesto que el genoma de rata todavía carece de anotación para regiones reguladoras, incorpora promotores redundante, las islas de CpG, Isla costas11y previamente identificadas secuencias ricas en GC en la rata metil-Seq plataforma12.
Para evaluar el éxito del diseño e implementación de la plataforma SureSelect enriquecimiento de Target (genéricamente conocido como metil-Seq) para el genoma de la rata, se empleó un modelo de estrés crónico variable (CVS)13 para identificar diferencialmente metilados ratas regiones entre los animales tensionados y unstressed. Nuestro diseño de la plataforma, el protocolo y la aplicación pueden ser útiles para investigadores que quieran realizar una investigación completa e imparcial epigenética en un organismo cuya secuencia genómica ya está disponible pero sigue siendo mal anotado.
Protocolo
Todos los experimentos fueron terminados de acuerdo y conformidad con todos lineamientos regulatorios e institucionales pertinentes, incluyendo el cuidado de Animal institucional y Comité de uso en la escuela de Medicina Johns Hopkins.
1. los animales
- Obtener ratas de Sprague-Dawley macho adolescentes en 4 semanas de edad. Los animales en jaulas de la rata de policarbonato en una temperatura de la casa- y sala de control de humedad en un ciclo oscuro de luz, 12 h 12 h con inicio ligero en h. 0600 proporcionan los animales con acceso ad libitum al agua.
- Permitir que las ratas se adapte durante 1 semana reducir el estrés asociado con el transporte. Casa par los animales (N = 16) para evitar tensión de aislamiento y en 5 semanas de edad, comenzar la crónica variable estrés régimen (CVS) durante 3 semanas.
2. crónicas de la tensión Variable
- Administrar el régimen CVS una vez en la mañana (9-11:00) y una vez en la tarde (1 – 15:00) en tiempos irregulares para mantener la rutina impredecible. Incorporan factores estresantes suaves durante la noche. El CVS régimen incluye: 1) 3 h de un cilindro de sujeción; 2) 10 min nado; Incline la jaula de 3) 3 h 4) h 1 lenta agitación plataforma; y 5) 1 h en la sala fría de 4 ° C.
Nota: Factores estresantes durante la noche incluyen sociales hacinamiento (5 por jaula), aislamiento social, ropa de cama húmeda, restricción alimentaria y luces encendidas. Un programa semanal típico del régimen de estrés se proporciona en la tabla 1.
3. los ensayos
-
Determinar los niveles de corticosterona (CORT) usando muestras de sangre (50 mL) de cola recogida al mismo tiempo (9:00) dos veces por semana durante todo el experimento, antes del régimen de CVS para establecer los niveles hormonales basales (día 0), una vez en el centro de la CVS semanal (días 4,11 y 18), después cada 7 días de CVS (días 7 y 14) y en la conclusión del CVS (día 21). Obtener muestras de sangre antes del régimen de estrés diario.
- Recoger una muestra de sangre del tronco final durante la eutanasia (día 25) para RIA y la extracción de ADN genómica.
- Centrifugar todas las muestras de sangre (600 x g, 4 ° C, 10 minutos) para separar el plasma de las células sanguíneas. Pipetee hacia fuera del plasma (sobrenadante) y almacenar las muestras a-80 ° C.
- Descongelar y utilizar el plasma para determinar niveles CORT por el radioinmunoanálisis (RIA). Asegúrese de que la semana 3 plasma CORT los niveles son elevados en los animales estresados para verificar la robustez de la régimen de estrés.
4. comportamiento
- Después del régimen de CVS (días 23 – 24), evaluar cada animal para ansiedad-como comportamiento en la elevada más laberinto (EPM)14.
- Utilizando una cámara de vídeo, registro de los animales en el aparato EPM para 300 s y cuenta el tiempo invertido en el centro, cierra los brazos y brazos abiertos.
5. diseño de la rata metil-Seq
- Utilizando el Browser del genoma de UCSC, obtener coordenadas genómicas no redundante (rata rn4 Asamblea de noviembre de 2004) para las islas de CpG y costas de la isla (± 1 kb que flanquean las islas de CpG), promotores (± 1 kb de cada TSS) de cada gen RefSeq y otras secuencias que pueden estar disponibles en literatura relevante.
Nota: Para la rata metil-Seq, adicionales secuencias ricas en GC desde una plataforma de metilación en arreglos de discos anteriores se agregó12. Regiones mayores a 5 kbps, alternas regiones de 500 bps se muestrearon seguido de 1 kbps que fueron omitidos. El diseño de metil-Seq rata final consiste en 111 Mbps, GPC 2,3 millones; y un tamaño promedio de la región de 594 bps. Objetivos 228.800 loci únicos. - Entrar en una lista compilada de coordenadas genómicas en un software de diseño de captura de destino disponibles comercialmente para el diseño de la sonda apropiada.
6. construcción de la biblioteca de metil-Seq de rata de la DNA Genomic
Nota: Para eliminar los efectos de la hornada, procesar varias muestras al mismo tiempo y aumentar por consiguiente la mezcla principal. Extracto de ADN utilizando un kit de extracción de ADN disponible en el mercado. Método basado en la columna o la precipitación tanto rendimiento calidad de ADN genómico (relación 260/280 ~ 1.8). No se recomienda el uso de métodos basados en fenol. Procederá a la elución o Resuspender el DNA en buffer de baja TE (10 mM TE, 0.1 mM EDTA, pH 8.0).
- Preparación de la muestra
Nota: Para cada paso usando DNA-que atan bolas magnéticas, asegúrese de que los granos están aclimatados a la temperatura ambiente durante al menos 30 minutos y bien mezclados antes de usar.- ADN del esquileo
- Utilice un Fluorímetro para determinar la concentración inicial de ADN bicatenario de cada muestra. Diluir > 1 μg del gDNA a 50 μL con tampón de baja TE (10 mM TE, 0.1 mM EDTA, pH 8.0) en tubos de microcentrífuga de ADN bajo.
- Muestras utilizando un sonicador isotérmico del esquileo (10% ciclo de trabajo, de intensidad 5, 200 ciclos por explosión, 6 ciclos de 60 s, barrido de frecuencia, 4 ° C).
- Evaluar la calidad de la DNA usando un sistema basado en la electroforesis que mide la cantidad y del tamaño de ADN.
Nota: La cantidad de ADN recomendada es 1 μg o 3 μg. Si no hay material de partida, la más baja cantidad de entrada debe ser > 500 ng, como menos cantidad afectará negativamente la cantidad y calidad de las bibliotecas generadas.
- Reparación ADN termina.
- Utilizar la rata metil-Seq kit para preparar la reparación final Master Mix en el hielo. Añadir 52 μl de mezcla a cada muestra e incubar en un termociclador sin una tapa caliente (20 ° C por 30 min, hold de 4 ° C).
Reparación final Master Mix (por muestra):
35,2 μl de agua
10 μl de tampón de reparación final de (10 x)
1.6 μl de dNTP Mix
1 μl de T4 DNA polimerasa
2 μl de ADN polimerasa de Klenow
2.2 μl de T4 Polinucleótido kinasa - Purificar las muestras usando 180 μl de bolas magnéticas del ADN y 400 μL de etanol al 70% recién preparado por muestra. Añadir 180 μl de granos a cada muestra e incubar 5 min a temperatura ambiente. Granos de la pelotilla, retirar el sobrenadante y resuspender el pellet en 200 μL de etanol al 70%. Eliminar el etanol y repita una vez lavado.
- Utilice una placa magnética para pellets granos y quitar tanto etanol como sea posible. Secar a 37 ° C protección para 3 – 5 min hasta que el sedimento de grano está completamente seco. Resuspender en 44 μl de agua libre de nucleasas y recoger aproximadamente 42 μl del sobrenadante.
Punto: Después de reparación del ADN, las muestras pueden ser selladas y almacenadas a-20 ° C.
- Utilizar la rata metil-Seq kit para preparar la reparación final Master Mix en el hielo. Añadir 52 μl de mezcla a cada muestra e incubar en un termociclador sin una tapa caliente (20 ° C por 30 min, hold de 4 ° C).
- Extremos de adenilato 3'.
- Preparar el Adenylation Master Mix en el hielo. Añadir 9 μl mezclar a cada muestra e incubar en un termociclador sin una tapa caliente (37 ° C por 30 min, hold de 4 ° C).
Adenylation Master Mix (por muestra):
5 μl de tampón de Klenow
1 μl de dATP
3 μl de DNA polimerasa de Klenow - Purificar las muestras usando 90 μl de bolas magnéticas del ADN y 400 μL de etanol al 70% recién preparado por muestra. Agregar 90 μl de granos a cada muestra e incubar 5 min a temperatura ambiente. Granos de la pelotilla, retirar el sobrenadante y resuspender el pellet en 200 μL de etanol al 70%. Eliminar el etanol y repita una vez lavado.
- Utilice una placa magnética para pellets granos y quitar tanto etanol como sea posible. Secar a 37 ° C protección para 3 – 5 min hasta que el sedimento de grano está completamente seco. Resuspender en 35 μl de agua libre de nucleasas y recoger aproximadamente 33.5 μl del sobrenadante.
- Preparar el Adenylation Master Mix en el hielo. Añadir 9 μl mezclar a cada muestra e incubar en un termociclador sin una tapa caliente (37 ° C por 30 min, hold de 4 ° C).
- Ligar el adaptador metilado.
- Preparar mezcla maestra de ligadura en hielo y añadir 16,5 μl de mezcla a cada muestra. Incubar en un termociclador sin una tapa caliente (20 ° C durante 15 min, hold de 4 ° C).
Ligadura de Master Mix (por muestra):
2,5 μl de agua
2,5 μl de metil-Seq metilado adaptador
10 μl de tampón de T4 ADN ligasa (5 x)
1.5 μl de T4 ADN ligasa - Purificar las muestras usando 90 μl de bolas magnéticas del ADN y 400 μL de etanol al 70% recién preparado por muestra. Agregar 90 μl de granos a cada muestra e incubar 5 min a temperatura ambiente. Granos de pellets, eliminar el sobrenadante y resuspender el pellet en 200 μL de etanol al 70%. Eliminar el etanol y repita una vez lavado.
- Utilice una placa magnética para pellets granos y quitar tanto etanol como sea posible. Secar a 37 ° C protección para 3 – 5 min hasta que el sedimento de grano está completamente seco. Resuspender en 22 μL de agua libre de nucleasas y recoger aproximadamente 22 μL del sobrenadante. Evaluar la calidad con un equipo bioanalyzer.
Nota: Si la cantidad total de ADN es inferior a 500 ng, corte y procesos ADN adicional antes de proceder con los pasos subsiguientes. Si el tamaño medio del ADN no aumenta por más de 30 bps, verifique para asegurarse de que los reactivos son nuevos, como la polimerasa de la DNA de T4, Klenow y T4 ligasa puede ser viejo.
Punto: Después de ligar el adaptador metilado, muestras pueden ser selladas y almacenadas a-20 ° C.
- Preparar mezcla maestra de ligadura en hielo y añadir 16,5 μl de mezcla a cada muestra. Incubar en un termociclador sin una tapa caliente (20 ° C durante 15 min, hold de 4 ° C).
- ADN del esquileo
- Hibridación
- Transferir las muestras a tubos de microcentrífuga de ADN bajo y utilizar un concentrador de vacío caliente para reducir el volumen de la muestra a menos de 3,4 μL. reconstituir las muestras μl 3.4.
Nota: Concentrado de las muestras aproximadamente ~ 3 μL para muestras se extraen de concentrador de vacío antes de todo el líquido se evapora. - Preparar el tampón de hibridación a temperatura ambiente y mezcla de metil-Seq bloque sobre hielo. Añadir 5.6 μl de mezcla de metil-Seq bloque a cada muestra e incubar en termociclador (95 ° C por 5 min, 65 ° C por 2 min, hold de 65 ° C).
Tampón de hibridación (por muestra):
6.63 μl de metil-Seq Hyb 1
0.27 μl de metil-Seq Hyb 2
2.65 μl de metil-Seq Hyb 3
3.45 μl de metil-Seq Hyb 4
Metil-Seq bloque Mix (por muestra):
2,5 μl de metil-Seq índice bloque 1
2,5 μl de metil-Seq bloque 2
0,6 μl de metil-Seq bloque 3 - Preparar Rnasa bloque de mezcla y la mezcla de hibridación captura biblioteca. Añadir 20 μl de captura biblioteca hibridación mezcla a cada muestra e incubar a 65 ° C durante al menos 16 h.
Rnasa bloque Mix (por muestra):
0,5 μl de Rnasa bloque
1.5 μl de agua
Captura de biblioteca hibridación mezcla (por muestra):
13 μl de tampón de hibridación
2 μl de Rnasa bloque Mix
5 μl de rata metil-Seq captura biblioteca
Nota: mantener reacciones a 65 ° C cuando se agrega la mezcla de hibridación para evitar atascamiento no específico. - Alícuota de 50 μl de granos magnéticos estreptavidina por muestra en un tubo de tiras de 8 pozos nuevos. Lavar los granos con 200 μL de tampón de Binding de metil-Seq. Usar placa magnética para pellets granos y eliminar sobrenadante entre cada lavado para un total de 3 lavados. Después del último lavado, resuspender estreptavidina granos en 200 μL de tampón de Binding de metil-Seq.
- Añadir muestras a 200 μL de granos magnéticos estreptavidina lavado e incubar a temperatura ambiente durante 30 min con un mezclador rotatorio. Mientras se mezcla, alícuota 200 μL de metil-Seq de tampón de lavado 2 en triplicado pocillos de una placa de 96 pocillos por muestra y el lugar en un termociclador para pre-calentar a 65 ° C.
- Después de la incubación de la pelotilla de estreptavidina granos magnéticos con placa magnética y resuspender los granos en 200 μL tampón de lavado del metil-Seq 1. Incubar por 15 min a temperatura ambiente. Use una placa magnética para pellets y descartar el sobrenadante.
- Lavar granos 3 veces con metil-Seq tampón de lavado 2: Resuspender el precipitado de grano en 200 μL de tampón lavar de 2 (previamente calentado en el paso 6.2.5.), incubar granos en termociclador (65 ° C, 10 min) y granos de la pelotilla. Desechar el sobrenadante después de cada lavado con un plato magnético.
Nota: Mantener reacciones de hibridación a 65 º C al añadir el tampón de lavado 2 para evitar la fijación no específica. - Añadir 20 μl de tampón de elución de metil-Seq a los granos lavados e incubar a temperatura ambiente durante 20 min uso una placa magnética a granos pellets y traslado de sobrenadante a un tubo nuevo de la tira. Deseche los granos.
Nota: Al mismo tiempo de incubación, preparar reactivo de conversión de bisulfito.
- Transferir las muestras a tubos de microcentrífuga de ADN bajo y utilizar un concentrador de vacío caliente para reducir el volumen de la muestra a menos de 3,4 μL. reconstituir las muestras μl 3.4.
- Conversión de bisulfito
Nota: Realizar conversión de bisulfito de la ssDNA eluída con reactivos apropiados y las instrucciones de un kit de conversión de bisulfito comercialmente disponibles.- Agregar 130 μl de reactivo de conversión bisulfito preparado al sobrenadante del paso anterior. Dividir cada una de las reacciones de μl 150 igualmente en dos pozos. Incubar en un termociclador (64 ° C por 2,5 h, bodega de 4 ° C).
Nota: La reacción de 150 μL se divide igualmente en dos pozos separados para asegurar la temperatura homogénea. Después de incubar durante 2.5 h, inmediatamente proceder al siguiente paso. - Se unen las muestras spin columnas añadir 600 μl de tampón de Binding y lavar una vez con 100 μl de tampón de lavado. Centrifugar columnas (15.000 x g, 1 min) entre todos los pasos de conversión bisulfito y deseche el flujo a través.
- Desulphonate las muestras mediante la adición de 200 μL de tampón de Desulphonation a las columnas. Incubar a temperatura ambiente durante 15-20 min repetir centrifugación y deseche el flujo a través.
- Lavar las columnas dos veces con 200 μL de tampón de lavado. Eluir cada muestra añadiendo 10 μl de tampón de elución a la columna, incubar 3 min a temperatura ambiente y centrifugar (15.000 x g, 1 min). Repita el paso de elución para un total de 20 μl.
- Preparar la reacción de PCR Master Mix 1 en hielo. Añadir 82 μl de la mezcla a cada muestra. Incubar en un termociclador con el siguiente programa.
PCR reacción de Master Mix 1 (por muestra):
30 μl de agua
50 μl de metil-Seq PCR Master Mix
1 μl de metil-Seq PCR1 Primer F
1 μl de metil-Seq PCR1 Primer R
Programa del termociclador:
Etapa 1, 1 ciclo: 95 ° C 2 min
Etapa 2, 8 ciclos: 95 ° C 30 s, 60 ° C 30 s, 72 ° C 30 s
Etapa 3, 1 ciclo: 72 º C 7 min.
Etapa 4, 1 ciclo: 4 ° C espera - Purificar las muestras usando 180 μl de bolas magnéticas del ADN y 400 μL de etanol al 70% recién preparado por muestra. Añadir 180 μl de granos a cada muestra e incubar 5 min a temperatura ambiente. Granos de la pelotilla, retirar el sobrenadante y resuspender el pellet en 200 μL de etanol al 70%. Eliminar el etanol y repita una vez lavado.
- Utilice una placa magnética para pellets granos y quitar tanto etanol como sea posible. Secar a 37 ° C protección para 3 – 5 min hasta que el sedimento de grano está completamente seco. Resuspender en 21 μl de agua libre de nucleasas y recoger aproximadamente 19.5 μl del sobrenadante.
- Agregar 130 μl de reactivo de conversión bisulfito preparado al sobrenadante del paso anterior. Dividir cada una de las reacciones de μl 150 igualmente en dos pozos. Incubar en un termociclador (64 ° C por 2,5 h, bodega de 4 ° C).
- Indexación de direcciones
- Preparar la reacción de PCR Master Mix 2 en el hielo. Añadir 25,5 μl Master Mix 2 para cada muestra. Añadir 5 μl de comercial indexación cartillas para muestras individuales e incubar en un termociclador.
PCR reacción de Master Mix 2 (por muestra):
25 μl metil-Seq PCR Master Mix
Cartilla de indexación común de 0,5 μl metil-Seq
Programa del termociclador:
Etapa 1, 1 ciclo: 95 ° C 2 min
Etapa 2, 6 ciclos de: 95 ° C 30 s, 60 ° C 30 s, 72 ° C 30 s
Etapa 3, 1 ciclo: 72 º C 7 min.
Etapa 4, 1 ciclo: 4 ° C espera
Nota: Ciclos adicionales (2-3) pueden ser necesarios si la concentración de ADN inicial está por debajo de los valores recomendados. - Purificar las muestras usando 90 μl de bolas magnéticas del ADN y 400 μL de etanol al 70% recién preparado por muestra. Agregar 90 μl de granos a cada muestra e incubar 5 min a temperatura ambiente. Granos de la pelotilla, retirar el sobrenadante y resuspender el pellet en 200 μL de etanol al 70%. Eliminar el etanol y repita una vez lavado.
- Utilice una placa magnética para pellets granos y quitar tanto etanol como sea posible. Secar a 37 ° C protección para 3 – 5 min hasta que el sedimento de grano está completamente seco. Resuspender en 24 μl de agua libre de nucleasas y recoger aproximadamente 24 μl de sobrenadante.
- Evaluar la concentración y tamaño de bp con los reactivos de detección de ADN de alta sensibilidad en un equipo bioanalyzer.
Nota: Si el equipo bioanalyzer no logra detectar la presencia de la biblioteca de ADN, repita los pasos de preparación con ADN adicional.
Punto de parada: después de la purificación, indexadas las muestras pueden ser selladas y almacenadas a-20 ° C. - Agrupación de las muestras para la plataforma adecuada secuenciación de próxima generación.
- Utilizando los datos de concentración del equipo bioanalyzer, que determina la molaridad de ADN basado en el tamaño de la biblioteca y la cantidad en un volumen dado, diluir con tampón TE baja (6.1.1.1) y combinar todas las muestras a una concentración final de 15 pM.
Nota: Un método más sensible de la cuantificación de la biblioteca es por PCR cuantitativa en tiempo real utilizando primers dirigidos a los adaptadores ligados. - Ejecutar ejemplos agrupados en el número de carriles que son suficientes para 4 muestras por carril en un secuenciador de última generación.
Nota: por ejemplo, si 16 muestras de biblioteca han sido indexadas y combinado único, correr a las bibliotecas de más de 4 carriles, equivalentes a 4 muestras por carril.
- Utilizando los datos de concentración del equipo bioanalyzer, que determina la molaridad de ADN basado en el tamaño de la biblioteca y la cantidad en un volumen dado, diluir con tampón TE baja (6.1.1.1) y combinar todas las muestras a una concentración final de 15 pM.
- Preparar la reacción de PCR Master Mix 2 en el hielo. Añadir 25,5 μl Master Mix 2 para cada muestra. Añadir 5 μl de comercial indexación cartillas para muestras individuales e incubar en un termociclador.
7. la secuencia en un secuenciador de última generación
- Enviar las muestras a la base institucional de la secuencia para el agrupamiento de la biblioteca de metil-Seq, seguida por secuenciación en una máquina de secuenciación de próxima generación.
8. análisis para identificar DMRs
- Implementar Bismark15, que invoca la pajarita 2.0 como una secuencia interna alineador16,17, para alinear el raw Lee entrada convertido a bisulfito, plus-filamento del genoma. Después de la alineación, utilice el Bismark_methylation_extractor para realizar controles de calidad y asignar un valor estimado de la metilación a cada GPC.
- Generar una lista de DMRs con el BS-Seq paquete18 en Bioconductor. Filtro el DMRs basado en tener mayor que 3 GPC consecutivos y valor P < 0.05.
Nota: Generar una lista DMR que incluye coordenadas genómicas, distancia a los genes RefSeq más cercano, el número de GPC dentro de cada DMR, promedio % valor de la metilación CpG en el DMR para los grupos de dos comparación (por ejemplo, tensionado y unstressed), el valor de P, y el valor FDR (tarifa falsa del descubrimiento). Utilice la lista DMR, i.e., coordenadas genómicas, para el diseño de cartillas de Pirosecuenciación para validación.
9. validación por pirosecuenciación de bisulfito
-
Primer diseño
- Diseño de primers para PCR de bisulfito y pirosecuenciación. Diseño de dos sistemas de cartillas de la polimerización en cadena (exterior y anidadas) que la PCR anidada amplificará 150 – 400 bps de un DMR.
Nota: en general, diseño de primers son bases al menos 24 de largo con al menos 4-5 no consecutivos G (C para la cartilla reversa) a cuenta para reducir recocido temperatura de pérdida de la complejidad de la secuencia. Uno de los iniciadores anidados será marcado con biotina y purificadas por HPLC. Sin embargo, imprimaciones estándar deben solicitarse primero optimizar el paso de la polimerización en cadena por resolver las reacciones en un gel de agarosa.- Diseño de la pirosecuenciación ensayo primer modo que dirige la biotinilado complementario filamento sólo 1 – 2 bases río arriba de la GPC a ensayarse. Diseño múltiples cebadores de pirosecuenciación como sea necesario, como cada primer de pirosecuenciación fiable puede ensayo 30 bps aguas abajo.
- Para el Rt1-m4, utilice lo siguiente:
rRT1M4 exterior-F TGTAYGATTTTGGTTATYGTAAAT
rRT1M4 exterior-R AACTTACAAATTTCACCAACTCA
rRT1M4 anidados – F GTGGGTTAYGTGGATAATATATAG
rRT1M4 anidados-R AATCACTTACCATTCTCTCTCTAACTA
rRT1M4 Pyro1 TAYGTGGATAATATATAGAT
rRT1M4 Pyro2 GATAGTTATTTGGYGAGTTAG
rRT1M4 Pyro3 GAGTATTTGGAGGAGTTGAT
rRT1M4 Pyro4 GGATTTTAATATTTGGT
- Diseño de primers para PCR de bisulfito y pirosecuenciación. Diseño de dos sistemas de cartillas de la polimerización en cadena (exterior y anidadas) que la PCR anidada amplificará 150 – 400 bps de un DMR.
-
Utilice un kit disponible comercialmente para la conversión de bisulfito del gDNA sangre de rata.
Nota: Los pasos de conversión de bisulfito se han adaptado del kit disponible comercialmente con las siguientes modificaciones: en el paso 1, agregar 50-100 ng de sangre gDNA y diluir con agua a 20 μl. En el paso 9, eluir 20 μl por muestra.- Preparar el reactivo de conversión de bisulfito según protocolo del fabricante y combina con gDNA diluido. Incubar en termociclador (64 ° C por 2,5 h, bodega de 4 ° C).
- Añadir tampón de Binding a gDNA convertida en columnas spin y centrifugar (15.000 x g, 1 min). Columnas de lavado una vez añada Desulphonation Buffer a las columnas e incuban por 15 min a temperatura ambiente. Centrífuga (15.000 x g, 1 min).
- Lavar la columna con tampón de lavado y centrífuga (15.000 x g, 1 min). Repetir el lavado con centrifugación (15.000 x g, 2 minutos). Añadir 20 μl tampón de elución y centrífuga (15.000 x g, 1 min) a fin de eluir.
-
Amplificación por PCR
- Preparar el exterior PCR Master Mix. Añadir 21,5 μl de la Master Mix a 3,5 μl convertido a bisulfito gDNA y ejecutar programa del termociclador.
Exterior PCR Master Mix:
16.25 μl de agua
2,5 μl de tampón de polimerasa [10 x]
0,5 μl de dNTP (10 mM)
1 μl de cebador Forward [0.1 μm]
1 μl de cebador reverso [0.1 μm]
0.25 μl de Taq ADN polimerasa [5000 U/mL].
Programa del termociclador:
Etapa 1, 1 ciclo: 94 º C 4 min.
Etapa 2, 47 ciclos: 94 ° C 1 min, 53 ° C 30 s, 72 ° C 1 min
Etapa 3, 1 ciclo: 72 ° C 8 min, 4 ° C espera - Preparar anidado PCR Master Mix. Añadir 23 μl de la Master Mix a 2 μl de la muestra de PCR fuera y repetir el programa exterior de Termociclador PCR. Evaluar la calidad del producto PCR mediante electroforesis en gel (1 buffer x TAE, gel de agarosa al 1%).
Anidados PCR Master Mix:
17,75 μl de agua
2,5 μl de tampón de polimerasa [10 x]
0,5 μl de dNTP (10 mM)
1 μl de cebador Forward [0.1 μm]
1 μl de cebador reverso [0.1 μm]
0.25 μl de Taq ADN polimerasa [5000 U/mL]
Nota: Para PCR anidada, el avance o la cartilla reversa debe ser biotinilado.
- Preparar el exterior PCR Master Mix. Añadir 21,5 μl de la Master Mix a 3,5 μl convertido a bisulfito gDNA y ejecutar programa del termociclador.
-
Pirosecuenciación
- Hacer una mezcla maestra que contiene 38 μl de tampón de Binding, 35 μl de agua y 2 μl de sepharose recubiertas de estreptavidina granos por muestra. En una placa de 96 pocillos, agregar 75 μl de la mezcla principal y 5 μl del producto PCR anidada. Agitar en un agitador de placa durante 15-60 minutos.
- Agitando, añadir 12 μl de cebador (0,5 μm, diluido en buffer recocido) en los pocillos de una placa de ensayo de pirosecuenciación.
- Después de agitar, realice los pasos de lavado utilizando tampones de lavado atascamiento reacción. Coloque la herramienta vacío en la cubeta llena de agua luego recoger muestras de la placa. Sumerja la herramienta vacío en canales lleno hasta la mitad con etanol al 70%, NaOH (0,2 M) y tampón de Tris acetato (10 mM, pH 7,4). Desconecte de la herramienta de vacío vacío y lugar en HS ensayo placa de transferencia de granos.
- Coloque la placa en el bloque de calor e incubar a 80 ° C por 2 minutos deje que la placa se enfríe durante 5 minutos, luego comenzar programa de pyro.
Resultados
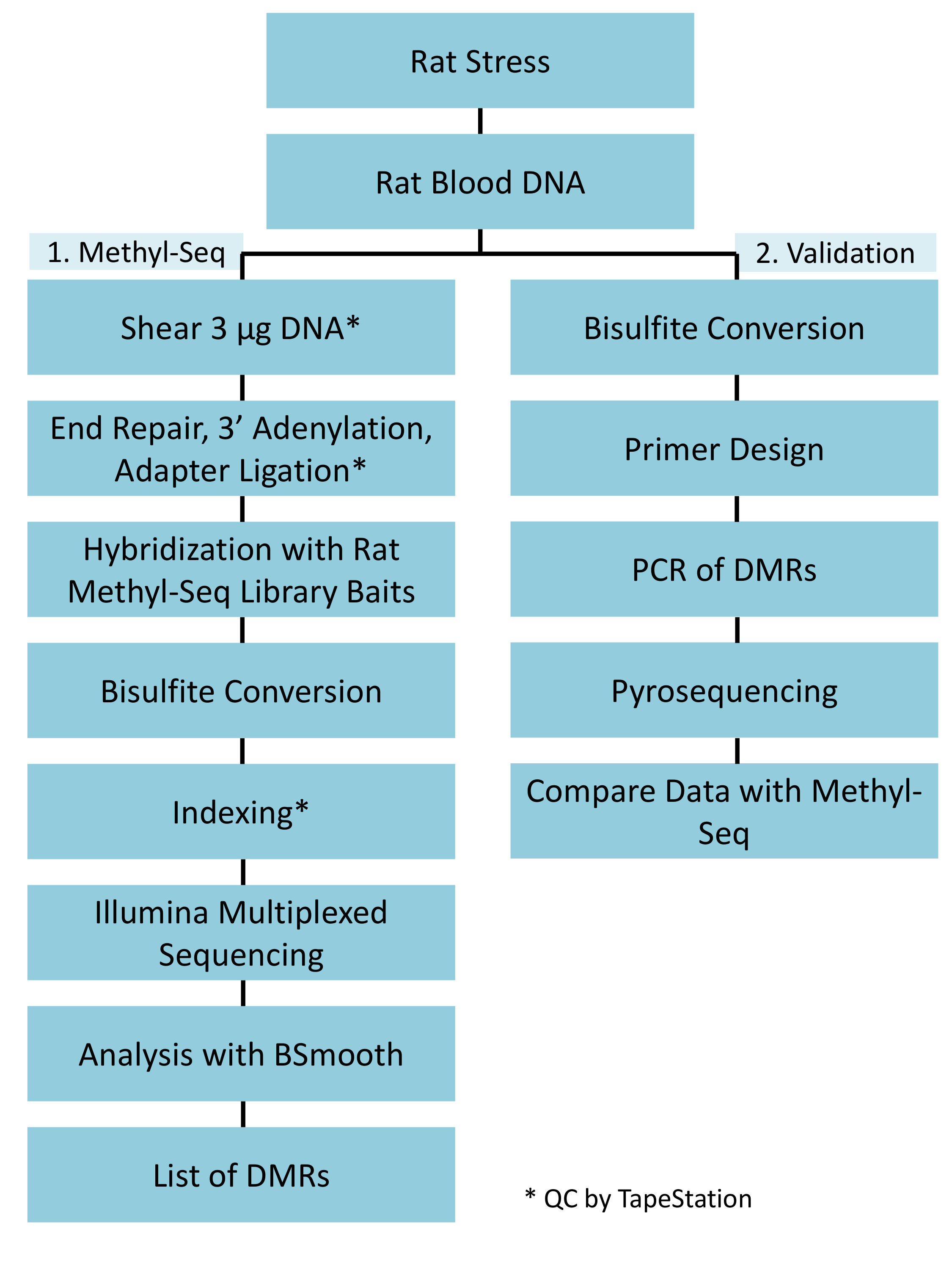
Una implementación exitosa de la plataforma de metil-Seq rata depende de varios criterios. Figura 1 muestra el flujo de trabajo general del estudio y destaca pasos específicos control de calidad (QC) que se necesitan antes de seguir adelante. Uno de los primeros factores a considerar es la robustez del modelo animal y el régimen de estrés, que determinan la magnitud de cambios epigenéticos que se producen en el metiloma. Puesto que nuestro trabajo animal está basada en nuestra observación anterior de que la exposición de corticosterona (CORT) puede conducir a cambios en la metilación de ADN19,20, nuestro régimen de estrés crónico variable (CV) debe ser de suficiente rigor para producir hizo hincapié en las ratas con niveles elevados de plasma CORT. Un típico régimen semanal de CVS se muestra en la tabla 1 y consistió de estresores diarios en la mañana, por la tarde, y durante la noche que cambian constantemente para evitar la habituación y disminuida respuesta de estrés. Durante el régimen de 3 semanas, los animales estresados exhibieron niveles significativamente elevados de plasma media CORT [Control de los días 4 y 21,: 32,7 3.7 ng/mL, estrés: 103,0 11.9 ng/mL (media SEM), P = 2.2 x 10-4, figura 2A] sobre los de unstressed, control de animales. Consistentemente, estos animales también mostraron mayor ansiedad-como comportamiento en la elevada y laberinto (EPM), como se indica por el significativamente más tiempo en los brazos cerrados del EPM y menos tiempo en los brazos abiertos (figura 2B). Estos resultados demuestran que la exposición CVS condujo a endocrinos significativos y cambios de comportamiento, que nos lleva a investigar si estos cambios se asociaron con firmas de metilación de ADN específicas.
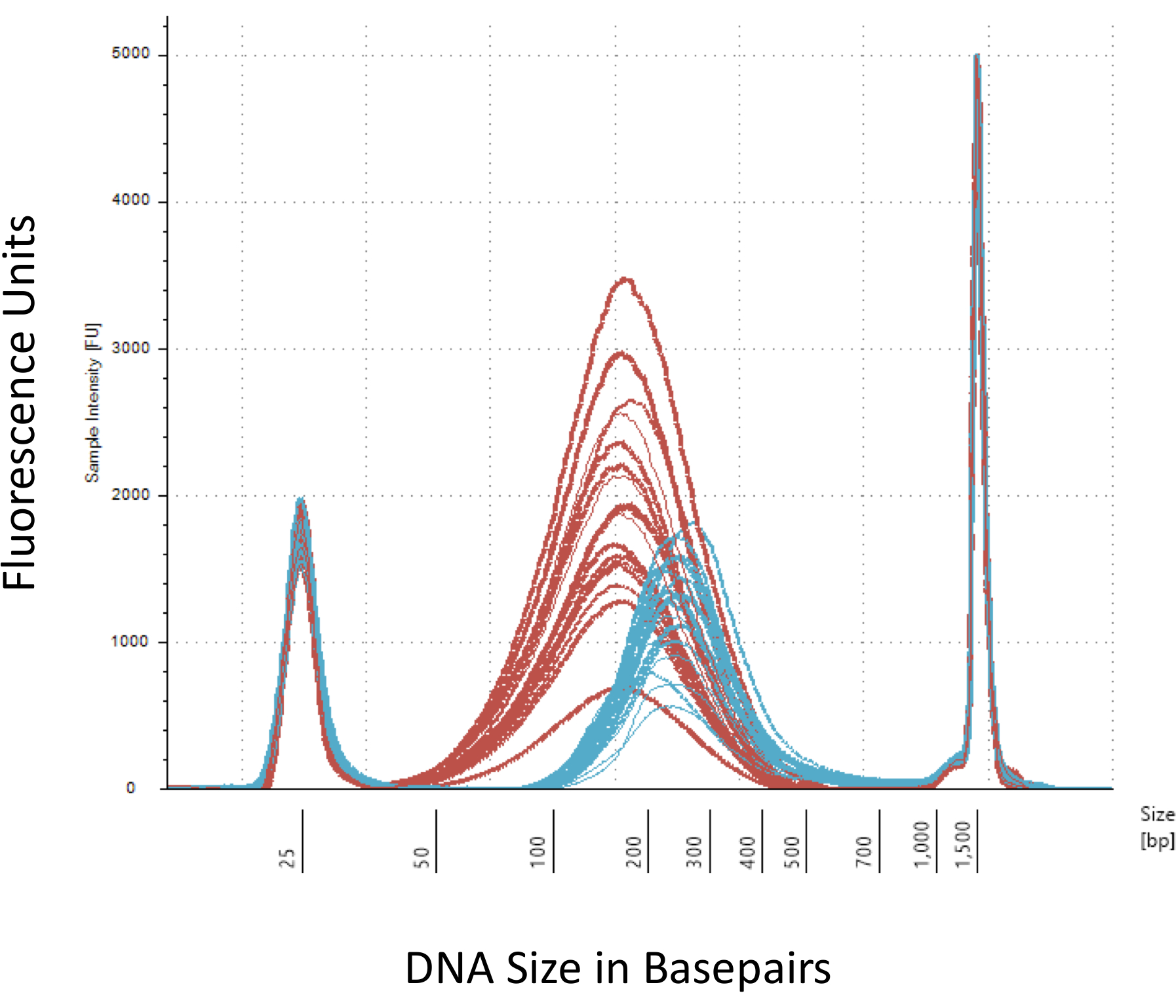
Destacamos varios puntos que son cruciales para la exitosa construcción de la biblioteca de metil-Seq. A partir de una cantidad suficiente de ADN es necesario, como la sonicación, múltiples lavado/purificación, enriquecimiento de destino, y pasos de conversión de bisulfito sucesivamente reducen la cantidad de DNA en la biblioteca acabada. Aunque varios pasos de amplificación de PCR aliviar la pérdida de la plantilla de la DNA, excesivo número de ciclo PCR puede introducir mayor Lee duplicado. Para el actual estudio de rata metil-Seq, 2 g de sangre gDNA por rata fue utilizado. Tomamos nota de que las bibliotecas metil-Seq se pueden hacer con a partir de cantidad de ADN 500 ng. Más pequeños a partir de material permite generar bibliotecas de ADN aislado por FACS (clasificación celular activado por fluorescencia) o golpes, la aguja aunque hay mayor riesgo de producir una cantidad insuficiente de bibliotecas para la secuencia posterior. Control de calidad se realiza por electroforesis de 1 L de la muestra en un equipo bioanalyzer, que proporciona el peso molecular de ADN, cantidad y molaridad. Tres pasos críticos que requieren el uso del equipo bioanalyzer son: 1) tras el paso de sonicación para asegurar suficiente corte de ADN (~ 170 bp, rojo, figura 3) 2) siguiente adaptador ligadura paso indicada por un cambio en el tamaño medio del ADN esquilado (~ 200 bp, azul, figura 3) para su posterior amplificación por PCR; y 3) siguiente paso de purificación final biblioteca para garantizar la cantidad y el tamaño de la biblioteca para la secuencia.
Los paquetes de R BSSeq y BSmooth en Bioconductor fueron utilizados para el análisis del bisulfito de secuenciación de datos18. Incluyen herramientas y métodos para alinear la Lee de la secuencia, realizar control de calidad, e identificar diferencialmente metiladas regiones (DMRs). BSmooth software invoca Bowtie 2.016,17 como un alineador de la secuencia interna para obtener resúmenes de medición de nivel de CpG, por alineación de raw Lee entrada a convertido a bisulfito de secuencias genómicas. Lee alineado entonces se filtra a través de procedimientos de control de calidad riguroso que pretenden identificar la secuencia sistemática y errores de base de llamadas que pueden sesgar el análisis descendentes. Una serie de diagramas se generan para visualmente facilitar en este proceso de filtrado. Indicadores de secuencia se generan también a información relevante del documento como el número de alineados Lee, % blanco y por cobertura de GPC, entre otros (tabla 2). Una vez que se filtran los datos, se realiza un algoritmo de suavizado/normalización, donde cada GPC se asigna un valor estimado de la metilación basado en control de calidad todos Lee de cada muestra y las estimaciones de los vecinos de CpGs para asegurar más exacto llamar de la metilación Estado incluso en casos donde es baja la cobertura de la secuencia. Este valor proporciona una estimación suavizada de la probabilidad de la metilación en cada sitio de CpG. Al comparar la media de las estimaciones de metilación alisado de cada muestra entre los dos grupos de tratamiento y clasificación regiones genómicas de los más significativamente diferentes al menos, se genera una lista de DMRs (tabla 3).
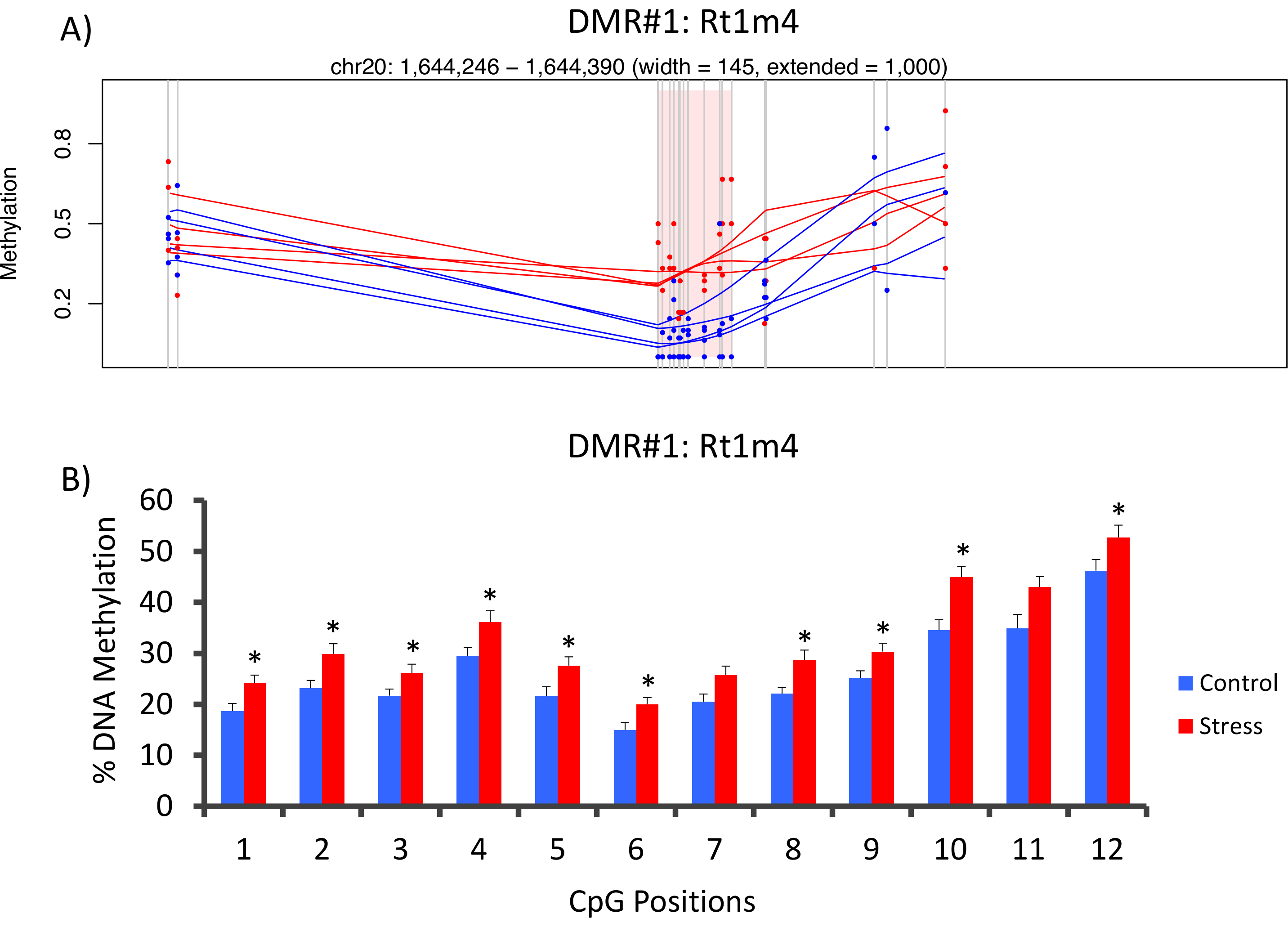
La parte superior con que DMR entre grupos de tensionado y unstressed se encontraba en el promotor del gen de histocompatibilidad rata Rt1-m4, tensionado animales exhibiendo niveles más altos de metilación a través de todas las GPC que unstressed animales (Figura 4A). Para confirmar la implementación exitosa de la plataforma de metil-Seq y el análisis de datos, se diseñaron cartillas contra el DMR y niveles de metilación de ADN en la cohorte completa de animales tensionados y unstressed (8 ordenados por metil-Seq y 8 no ordenados) se evaluaron por pirosecuenciación de bisulfito. Resultados demuestran un aumento significativo en la metilación del ADN en 10 de los 12 CpGs ensayaron (5.1 – 10.4 cambio en % de metilación, P < 0.037, Figura 4B). Análisis del camino KEGG fue realizada en todo el DMRs nominalmente significativos para identificar vías asociadas con el estrés. Constantemente, vías DMR asociado implicado enfermedades asociadas con la exposición a estrés crónico, como diabetes, enfermedades cardiovasculares y cáncer (tabla 4). 21 , 22 , 23 para demostrar una asociación entre los datos de la epigenéticos y el grado de exposición al estrés, los niveles de metilación CpG-10 fueron comparados con el nivel medio de la CORT de 3 semanas para cada animal. Los resultados mostraron una modesta correlación entre los datos de endocrino y metilación (R2= 0.54, P = 0.001, figura 5).

Figura 1: flujo de trabajo general esquemática para la plataforma de rata metil-Seq. G uno de la DNA genomic extraída de la sangre de tensionado y ratas de control se procesa en primer lugar para la construcción de las bibliotecas de metil-Seq de la secuencia, el análisis y la identificación de objetivos. Otros 100 ng de ADN se utiliza para la validación independiente de los objetivos identificados epigenéticos por pirosecuenciación de bisulfito. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 2: exposición a estrés crónico variable (CVS) conduce a los cambios endocrinos y del comportamiento en ratas. (A) múltiples muestreos de corticosterona (CORT) demuestran la robustez de la semana 3 régimen de CVS. Muestras de sangre se recolectaron en la mañana antes del régimen de estrés diario. (B) animales Stressed pasaron más tiempo en los brazos cerrados y menos tiempo en los brazos abiertos de la elevada más laberinto (EPM). Se muestran los boxplots con punto de datos para cada animal. Se realizó prueba T de Student para la significación estadística. * P < 0.05, ** P < 0.01, y *** P < 0.001. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 3: cuantificación de rata esquilado y adaptador ligarse DNA en un equipo bioanalyzer. Las curvas rojas y azules muestran la cantidad y tamaño de la DNA genomic (rojo) después de esquilar en un sonicador isotérmico y ligadura del adaptador, respectivamente. Cada línea representa una muestra y el rojo y azul curvas reflejan tanto la pérdida de ADN durante los pasos de varios (final de reparación, 3'-adenylation y limpieza de muestra) y aumentan de tamaño de bp por la ligación de los adaptadores. Afilados picos 25 bp y bp 1500 son marcadores estándar que se han añadido al buffer de carga. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 4: cambios epigenéticos inducidos por la CVS son detectados por rata metil-SEQ (A) análisis de la rata metil-Seq datos implicado el promotor del gen de la Rt1m4 como una región diferencialmente metilada (DMR) entre estresada (rojo) y las ratas de control (azul). La salida gráfica para Rt1m4 DMR (región sombreada rosa) muestra cada GPC (línea gris vertical), las cuatro muestras en cada grupo (líneas en rojas o azules) y los niveles de metilación % para cada animal (punto rojo o azul). (B) doce GPC dentro de lo DMR fueron validados por pirosecuenciación de bisulfito. Los gráficos de barras se representan como media SEM, y se realizó prueba T de Student para la significación estadística. * P < 0.05. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 5: Análisis de regresión lineal mostró una modesta correlación entre % ADN metilación CpG-10 de Rt1m4 y la 3 semana significan plasma niveles CORT de ambos destacó y controlan de animales (N = 16). Datos de animales estresados son representados por círculos rojo. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
| Semana | Día 1 | Día 2 | Día 3 | Día 4 | Día 5 | Día 6 | Día 7 |
| AM | Sistema de seguridad para | Nadar | Cuarto frío | Nadar | Sistema de seguridad para | Coctelera | Nadar |
| PM | Coctelera | Inclinación de la jaula | Sistema de seguridad para | Coctelera | Cuarto frío | Sistema de seguridad para | Cuarto frío |
| Durante la noche | Restringir alimentos | Mojado de cama | Aislamiento | Luz en | Apretadura | Luz en | Mojado de cama |
Tabla 1: Un programa semanal típico del régimen de tensión variable crónica (CVS).
| Secuencia de mediciones | Tensión1 | Control1 |
| (n = 4) | (n = 4) | |
| Junto de lecturas finales (por) | 89,290,397 | 80,165,674 |
| Final pares asignado únicamente Lee (UMPER) | 39,200,255 | 35,013,406 |
| Eficiencia de asignación de tipo de alineación (UMPER por) | 44% | 44% |
| Lee duplicado (% de UMPER) | 73% | 65% |
| UMPER reduplicada | 10,481,031 | 12,306,018 |
| Promedio de cobertura de profundidad de lectura (x) (ARDC) | 6 x | 6 x |
| GPC (N) | 12,056,878 | 12,056,878 |
| ARDC (x) de la GPC | 2 x | 2 x |
| GPC con al menos 10 Lee (N) | 481.383 | 595.850 |
| ARDC (X) de la GPC con Lee por lo menos 10 | 19 | 19 |
| En la GPC objetivo (traslapo completo con sonda regiones de destino) | 1.923.872 | 2.007.638 |
| Objetivo ARDC (x) de la GPC | 7 x | 8 x |
| En la GPC objetivo con al menos 10 Lee (N) | 428.249 | 531.419 |
| En destino ARDC (x) de la GPC con Lee por lo menos 10 | 38: | 38: |
| En blanco (por traslapo de 1 o más pares con sonda regiones de destino) (UMPER) | 8.277.715 | 9.369.523 |
| % En blanco (de UMPER reduplicada) | 78% | 77% |
| En blanco (Bases totales asignados) Mb | 125 mb | 128 mb |
| En destino leer media profundidad cobertura (x) (ARDC) | 9 x | 30: |
| 1 Secuencia de mediciones basadas en promedios de sujetos en cada grupo |
Tabla 2: Secuencias métricas de la plataforma de metil-Seq de rata.
| Chr | Inicio | final | gene | distancia | areaStat | meanDiff | estrés | control | Dirección |
| chr20 | 1.644.246 | 1.644.390 | RT1-M4 | in_gene | 93.03 | 0.22 | 0.33 | 0.11 | ganancia |
| chr5 | 160,361,352 | 160,361,564 | LOC690911 | in_gene | -70.75 | : -0,19 | 0,72 | 0.91 | pérdida |
| chr3 | 61,138,281 | 61,138,330 | RGD1564319 | 265569 | 61.79 | 0.21 | 0.94 | 0,72 | ganancia |
| CHR2 | 143,064,811 | 143,065,010 | Ufm1 | 8569 | -59.48 | -0,11 | 0.13 | 0.24 | pérdida |
| chr7 | 30,764,111 | 30,764,284 | Ntn4 | in_gene | 57.04 | 0.21 | 0.94 | 0.73 | ganancia |
| chr17 | 12,469,112 | 12,469,218 | Idnk | 41996 | -50.91 | -0,13 | 0.74 | 0.88 | pérdida |
| chr7 | 47,101,725 | 47,101,930 | Pawr | in_gene | -50.54 | -0,12 | 0,64 | 0,76 | pérdida |
| chr5 | 76,111,248 | 76,111,822 | Txndc8 | 151703 | -50.38 | -0,11 | 0.85 | 0.96 | pérdida |
| chr11 | 80,640,132 | 80,640,356 | Dgkg | in_gene | -50.07 | -0.16 | 0.73 | 0,89 | pérdida |
| chr8 | 71,759,248 | 71,759,411 | Mir190 | 210226 | -47.84 | -0,17 | 0.58 | 0.75 | pérdida |
Tabla 3: Top 10 diferencialmente metiladas regiones. Para cada DMR, la tabla de salida muestra desde la izquierda a la columna de la derecha: Localización cromosómica (chr), coordina (inicio y fin), nombre de gen, distancia de la transcripción comienza sitio, estadísticas de área diferencial entre destacó y controlan de grupos (areaStat), significa hizo hincapié en los niveles de metilación media en cada DMR para metilación diferencial (meanDiff), y grupos de control (control de estrés) y la dirección de la metilación de controles.
| Términos del camino KEGG | Conde de gene | % | Valor de P | Benjamini |
| Diabetes | ||||
| Diabetes mellitus tipo II | 12 | 0.1 | 3.6 x 10-4 | 9.8 x 10-3 |
| Enfermedades cardiovasculares | ||||
| Contracción del músculo liso vascular | 18 | 0.1 | 1.6 x 10-3 | 3.6 x 10-2 |
| Cardiomiopatía ventricular correcta arritmogénica (ARVC) | 13 | 0.1 | 4.0 x 10-3 | 7.1 x 10-2 |
| Miocardiopatía dilatada | 14 | 0.1 | 7.6 x 10-3 | 1,2 x 10-1 |
| Función de la neurona | ||||
| Potenciación a largo plazo | 11 | 0.1 | 1.5 x 10-2 | 1.4 x 10-1 |
| Señalización | ||||
| Vía de señalización de MAPK | 35 | 0.2 | 2.4 x 10-4 | 9.9 x 10-3 |
| Vía de señalización de calcio | 22 | 0.1 | 1,2 x 10-2 | 1.4 x 10-1 |
| Vía de señalización de quimioquinas | 21 | 0.1 | 1,2 x 10-2 | 1.3 x 10-1 |
| Cáncer | ||||
| En el cáncer de las vías | 42 | 0.3 | 4.1 x 10-5 | 3.4 x 10-3 |
| Glioma de | 15 | 0.1 | 4.4 x 10-5 | 2.4 x 10-3 |
| Cáncer de pulmón de células no pequeñas | 10 | 0.1 | 7,9 x 10-3 | 1.1 x 10-1 |
| Cáncer colorrectal | 13 | 0.1 | 8.4 x 10-3 | 1.1 x 10-1 |
| Leucemia mieloide crónica | 12 | 0.1 | 1,2 x 10-2 | 1.3 x 10-1 |
Tabla 4: Análisis de camino KEGG de DMRs identificado de la rata metil-SS.
Discusión
En este estudio, diseñó e implementó la plataforma de metil-Seq para el genoma de la rata. Demostrando su utilidad con un modelo de rata de estrés, hemos demostrado que la tubería experimental y analítica puede proporcionar regiones diferencialmente metiladas entre dos grupos de comparación.
Para asegurar una implementación exitosa de la plataforma, varios pasos críticos necesitan ser observados. Primero, inicial, cantidad y calidad de ADN tiene un impacto significativo en la calidad y cantidad de la última biblioteca de metil-Seq. Utilizamos un fluorómetro, en lugar de un espectrofotómetro, para asegurar que nuestra medición de ADN refleja la cantidad de ADN de doble hebra presentes. El equipo bioanalyzer fue utilizado para medir el tamaño molecular y la cantidad de ADN tras el corte y después de la ligadura del adaptador. Verificar el tamaño molecular 'shift' entre estos pasos es crucial para confirmar la presencia de adaptadores a los extremos de cada fragmento de ADN que será sometido a PCR mediada por el adaptador en los pasos posteriores. La cantidad de DNA restante al final del paso de la ligadura de adaptador también es importante, desde menos de 100 ng del producto biblioteca es necesario en este paso para garantizar la suficiente cantidad tras los pasos de conversión de enriquecimiento y bisulfito de destino. Una medición final de alta sensibilidad fue realizada en la biblioteca de metil-Seq construida para que la biblioteca puede diluirse adecuadamente para el agrupamiento posterior en el secuenciador de última generación. Por último, bisulfito pirosecuenciación fue empleado como un método altamente cuantitativa, independiente para evaluar la exactitud de la tubería analítica. La validación final usando las muestras originales y replicación con animales adicionales son pasos cruciales para que el experimento puede detectar biológicamente importantes cambios en la metilación del ADN.
También incluimos varias directrices en caso de desviación del protocolo o si se encuentran problemas. En primer lugar, es posible perder demasiado ADN durante la reparación final, adaptador ligadura o pasos de purificación grano magnético. Por otra parte, a partir de cantidades de DNA puede ser pequeño (< 200 ng) debido a la disponibilidad limitada del tejido ADN o aplicación de varios métodos de enriquecimiento tales como de fluorescencia activada célula clasificación. Aumentar el número de ciclo durante los pasos de amplificación de la dos biblioteca puede ser capaz de compensar la pérdida excesiva de ADN o baja a partir de la cantidad de ADN en el protocolo de construcción de la biblioteca. Sin embargo, no más de un 2-3 ciclos se recomiendan, como plantilla excesiva amplificación es probable que conduzca a un aumento en el número de duplicados dice ser secuenciado. Estos duplicados se excluyen durante la etapa de alineamiento para evitar sesgo en los cálculos del porcentaje de metilación. En segundo lugar, si el tamaño medio del ADN no aumenta por más de 30 bps, verifique para asegurarse de que los reactivos son nuevos, como la polimerasa de la DNA de T4, Klenow y T4 ligasa puede ser viejo. Reactivos de recambio disponibles en el mercado pueden ser utilizados.
Además, es posible que el DMRs previstas no pueden validar por pirosecuenciación, donde las diferencias de la metilación de ADN no existen o son significativamente menores que las predichas por el análisis. Mala validación de regiones de candidato es un problema muy común para muchos análisis de genoma, como cuando pirosecuenciación resultados no confirman la metilación diferencial o el tamaño del efecto es mucho menor que la predicha por el análisis. BSmooth es un paquete analítico "suaviza" los niveles de metilación a través de una ventana de múltiples GPC. Para el experimento actual, el BSmooth implicado un DMR cuyos niveles de metilación se validaron por pirosecuenciación de bisulfito. Sin embargo, es probable que habrá discrepancias entre los niveles de metilación predichos por BSmooth y los verificados por pirosecuenciación. Las discrepancias surgen de la función de suavizado que estima los valores promedio de metilación a través de la GPC en un DMR, incluyendo consecutivos GPC que puede diferir en más del 50% en la metilación del ADN o GPC cuyos valores de metilación se excluyeron debido a subumbrales leer a profundidad. Paquetes de R como MethylKit24 pueden utilizarse para identificar ventanas más pequeñas de GPC o incluso sola GPC cuyos niveles de metilación se correlacionan fuertemente con aquellos validados por pirosecuenciación. Diferentes paquetes de aplicación y prueba sus regiones previstas o GPC de metilación diferencial por pirosecuenciación asegurará la solidez de los datos. Alternativamente, bibliotecas de metil-Seq originales pueden reordenar y añadir a archivos de lectura para aumentar la profundidad de lectura. Determinación de los niveles de metilación son semi cuantitativa y dictado por el número de lecturas [(# of CpGs) / (# de TpGs + GPC)], aumento de la profundidad de lectura para una GPC dada aumentarán la precisión de su valor de metilación por ciento. En este estudio, se consideraron sólo GPC cuyos valores de metilación se determinaron por Lee por lo menos diez y alcanzó una cobertura Lea general de 19 x de cada GPC.
La plataforma de metil-Seq de rata no es sin sus limitaciones. Mientras que es más rentable que la secuenciación del genoma completo de bisulfito, es considerablemente más caro que otros métodos. Sin embargo, la mayor parte del costo fue para comprar pistas en el secuenciador y no para el sistema de captura. Dependiendo de la profundidad de lectura necesaria, con comparaciones Cruz-tejido que requieren menos debido a grande (25-70%) las diferencias12 en la metilación del ADN, el costo puede reducirse por multiplexación más muestras por carril y el uso de una plataforma de mayor capacidad. Además, la preparación de la muestra es más lento que otros métodos. Mientras que es similar a otros enfoques de pulldown que incorporan la última generación de la secuencia, los pasos de conversión y purificación de bisulfito añadido añadir a la carga de trabajo. En general, la plataforma de metil-Seq es una alternativa rentable a la secuenciación del genoma completo y ofrece resolución de base par a más de 2,3 millones de GPC, que es mucho más que por las plataformas de microarreglos. Hasta la fecha, el disponible en el comercio humano y ratón se han utilizado plataformas metil-Seq para documentar cambios dependientes del alcohol en el cerebro de macacos25,26, genes del desarrollo neurológico en el cerebro de ratón9y sangre-cerebro objetivos de glucocorticoides10. Además, la capacidad de regiones específicas, independientemente del reconocimiento de la secuencia de destino por enzimas de restricción, es una plataforma ideal para las comparaciones entre especies. Para este estudio, se diseñó la plataforma metil-Seq para la rata, que se llevan a cabo muchos experimentos farmacológicos, metabólicos y de comportamiento sin el beneficio de una herramienta de methylomic de todo el genoma. Nuestros datos muestran que puede ser utilizado para detectar DMRs en un modelo de rata de estrés y correlacionó otros parámetros fisiológicos como los niveles del plasma total CORT.
La plataforma de metil-Seq es ideal para los experimentos epigenéticos en animales con genomas secuenciados que pueden no tener suficiente evidencia experimental que documentan las regiones reguladoras. Cuando tales regiones son disponibles, regiones adicionales pueden diseñado y adjunto a la versión actual. Además, la plataforma es ideal para genómica comparativa, puesto que el enriquecimiento de destino no está limitado por el reconocimiento de la enzima de la restricción. Por ejemplo, la región del promotor de cualquier gen de interés puede ser capturada sin importar si alberga un sitio de restricción específicas. Del mismo modo, se pueden capturar cualquier regiones reguladoras, como los identificados en ratón o en los seres humanos, que se conservan en el genoma de interés.
Divulgaciones
El manuscrito forma parte de un premio de concurso de Agilent Technologies.
Agradecimientos
Este estudio fue financiado por los NIH grant MH101392 (RSL) y apoyo de las siguientes fundaciones y premios: un premio al investigador joven NARSAD, Margaret Ann precio investigador fondo, el fondo James Wah humor trastornos erudito por la Charles T. Bauer Foundation, Baker Fundación y la Fundación del partido de proyecto (RSL).
Materiales
| Name | Company | Catalog Number | Comments |
| Radioimmuno assay (RIA) | MP Biomedicals | 7120126 | Corticosterone, 125I labeled |
| Master Pure DNA Purification Kit | Epicentre/Illumina | MC85200 | |
| Thermal-LOK 2-Position Dry Heat Bath | USA Scientific | 2510-1102 | Used with 1.5 mL tubes |
| Vortex Genie 2 | Fisher | 12-812 | Vortex Mixer |
| Ethyl alcohol, Pure | Sigma-Aldrich | E7023 | 100% Ethanol, molecular grade |
| Centrifuge 5424 R | Eppendorf | - | Must be capable of 20,000 x g |
| Qubit 2.0 | ThermoFisher Scientific | Q32866 | Fluorometer |
| Qubit dsDNA BR Assay Kit | ThermoFisher Scientific | Q32850 | |
| Qubit dsDNA HS Assay Kit | ThermoFisher Scientific | Q32851 | High sensitivity DNA detection reagents |
| Qubit Assay Tubes | ThermoFisher Scientific | Q32856 | |
| SureSelectXT Rat Methyl-Seq Reagent Kit | Agilent Technologies | G9651A | Reagents for preparing the Methyl-Seq library |
| SureSelect Rat Methyl-Seq Capture Library | Agilent Technologies | 931143 | RNA baits for enrichment of rat targets |
| IDTE, pH 8.0 | IDT DNA | 11-05-01-09 | 10 mM TE, 0.1 mM EDTA |
| DNA LoBind Tube 1.5 mL | Eppendorf | 22431021 | |
| Covaris E-series or S-series | Covaris | - | Isothermal sonicator |
| microTUBE AFA Fiber Pre-Slit Snap-Cap 6x16mm (25) | Covaris | 520045 | |
| Water, Ultra Pure (Molecular Biology Grade) | Quality Biological | 351-029-721 | |
| Veriti 96 Well-Thermal Cycler | Applied Biosystems | 4375786 | |
| AMPure XP Beads | Beckman Coulter | A63880 | DNA-Binding magnetic beads |
| 96S Super Magnet | ALPAQUA | A001322 | Magnetic plate for purification steps |
| 2200 TapeStation | Agilent Technologies | G2965AA | Electrophresis-based bioanalyzer |
| D1000 ScreenTape | Agilent Technologies | 5067-5582 | |
| D1000 ScreenTape High Sensitivity | Agilent Technologies | 5067-5584 | |
| D1000 Reagents | Agilent Technologies | 5067-5583 | |
| D1000 Reagents High Sensitivity | Agilent Technologies | 5067-5585 | |
| DNA110 SpeedVac | ThermoFisher Scientific | - | Vacuum Concentrator |
| Dynabeads MyOne Streptavidin T1 magnetic beads | Invitrogen | 65601 | Streptavidin magnetic beads |
| Labquake Tube Rotator | ThermoFisher Scientific | 415110Q | Nutator Mixer is also acceptable |
| EZ DNA Methylation-Gold Kit | Zymo Research | D5006 | Bisulfite conversion kit. Contains Binding, Wash, Desulphonation, and Elution buffers |
| Illumina Hi-Seq 2500 | Illumina | - | Next-generation sequencing machine |
| PCR and Pyrosequencing Primers | IDT DNA | Variable | |
| Taq DNA Polymerase with ThermoPol Buffer - 2,000 units | New England BioLabs | M0267L | |
| Deoxynucleotide (dNTP) Solution Set | New England BioLabs | N0446S | |
| Pyromark MD96 | QIAGEN | - | Pyrosequencing machine |
| Ethyl Alcohol 200 Proof | Pharmco-Aaper | 111000200 | 70% Ethanol solution |
| Sodium Hydroxide Pellets | Sigma-Aldrich | 221465 | 0.2 M NaOH denature buffer solution |
| Tris (Base) from J.T. Baker | Fisher Scientific | 02-004-508 | 10 mM Tris Acetate Buffer wash buffer solution |
| PyroMark Gold Q96 Reagents (50x96) | QIAGEN | 972807 | Reagents required for pyrosequencing |
| PyroMark Annealing Buffer | QIAGEN | 979009 | |
| PyroMark Binding Buffer (200 mL) | QIAGEN | 979006 | |
| Streptavidin Sepharose High Performance Beads | GE Healthcare | 17-5113-01 | Streptavidin-coated sepharose beads |
| PyroMark Q96 HS Plate | QIAGEN | 979101 | Pyrosequencing assay plate |
| Eppendorf Thermomixer R | Fisher Scientific | 05-400-205 | Plate mixer. 96-well block sold separately (cat. No 05-400-207) |
| SureDesign Website | Agilent Technologies | - | Target capture design software (https://earray.chem.agilent.com/suredesign/) |
| UCSC Genome Browser | University of California Santa Cruz | - | rat Nov 2004 rn4 assembly |
| Agilent Methyl-Seq Protocol | Agilent Technologies | - | https://www.agilent.com/cs/library/usermanuals/public/G7530-90002.pdf |
Referencias
- Barski, A., et al. High-resolution profiling of histone methylations in the human genome. Cell. 129 (4), 823-837 (2007).
- Meissner, A., et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 454 (7205), 766-770 (2008).
- Bibikova, M., et al. High density DNA methylation array with single CpG site resolution. Genomics. 98 (4), 288-295 (2011).
- Naumov, V. A., et al. Genome-scale analysis of DNA methylation in colorectal cancer using Infinium HumanMethylation450 BeadChips. Epigenetics. 8 (9), 921-934 (2013).
- Wockner, L. F., et al. Genome-wide DNA methylation analysis of human brain tissue from schizophrenia patients. Translational Psychiatry. 4, e339 (2014).
- Meissner, A., et al. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Research. 33 (18), 5868-5877 (2005).
- Smith, Z. D., Gu, H., Bock, C., Gnirke, A., Meissner, A. High-throughput bisulfite sequencing in mammalian genomes. Methods. 48 (3), 226-232 (2009).
- Slieker, R. C., et al. Identification and systematic annotation of tissue-specific differentially methylated regions using the Illumina 450k array. Epigenetics Chromatin. 6 (1), 26 (2013).
- Hing, B., et al. Adaptation of the targeted capture Methyl-Seq platform for the mouse genome identifies novel tissue-specific DNA methylation patterns of genes involved in neurodevelopment. Epigenetics. 10 (7), 581-596 (2015).
- Seifuddin, F., et al. Genome-wide Methyl-Seq analysis of blood-brain targets of glucocorticoid exposure. Epigenetics. 12 (8), 637-652 (2017).
- Irizarry, R. A., et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nature Genetics. 41 (2), 178-186 (2009).
- Lee, R. S., et al. Adaptation of the CHARM DNA methylation platform for the rat genome reveals novel brain region-specific differences. Epigenetics. 6 (11), 1378-1390 (2011).
- Jankord, R., et al. Stress vulnerability during adolescent development in rats. Endocrinology. 152 (2), 629-638 (2011).
- Pellow, S., Chopin, P., File, S. E., Briley, M. Validation of open:closed arm entries in an elevated plus-maze as a measure of anxiety in the rat. Journal of Neuroscience Methods. 14 (3), 149-167 (1985).
- Krueger, F., Andrews, S. R. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 27 (11), 1571-1572 (2011).
- Langmead, B., Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nature Methods. 9 (4), 357-359 (2012).
- Langmead, B., Trapnell, C., Pop, M., Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biology. 10 (3), R25 (2009).
- Hansen, K. D., Langmead, B., Irizarry, R. A. BSmooth: from whole genome bisulfite sequencing reads to differentially methylated regions. Genome Biology. 13 (10), R83 (2012).
- Lee, R. S., et al. Chronic corticosterone exposure increases expression and decreases deoxyribonucleic acid methylation of Fkbp5 in mice. Endocrinology. 151 (9), 4332-4343 (2010).
- Lee, R. S., et al. A measure of glucocorticoid load provided by DNA methylation of Fkbp5 in mice. Psychopharmacology. , (2011).
- Bose, M., Olivan, B., Laferrere, B. Stress and obesity: the role of the hypothalamic-pituitary-adrenal axis in metabolic disease. Current Opinion in Endocrinology, Diabetes, and Obesity. 16 (5), 340-346 (2009).
- Brydon, L., Magid, K., Steptoe, A. Platelets, coronary heart disease, and stress. Brain, Behavior, and Immunity. 20 (2), 113-119 (2006).
- McKlveen, J. M., et al. Chronic Stress Increases Prefrontal Inhibition: A Mechanism for Stress-Induced Prefrontal Dysfunction. Biological Psychiatry. 80 (10), 754-764 (2016).
- Akalin, A., et al. methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biology. 13 (10), R87 (2012).
- Cervera-Juanes, R., Wilhelm, L. J., Park, B., Grant, K. A., Ferguson, B. Alcohol-dose-dependent DNA methylation and expression in the nucleus accumbens identifies coordinated regulation of synaptic genes. Translational Psychiatry. 7 (1), e994 (2017).
- Cervera-Juanes, R., Wilhelm, L. J., Park, B., Grant, K. A., Ferguson, B. Genome-wide analysis of the nucleus accumbens identifies DNA methylation signals differentiating low/binge from heavy alcohol drinking. Alcohol. 60, 103-113 (2017).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoExplorar más artículos
This article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados