
Method Article
Una piattaforma di metil-Seq ratto per identificare i cambiamenti epigenetici connessi con l'esposizione di sforzo
In questo articolo
Riepilogo
Qui, descriviamo il protocollo e l'implementazione di metil-Seq, una piattaforma di epigenomic, utilizzando un modello del ratto per identificare i cambiamenti epigenetici associati all'esposizione a stress cronico. I risultati dimostrano che la piattaforma di metil-Seq ratto è in grado di rilevare differenze di metilazione che derivano dall'esposizione di sforzo in ratti.
Abstract
Genomi di una più ampia varietà di animali, appena disponibili, c'è un crescente bisogno di strumenti che possono catturare i cambiamenti epigenetici dinamici in questi modelli animali. Il topo è un particolare modello animale, dove uno strumento epigenetico possa integrare molti studi farmacologici e comportamentali per fornire informazioni meccanicistiche penetranti. A tal fine, abbiamo adattato il SureSelect Target Capture System (denominate metil-Seq) per il ratto, che può valutare i livelli di metilazione del DNA in tutto il genoma di topo. Il design di ratto mirato promotori, isole CpG, coste dell'isola e regioni ricche GC da tutti RefSeq geni.
Per implementare la piattaforma su un esperimento di ratto, ratti Sprague Dawley maschii sono stati esposti allo sforzo cronico variabile per 3 settimane, dopo di che sono stati raccolti campioni di sangue per estrazione del DNA genomico. Metil-Seq librerie furono costruite dai campioni di DNA di ratto dalla tosatura, adattatore legatura, arricchimento di destinazione, conversione di bisolfito e multiplexing. Le librerie sono state sequenziate su una piattaforma di sequenziamento di nuova generazione e le letture in sequenza sono state analizzate per identificare DMRs tra DNA dei ratti stressati e atone. Candidato ideale DMRs indipendentemente sono state convalidate da bisolfito pyrosequencing per confermare l'affidabilità della piattaforma.
I risultati dimostrano che la piattaforma di metil-Seq ratto è un utile strumento epigenetico che può acquisire le modifiche di metilazione indotte tramite l'esposizione allo stress.
Introduzione
Gli avanzamenti in sequenza ad alta velocità hanno condotto ad una ricchezza delle sequenze genomic per modello e organismi non-modello. La disponibilità di tali sequenze ha facilitato la ricerca in genetica, la genomica comparativa e trascrittomica. Per esempio, sequenze genomiche disponibili sono altamente utile per allineare i dati di sequenziamento da esperimenti di ChIP-Seq che arricchiscono DNA sulla base della sua associazione con istone modifiche1o del bisolfuro, che misura la metilazione del DNA di rilevamento di uracile formata da conversione di bisolfito di unmethylated cytosines2. Tuttavia, ci sono stati ritardi nell'implementazione di piattaforme di epigenomic che incorporano dati di sequenziamento genomico disponibile nel loro disegno a causa della mancanza di dati con annotazioni di sequenze regolatrici specie-specifici che possono influenzare la funzione del gene.
In particolare, la metilazione del DNA è uno dei più ampiamente studiati modificazioni epigenetiche sul DNA che possono sfruttare dati genomici disponibili per la costruzione di una piattaforma di methylomic. Un esempio è una piattaforma basata su array per la methylome umana3, che è stato ampiamente utilizzato in varie discipline da oncologia psichiatria4,5. Purtroppo, simili piattaforme per modelli animali non-umani sono scarsi, come non ci è praticamente nessuna piattaforma ampiamente usato che hanno approfittato della sequenza genomica nella loro progettazione iniziale.
Un metodo comune per valutare il paesaggio di methylomic di modelli animali non-umani è rappresentazione ridotta bisolfito sequenziamento (RRBS)6. Questo approccio supera il costo dell'intero genoma del bisolfuro che, fornendo al contempo un paesaggio completo methylomic, fornisce una lettura-profondità copertura inferiore a causa di costo e informazioni funzionali limitate nelle grandi zone di gene-poveri del genoma2 . RRBS comporta la raccolta di limitazione e selezione della dimensione del DNA genomico a arricchire per altamente sequenze GC-ricche come le isole di CpG che sono comunemente trovati nelle vicinanze di promotori di geni e pensati per svolgere un ruolo nel gene regolamento7. Mentre il metodo RRBS è stato utilizzato in una serie di importanti studi, sua dipendenza da enzimi di restrizione non è senza limiti e sfide notevoli. Per esempio, arricchimento delle sequenze di GC-rich in RRBS è interamente dipendente dalla presenza di specifiche sequenze di riconosciuta dall'enzima di restrizione e selezione dimensione successiva tramite l'elettroforesi. Ciò significa che eventuali aree genomiche che non contengono questi siti di restrizione vengono esclusi durante la selezione della dimensione. Inoltre, i confronti cross-specie sono difficili a meno che gli stessi siti di restrizione sono presenti nei loci stessi tra le diverse specie.
Un approccio per superare le limitazioni del RRBS è quello di utilizzare un metodo di arricchimento che si avvale della sequenza genomica pubblicata nella progettazione della piattaforma. La piattaforma umana basata su array utilizza sonde primer progettati contro specifici CpGs per estensione di allele-specifico (CG vs TG dopo la conversione di bisolfito) destinazione ricottura e primer. Il suo design riflette non solo la sequenza genomic umano disponibile, ma nelle regioni regolatrici verificato sperimentalmente acquisite da più linee di indagine, ad esempio ENCODE ed ENSEMBL8. Nonostante il suo ampio uso nelle indagini di methylomic umano, una piattaforma simile non esiste per modelli animali. Inoltre, il formato basato su matrice impone costrizioni significative la superficie disponibile per il posizionamento della sonda. In ultimi anni, gli sforzi sono stati effettuati per combinare la specificità di destinazione accordata dal design della sonda di cattura e la caratteristica di alto-rendimento di sequenziamento di nuova generazione. Un tale sforzo ha provocato il sistema di arricchimento di sequenziamento-basata su target per il genoma del mouse (mouse metil-Seq), che è stato usato per identificare le differenze specifiche del cervello o indotta da glucocorticoidi nella metilazione9,10. Piattaforme simili per altri modello e non-modelli animali sono necessari per facilitare la ricerca di epigenomic in questi animali.
Qui, noi dimostrare l'attuazione di questa nuova piattaforma per condurre analisi methylomic sul ratto. Il ratto ha servito come un importante modello animale in farmacologia, metabolismo, neuroendocrinologia e comportamento. Ad esempio, c'è una crescente necessità di comprendere i meccanismi di fondo che danno luogo a tossicità dei farmaci, l'obesità, risposta allo stress o la tossicodipendenza. Una piattaforma di alto-rendimento in grado di catturare i cambiamenti methylomic sono associati con queste condizioni sarebbe aumentare la nostra comprensione dei meccanismi. Poiché il genoma di topo manca ancora annotazione per regioni regolative, abbiamo incorporato promotori non ridondante, isole CpG, isola shores11e identificati in precedenza sequenze GC-rich in ratto metil-Seq piattaforma12.
Per valutare la corretta progettazione e implementazione della piattaforma SureSelect destinazione arricchimento (genericamente come metil-Seq) per il genoma del ratto, abbiamo impiegato un modello del ratto di stress cronico variabile (CVS)13 per identificare differenzialmente metilato regioni tra animali atone e stressati. La nostra piattaforma di progettazione, protocollo e implementazione può essere utile per gli investigatori che vogliono condurre un'indagine completa ed imparziale epigenetica su un organismo cui sequenza genomica è già disponibile ma rimane scarsamente con annotazioni.
Protocollo
Tutti gli esperimenti sono stati completati in conformità e rispetto tutti i pertinenti orientamenti normativi ed istituzionali, tra cui il Comitato di utilizzo presso la Johns Hopkins School of Medicine e istituzionali Animal Care.
1. gli animali
- Ottenere maschi ratti Sprague-Dawley adolescenti a 4 settimane di età. Gli animali in gabbie di ratto in policarbonato in una temperatura della casa- e camera di umidità controllata su un 12 h, 12 h scuro ciclo di luce con luce inizio alle h. 0600 fornire agli animali con ad libitum accesso all'acqua.
- Consentire ratti di acclimatare per 1 settimana per ridurre lo stress associato con trasporto. Coppia-casa degli animali (N = 16) per precludere sforzo di isolamento e a 5 settimane di età, iniziano la variabile cronica stress regime (CVS) per 3 settimane.
2. cronico Stress variabile
- Amministrare il regime CVS al mattino (9-11) e una volta al pomeriggio (1 – 15) a volte irregolari per mantenere la routine imprevedibile. Incorporare una notte mite fattori di stress. Il CVS regime comprende: 1) 3 h in un cilindro di contenimento; 2) 10 min nuotata; gabbia di h 3) 3 inclinazione 4) 1h lento scuotendo la piattaforma; e 5) 1h nella cella frigorifera a 4 ° C.
Nota: Durante la notte fattori di stress includono sociale affollamento (5 a gabbia), isolamento sociale, biancheria da letto bagnato, restrizione alimentare e luci accese. Nella tabella 1viene fornito un programma settimanale tipico del regime di sforzo.
3. endocrini saggi
-
Determinare i livelli di corticosterone (CORT) utilizzando i prelievi di sangue (~ 50 mL) coda raccolti allo stesso tempo (9) due volte alla settimana in tutto l'esperimento, prima del regime CVS per stabilire i livelli di ormone di baseline (giorno 0), una volta durante la metà del download CVS (giorni 4,11 e 18), dopo ogni 7 giorni di CVS (giorni 7 e 14) e a conclusione del CVS (giorno 21). Raccogliere campioni di sangue prima del regime lo stress quotidiano.
- Raccogliere un campione di sangue finale tronco durante l'eutanasia (giorno 25) per RIA ed estrazione di DNA genomico.
- Centrifugare tutti i campioni di sangue (600 x g, 4 ° C, 10 min) per separare il plasma dalle cellule del sangue. Dispensare fuori il plasma (surnatante) e conservare i campioni a-80 ° C.
- Scongelare e utilizzare il plasma per determinare i livelli di CORT dalla radioimmunoanalisi (RIA). Garantire che i livelli CORT del plasma di 3 settimana sono elevati negli animali sollecitati per verificare la robustezza del regime di sforzo.
4. comportamento
- Dopo il regime CVS (giorni 23 – 24), valutare ogni animale per ansia-come comportamento su elevato più labirinto (EPM)14.
- Utilizzando una videocamera, record gli animali sull'apparato EPM per 300 s e il tempo trascorso nel centro, il Punteggio chiuso le braccia e a braccia aperte.
5. progettazione di ratto metil-Seq
- Utilizzando il Browser del genoma di UCSC, ottenere coordinate genomiche non ridondante (ratto Nov 2004 rn4 assembly) per isole CpG e coste dell'isola (± 1 kb che fiancheggiano isole CpG), promotori (± 1 kb di ogni TSS) di ogni gene RefSeq e altre sequenze che possono essere disponibili da letteratura relativa.
Nota: Per il ratto metil-Seq, ulteriori sequenze di GC-ricche da una piattaforma basata su array metilazione precedente è stato aggiunto12. Per le regioni di maggiore di 5 kbps, alterne regioni di bps 500 sono state campionate seguita da 1 kbps che sono stati ignorati. Il design di metil-Seq ratto finale è costituito da 111 Mbps, CpGs 2,3 milioni; e una dimensione media regione di 594 bps. Esso si rivolge 228.800 loci unici. - Immettere un elenco compilato di genomiche coordinate in un software di progettazione di cattura di destinazione disponibili in commercio per design appropriato della sonda.
6. costruzione della biblioteca di metil-Seq ratto da DNA Genomic
Nota: Per eliminare effetti di batch, elaborare campioni multipli allo stesso tempo e scalare il mix master di conseguenza. Estrazione del DNA utilizzando un kit di estrazione del DNA commercialmente disponibile. Metodi basati su colonna o precipitazione sia restituiscono del DNA di genomic di alta qualità (rapporto di 260/280 ~ 1,8). Uso di metodi basati su fenolo non sono raccomandati. Eluire o risospendere il DNA in buffer di TE basso (10 mM TE, 0,1 mM EDTA, pH 8.0).
- Preparazione del campione
Nota: Per ogni passo usando i branelli magnetici DNA-legantesi, assicurarsi che le perle sono acclimatate alla temperatura ambiente per almeno 30 min e ben miscelate prima dell'uso.- Taglio del DNA
- Utilizzare un fluorimetro per determinare la concentrazione di DNA double-stranded iniziale di ciascun campione. Diluire > 1 µ g di gDNA a 50 µ l con buffer di TE basso (10 mM TE, 0,1 mM EDTA, pH 8.0) in basso per microcentrifuga DNA-legantesi.
- Taglio campioni usando un sonicatore isotermica (10% Duty Cycle, 5 intensità, 200 cicli al Burst, 6 cicli di 60 s, spazzamento di frequenza, 4 ° C).
- Valutare la qualità di DNA usando un sistema basato su elettroforesi che misura la quantità e la dimensione del DNA.
Nota: La quantità di DNA consigliata è di 1 µ g, o 3 µ g. Se c'è un limitato materiale di partenza, l'importo più basso input deve essere > 500 ng, come gli importi più bassi pregiudicherebbero la quantità e la qualità delle librerie generate.
- Riparazione DNA si conclude.
- Utilizzare il ratto metil-Seq kit per preparare la fine-riparazione Master Mix sul ghiaccio. Aggiungere 52 µ l di miscela per ogni campione e incubare in un termociclatore privo di coperchio riscaldato (20 ° C per 30 min, attesa di 4 ° C).
Fine-riparazione Master Mix (per esempio):
35,2 µ l di acqua
10 µ l di tampone di fine riparazione (10x)
1,6 µ l di dNTP Mix
1 µ l di T4 DNA polimerasi
2 µ l di DNA polimerasi di Klenow
2,2 µ l della chinasi di polinucleotide T4 - Purificare i campioni utilizzando 180 µ l di biglie magnetiche DNA-legantesi e 400 µ l di etanolo al 70% appena preparata per campione. Aggiungere 180 µ l di perline per ogni campione e incubare per 5 min a temperatura ambiente. A pellet perline, rimuovere il surnatante e risospendere il pellet in 200 µ l di etanolo al 70%. Eliminare l'etanolo e ripetere il lavaggio una volta.
- Utilizzare una piastra magnetica per pellet perline e rimuovere quanto più etanolo come possibile. A secco in un a 37 ° C termoblocco per 3 – 5 min fino a quando il pellet perlina è completamente a secco. Risospendere in 44 µ l di acqua priva di nucleasi e raccogliere circa 42 µ l del surnatante.
Punto di sosta: Al termine della riparazione del DNA, i campioni possono essere sigillati e conservati a-20 ° C.
- Utilizzare il ratto metil-Seq kit per preparare la fine-riparazione Master Mix sul ghiaccio. Aggiungere 52 µ l di miscela per ogni campione e incubare in un termociclatore privo di coperchio riscaldato (20 ° C per 30 min, attesa di 4 ° C).
- Adenilato 3' estremità.
- Preparare la miscela di poliadenilazione Master sul ghiaccio. Aggiungere 9 µ l mix per ogni campione e incubare in un termociclatore privo di coperchio riscaldato (37 ° C per 30 min, attesa di 4 ° C).
Poliadenilazione Master Mix (per esempio):
5 µ l di tampone di Klenow
1 µ l di dATP
3 µ l di DNA polimerasi di Klenow - Purificare i campioni utilizzando 90 µ l di biglie magnetiche DNA-legantesi e 400 µ l di etanolo al 70% appena preparata per campione. Aggiungere 90 µ l di perline per ogni campione e incubare per 5 min a temperatura ambiente. A pellet perline, rimuovere il surnatante e risospendere il pellet in 200 µ l di etanolo al 70%. Eliminare l'etanolo e ripetere il lavaggio una volta.
- Utilizzare una piastra magnetica per pellet perline e rimuovere quanto più etanolo come possibile. A secco in un a 37 ° C termoblocco per 3 – 5 min fino a quando il pellet perlina è completamente a secco. Risospendere in 35 µ l di acqua priva di nucleasi e raccogliere circa 33,5 µ l del surnatante.
- Preparare la miscela di poliadenilazione Master sul ghiaccio. Aggiungere 9 µ l mix per ogni campione e incubare in un termociclatore privo di coperchio riscaldato (37 ° C per 30 min, attesa di 4 ° C).
- Legare l'adattatore metilato.
- Preparare la legatura Master Mix sul ghiaccio e aggiungere 16,5 µ l di miscela per ogni campione. Incubare in un termociclatore privo di coperchio riscaldato (20 ° C per 15 min, attesa di 4 ° C).
La legatura Master Mix (per esempio):
2,5 µ l di acqua
2,5 µ l di metil-Seq metilato adattatore
10 µ l di tampone di T4 DNA ligasi (5x)
1,5 µ l di T4 DNA ligasi - Purificare i campioni utilizzando 90 µ l di biglie magnetiche DNA-legantesi e 400 µ l di etanolo al 70% appena preparata per campione. Aggiungere 90 µ l di perline per ogni campione e incubare per 5 min a temperatura ambiente. A pellet perline, rimuovere il surnatante e risospendere il pellet in 200 µ l di etanolo al 70%. Eliminare l'etanolo e ripetere il lavaggio una volta.
- Utilizzare una piastra magnetica per pellet perline e rimuovere quanto più etanolo come possibile. A secco in un a 37 ° C termoblocco per 3 – 5 min fino a quando il pellet perlina è completamente a secco. Risospendere in 22 µ l di acqua priva di nucleasi e raccogliere circa 22 µ l del surnatante. Valutare la qualità utilizzando un bioanalyzer.
Nota: Se l'importo totale del DNA è meno di 500 ng, cesoia e processo di DNA aggiuntive prima di procedere con i passaggi successivi. Se la dimensione media di DNA non aumenta da più di 30 bps, controllare che i reagenti siano nuovi, come T4 DNA polimerasi, Klenow, e/o T4 ligasi potrebbe essere vecchia.
Punto di sosta: Dopo legatura adattatore metilato, campioni possono essere sigillati e conservati a-20 ° C.
- Preparare la legatura Master Mix sul ghiaccio e aggiungere 16,5 µ l di miscela per ogni campione. Incubare in un termociclatore privo di coperchio riscaldato (20 ° C per 15 min, attesa di 4 ° C).
- Taglio del DNA
- Ibridazione
- Trasferire i campioni a basso microcentrifuga DNA-legantesi e utilizzare un concentratore a vuoto riscaldato per ridurre il volume di campione di meno di 3,4 µ l. ricostituire i campioni a 3,4 µ l.
Nota: Concentrato i campioni a circa ~ 3 µ l per assicurare i campioni vengono rimossi dal concentratore a vuoto prima di tutto il liquido evapora. - Preparare il tampone di ibridazione a temperatura ambiente e metil-Seq blocco Mix sul ghiaccio. µ L 5,6 di metil-Seq blocco Mix per ogni campione e incubare in termociclatore (95 ° C per 5 min, 65 ° C per 2 min, attesa di 65 ° C).
Tampone di ibridazione (per esempio):
6.63 µ l di Ibrid. metil-Seq 1
0,27 µ l di metil-Seq Hyb 2
2,65 µ l di metil-Seq Hyb 3
3,45 µ l di metil-Seq Hyb 4
Metil-Seq Mix di blocco (per esempio):
2,5 µ l di metil-Seq indicizzazione blocco 1
2,5 µ l di metil-Seq blocco 2
0,6 µ l di metil-Seq blocco 3 - Preparare la RNasi blocco Mix e la miscela di ibridazione Capture Library. Aggiungere 20 µ l di miscela di ibridazione biblioteca Capture per ogni campione e incubare a 65 ° C per almeno 16 h.
RNasi Mix di blocco (per esempio):
0,5 µ l di RNAsi blocco
1,5 µ l di acqua
Acquisizione di miscela di ibridazione di libreria (per esempio):
13 µ l di tampone di ibridazione
2 µ l di RNAsi blocco Mix
5 µ l di ratto metil-Seq catturare biblioteca
Nota: tenere le reazioni a 65 ° C quando si aggiunge la miscela di ibridazione per impedire il legame non specifico. - Aliquotare e 50 µ l di streptavidina biglie magnetiche per campione in una nuova provetta 8 pozzetti striscia. Lavare perline con 200 µ l di tampone di associazione di metil-Seq. Utilizzare piastra magnetica per pellet perline e rimuovere il surnatante tra ogni lavaggio per un totale di 3 lavaggi. Dopo il lavaggio finale, risospendere streptavidina perline in 200 µ l di tampone di associazione di metil-Seq.
- Aggiungere campioni a 200 µ l di biglie magnetiche lavato streptavidina e incubare a temperatura ambiente per 30 minuti utilizzando un miscelatore rotante. Durante la miscelazione, aliquota 200 µ l di tampone di lavaggio metilico-Seq 2 in triplice copia pozzetti di una piastra a 96 pozzetti per campione e posto in un termociclatore per preriscaldare a 65 ° C.
- Dopo l'incubazione, pellet streptavidina biglie magnetiche utilizzando piastra magnetica e risospendere le sfere in 200 µ l tampone di lavaggio di metil-Seq 1. Incubare per 15 min a temperatura ambiente. Utilizzare una piastra magnetica per pellet ed eliminare il surnatante.
- Lavare perline 3 volte con tampone di lavaggio metil-Seq 2: perlina risospendere in 200 µ l di tampone di lavaggio 2 (pre-riscaldati al punto 6.2.5.), incubare perline in termociclatore (65 ° C, 10 min) e a pellet di perline. Gettare il surnatante dopo ogni lavaggio utilizzando una piastra magnetica.
Nota: Mantenere le reazioni di ibridazione a 65 ° C durante l'aggiunta di Wash Buffer 2 per evitare che il legame non specifico. - Aggiungere 20 µ l di tampone di eluizione di metil-Seq ai talloni lavati e incubare a temperatura ambiente per 20 min. uso una piastra magnetica a pellet perline e trasferimento surnatante in un nuovo tubo di striscia. Scartare le perline.
Nota: Durante l'incubazione, preparazione del reagente di conversione di bisolfito.
- Trasferire i campioni a basso microcentrifuga DNA-legantesi e utilizzare un concentratore a vuoto riscaldato per ridurre il volume di campione di meno di 3,4 µ l. ricostituire i campioni a 3,4 µ l.
- Conversione di bisolfito
Nota: Eseguire la conversione di bisolfito del ssDNA eluito utilizzando reagenti appropriati e le istruzioni da un kit di conversione disponibili in commercio bisolfito.- Aggiungere il 130 µ l preparati bisolfito conversione reagente al supernatante dal passaggio precedente. Dividere ciascuna delle 150 reazioni µ l equamente in due pozzi. Incubare in un termociclatore (64 ° C per 2,5 h, presa di 4 ° C).
Nota: La reazione di 150 µ l è diviso equamente in due pozzetti separati per garantire la temperatura omogenea. Dopo incubazione per 2,5 h, procedere immediatamente alla fase successiva. - Associare i campioni di spin colonne aggiungendo 600 µ l di Binding Buffer e lavare una volta con 100 µ l di tampone di lavaggio. Centrifugare le colonne (15.000 x g, 1 min) tra tutti i passaggi di conversione di bisolfito e gettare flusso attraverso.
- Campioni di Desulphonate con l'aggiunta di 200 µ l di tampone di Desulphonation alle colonne. Incubare a temperatura ambiente per 15-20 min. Repeat centrifugazione e scartare flusso attraverso.
- Lavare le colonne due volte con 200 µ l di tampone di lavaggio. Eluire ogni campione aggiungendo 10 µ l di tampone di eluizione per la colonna, incubando per 3 min a temperatura ambiente e la centrifugazione (15.000 x g, 1 min). Ripetere il passaggio di eluizione per un totale di 20 µ l.
- Preparare la reazione di PCR Master Mix 1 sul ghiaccio. Aggiungere 82 µ l di miscela per ogni campione. Incubare in un termociclatore con il seguente programma.
Reazione di PCR Master Mix 1 (per esempio):
30 µ l di acqua
50 µ l di metil-Seq PCR Master Mix
1 µ l di Primer di metil-Seq PCR1 F
1 µ l di Primer di metil-Seq PCR1 R
Termociclatore programma:
Fase 1, 1 ciclo: 95 ° C 2 min
Fase 2, 8 cicli: 95 ° C 30 s, 60 ° C 30 s, 72 ° C 30 s
Fase 3, 1 ciclo: 72 ° C 7 min
Fase 4, 1 ciclo: 4 ° C Hold - Purificare i campioni utilizzando 180 µ l di biglie magnetiche DNA-legantesi e 400 µ l di etanolo al 70% appena preparata per campione. Aggiungere 180 µ l di perline per ogni campione e incubare per 5 min a temperatura ambiente. A pellet perline, rimuovere il surnatante e risospendere il pellet in 200 µ l di etanolo al 70%. Eliminare l'etanolo e ripetere il lavaggio una volta.
- Utilizzare una piastra magnetica per pellet perline e rimuovere quanto più etanolo come possibile. A secco in un a 37 ° C termoblocco per 3 – 5 min fino a quando il pellet perlina è completamente a secco. Risospendere in 21 µ l di acqua priva di nucleasi e raccogliere circa 19,5 µ l del surnatante.
- Aggiungere il 130 µ l preparati bisolfito conversione reagente al supernatante dal passaggio precedente. Dividere ciascuna delle 150 reazioni µ l equamente in due pozzi. Incubare in un termociclatore (64 ° C per 2,5 h, presa di 4 ° C).
- L'indicizzazione
- Preparare la reazione di PCR Master Mix 2 sul ghiaccio. µ L 25,5 Master Mix 2 per ogni campione. Aggiungere 5 µ l commerciale indicizzazione primer ai singoli campioni e incubare in un termociclatore.
Reazione di PCR Master Mix 2 (per esempio):
25 µ l metil-Seq PCR Master Mix
0,5 µ l metil-Seq comune indicizzazione Primer
Termociclatore programma:
Fase 1, 1 ciclo: 95 ° C 2 min
Fase 2, 6 cicli: 95 ° C 30 s, 60 ° C 30 s, 72 ° C 30 s
Fase 3, 1 ciclo: 72 ° C 7 min
Fase 4, 1 ciclo: 4 ° C Hold
Nota: Ulteriori cicli (2-3) possono essere necessarie se la concentrazione di DNA iniziale è sotto i valori consigliati. - Purificare i campioni utilizzando 90 µ l di biglie magnetiche DNA-legantesi e 400 µ l di etanolo al 70% appena preparata per campione. Aggiungere 90 µ l di perline per ogni campione e incubare per 5 min a temperatura ambiente. A pellet perline, rimuovere il surnatante e risospendere il pellet in 200 µ l di etanolo al 70%. Eliminare l'etanolo e ripetere il lavaggio una volta.
- Utilizzare una piastra magnetica per pellet perline e rimuovere quanto più etanolo come possibile. A secco in un a 37 ° C termoblocco per 3 – 5 min fino a quando il pellet perlina è completamente a secco. Risospendere in 24 µ l di acqua priva di nucleasi e raccogliere circa 24 µ l del surnatante.
- Valutare la concentrazione e la bp dimensione utilizzando i reagenti di rilevazione del DNA di alto-sensibilità su un bioanalyzer.
Nota: Se il bioanalyzer non riesce a rilevare la presenza della libreria del DNA, ripetere i passaggi di preparazione con ulteriore DNA.
Punto di arresto: dopo purificazione, indicizzati campioni possono essere sigillati e conservati a-20 ° C. - Pool di campioni per la piattaforma appropriata sequenziamento di nuova generazione utilizzata.
- Utilizzando i dati di concentrazione da bioanalyzer, che determina la molarità di DNA basato su libreria dimensioni e quantità in un dato volume, diluire con buffer di TE basso (6.1.1.1) e combinare tutti i campioni ad una concentrazione finale di 15 pM.
Nota: Un metodo più sensibile di quantificare la libreria è mediante PCR quantitativa in tempo reale, utilizzando primers che le schede di legato di destinazione. - Eseguire campioni riuniti sul numero di corsie che sono sufficienti per 4 campioni per corsia su un sequencer di prossima generazione.
Nota: per esempio, se 16 campioni di libreria sono stati indicizzati in modo univoco e combinati, eseguire le librerie oltre 4 corsie, equivalente a 4 campioni per corsia.
- Utilizzando i dati di concentrazione da bioanalyzer, che determina la molarità di DNA basato su libreria dimensioni e quantità in un dato volume, diluire con buffer di TE basso (6.1.1.1) e combinare tutti i campioni ad una concentrazione finale di 15 pM.
- Preparare la reazione di PCR Master Mix 2 sul ghiaccio. µ L 25,5 Master Mix 2 per ogni campione. Aggiungere 5 µ l commerciale indicizzazione primer ai singoli campioni e incubare in un termociclatore.
7. impostazione della sequenza su un Sequencer di prossima generazione
- Inviare i campioni al nucleo istituzionale sequenziamento per il clustering della biblioteca metil-Seq, seguita dall'ordinamento su una macchina di sequenziamento di nuova generazione.
8. analisi per identificare DMRs
- Implementare il Bismark15, che richiama Bowtie 2.0 come una sequenza interna allineatore16,17, per allineare letture raw inpue al genoma bisolfito-convertito, plus-strand. A seguito di allineamento, utilizzare il Bismark_methylation_extractor per eseguire il controllo di qualità e assegnare un valore stimato di metilazione a ciascun CpG.
- Generare un elenco di DMRs con il pacchetto di BS-Seq18 in Bioconductor. Filtro la DMRs basato su avendo maggiore di 3 consecutivi CpGs e valore-P < 0.05.
Nota: Generare un elenco DMR che include Coordinate genomiche, distanza al gene RefSeq più vicino, numero CpGs all'interno di ogni DMR, media % valore di metilazione di CpG attraverso il DMR per i gruppi di due confronto (per esempio, ha sottolineato vs non accentata), il P-valore, e il valore FDR (tasso di falsi scoperta). Utilizzare l'elenco DMR, vale a dire., coordinate genomiche, per disegnare primers pyrosequencing per la convalida.
9. convalida di bisolfito Pyrosequencing
-
Disegno dell'iniettore
- Disegno primer per bisolfito di PCR e pirosequenziamento. Progettare due insiemi degli iniettori PCR (esterno e annidati) in modo che la PCR annidata amplificherà 150 – 400 bps di un DMR.
Nota: In generale, progettato gli iniettori sono almeno 24 basi lungo con almeno 4 – 5 non consecutivi G (C per il primer reverse) al conto per ridotta temperatura da perdita di complessità di sequenza di ricottura. Uno degli iniettori annidati sarà biotina e purificato per HPLC. Tuttavia, primer standard dovrebbe essere ordinato prima per ottimizzare la fase PCR risolvendo le reazioni su un gel di agarosio.- Progettazione del pirosequenziamento dosaggio primer affinché esso si rivolge il biotinilati complementari filo solo 1 – 2 basi a Monte di CpGs deve essere analizzato. Design più pyrosequencing primer come necessario, come ogni primer pyrosequencing può attendibilmente dosaggio 30 bps a valle.
- Per Rt1-m4, utilizzare le seguenti operazioni:
rRT1M4 all'esterno – F TGTAYGATTTTGGTTATYGTAAAT
rRT1M4 all'esterno – R AACTTACAAATTTCACCAACTCA
rRT1M4 Nested – F GTGGGTTAYGTGGATAATATATAG
rRT1M4 Nested – R AATCACTTACCATTCTCTCTCTAACTA
rRT1M4 Pyro1 TAYGTGGATAATATATAGAT
rRT1M4 Pyro2 GATAGTTATTTGGYGAGTTAG
rRT1M4 Pyro3 GAGTATTTGGAGGAGTTGAT
rRT1M4 Pyro4 GGATTTTAATATTTGGT
- Disegno primer per bisolfito di PCR e pirosequenziamento. Progettare due insiemi degli iniettori PCR (esterno e annidati) in modo che la PCR annidata amplificherà 150 – 400 bps di un DMR.
-
Utilizzare un kit disponibile in commercio per la conversione di bisolfito di ratto sangue gDNA.
Nota: La procedura di conversione di bisolfito è stata adattata dal kit disponibile in commercio con le seguenti modifiche: nel passaggio 1, aggiungere 50 – 100 ng di sangue gDNA e diluire con acqua a 20 µ l. Nel passaggio 9, eluire 20 µ l per campione.- Preparazione del reagente di conversione di bisolfito secondo il protocollo del produttore e si combinano con gDNA diluito. Incubare in termociclatore (64 ° C per 2,5 h, presa di 4 ° C).
- Aggiungere Binding Buffer gDNA convertito in spin colonne e centrifugare (15.000 x g, 1 min). Colonne di lavaggio una volta poi aggiungere Desulphonation Buffer alle colonne e incubare per 15 min a temperatura ambiente. Centrifuga (15.000 x g, 1 min).
- Lavare la colonna con tampone di lavaggio e centrifuga (15.000 x g, 1 min). Ripetere il passaggio di lavaggio con centrifugazione (15.000 x g, 2 min). Aggiungere 20 µ l tampone di eluizione e centrifugare (15.000 x g, 1 min) per eluire.
-
Amplificazione di PCR
- Preparare esterno PCR Master Mix. Aggiungere 21,5 µ l di Master Mix di 3,5 µ l bisolfito-convertito gDNA ed eseguire programma termociclatore.
Esterno PCR Master Mix:
16.25 µ l di acqua
2,5 µ l di tampone di polimerasi [10 x]
0,5 µ l di dNTP [10 mM]
1 µ l di Primer Forward [0,1 µM]
1 µ l di Primer Reverse [0,1 µM]
0,25 µ l di Taq DNA polimerasi [5000 U/mL].
Programma del termociclatore:
Fase 1, 1 ciclo: 94 ° C 4 min
Fase 2, 47 cicli: 94 ° C 1 min, 53 ° C 30 s, 72 ° C 1 min
Fase 3, 1 ciclo: 72 ° C 8 min, 4 ° C Hold - Preparare la PCR annidata Master Mix. Aggiungere 23 µ l di Master Mix 2 µ l di campione da esterno PCR e ripetere il programma esterno del termociclatore PCR. Valutare la qualità del prodotto PCR mediante elettroforesi su gel (1 x TAE buffer, gel di agarosio 1%).
Nested PCR Master Mix:
17,75 µ l di acqua
2,5 µ l di tampone di polimerasi [10 x]
0,5 µ l di dNTP [10 mM]
1 µ l di Primer Forward [0,1 µM]
1 µ l di Primer Reverse [0,1 µM]
0,25 µ l di Taq DNA polimerasi [5000 U/mL]
Nota: Per la PCR annidata, inoltra o il reverse primer deve essere biotinilati.
- Preparare esterno PCR Master Mix. Aggiungere 21,5 µ l di Master Mix di 3,5 µ l bisolfito-convertito gDNA ed eseguire programma termociclatore.
-
Pyrosequencing
- Fare un mix master contenente 38 µ l di tampone di Binding, 35 µ l di acqua e 2 µ l di perline rivestite con streptavidin sepharose per campione. In una piastra a 96 pozzetti, aggiungere 75 µ l di master mix e 5 µ l di prodotto PCR annidata. Agitare su un agitatore per piastre per 15-60 min.
- Durante l'agitazione, aggiungere 12 µ l di primer (0,5 µM, diluito in tampone di ricottura) nei pozzetti di una piastra di dosaggio pirosequenziamento.
- Dopo l'agitazione, eseguire le fasi di lavaggio utilizzando associazione reazione wash buffer. Posto vuoto strumento nella vasca riempita con acqua quindi raccogliere campioni dalla piastra. Immergere il vuoto strumento in trogoli riempito a metà contenente etanolo al 70%, NaOH (0,2 M) e tampone di acetato di Tris (10 mM, pH 7.4). Scollegare dal strumento vuoto vuoto e posto nel HS saggio piastra per trasferire perline.
- Collocare la piastra sul blocco di calore ed incubare a 80 ° C per 2 min piastra Consenti a raffreddare per 5 min poi cominciare il programma di pyro.
Risultati
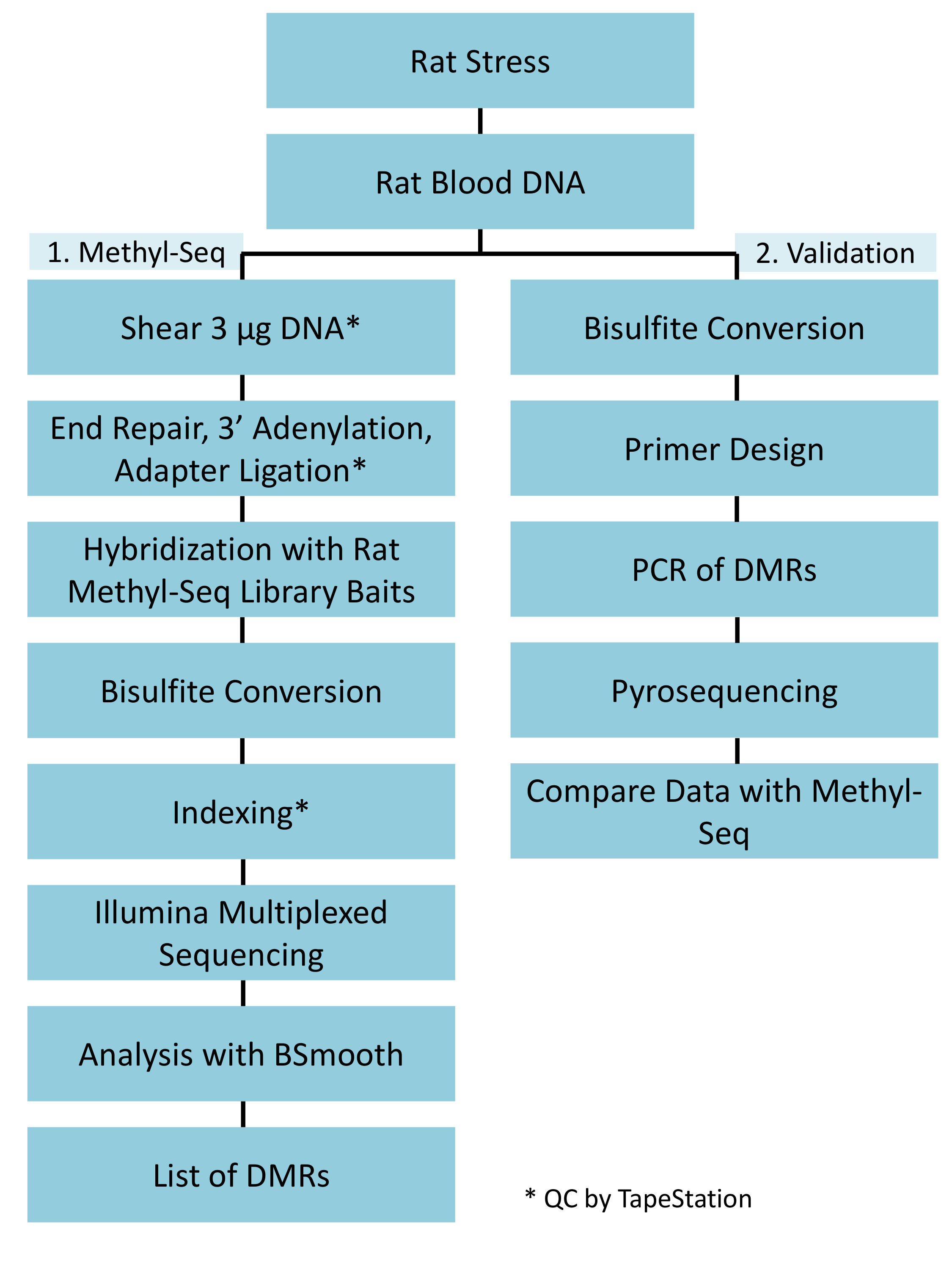
Un'implementazione corretta della piattaforma metil-Seq ratto è dipende da diversi criteri. Figura 1 Mostra il flusso di lavoro globale dello studio e mette in evidenza passi specifici (controllo qualità) che sono necessari prima di andare avanti. Uno dei primi fattori da considerare è la robustezza del modello animale ed il regime di sforzo, che determinano la grandezza dei cambiamenti epigenetici che si verificano in tutto il methylome. Poiché il nostro lavoro animale è basata sulla nostra osservazione precedente che esposizione di corticosterone (CORT) può portare a cambiamenti nella metilazione del DNA19,20, il nostro regime di stress cronico variabile (CVS) doveva essere di rigore sufficienti per produrre ha sottolineato ratti con i livelli elevati del plasma CORT. Un regime settimanale tipico di CVS è indicato nella tabella 1 e consisteva di sforzo quotidiani al mattino, pomeriggio, e durante la notte che vengono costantemente modificati per evitare assuefazione e diminuita risposta allo stress. Durante il regime di 3 settimane, gli animali stressati hanno esibito i livelli significativamente elevati di plasmatica CORT [giorni 4 – 21, controllano: 32,7 3,7 ng/mL, Stress: 103,0 11,9 ng/mL (media SEM), P = 2,2 x 10-4, Figura 2A] sopra quelli di unstressed, animali di controllo. Coerentemente, questi animali inoltre hanno mostrati una maggiore ansia-come comportamento su elevato e labirinto (EPM), come indicato da molto più tempo trascorso tra le braccia chiuse di EPM e meno tempo a braccia aperte (Figura 2B). Questi risultati dimostrano che l'esposizione CVS hanno portato a significativo endocrino e cambiamenti comportamentali, che ci porta a indagare se questi cambiamenti sono stati associati con le firme di metilazione di DNA specifiche.
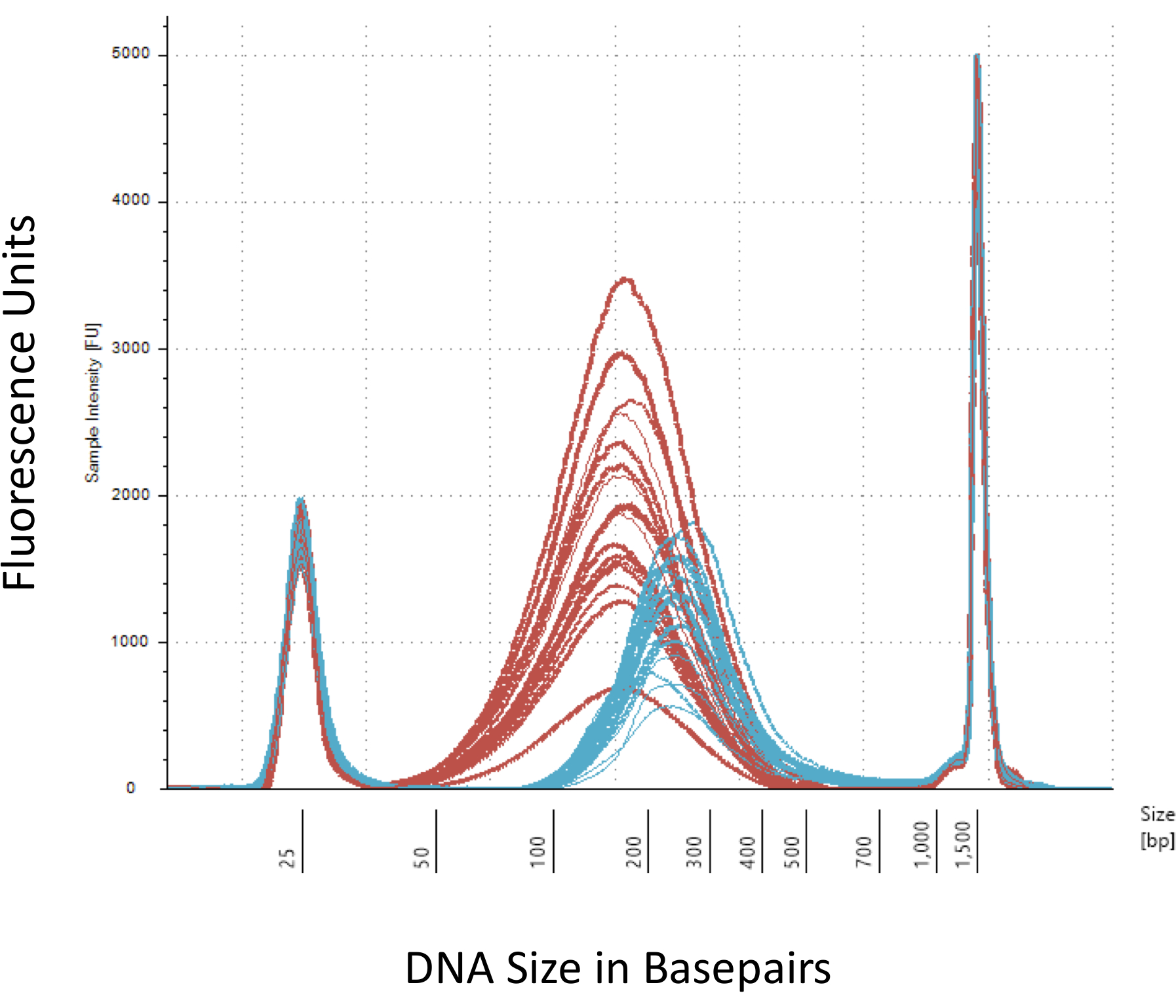
Diamo risalto a diversi posti di blocco che sono cruciali per la costruzione di successo della libreria metil-Seq. A partire con una quantità sufficiente di DNA è necessario, come sonicazione, più lavaggio/purificazione, arricchimento di destinazione, e passaggi di conversione bisolfito successivamente riducono la quantità di DNA nella libreria finito. Anche se diversi passaggi di amplificazione di PCR alleviare la perdita della mascherina del DNA, eccessivo numero di ciclo PCR può introdurre maggiore letture duplicate. Per lo studio corrente ratto metil-Seq, 2 g di sangue gDNA per ratto è stato utilizzato. Notiamo che librerie di metil-Seq possono essere fatta con quantità di DNA di partenza à partir 500 ng. Più piccolo materiale di partenza permette agli utenti di generare librerie da DNA isolato da FACS (ordinamento di fluorescenza-attivato delle cellule) o dell'ago di pugni, anche se c'è rischio aumentato di produrre una quantità insufficiente di librerie per la sequenza successiva. QC viene eseguita tramite l'elettroforesi di 1 L del campione su un bioanalyzer, che fornisce il peso molecolare, la quantità e la molarità di DNA. Tre passaggi critici che richiedono l'uso di bioanalyzer sono: 1) dopo il passaggio di sonicazione per garantire sufficiente tosatura del DNA (~ 170 bp, rosso, Figura 3); 2) a seguito di passaggio di legatura adattatore indicato da un cambiamento nella dimensione media del DNA tranciata (~ 200 bp, blu, Figura 3) per garantire la loro successiva amplificazione mediante PCR; e 3) dopo il passaggio di purificazione finale libreria per garantire la quantità e le dimensioni della libreria per il sequenziamento.
I pacchetti di R BSSeq e BSmooth in Bioconductor sono stati usati per analizzare il bisolfito sequenziamento dati18. Essi comprendono strumenti e metodi per allineare le letture di sequenza, esegue il controllo di qualità, e identificare differenzialmente metilato regioni (DMRs). BSmooth software richiama Bowtie 2.016,17 come un allineatore sequenza interna per ottenere riepiloghi di misurazione CpG-livello, tramite l'allineamento di raw inpue letture a sequenze genomic bisolfuro-convertito. Le letture allineate vengono quindi filtrate attraverso le procedure di controllo di qualità rigoroso che cercano di individuare sequenziamento sistematico ed errori di base chiamata che possono comportare un'asimmetria analisi a valle. Una serie di trame vengono generati per aiutare visivamente in questo processo di filtraggio. Sequenziamento metriche vengono generati anche per documentare le pertinenti informazioni quali il numero di letture allineate, destinazione % e a copertura di CpG, tra gli altri (tabella 2). Una volta che i dati vengono filtrati, un algoritmo di smussatura/normalizzazione viene eseguito, dove ogni CpG è assegnato un valore stimato di metilazione basato su QC tutti legge da ogni campione e stima dalla vicina CpGs per garantire più precisa vocazione di metilazione stato anche nei casi dove la copertura di sequenza è bassa. Questo valore fornisce una levigata stima della probabilità di metilazione in ogni sito di CpG. Confrontando la media delle stime levigata metilazione di ciascun campione tra i due gruppi di trattamento e classifica regioni genomiche dal più significativamente differente al minore, viene generato un elenco di DMRs (tabella 3).
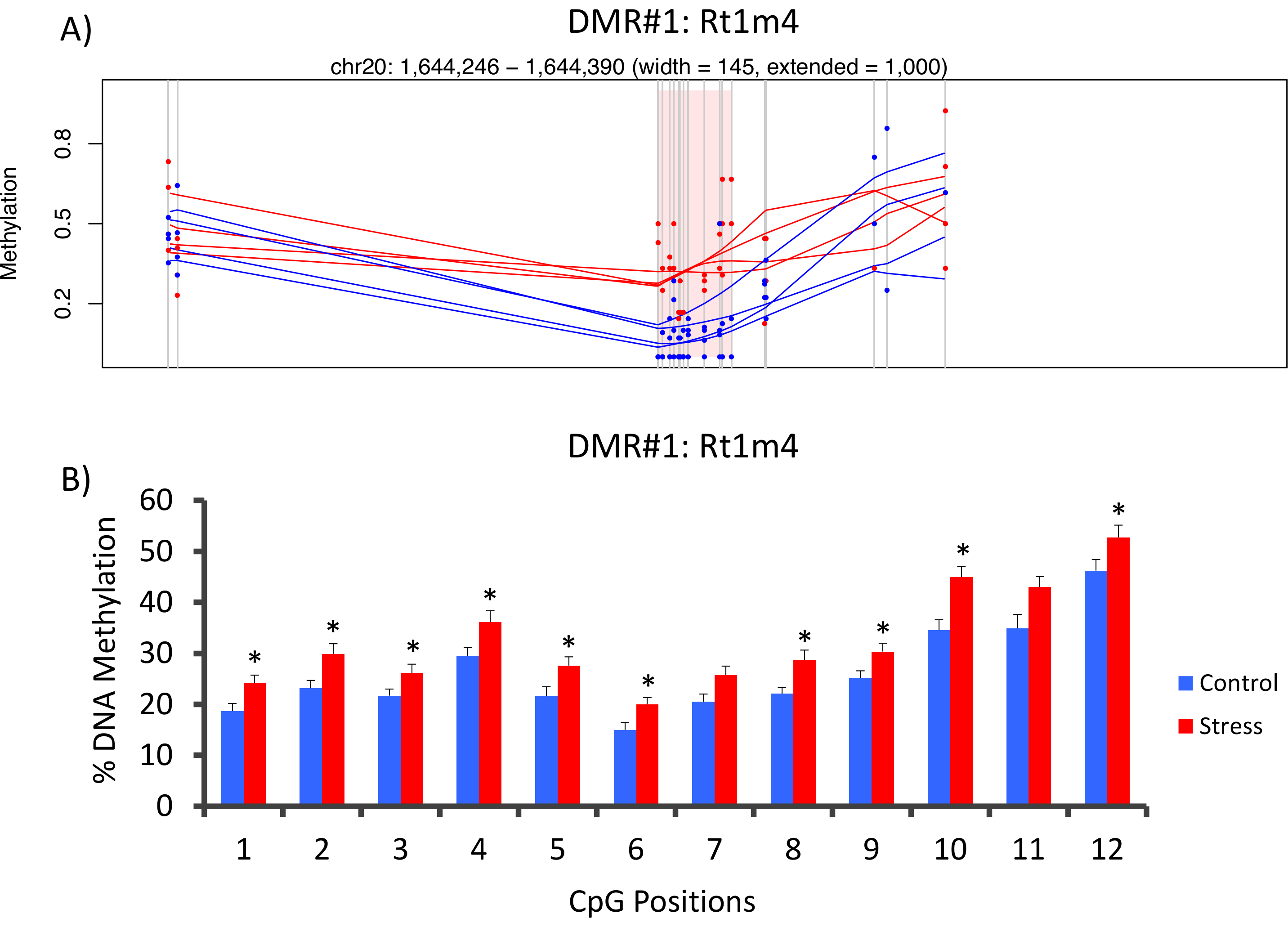
Parte superiore che DMR tra gruppi sollecitate e unstressed era situato nel promotore del gene di istocompatibilità ratto Rt1-m4, con sottolineato animali che esibiscono i livelli elevati di metilazione attraverso tutti i CpGs rispetto agli animali non accentati (Figura 4A). Per confermare la riuscita implementazione della piattaforma metil-Seq e l'analisi dei dati, gli iniettori sono stati progettati contro il DMR e i livelli di metilazione del DNA di sangue nella intera coorte degli animali stressati e unstressed (8 sequenziato da metil-Seq e 8 non in sequenza) sono stati valutati da bisolfito pirosequenziamento. I risultati dimostrano un aumento significativo nella metilazione del DNA attraverso 10 fuori il 12 CpGs analizzati (variazione 5.1 – 10.4% metilazione, P < 0,037, Figura 4B). KEGG pathway analisi è stata effettuata su tutte le dimensioni nominali significativo DMRs per identificare vie connesse con lo sforzo. Coerentemente, DMR-collegata vie implicati malattie connesse con l'esposizione di stress cronico, come diabete, malattie cardiovascolari e cancro (tabella 4). 21 , 22 , 23 per dimostrare un'associazione tra i dati epigenetici e il grado di esposizione allo stress, i livelli di metilazione CpG-10 sono stati confrontati con i livelli medi di CORT 3-settimana per ogni animale. Risultati hanno mostrato una modesta correlazione tra i dati di sistema endocrino e metilazione (R2= 0,54, P = 0,001, Figura 5).

Figura 1: flusso di lavoro nel complesso schematico per la piattaforma di ratto metil-Seq. Un g di DNA genomic estratto dal sangue del sollecitato e ratti di controllo viene elaborato in primo luogo per la costruzione le librerie di metil-Seq per sequenziamento, analisi e identificazione del target. Un altro 100 ng di DNA è utilizzato per la convalida indipendente degli obiettivi identificati epigenetici di bisolfito pirosequenziamento. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 2: l'esposizione cronica a stress variabile (CVS) conduce ai cambiamenti endocrini e comportamentistici in ratti. (A) più campionamenti di corticosterone (CORT) dimostrano la robustezza della settimana 3 regime di CVS. I campioni di sangue sono stati raccolti al mattino prima del regime lo stress quotidiano. (B) gli animali stressato trascorso più tempo tra le braccia chiuse e meno tempo a braccia aperte del elevato più labirinto (EPM). BoxPlot con punto di dati per ogni animale sono mostrati. Test T di Student è stato effettuato per la significatività statistica. * P < 0,05, * * P < 0.01, e * * * P < 0,001. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 3: quantificazione del ratto tranciata e adattatore-legati del DNA su un bioanalyzer. Le curve rosse e blue mostrano la quantità e le dimensioni del DNA genomico (rosso) seguendo la tosatura in un sonicatore isotermici e legatura adattatore, rispettivamente. Ogni riga rappresenta un campione e il rosso e blue curve riflettono sia la perdita di DNA durante le varie fasi (fine-riparazione, 3'-poliadenilazione e pulitura del campione) e aumentano di dimensione bp dovuto la legatura degli adattatori. Taglienti punte a 25 bp e bp 1500 sono marcatori standard che sono stati aggiunti al buffer di caricamento. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 4: cambiamenti epigenetici indotti da CVS vengono rilevati dal ratto metil-segg. (A) analisi del ratto metil-Seq dati implicati il promotore del gene di Rt1m4 come una regione differenzialmente metilata (DMR) tra stressato (rosso) e ratti di controllo (blu). L'output grafico per Rt1m4 DMR (rosa area ombreggiata) consente di visualizzare ogni CpG (linea grigia verticale), i quattro campioni in ciascun gruppo (righe rosse o blu) e i livelli di metilazione % per ciascun animale (punto rosso o blu). (B) dodici CpGs entro il DMR sono state convalidate da bisolfito pirosequenziamento. I grafici a barre sono rappresentati come media SEM, e T-test di uno studente è stato eseguito per la significatività statistica. * P < 0.05. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 5: analisi di regressione lineare ha mostrato una modesta correlazione tra % DNA metilazione di CpG-10 del Rt1m4 e la settimana 3 del plasma medio livelli CORT di entrambi ha sottolineato e controllare gli animali (N = 16). I dati di animali sollecitati sono rappresentati da cerchi rossi. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}
| Settimana | 1 ° giorno | 2 ° giorno | 3 ° giorno | 4 ° giorno | Giorno 5 | 6 ° giorno | Giorno 7 |
| AM | Sistema di ritenuta | Nuotare | Camera fredda | Nuotare | Sistema di ritenuta | Shaker | Nuotare |
| PM | Shaker | Tilt di gabbia | Sistema di ritenuta | Shaker | Camera fredda | Sistema di ritenuta | Camera fredda |
| Pernottamento | Limitare l'alimento | Biancheria da letto di bagnato | Isolamento | Luce su | Affollamento | Luce su | Biancheria da letto di bagnato |
Tabella 1: Un programma settimanale tipico del regime di sforzo variabile cronico (CVS).
| Metriche di sequenziamento | Lo stress1 | Controllo1 |
| (n = 4) | (n = 4) | |
| Accoppiato letture di fine (al) | 89,290,397 | 80,165,674 |
| Accoppiati in modo univoco mappato fine legge (UMPER) | 39,200,255 | 35,013,406 |
| L'efficienza di tasso/mappatura di allineamento (UMPER / a) | 44% | 44% |
| Duplicati letture (% UMPER) | 73% | 65% |
| UNZIONE deduplicati | 10,481,031 | 12,306,018 |
| Lettura profondità copertura media (x) (ARDC) | 6 x | 6 x |
| CpGs (N) | 12,056,878 | 12,056,878 |
| ARDC (x) di CpGs | 2 x | 2 x |
| CpGs con almeno 10 letture (N) | 481.383 | 595.850 |
| ARDC (X) di CpGs con almeno 10 letture | 19 | 19 |
| Il Target CpGs (completa sovrapposizione con sonda regioni di destinazione) | 1.923.872 | 2.007.638 |
| Il Target ARDC (x) di CpGs | 7 x | 8 x |
| Il Target CpGs con almeno 10 letture (N) | 428.249 | 531.419 |
| Nella destinazione ARDC (x) di CpGs con almeno 10 legge | 18 x | 18 x |
| Il Target (PER con 1 o più coppie di basi sovrapposizione con sonda regioni Target) (UMPER) | 8.277.715 | 9.369.523 |
| % Sul bersaglio (di unzione deduplicati) | 78% | 77% |
| Il Target (basi totali mappati) Mb | 125 mb | 128 mb |
| In copertura di profondità media di lettura di obiettivo (x) (ARDC) | 9 x | 10 x |
| 1 Sequenziamento metriche basate su medie tra i soggetti in ciascun gruppo |
Tabella 2: Sequenziamento metriche ottenute dalla piattaforma di metil-Seq del ratto.
| Chr | inizio | fine | gene | distanza | areaStat | meanDiff | stress | controllo | direzione |
| chr20 | 1.644.246 | 1.644.390 | RT1-M4 | in_gene | 93.03 | 0.22 | 0,33 | 0,11 | guadagno |
| chr5 | 160,361,352 | 160,361,564 | LOC690911 | in_gene | -70.75 | -0,19 | 0,72 | 0.91 | perdita |
| chr3 | 61,138,281 | 61,138,330 | RGD1564319 | 265569 | 61.79 | 0.21 | 0.94 | 0,72 | guadagno |
| chr2 | 143,064,811 | 143,065,010 | Ufm1 | 8569 | -59.48 | -0.11 | 0.13 | 0,24 | perdita |
| chr7 | 30,764,111 | 30,764,284 | Ntn4 | in_gene | 57.04 | 0.21 | 0.94 | 0,73 | guadagno |
| chr17 | 12,469,112 | 12,469,218 | Idnk | 41996 | -50.91 | -0,13 | 0,74 | 0.88 | perdita |
| chr7 | 47,101,725 | 47,101,930 | PAWR | in_gene | -50.54 | -0.12 | 0,64 | 0,76 | perdita |
| chr5 | 76,111,248 | 76,111,822 | Txndc8 | 151703 | -50.38 | -0.11 | 0.85 | 0,96 | perdita |
| chr11 | 80,640,132 | 80,640,356 | DGKG | in_gene | -50.07 | -0.16 | 0,73 | 0.89 | perdita |
| chr8 | 71,759,248 | 71,759,411 | Mir190 | 210226 | -47.84 | -0,17 | 0,58 | 0,75 | perdita |
Tabella 3: Top 10 differenzialmente metilato regioni. Per ogni DMR, tabella di output Mostra da sinistra a destra: posizione cromosomica (chr), coordina (inizio/fine), nome del gene, distanza dalla trascrizione avviare sito, statistiche dell'area differenziale tra gruppi (areaStat) di controllo e stressato, significa differenziali di metilazione (meanDiff), i livelli di metilazione media attraverso ogni DMR per stressati e gruppi di controllo (controllo, dello stress) e la direzione di metilazione modificare da controlli.
| KEGG Pathway termini | Conteggio di gene | % | Valore di P | Benjamini |
| Diabete | ||||
| Diabete mellito di tipo II | 12 | 0.1 | 3.6 x 10-4 | 9,8 x 10-3 |
| Malattia cardiovascolare | ||||
| Contrazione del muscolo liscio vascolare | 18 | 0.1 | 1.6 x 10-3 | 3.6 x 10-2 |
| Cardiomiopatia aritmogena del ventricolo destro (ARVC) | 13 | 0.1 | 4,0 x 10-3 | 7.1 x 10-2 |
| Cardiomiopatia dilatativa | 14 | 0.1 | 7,6 x 10-3 | 1.2 x 10-1 |
| Funzione del neurone | ||||
| Potenziamento a lungo termine | 11 | 0.1 | 1.5 x 10-2 | 1.4 x 10-1 |
| Segnalazione | ||||
| Via di segnalazione MAPK | 35 | 0.2 | 2.4 x 10-4 | 9,9 x 10-3 |
| Via di segnalazione del calcio | 22 | 0.1 | 1.2 x 10-2 | 1.4 x 10-1 |
| Via di segnalazione di chemokine | 21 | 0.1 | 1.2 x 10-2 | 1.3 x 10-1 |
| Cancro | ||||
| Percorsi in cancro | 42 | 0.3 | 4.1 x 10-5 | 3.4 x 10-3 |
| Glioma | 15 | 0.1 | 4.4 x 10-5 | 2.4 x 10-3 |
| Carcinoma polmonare non a piccole cellule | 10 | 0.1 | 7,9 x 10-3 | 1.1 x 10-1 |
| Cancro colorettale | 13 | 0.1 | 8,4 x 10-3 | 1.1 x 10-1 |
| Leucemia mieloide cronica | 12 | 0.1 | 1.2 x 10-2 | 1.3 x 10-1 |
Tabella 4: Analisi KEGG Pathway di DMRs identificato dal ratto metil-segg.
Discussione
In questo studio, abbiamo progettato e implementato la piattaforma di metil-Seq per il genoma del ratto. Dimostrando la sua utilità con un modello del ratto di stress, abbiamo dimostrato che la pipeline sperimentale e analitica può fornire differenzialmente metilate regioni tra due gruppi di confronto.
Per garantire un'implementazione corretta della piattaforma, alcuni passaggi critici devono essere osservati. Primo, prima la quantità e la qualità di DNA ha un impatto significativo sulla qualità e quantità della biblioteca metil-Seq finale. Abbiamo utilizzato un fluorimetro, piuttosto che uno spettrofotometro, per garantire che la nostra misura di DNA riflette la quantità di DNA double-stranded presente. Il bioanalyzer fu utilizzato per misurare la dimensione molecolare e la quantità di DNA dopo la tosatura e dopo la legatura adattatore. Verificare la dimensione molecolare "Maiusc" tra questi passaggi è fondamentale per confermare la presenza di adattatori alle estremità di ogni frammento di DNA che saranno sottoposti a PCR adattatore-mediata nei passaggi successivi. La quantità di DNA esogeno che rimane alla fine del passaggio di legatura del adattatore anche è importante, poiché almeno 100 ng del prodotto biblioteca è necessario in questa fase per garantire quantità sufficiente è disponibile dopo la procedura di conversione di arricchimento e bisolfito di destinazione. Una misura finale di alto-sensibilità è stata eseguita la libreria di metil-Seq costruita così che la libreria può essere diluita correttamente per il clustering successive del sequenziatore di nuova generazione. Infine, bisolfito pyrosequencing è stato impiegato come metodo altamente-quantitativa, indipendente per valutare l'accuratezza della pipeline analitica. La validazione finale utilizzando i campioni originale e la replica utilizzando animali addizionali sono passi fondamentali per garantire che l'esperimento può rilevare biologicamente significativi cambiamenti nella metilazione del DNA.
Abbiamo anche diverse linee guida in caso di deviazione dal protocollo o se si verificano problemi. In primo luogo, è possibile perdere troppo DNA durante il fine-riparazione, legatura di adattatore o fasi di purificazione di biglie magnetiche. In alternativa, potrebbe essere piccolo a partire quantità di DNA (< 200 ng) a causa della disponibilità limitata dei tessuti/DNA o implementazione dei vari metodi di arricchimento come ordinamento di attivate la fluorescenza delle cellule. Aumentando il numero di ciclo durante le fasi di amplificazione due libreria può essere in grado di compensare la perdita eccessiva di DNA o bassa a partire quantità di DNA in tutto il protocollo di costruzione di libreria. Tuttavia, non più di un ulteriore 2-3 cicli sono raccomandati, come amplificazione eccessiva modello rischia di condurre ad un aumento del numero di duplicati letture essere sequenziato. Questi duplicati vengono esclusi durante il passo di allineamento per evitare pregiudizi nei calcoli di percentuale di metilazione. In secondo luogo, se la dimensione media di DNA non aumenta da più di 30 bps, controllare che i reagenti siano nuovi, come T4 DNA polimerasi, Klenow, e/o T4 ligasi potrebbe essere vecchia. Reagenti di ricambio disponibili in commercio possono essere utilizzati.
Inoltre, è possibile che il predetto DMRs potrebbero non convalidare da pyrosequencing, dove le differenze di metilazione del DNA non esistono o sono significativamente inferiori a quelli previsti dall'analisi. Validazione poveri dei paesi candidati è un problema troppo comune per molte analisi del genoma, ad esempio quando pyrosequencing risultati non confermano differenziali di metilazione o la dimensione dell'effetto è molto più piccola rispetto a quanto previsto dall'analisi. BSmooth è un pacchetto analitico che "leviga" i livelli di metilazione attraverso una finestra di CpGs multiple. Per l'esperimento corrente, BSmooth implicato un DMR in cui i livelli di metilazione sono stati convalidati da bisolfito pirosequenziamento. Tuttavia, ci sarà probabilmente discrepanze tra i livelli di metilazione preveduti di BSmooth e quelli verificati da pirosequenziamento. Le discrepanze derivano dalla funzione di smussatura che stima i valori medio metilazione in tutti il CpGs entro un DMR, tra cui consecutivi CpGs che possono differire nella metilazione del DNA di più del 50% o CpGs i cui valori di metilazione sono stati esclusi a causa sotto soglia legge profondità. R-pacchetti come MethylKit24 possono essere utilizzati per identificare le finestre più piccole di CpGs o anche singole CpGs cui i livelli di metilazione fortemente correlano con quelli convalidati da pirosequenziamento. Implementazione di pacchetti diversi e test loro regioni previste o CpGs di differenziali di metilazione di pyrosequencing garantirà la robustezza dei dati. In alternativa, originale metil-Seq librerie possono essere Live e aggiunte ai file di lettura per aumentare la profondità di lettura. Poiché la determinazione dei livelli di metilazione sono semi-quantitativa e dettati dal numero di letture [(# of CpGs) / (n # di TpGs + CpGs)], aumentando la profondità di lettura per un determinato CpG aumenta la precisione del suo valore di percentuale di metilazione. In questo studio, abbiamo considerato solo CpGs i cui valori di metilazione sono stati determinati da almeno dieci letture e raggiunto una copertura di lettura complessiva di 19 x per ogni CpG.
La piattaforma di metil-Seq del ratto non è priva di limiti. Mentre è più conveniente che l'intero genoma del bisolfuro, è notevolmente più costoso rispetto ad altri metodi. Tuttavia, la maggior parte del costo era per l'acquisto di corsie sul sequencer e non per il sistema di acquisizione. A seconda della profondità di lettura necessaria, con confronti di croce-tessuto che richiedono meno a causa di differenze di grandi (25 – 70%)12 nella metilazione del DNA, il costo può essere ridotto più campioni per corsia di multiplexing e utilizzando una piattaforma di maggiore capacità. Inoltre, la preparazione del campione è più laborioso rispetto ad altri metodi. Sebbene sia simile ad altri approcci di pulldown che incorporano il sequenziamento di nuova generazione, i passaggi di conversione e purificazione di bisolfito aggiunto aggiungere il carico di lavoro. Nel complesso, la piattaforma di metil-Seq è un'alternativa conveniente all'intero genoma sequenziamento e fornisce la risoluzione di base-accoppiamenti a CpGs più di 2,3 milioni, che è considerevolmente più di quelli analizzati da piattaforme basate su microarray. Ad oggi, l'umani disponibili in commercio e il mouse metil-Seq piattaforme sono stati usati per documentare le modifiche alcool-dipendenti nel macaco cervello25,26, neurodevelopmental geni nel cervello di topo9ed emato-encefalica obiettivi di glucocorticoidi10. Inoltre, la possibilità di indirizzare specifiche regioni indipendentemente dal riconoscimento di sequenza di enzimi di restrizione rende una piattaforma ideale per i confronti inter-specie. Per questo studio, abbiamo progettato la piattaforma di metil-Seq per il ratto, per cui molti esperimenti farmacologici, metabolici e comportamentali vengono eseguiti senza il beneficio di un genoma methylomic strumento. I nostri dati indicano che può essere utilizzato per rilevare DMRs in un modello del ratto di stress correlato altri parametri fisiologici quali livelli plasmatici totali di CORT.
La piattaforma di metil-Seq è ideale per epigenetici esperimenti in animali con genomi sequenziati che potrebbero non avere abbastanza prove sperimentali che documentano regioni regolatorie. Quando tali regioni sono resi disponibili, altre regioni possono essere personalizzati e associate alla versione corrente. Inoltre, la piattaforma è ideale per la genomica comparativa, poiché l'arricchimento di destinazione non è vincolato al riconoscimento degli enzimi di limitazione. Per esempio, la regione del promotore di qualsiasi gene di interesse può essere catturato indipendentemente dal fatto se ospita un sito di restrizione specifici. Allo stesso modo, qualsiasi regioni regolatorie, quali quelle individuate nel mouse o esseri umani, che si conservano nel genoma di interesse possono essere catturati.
Divulgazioni
Il manoscritto è parte di un premio di concorso da Agilent Technologies.
Riconoscimenti
Questo studio è stato finanziato dal NIH grant MH101392 (RSL) e sostegno dei seguenti premi e fondazioni: un NARSAD Young Investigator Award, Margaret Ann prezzo investigatore, sul fondo di studioso James disordini di umore Wah via Charles T. Bauer Foundation, Baker Fondazione e la Fondazione di partita di progetto (RSL).
Materiali
| Name | Company | Catalog Number | Comments |
| Radioimmuno assay (RIA) | MP Biomedicals | 7120126 | Corticosterone, 125I labeled |
| Master Pure DNA Purification Kit | Epicentre/Illumina | MC85200 | |
| Thermal-LOK 2-Position Dry Heat Bath | USA Scientific | 2510-1102 | Used with 1.5 mL tubes |
| Vortex Genie 2 | Fisher | 12-812 | Vortex Mixer |
| Ethyl alcohol, Pure | Sigma-Aldrich | E7023 | 100% Ethanol, molecular grade |
| Centrifuge 5424 R | Eppendorf | - | Must be capable of 20,000 x g |
| Qubit 2.0 | ThermoFisher Scientific | Q32866 | Fluorometer |
| Qubit dsDNA BR Assay Kit | ThermoFisher Scientific | Q32850 | |
| Qubit dsDNA HS Assay Kit | ThermoFisher Scientific | Q32851 | High sensitivity DNA detection reagents |
| Qubit Assay Tubes | ThermoFisher Scientific | Q32856 | |
| SureSelectXT Rat Methyl-Seq Reagent Kit | Agilent Technologies | G9651A | Reagents for preparing the Methyl-Seq library |
| SureSelect Rat Methyl-Seq Capture Library | Agilent Technologies | 931143 | RNA baits for enrichment of rat targets |
| IDTE, pH 8.0 | IDT DNA | 11-05-01-09 | 10 mM TE, 0.1 mM EDTA |
| DNA LoBind Tube 1.5 mL | Eppendorf | 22431021 | |
| Covaris E-series or S-series | Covaris | - | Isothermal sonicator |
| microTUBE AFA Fiber Pre-Slit Snap-Cap 6x16mm (25) | Covaris | 520045 | |
| Water, Ultra Pure (Molecular Biology Grade) | Quality Biological | 351-029-721 | |
| Veriti 96 Well-Thermal Cycler | Applied Biosystems | 4375786 | |
| AMPure XP Beads | Beckman Coulter | A63880 | DNA-Binding magnetic beads |
| 96S Super Magnet | ALPAQUA | A001322 | Magnetic plate for purification steps |
| 2200 TapeStation | Agilent Technologies | G2965AA | Electrophresis-based bioanalyzer |
| D1000 ScreenTape | Agilent Technologies | 5067-5582 | |
| D1000 ScreenTape High Sensitivity | Agilent Technologies | 5067-5584 | |
| D1000 Reagents | Agilent Technologies | 5067-5583 | |
| D1000 Reagents High Sensitivity | Agilent Technologies | 5067-5585 | |
| DNA110 SpeedVac | ThermoFisher Scientific | - | Vacuum Concentrator |
| Dynabeads MyOne Streptavidin T1 magnetic beads | Invitrogen | 65601 | Streptavidin magnetic beads |
| Labquake Tube Rotator | ThermoFisher Scientific | 415110Q | Nutator Mixer is also acceptable |
| EZ DNA Methylation-Gold Kit | Zymo Research | D5006 | Bisulfite conversion kit. Contains Binding, Wash, Desulphonation, and Elution buffers |
| Illumina Hi-Seq 2500 | Illumina | - | Next-generation sequencing machine |
| PCR and Pyrosequencing Primers | IDT DNA | Variable | |
| Taq DNA Polymerase with ThermoPol Buffer - 2,000 units | New England BioLabs | M0267L | |
| Deoxynucleotide (dNTP) Solution Set | New England BioLabs | N0446S | |
| Pyromark MD96 | QIAGEN | - | Pyrosequencing machine |
| Ethyl Alcohol 200 Proof | Pharmco-Aaper | 111000200 | 70% Ethanol solution |
| Sodium Hydroxide Pellets | Sigma-Aldrich | 221465 | 0.2 M NaOH denature buffer solution |
| Tris (Base) from J.T. Baker | Fisher Scientific | 02-004-508 | 10 mM Tris Acetate Buffer wash buffer solution |
| PyroMark Gold Q96 Reagents (50x96) | QIAGEN | 972807 | Reagents required for pyrosequencing |
| PyroMark Annealing Buffer | QIAGEN | 979009 | |
| PyroMark Binding Buffer (200 mL) | QIAGEN | 979006 | |
| Streptavidin Sepharose High Performance Beads | GE Healthcare | 17-5113-01 | Streptavidin-coated sepharose beads |
| PyroMark Q96 HS Plate | QIAGEN | 979101 | Pyrosequencing assay plate |
| Eppendorf Thermomixer R | Fisher Scientific | 05-400-205 | Plate mixer. 96-well block sold separately (cat. No 05-400-207) |
| SureDesign Website | Agilent Technologies | - | Target capture design software (https://earray.chem.agilent.com/suredesign/) |
| UCSC Genome Browser | University of California Santa Cruz | - | rat Nov 2004 rn4 assembly |
| Agilent Methyl-Seq Protocol | Agilent Technologies | - | https://www.agilent.com/cs/library/usermanuals/public/G7530-90002.pdf |
Riferimenti
- Barski, A., et al. High-resolution profiling of histone methylations in the human genome. Cell. 129 (4), 823-837 (2007).
- Meissner, A., et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 454 (7205), 766-770 (2008).
- Bibikova, M., et al. High density DNA methylation array with single CpG site resolution. Genomics. 98 (4), 288-295 (2011).
- Naumov, V. A., et al. Genome-scale analysis of DNA methylation in colorectal cancer using Infinium HumanMethylation450 BeadChips. Epigenetics. 8 (9), 921-934 (2013).
- Wockner, L. F., et al. Genome-wide DNA methylation analysis of human brain tissue from schizophrenia patients. Translational Psychiatry. 4, e339 (2014).
- Meissner, A., et al. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Research. 33 (18), 5868-5877 (2005).
- Smith, Z. D., Gu, H., Bock, C., Gnirke, A., Meissner, A. High-throughput bisulfite sequencing in mammalian genomes. Methods. 48 (3), 226-232 (2009).
- Slieker, R. C., et al. Identification and systematic annotation of tissue-specific differentially methylated regions using the Illumina 450k array. Epigenetics Chromatin. 6 (1), 26 (2013).
- Hing, B., et al. Adaptation of the targeted capture Methyl-Seq platform for the mouse genome identifies novel tissue-specific DNA methylation patterns of genes involved in neurodevelopment. Epigenetics. 10 (7), 581-596 (2015).
- Seifuddin, F., et al. Genome-wide Methyl-Seq analysis of blood-brain targets of glucocorticoid exposure. Epigenetics. 12 (8), 637-652 (2017).
- Irizarry, R. A., et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nature Genetics. 41 (2), 178-186 (2009).
- Lee, R. S., et al. Adaptation of the CHARM DNA methylation platform for the rat genome reveals novel brain region-specific differences. Epigenetics. 6 (11), 1378-1390 (2011).
- Jankord, R., et al. Stress vulnerability during adolescent development in rats. Endocrinology. 152 (2), 629-638 (2011).
- Pellow, S., Chopin, P., File, S. E., Briley, M. Validation of open:closed arm entries in an elevated plus-maze as a measure of anxiety in the rat. Journal of Neuroscience Methods. 14 (3), 149-167 (1985).
- Krueger, F., Andrews, S. R. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 27 (11), 1571-1572 (2011).
- Langmead, B., Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nature Methods. 9 (4), 357-359 (2012).
- Langmead, B., Trapnell, C., Pop, M., Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biology. 10 (3), R25 (2009).
- Hansen, K. D., Langmead, B., Irizarry, R. A. BSmooth: from whole genome bisulfite sequencing reads to differentially methylated regions. Genome Biology. 13 (10), R83 (2012).
- Lee, R. S., et al. Chronic corticosterone exposure increases expression and decreases deoxyribonucleic acid methylation of Fkbp5 in mice. Endocrinology. 151 (9), 4332-4343 (2010).
- Lee, R. S., et al. A measure of glucocorticoid load provided by DNA methylation of Fkbp5 in mice. Psychopharmacology. , (2011).
- Bose, M., Olivan, B., Laferrere, B. Stress and obesity: the role of the hypothalamic-pituitary-adrenal axis in metabolic disease. Current Opinion in Endocrinology, Diabetes, and Obesity. 16 (5), 340-346 (2009).
- Brydon, L., Magid, K., Steptoe, A. Platelets, coronary heart disease, and stress. Brain, Behavior, and Immunity. 20 (2), 113-119 (2006).
- McKlveen, J. M., et al. Chronic Stress Increases Prefrontal Inhibition: A Mechanism for Stress-Induced Prefrontal Dysfunction. Biological Psychiatry. 80 (10), 754-764 (2016).
- Akalin, A., et al. methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biology. 13 (10), R87 (2012).
- Cervera-Juanes, R., Wilhelm, L. J., Park, B., Grant, K. A., Ferguson, B. Alcohol-dose-dependent DNA methylation and expression in the nucleus accumbens identifies coordinated regulation of synaptic genes. Translational Psychiatry. 7 (1), e994 (2017).
- Cervera-Juanes, R., Wilhelm, L. J., Park, B., Grant, K. A., Ferguson, B. Genome-wide analysis of the nucleus accumbens identifies DNA methylation signals differentiating low/binge from heavy alcohol drinking. Alcohol. 60, 103-113 (2017).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati