
Method Article
A Rat Methyl-Seq Platform to Identify Epigenetic Changes Associated with Stress Exposure
In This Article
Summary
Here, we describe the protocol and implementation of Methyl-Seq, an epigenomic platform, using a rat model to identify epigenetic changes associated with chronic stress exposure. Results demonstrate that the rat Methyl-Seq platform is capable of detecting methylation differences that arise from stress exposure in rats.
Abstract
As genomes of a wider variety of animals become available, there is an increasing need for tools that can capture dynamic epigenetic changes in these animal models. The rat is one particular model animal where an epigenetic tool can complement many pharmacological and behavioral studies to provide insightful mechanistic information. To this end, we adapted the SureSelect Target Capture System (referred to as Methyl-Seq) for the rat, which can assess DNA methylation levels across the rat genome. The rat design targeted promoters, CpG islands, island shores, and GC-rich regions from all RefSeq genes.
To implement the platform on a rat experiment, male Sprague Dawley rats were exposed to chronic variable stress for 3 weeks, after which blood samples were collected for genomic DNA extraction. Methyl-Seq libraries were constructed from the rat DNA samples by shearing, adapter ligation, target enrichment, bisulfite conversion, and multiplexing. Libraries were sequenced on a next-generation sequencing platform and the sequenced reads were analyzed to identify DMRs between DNA of stressed and unstressed rats. Top candidate DMRs were independently validated by bisulfite pyrosequencing to confirm the robustness of the platform.
Results demonstrate that the rat Methyl-Seq platform is a useful epigenetic tool that can capture methylation changes induced by exposure to stress.
Introduction
Advances in high-throughput sequencing have led to a wealth of genomic sequences for both model and non-model organisms. The availability of such sequences has facilitated research in genetics, comparative genomics, and transcriptomics. For instance, available genomic sequences are highly useful for aligning sequencing data from ChIP-Seq experiments that enrich DNA based on its association with histone modifications1, or bisulfite sequencing, which measures DNA methylation by detecting uracil formed from bisulfite conversion of unmethylated cytosines2. However, there have been delays in the implementation of epigenomic platforms that incorporate available genomic sequencing data in their design due to a lack of annotated data of species-specific regulatory sequences that can influence gene function.
In particular, DNA methylation is one of the most widely studied epigenetic modifications on DNA that can leverage available genomic data for building a methylomic platform. One such example is an array-based platform for the human methylome3, which has been widely used in various disciplines from oncology to psychiatry4,5. Unfortunately, similar platforms for non-human animal models are scarce, as there are virtually no widely-used platforms that have taken advantage of the genomic sequence in their initial design.
A common method to assess the methylomic landscape of non-human animal models is reduced representation bisulfite sequencing (RRBS)6. This approach overcomes the cost of whole-genome bisulfite sequencing that, while providing a comprehensive methylomic landscape, provides lower read-depth coverage due to cost and limited functional information in large gene-poor areas of the genome2. RRBS involves restriction digest and size-selection of genomic DNA to enrich for highly GC-rich sequences such as CpG islands that are commonly found near gene promoters and thought to play a role in gene regulation7. While the RRBS method has been used in a number of important studies, its reliance on restriction enzymes is not without notable challenges and limitations. For instance, enrichment of GC-rich sequences in RRBS is entirely dependent on the presence of specific sequences recognized by the restriction enzyme and subsequent size selection by electrophoresis. This means that any genomic areas that do not contain these restriction sites are excluded during size selection. Also, cross-species comparisons are challenging unless the same restriction sites are present in the same loci among the different species.
One approach to overcoming the limitations of RRBS is to use an enrichment method that takes advantage of the published genomic sequence in the design of the platform. The array-based human platform uses primer probes designed against specific CpGs for allele-specific (CG vs. TG after bisulfite conversion) target annealing and primer extension. Its design reflects not only the available human genomic sequence, but experimentally-verified regulatory regions acquired from multiple lines of inquiry, such as ENCODE and ENSEMBL8. Despite its wide use in human methylomic investigations, a similar platform does not exist for model animals. In addition, the array-based format places significant constraint on the surface area available for probe placement. In the past several years, efforts have been made to combine the target-specificity afforded by capture probe design and the high-throughput feature of next-generation sequencing. Such an endeavor has resulted in the sequencing-based target enrichment system for the mouse genome (mouse Methyl-Seq), which was used to identify brain-specific or glucocorticoid-induced differences in methylation9,10. Similar platforms for other model and non-model animals are needed to facilitate epigenomic research in these animals.
Here, we demonstrate the implementation of this novel platform to conduct methylomic analysis on the rat. The rat has served as an important animal model in pharmacology, metabolism, neuroendocrinology, and behavior. For example, there is an increasing need to understand the underlying mechanisms that give rise to drug toxicity, obesity, stress response, or drug addiction. A high-throughput platform capable of capturing methylomic changes associated with these conditions would increase our understanding of the mechanisms. Since the rat genome still lacks annotation for regulatory regions, we incorporated non-redundant promoters, CpG islands, island shores11, and previously identified GC-rich sequences into the rat Methyl-Seq platform12.
To assess successful design and implementation of the SureSelect Target Enrichment (generically referred to as Methyl-Seq) platform for the rat genome, we employed a rat model of chronic variable stress (CVS)13 to identify differentially methylated regions between unstressed and stressed animals. Our platform design, protocol, and implementation may be useful for investigators who may want to conduct a comprehensive and unbiased epigenetic investigation on an organism whose genomic sequence is already available but remains poorly annotated.
Protocol
All experiments were completed in accordance and compliance with all relevant regulatory and institutional guidelines, including the Institutional Animal Care and Use Committee at the Johns Hopkins School of Medicine.
1. Animals
- Obtain male adolescent Sprague-Dawley rats at 4 weeks of age. House the animals in polycarbonate rat cages in a temperature-and humidity-controlled room on a 12 h light, 12 h dark cycle with light onset at 0600 h. Provide the animals with ad libitum access to water.
- Allow rats to acclimate for 1 week to reduce stress associated with transportation. Pair-house the animals (N=16) to preclude isolation stress, and at 5 weeks of age, begin the chronic variable stress (CVS) regimen for 3 weeks.
2. Chronic Variable Stress
- Administer the CVS regimen once in the morning (9-11 AM) and once in the afternoon (1–3 PM) at irregular times to keep the routine unpredictable. Incorporate overnight mild stressors. The CVS regimen includes: 1) 3 h in a restraint cylinder; 2) 10 min swim; 3) 3 h cage tilt 4) 1 h slow shaking platform; and 5) 1 h in the 4 °C cold room.
Note: Overnight stressors include social crowding (5 per cage), social isolation, wet bedding, food restriction, and lights-on. A typical weekly schedule of the stress regimen is provided in Table 1.
3. Endocrine Assays
- Determine levels of corticosterone (CORT) using tail blood (~50 μL) samplings collected at the same time (9 AM) twice per week throughout the experiment, prior to CVS regimen to establish baseline hormone levels (Day 0), once during the middle of the weekly CVS (Days 4,11, and 18), after every 7 days of CVS (Days 7 and 14), and at the conclusion of CVS (Day 21). Collect blood samples prior to the daily stress regimen.
- Collect one final trunk blood sample during euthanasia (Day 25) for RIA and genomic DNA extraction.
- Centrifuge all blood samples (600 x g, 4 °C, 10 min) to separate the plasma from the blood cells. Pipet out the plasma (supernatant) and store the samples at -80 °C.
- Thaw and use the plasma to determine CORT levels by radioimmunoassay (RIA). Ensure that the 3 week plasma CORT levels are elevated in the stressed animals to verify the robustness of the stress regimen.
4. Behavior
- After the CVS regimen (Days 23–24), assess each animal for anxiety-like behavior on the elevated plus maze (EPM)14.
- Using a video camera, record the animals on the EPM apparatus for 300 s and score the time spent in the center, closed arms, and open arms.
5. Design of the Rat Methyl-Seq
- Using the UCSC Genome Browser, obtain non-redundant genomic coordinates (rat Nov 2004 rn4 assembly) for CpG islands and island shores (± 1 kb flanking CpG islands), promoters (± 1 kb of each TSS) of each RefSeq gene, and other sequences that may be available from relevant literature.
Note: For the rat Methyl-Seq, additional GC-rich sequences from a previous array-based methylation platform was added12. For regions greater than 5 kbps, alternating regions of 500 bps were sampled followed by 1 kbps that were skipped. The final rat Methyl-Seq design consists of 111 Mbps, 2.3 million CpGs; and an average region size of 594 bps. It targets 228,800 unique loci. - Enter a compiled list of genomic coordinates into a commercially-available target capture design software for appropriate probe design.
6. Construction of the Rat Methyl-Seq Library from Genomic DNA
NOTE: To eliminate batch effects, process multiple samples at the same time, and scale up the master mixes accordingly. Extract DNA using a commercially available DNA extraction kit. Column- or precipitation-based methods both yield high-quality genomic DNA (260/280 ratio ~1.8). Use of phenol-based methods are not recommended. Elute or resuspend DNA in Low TE buffer (10 mM TE, 0.1 mM EDTA, pH 8.0).
- Sample Preparation
NOTE: For every step using DNA-binding magnetic beads, make sure the beads are acclimated to room temperature for at least 30 min and well mixed before use.- Shear DNA
- Use a fluorometer to determine initial double-stranded DNA concentration of each sample. Dilute >1 µg of gDNA to 50 µL with Low TE buffer (10 mM TE, 0.1 mM EDTA, pH 8.0) in low DNA-binding microcentrifuge tubes.
- Shear samples using an isothermal sonicator (10% Duty Cycle, 5 Intensity, 200 Cycles per Burst, 6 cycles of 60 s, Frequency sweeping, 4 °C).
- Assess quality of DNA using an electrophoresis-based system that measures DNA size and quantity.
NOTE: The DNA amount recommended is 1 µg, or 3 µg. If there is limited starting material, the lowest input amount should be >500 ng, as lower amounts will adversely affect the quantity and quality of the libraries generated.
- Repair DNA ends.
- Use the rat Methyl-Seq kit to prepare the End-repair Master Mix on ice. Add 52 µL of mix to each sample and incubate in a thermal cycler without a heated lid (20 °C for 30 min, 4 °C hold).
End-repair Master Mix (per sample):
35.2 µL of Water
10 µL of End Repair Buffer (10x)
1.6 µL of dNTP Mix
1 µL of T4 DNA Polymerase
2 µL of Klenow DNA Polymerase
2.2 µL of T4 Polynucleotide Kinase - Purify samples using 180 µL of DNA-binding magnetic beads and 400 µL of freshly prepared 70% ethanol per sample. Add 180 µL of beads to each sample and incubate for 5 min at room temperature. Pellet beads, remove supernatant and resuspend pellet in 200 µL of 70% ethanol. Remove ethanol and repeat wash once.
- Use a magnetic plate to pellet beads and remove as much ethanol as possible. Dry in a 37 °C heatblock for 3–5 min until the bead pellet is completely dry. Resuspend in 44 µL of nuclease-free water and collect approximately 42 µL of supernatant.
Stopping Point: After repairing DNA ends, samples may be sealed and stored at -20 °C.
- Use the rat Methyl-Seq kit to prepare the End-repair Master Mix on ice. Add 52 µL of mix to each sample and incubate in a thermal cycler without a heated lid (20 °C for 30 min, 4 °C hold).
- Adenylate the 3’ ends.
- Prepare Adenylation Master Mix on ice. Add 9 µL mix to each sample and incubate in a thermal cycler without a heated lid (37 °C for 30 min, 4 °C hold).
Adenylation Master Mix (per sample):
5 µL of Klenow buffer
1 µL of dATP
3 µL of Klenow DNA Polymerase - Purify samples using 90 µL of DNA-binding magnetic beads and 400 µL of freshly prepared 70% ethanol per sample. Add 90 µL of beads to each sample and incubate for 5 min at room temperature. Pellet beads, remove supernatant and resuspend pellet in 200 µL of 70% ethanol. Remove ethanol and repeat wash once.
- Use a magnetic plate to pellet beads and remove as much ethanol as possible. Dry in a 37 °C heatblock for 3–5 min until the bead pellet is completely dry. Resuspend in 35 µL of nuclease-free water and collect approximately 33.5 µL of supernatant.
- Prepare Adenylation Master Mix on ice. Add 9 µL mix to each sample and incubate in a thermal cycler without a heated lid (37 °C for 30 min, 4 °C hold).
- Ligate the methylated adapter.
- Prepare Ligation Master Mix on ice and add 16.5 µL of mix to each sample. Incubate in a thermal cycler without a heated lid (20 °C for 15 min, 4 °C hold).
Ligation Master Mix (per sample):
2.5 µL of Water
2.5 µL of Methyl-Seq Methylated Adapter
10 µL of T4 DNA Ligase Buffer (5x)
1.5 µL of T4 DNA Ligase - Purify samples using 90 µL of DNA-binding magnetic beads and 400 µL of freshly prepared 70% ethanol per sample. Add 90 µL of beads to each sample and incubate for 5 min at room temperature. Pellet beads, remove supernatant, and resuspend pellet in 200 µL of 70% ethanol. Remove ethanol and repeat wash once.
- Use a magnetic plate to pellet beads and remove as much ethanol as possible. Dry in a 37 °C heatblock for 3–5 min until the bead pellet is completely dry. Resuspend in 22 µL of nuclease-free water and collect approximately 22 µL of supernatant. Assess quality using a bioanalyzer.
Note: If the total amount of DNA is less than 500 ng, shear and process additional DNA prior to proceeding with the subsequent steps. If the average DNA size does not increase by more than 30 bps, check to ensure that the reagents are new, as T4 DNA polymerase, Klenow, and/or T4 ligase may be old.
Stopping Point: After ligating methylated adapter, samples may be sealed and stored at -20 °C.
- Prepare Ligation Master Mix on ice and add 16.5 µL of mix to each sample. Incubate in a thermal cycler without a heated lid (20 °C for 15 min, 4 °C hold).
- Shear DNA
- Hybridization
- Transfer samples to low DNA-binding microcentrifuge tubes and use a heated vacuum concentrator to reduce sample volume to less than 3.4 µL. Reconstitute samples to 3.4 µL.
NOTE: Concentrate the samples to approximately ~3 µL to ensure samples are removed from vacuum concentrator before all liquid evaporates. - Prepare hybridization buffer at room temperature and Methyl-Seq Block Mix on ice. Add 5.6 µL of Methyl-Seq Block Mix to each sample and incubate in thermal cycler (95 °C for 5 min, 65 °C for 2 min, 65 °C hold).
Hybridization Buffer (per sample):
6.63 µL of Methyl-Seq Hyb 1
0.27 µL of Methyl-Seq Hyb 2
2.65 µL of Methyl-Seq Hyb 3
3.45 µL of Methyl-Seq Hyb 4
Methyl-Seq Block Mix (per sample):
2.5 µL of Methyl-Seq Indexing Block 1
2.5 µL of Methyl-Seq Block 2
0.6 µL of Methyl-Seq Block 3 - Prepare RNase Block Mix and the Capture Library hybridization mix. Add 20 µL of Capture Library Hybridization Mix to each sample and incubate at 65 °C for at least 16 h.
RNase Block Mix (per sample):
0.5 µL of RNase Block
1.5 µL of Water
Capture Library Hybridization Mix (per sample):
13 µL of Hybridization Buffer
2 µL of RNase Block Mix
5 µL of Rat Methyl-Seq Capture Library
NOTE: Keep reactions at 65 °C when adding Hybridization Mix to prevent non-specific binding. - Aliquot 50 µL of streptavidin magnetic beads per sample into a new 8-well strip tube. Wash beads with 200 µL of Methyl-Seq Binding Buffer. Use magnetic plate to pellet beads and remove supernatant between each wash for a total of 3 washes. After the final wash, resuspend streptavidin beads in 200 µL of Methyl-Seq Binding Buffer.
- Add samples to 200 µL of washed streptavidin magnetic beads and incubate at room temperature for 30 min using a rotating mixer. While mixing, aliquot 200 µL of Methyl-Seq Wash Buffer 2 into triplicate wells of a 96-well plate per sample and place in a thermal cycler to pre-warm to 65 °C.
- After incubation, pellet streptavidin magnetic beads using magnetic plate and resuspend the beads in 200 µL Methyl-Seq Wash Buffer 1. Incubate for 15 min at room temperature. Use a magnetic plate to pellet and discard supernatant.
- Wash beads 3 times with Methyl-Seq Wash Buffer 2: resuspend bead pellet in 200 µL of Wash Buffer 2 (pre-warmed in step 6.2.5.), incubate beads in thermal cycler (65 °C, 10 min), and pellet beads. Discard supernatant after each wash using a magnetic plate.
NOTE: Maintain hybridization reactions at 65 °C when adding Wash Buffer 2 to prevent non-specific binding. - Add 20 µL of Methyl-Seq Elution Buffer to the washed beads and incubate at room temperature for 20 min. Use a magnetic plate to pellet beads and transfer supernatant to a new strip tube. Discard the beads.
Note: While incubating, prepare bisulfite conversion reagent.
- Transfer samples to low DNA-binding microcentrifuge tubes and use a heated vacuum concentrator to reduce sample volume to less than 3.4 µL. Reconstitute samples to 3.4 µL.
- Bisulfite Conversion
NOTE: Perform bisulfite conversion of the eluted ssDNA using appropriate reagents and instructions from a commercially-available bisulfite conversion kit.- Add 130 µL prepared bisulfite conversion reagent to supernatant from previous step. Divide each of the 150 µL reactions equally into two wells. Incubate in a thermal cycler (64 °C for 2.5 h, 4 °C hold).
NOTE: The 150 µL reaction is divided equally into two separate wells to ensure homogenous temperature. After incubating for 2.5 h, immediately proceed to the next step. - Bind samples to spin columns by adding 600 µL of Binding Buffer and wash once with 100 µL of Wash Buffer. Centrifuge columns (15,000 x g, 1 min) between all bisulfite conversion steps and discard flow through.
- Desulphonate samples by adding 200 µL of Desulphonation Buffer to columns. Incubate at room temperature for 15 - 20 min. Repeat centrifugation and discard flow through.
- Wash columns twice with 200 µL of Wash Buffer. Elute each sample by adding 10 µL of Elution Buffer to the column, incubating for 3 min at room temperature, and centrifuging (15,000 x g, 1 min). Repeat elution step for a total of 20 µL.
- Prepare PCR reaction Master Mix 1 on ice. Add 82 µL of mix to each sample. Incubate in a thermal cycler with the following program.
PCR Reaction Master Mix 1 (per sample):
30 µL of Water
50 µL of Methyl-Seq PCR Master Mix
1 µL of Methyl-Seq PCR1 Primer F
1 µL of Methyl-Seq PCR1 Primer R
Thermal Cycler Program:
Stage 1, 1 cycle: 95 °C 2 min
Stage 2, 8 cycles: 95 °C 30 s, 60 °C 30 s, 72 °C 30 s
Stage 3, 1 cycle: 72 °C 7 min
Stage 4, 1 cycle: 4 °C Hold - Purify samples using 180 µL of DNA-binding magnetic beads and 400 µL of freshly prepared 70% ethanol per sample. Add 180 µL of beads to each sample and incubate for 5 min at room temperature. Pellet beads, remove supernatant and resuspend pellet in 200 µL of 70% ethanol. Remove ethanol and repeat wash once.
- Use a magnetic plate to pellet beads and remove as much ethanol as possible. Dry in a 37 °C heatblock for 3–5 min until the bead pellet is completely dry. Resuspend in 21 µL of nuclease-free water and collect approximately 19.5 µL of supernatant.
- Add 130 µL prepared bisulfite conversion reagent to supernatant from previous step. Divide each of the 150 µL reactions equally into two wells. Incubate in a thermal cycler (64 °C for 2.5 h, 4 °C hold).
- Indexing
- Prepare PCR reaction Master Mix 2 on ice. Add 25.5 µL Master Mix 2 to each sample. Add 5 µL commercial indexing primers to individual samples and incubate in a thermal cycler.
PCR Reaction Master Mix 2 (per sample):
25 µL Methyl-Seq PCR Master Mix
0.5 µL Methyl-Seq Common Indexing Primer
Thermal Cycler Program:
Stage 1, 1 cycle: 95 °C 2 min
Stage 2, 6 cycles: 95 °C 30 s, 60 °C 30 s, 72 °C 30 s
Stage 3, 1 cycle: 72 °C 7 min
Stage 4, 1 cycle: 4 °C Hold
NOTE: Additional cycles (2 - 3) may be necessary if the starting DNA concentration is below recommended values. - Purify samples using 90 µL of DNA-binding magnetic beads and 400 µL of freshly prepared 70% ethanol per sample. Add 90 µL of beads to each sample and incubate for 5 min at room temperature. Pellet beads, remove supernatant and resuspend pellet in 200 µL of 70% ethanol. Remove ethanol and repeat wash once.
- Use a magnetic plate to pellet beads and remove as much ethanol as possible. Dry in a 37 °C heatblock for 3–5 min until the bead pellet is completely dry. Resuspend in 24 µL of nuclease-free water and collect approximately 24 µL of supernatant.
- Assess concentration and bp size using the high-sensitivity DNA detection reagents on a bioanalyzer.
Note: If the bioanalyzer fails to detect the presence of the library DNA, repeat the preparation steps with additional DNA.
Stopping point: After purification, indexed samples may be sealed and stored at -20 °C. - Pooling Samples for the appropriate next-generation sequencing platform used.
- Using the concentration data from the bioanalyzer, which determines DNA molarity based on library size and quantity in a given volume, dilute with Low TE buffer (6.1.1.1) and combine all samples to a final concentration of 15 pM.
NOTE: A more sensitive method of quantifying the library is by quantitative real-time PCR using primers that target the ligated adapters. - Run pooled samples on the number of lanes that are sufficient for 4 samples per lane on a next-generation sequencer.
NOTE: For instance, if 16 library samples have been uniquely indexed and combined, run the libraries over 4 lanes, equivalent to 4 samples per lane.
- Using the concentration data from the bioanalyzer, which determines DNA molarity based on library size and quantity in a given volume, dilute with Low TE buffer (6.1.1.1) and combine all samples to a final concentration of 15 pM.
- Prepare PCR reaction Master Mix 2 on ice. Add 25.5 µL Master Mix 2 to each sample. Add 5 µL commercial indexing primers to individual samples and incubate in a thermal cycler.
7. Sequencing on a Next-generation Sequencer
- Send the samples to the institutional sequencing core for clustering of the Methyl-Seq library, followed by sequencing on a next-generation sequencing machine.
8. Analysis to Identify DMRs
- Implement Bismark15, which invokes Bowtie 2.0 as an internal sequence aligner16,17, to align raw input reads to bisulfite-converted, plus-strand genome. Following alignment, use the Bismark_methylation_extractor to perform quality control and assign an estimated methylation value to each CpG.
- Generate a list of DMRs with the BS-Seq package18 in Bioconductor. Filter the DMRs based on having greater than 3 consecutive CpGs and P-value <0.05.
NOTE: Generate a DMR list that includes genomic coordinates, distance to the nearest RefSeq gene, number of CpGs within each DMR, average% CpG methylation value across the DMR for the two comparison groups (e.g., stressed vs. unstressed), the P-value, and the FDR (false discovery rate) value. Use the DMR list, i.e., genomic coordinates, to design pyrosequencing primers for validation.
9. Validation by Bisulfite Pyrosequencing
- Primer Design
- Design primers for bisulfite PCR and pyrosequencing. Design two sets of PCR primers (outside and nested) so that the nested PCR will amplify 150–400 bps of a DMR.
NOTE: In general, designed primers are at least 24 bases long with at least 4–5 non-consecutive G’s (C’s for the reverse primer) to account for reduced annealing temperature from loss of sequence complexity. One of the nested primers will be biotin-labeled and HPLC-purified. However, standard primers should be ordered first to optimize the PCR step by resolving the reactions on an agarose gel.- Design the pyrosequencing assay primer so that it targets the complementary biotinylated strand just 1–2 bases upstream of the CpGs to be assayed. Design multiple pyrosequencing primers as necessary, as each pyrosequencing primer can reliably assay 30 bps downstream.
- For the Rt1-m4, use the following:
rRT1M4 Outside – F TGTAYGATTTTGGTTATYGTAAAT
rRT1M4 Outside – R AACTTACAAATTTCACCAACTCA
rRT1M4 Nested – F GTGGGTTAYGTGGATAATATATAG
rRT1M4 Nested – R AATCACTTACCATTCTCTCTCTAACTA
rRT1M4 Pyro1 TAYGTGGATAATATATAGAT
rRT1M4 Pyro2 GATAGTTATTTGGYGAGTTAG
rRT1M4 Pyro3 GAGTATTTGGAGGAGTTGAT
rRT1M4 Pyro4 GGATTTTAATATTTGGT
- Design primers for bisulfite PCR and pyrosequencing. Design two sets of PCR primers (outside and nested) so that the nested PCR will amplify 150–400 bps of a DMR.
- Use a commercially-available kit for bisulfite conversion of rat blood gDNA.
NOTE: The bisulfite conversion steps have been adapted from the commercially-available kit with the following modifications: In step 1, add 50–100 ng of blood gDNA and dilute with water to 20 µL. In step 9, elute 20 µL per sample.- Prepare bisulfite conversion reagent according to the manufacturer’s protocol and combine with diluted gDNA. Incubate in thermal cycler (64 °C for 2.5 h, 4 °C hold).
- Add Binding Buffer to converted gDNA in spin columns and centrifuge (15,000 x g, 1 min). Wash columns once then add Desulphonation Buffer to the columns and incubate for 15 min at room temperature. Centrifuge (15,000 x g, 1 min).
- Wash column with Wash Buffer and centrifuge (15,000 x g, 1 min). Repeat wash step with centrifugation (15,000 x g, 2 min). Add 20 µL Elution Buffer and centrifuge (15,000 x g, 1 min) to elute.
- PCR amplification
- Prepare Outside PCR Master Mix. Add 21.5 µL of Master Mix to 3.5 µL bisulfite-converted gDNA and run thermal cycler program.
Outside PCR Master Mix:
16.25 µL of Water
2.5 µL of Polymerase Buffer [10x]
0.5 µL of dNTP [10 mM]
1 µL of Forward Primer [0.1 µM]
1 µL of Reverse Primer [0.1 µM]
0.25 µL of Taq DNA Polymerase [5000 U/mL].
Thermal cycler program:
Stage 1, 1 cycle: 94 °C 4 min
Stage 2, 47 cycles: 94 °C 1 min, 53 °C 30 s, 72 °C 1 min
Stage 3, 1 cycle: 72 °C 8 min, 4 °C Hold - Prepare Nested PCR Master Mix. Add 23 µL of Master Mix to 2 µL of sample from outside PCR and repeat the outside PCR thermal cycler program. Assess PCR product quality through gel electrophoresis (1x TAE buffer, 1% agarose gel).
Nested PCR Master Mix:
17.75 µL of Water
2.5 µL of Polymerase Buffer [10x]
0.5 µL of dNTP [10 mM]
1 µL of Forward Primer [0.1 µM]
1 µL of Reverse Primer [0.1 µM]
0.25 µL of Taq DNA Polymerase [5000 U/mL]
Note: For nested PCR, either the forward or the reverse primer must be biotinylated.
- Prepare Outside PCR Master Mix. Add 21.5 µL of Master Mix to 3.5 µL bisulfite-converted gDNA and run thermal cycler program.
- Pyrosequencing
- Make a master mix containing 38 µL of Binding Buffer, 35 µL of water, and 2 µL of streptavidin-coated sepharose beads per sample. In a 96-well plate, add 75 µL of master mix and 5 µL of nested PCR product. Shake on a plate shaker for 15 - 60 min.
- While shaking, add 12 µL of primer (0.5 µM, diluted in annealing buffer) into the wells of a pyrosequencing assay plate.
- After shaking, perform wash steps using binding reaction wash buffers. Place vacuum tool in trough filled with water then collect samples from plate. Submerge vacuum tool in half-filled troughs containing 70% ethanol, NaOH (0.2 M), and Tris acetate buffer (10 mM, pH 7.4). Disconnect from vacuum and place vacuum tool in HS assay plate to transfer beads.
- Place plate on heat block and incubate at 80 °C for 2 min. Allow plate to cool for 5 min then begin pyro program.
Results
A successful implementation of the rat Methyl-Seq platform depends on several criteria. Figure 1 shows the overall workflow of the study and highlights specific quality control (QC) steps that are needed before moving forward. One of the first factors to consider is the robustness of the animal model and the stress regimen, which determine the magnitude of epigenetic changes that occur across the methylome. Since our animal work is predicated on our previous observation that corticosterone (CORT) exposure can lead to changes in DNA methylation19,20, our chronic variable stress (CVS) regimen needed to be of sufficient rigor to produce stressed rats with elevated plasma CORT levels. A typical weekly CVS regimen is shown in Table 1 and consisted of daily stressors in the morning, afternoon, and overnight that are constantly changed to prevent habituation and diminished stress response. Throughout the 3-week regimen, the stressed animals exhibited significantly elevated levels of mean plasma CORT [Days 4–21, Control: 32.7 3.7 ng/mL, Stress: 103.0 11.9 ng/mL (mean SEM), P = 2.2 x 10-4, Figure 2A] over those of unstressed, control animals. Consistently, these animals also showed greater anxiety-like behavior on the elevated plus maze (EPM), as indicated by the significantly more time spent in the closed arms of the EPM and less time in the open arms (Figure 2B). These results demonstrate that the CVS exposure led to significant endocrine and behavioral changes, leading us to investigate whether these changes were associated with specific DNA methylation signatures.
We emphasize several checkpoints that are crucial for the successful construction of the Methyl-Seq library. Starting with a sufficient quantity of DNA is necessary, as sonication, multiple wash/purification, target enrichment, and bisulfite conversion steps successively reduce the quantity of DNA in the finished library. Although several PCR amplification steps alleviate the loss of DNA template, excessive PCR cycle numbers can introduce higher duplicate reads. For the current rat Methyl-Seq study, 2 μg of blood gDNA per rat was used. We note that Methyl-Seq libraries can be made with starting DNA amount as low as 500 ng. Smaller starting material allows users to generate libraries from DNA isolated by FACS (fluorescence-activated cell sorting) or needle punches, although there is increased risk of producing an insufficient amount of libraries for subsequent sequencing. QC is performed by electrophoresis of 1 μL of the sample on a bioanalyzer, which provides DNA molecular weight, quantity, and molarity. Three critical steps that require the use of the bioanalyzer are: 1) following sonication step to ensure sufficient shearing of DNA (~170 bp, red, Figure 3); 2) following adapter ligation step indicated by a shift in the average size of the sheared DNA (~200 bp, blue, Figure 3) to ensure their subsequent amplification by PCR; and 3) following final library purification step to ensure the quantity and size of the library for sequencing.
The R-packages BSSeq and BSmooth in Bioconductor were used for analyzing the bisulfite sequencing data18. They include tools and methods for aligning the sequence reads, performing quality control, and identifying differentially methylated regions (DMRs). BSmooth software invokes Bowtie 2.016,17 as an internal sequence aligner to obtain CpG-level measurement summaries, by alignment of raw input reads to bisulfite-converted genomic sequences. The aligned reads are then filtered through rigorous quality control procedures that seek to identify systematic sequencing and base-calling errors that may skew downstream analyses. A series of plots are generated to visually aid in this process of filtering. Sequencing metrics are also generated to document relevant information such as number of aligned reads, % target, and per CpG coverage, among others (Table 2). Once the data are filtered, a smoothing/normalization algorithm is performed, where every CpG is assigned an estimated methylation value based on all QC reads from each sample and estimates from neighboring CpGs to ensure more accurate calling of methylation status even in cases where the sequence coverage is low. This value provides a smoothed estimate of the probability of methylation at each CpG site. By comparing the mean of the smoothed methylation estimates of each sample between the two treatment groups and ranking genomic regions from the most significantly different to least, a list of DMRs is generated (Table 3).
The top DMR between stressed and unstressed groups was located in the promoter of the rat major histocompatibility gene Rt1-m4, with stressed animals exhibiting higher methylation levels across all CpGs than unstressed animals (Figure 4A). To confirm successful implementation of the Methyl-Seq platform and the data analysis, primers were designed against the DMR, and blood DNA methylation levels in the entire cohort of stressed and unstressed animals (8 sequenced by Methyl-Seq and 8 not sequenced) were assessed by bisulfite pyrosequencing. Results demonstrate significant increase in DNA methylation across 10 out of the 12 CpGs assayed (5.1–10.4 change in % methylation, P <0.037, Figure 4B). KEGG pathway analysis was performed on all of the nominally significant DMRs to identify pathways associated with stress. Consistently, DMR-associated pathways implicated diseases associated with chronic stress exposure, such as diabetes, cardiovascular disease, and cancer (Table 4).21,22,23 To demonstrate an association between the epigenetic data and the degree of exposure to stress, methylation levels at CpG-10 were compared to the mean 3-week CORT levels for each animal. Results showed a modest correlation between the endocrine and methylation data (R2=0.54, P=0.001, Figure 5).

Figure 1: Overall schematic workflow for the rat Methyl-Seq platform. One μg of the genomic DNA extracted from the blood of stressed and control rats is first processed for constructing the Methyl-Seq libraries for sequencing, analysis, and target identification. Another 100 ng of DNA is used for independent validation of the identified epigenetic targets by bisulfite pyrosequencing. Please click here to view a larger version of this figure.
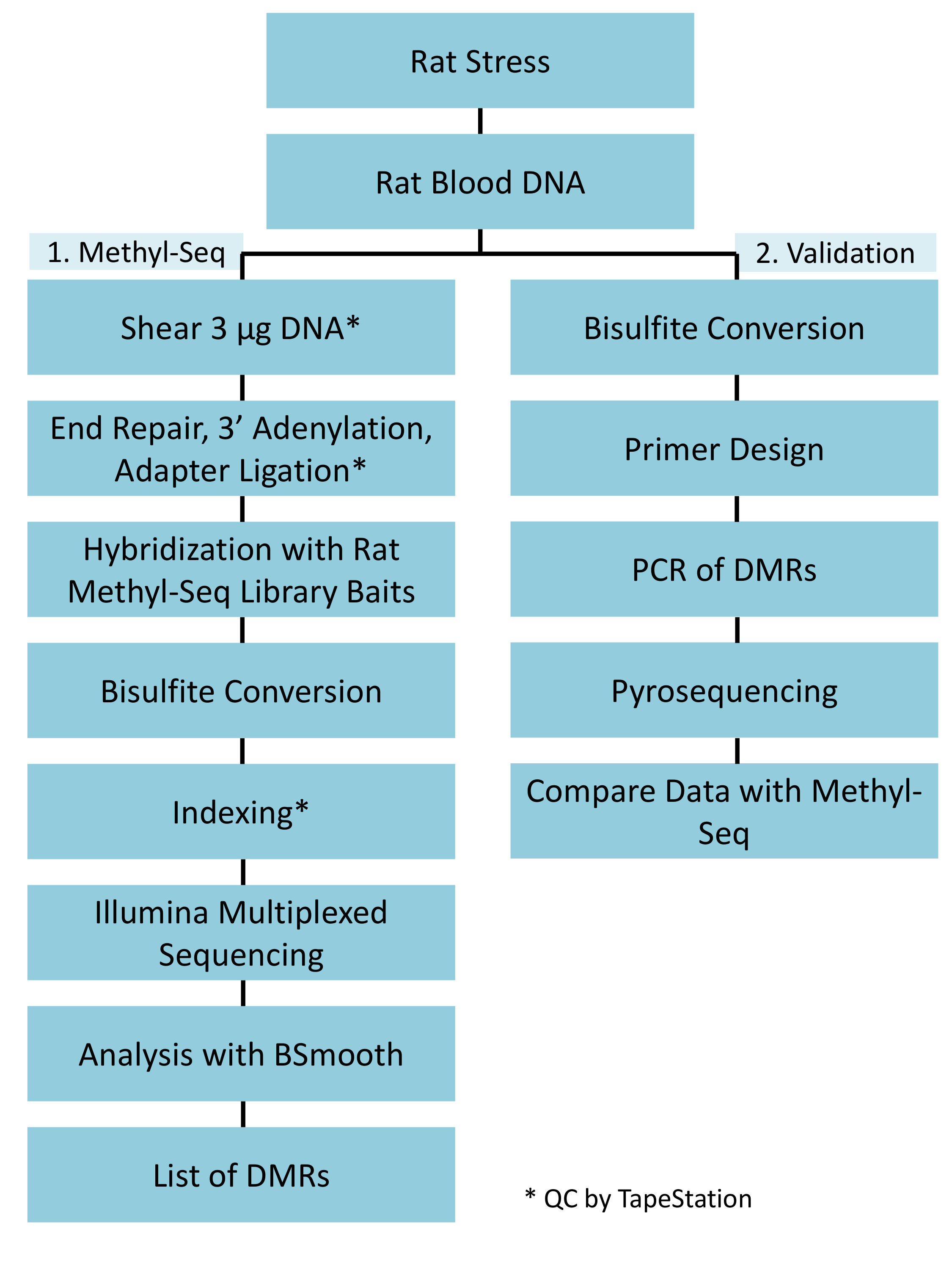{kind=link}

Figure 2: Exposure to chronic variable stress (CVS) leads to endocrine and behavioral changes in rats. (A) Multiple samplings of corticosterone (CORT) demonstrate the robustness of the 3 week CVS regimen. Blood samples were collected in the morning prior to the daily stress regimen. (B) Stressed animals spent more time in the closed arms and less time in the open arms of the elevated plus maze (EPM). Boxplots with data point for each animal are shown. Student's T-test was performed for statistical significance. *P<0.05, **P<0.01, and ***P<0.001. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Quantitation of sheared and adapter-ligated rat DNA on a bioanalyzer. The red and blue curves show the quantity and size of genomic DNA (red) following shearing in an isothermal sonicator and adapter ligation, respectively. Each line represents one sample and the red and blue curves reflect both loss of DNA during the several steps (end-repair, 3'-adenylation, and sample cleanup) and increase in bp size due to the ligation of the adapters. Sharp peaks at 25 bp and 1500 bp are standard markers that have been added to the loading buffer. Please click here to view a larger version of this figure.
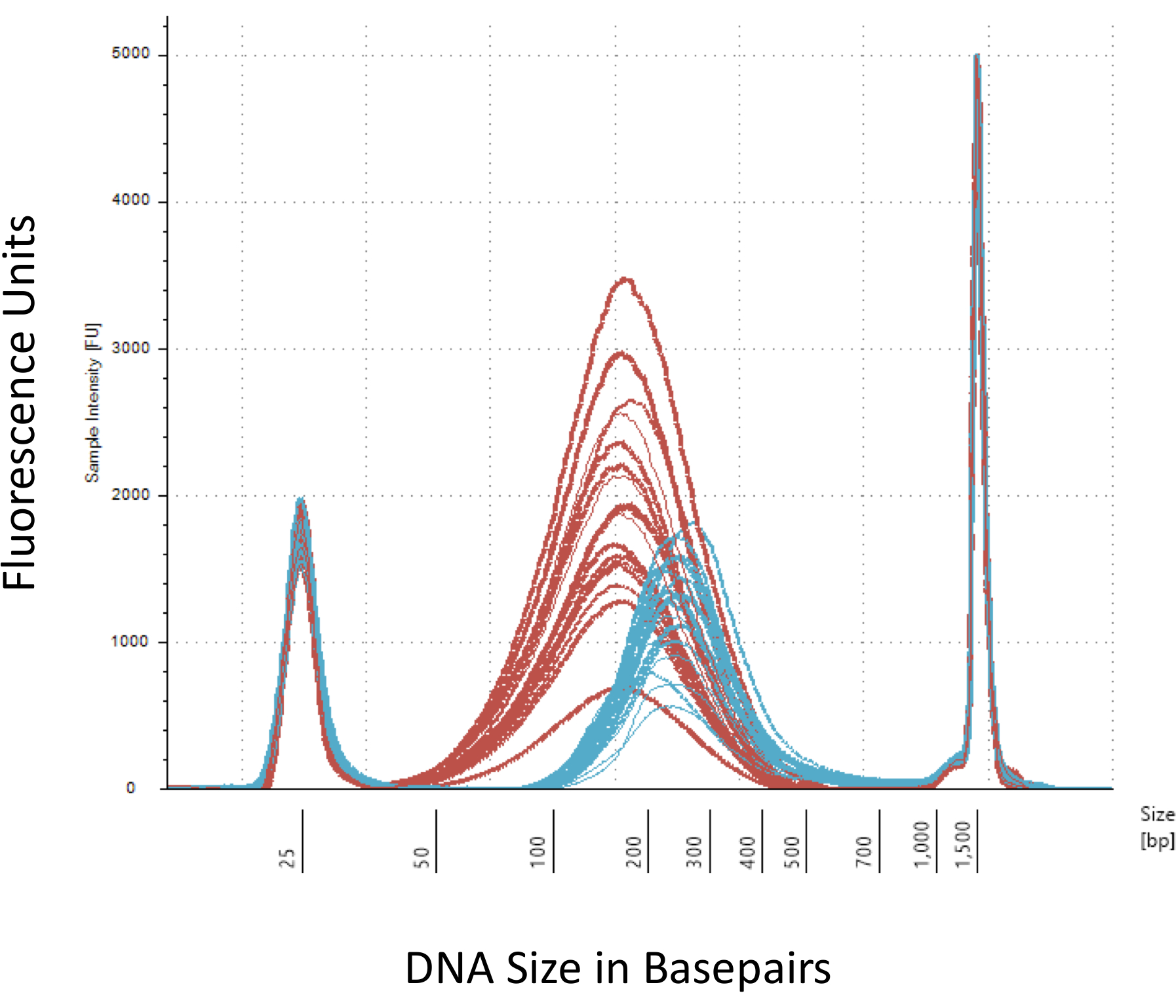{kind=link}

Figure 4: CVS-induced epigenetic changes are detected by rat Methyl-Seq. (A) Analysis of the rat Methyl-Seq data implicated the promoter of the gene Rt1m4 as a differentially methylated region (DMR) between stressed (red) and control (blue) rats. The graphical output for the Rt1m4 DMR (pink shaded region) displays each CpG (vertical gray line), the four samples in each group (red or blue lines), and the% methylation levels for each animal (red or blue dot). (B) Twelve CpGs within the DMR were validated by bisulfite pyrosequencing. The bar graphs are represented as mean SEM, and a Student's T-test was performed for statistical significance. *P<0.05. Please click here to view a larger version of this figure.
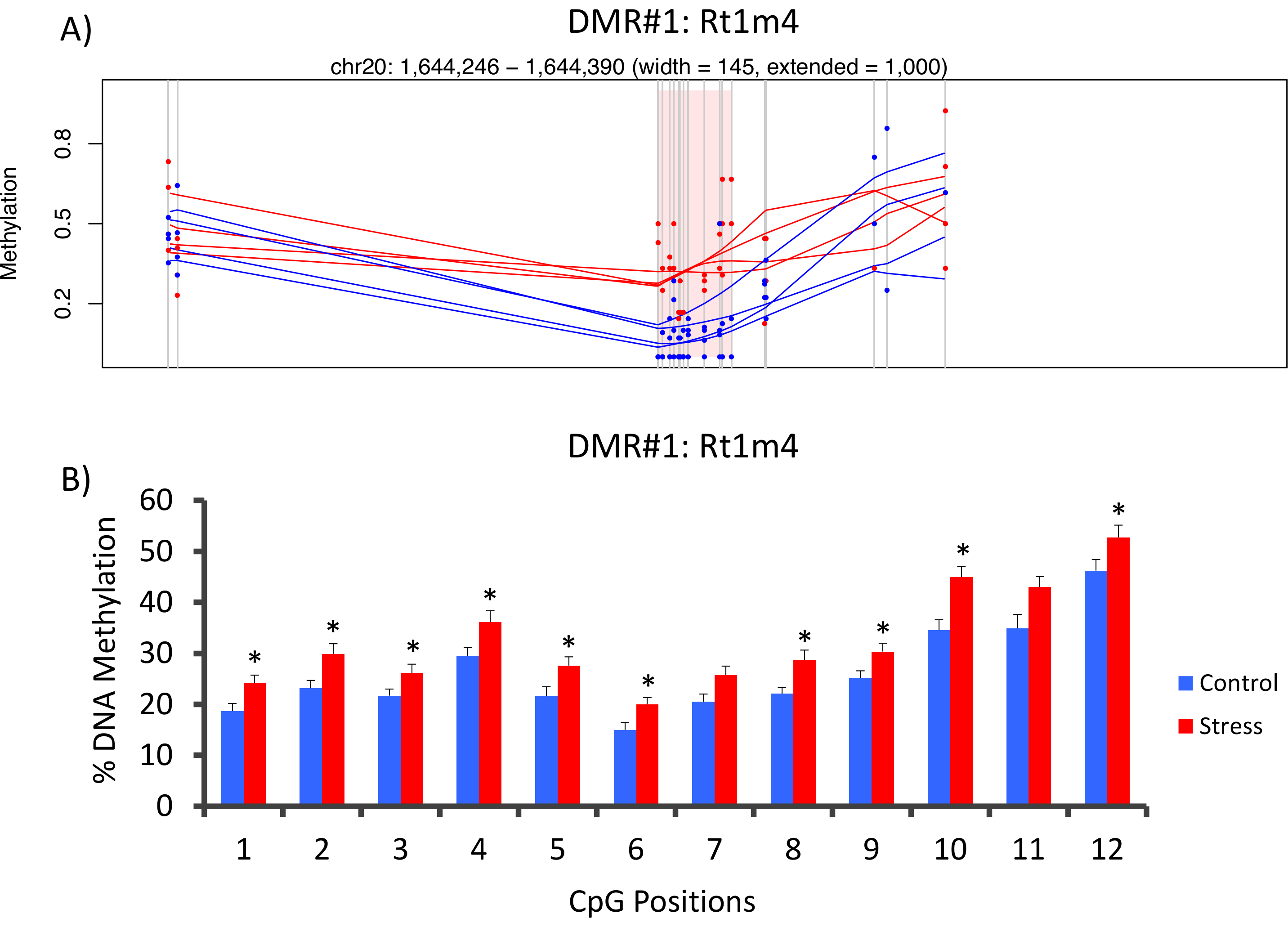{kind=link}

Figure 5: Linear regression analysis showed a modest correlation between % DNA methylation at CpG-10 of Rt1m4 and the 3 week mean plasma CORT levels of both stressed and control animals (N=16). Data from stressed animals are represented by red circles. Please click here to view a larger version of this figure.
{kind=link}
| Week | Day 1 | Day 2 | Day 3 | Day 4 | Day 5 | Day 6 | Day 7 |
| AM | Restraint | Swim | Cold Room | Swim | Restraint | Shaker | Swim |
| PM | Shaker | Cage Tilt | Restraint | Shaker | Cold Room | Restraint | Cold Room |
| Overnight | Food Restrict | Wet Bedding | Isolation | Light On | Crowding | Light On | Wet Bedding |
Table 1: A typical weekly schedule of the chronic variable stress regimen (CVS).
| Sequencing Metrics | Stress1 | Control1 |
| (n = 4) | (n = 4) | |
| Paired End Reads (PER) | 89,290,397 | 80,165,674 |
| Uniquely Mapped Paired End Reads (UMPER) | 39,200,255 | 35,013,406 |
| Alignment Rate/Mapping Efficiency (UMPER/PER) | 44% | 44% |
| Duplicate Reads (% of UMPER) | 73% | 65% |
| Deduplicated UMPER | 10,481,031 | 12,306,018 |
| Average Read Depth Coverage (x) (ARDC) | 6x | 6x |
| CpGs (N) | 12,056,878 | 12,056,878 |
| ARDC (x) of CpGs | 2x | 2x |
| CpGs with at least 10 reads (N) | 481,383 | 595,850 |
| ARDC (X) of CpGs with at least 10 reads | 19 | 19 |
| On Target CpGs (complete overlap with Probe Target Regions) | 1,923,872 | 2,007,638 |
| On Target ARDC (x) of CpGs | 7x | 8x |
| On Target CpGs with at least 10 reads (N) | 428,249 | 531,419 |
| On Target ARDC (x) of CpGs with at least 10 reads | 18x | 18x |
| On Target (PER with 1 or more Base Pair overlap with Probe Target Regions) (UMPER) | 8,277,715 | 9,369,523 |
| % On Target (of Deduplicated UMPER) | 78% | 77% |
| On Target (Total Bases Mapped) Mb | 125 Mb | 128 Mb |
| On Target Average Read Depth Coverage (x) (ARDC) | 9x | 10x |
| 1Sequencing metrics based on averages across subjects in each group |
Table 2: Sequencing metrics obtained from the rat Methyl-Seq platform.
| chr | start | end | gene | distance | areaStat | meanDiff | stress | control | direction |
| chr20 | 1,644,246 | 1,644,390 | RT1-M4 | in_gene | 93.03 | 0.22 | 0.33 | 0.11 | gain |
| chr5 | 160,361,352 | 160,361,564 | LOC690911 | in_gene | -70.75 | -0.19 | 0.72 | 0.91 | loss |
| chr3 | 61,138,281 | 61,138,330 | RGD1564319 | 265569 | 61.79 | 0.21 | 0.94 | 0.72 | gain |
| chr2 | 143,064,811 | 143,065,010 | Ufm1 | 8569 | -59.48 | -0.11 | 0.13 | 0.24 | loss |
| chr7 | 30,764,111 | 30,764,284 | Ntn4 | in_gene | 57.04 | 0.21 | 0.94 | 0.73 | gain |
| chr17 | 12,469,112 | 12,469,218 | Idnk | 41996 | -50.91 | -0.13 | 0.74 | 0.88 | loss |
| chr7 | 47,101,725 | 47,101,930 | Pawr | in_gene | -50.54 | -0.12 | 0.64 | 0.76 | loss |
| chr5 | 76,111,248 | 76,111,822 | Txndc8 | 151703 | -50.38 | -0.11 | 0.85 | 0.96 | loss |
| chr11 | 80,640,132 | 80,640,356 | Dgkg | in_gene | -50.07 | -0.16 | 0.73 | 0.89 | loss |
| chr8 | 71,759,248 | 71,759,411 | Mir190 | 210226 | -47.84 | -0.17 | 0.58 | 0.75 | loss |
Table 3: Top 10 differentially methylated regions. For each DMR, the output table shows from the left to right column: chromosomal location (chr), coordinates (start/end), gene name, distance from the transcription start site, differential area statistics between stressed and control groups (areaStat), mean differential methylation (meanDiff), mean methylation levels across each DMR for stressed and control groups (stress/control), and direction of methylation change from controls.
| KEGG Pathway Terms | Gene Count | % | P-value | Benjamini |
| Diabetes | ||||
| Type II diabetes mellitus | 12 | 0.1 | 3.6 x 10-4 | 9.8 x 10-3 |
| Cardiovascular Disease | ||||
| Vascular smooth muscle contraction | 18 | 0.1 | 1.6 x 10-3 | 3.6 x 10-2 |
| Arrhythmogenic right ventricular cardiomyopathy (ARVC) | 13 | 0.1 | 4.0 x 10-3 | 7.1 x 10-2 |
| Dilated cardiomyopathy | 14 | 0.1 | 7.6 x 10-3 | 1.2 x 10-1 |
| Neuron Function | ||||
| Long-term potentiation | 11 | 0.1 | 1.5 x 10-2 | 1.4 x 10-1 |
| Signaling | ||||
| MAPK signaling pathway | 35 | 0.2 | 2.4 x 10-4 | 9.9 x 10-3 |
| Calcium signaling pathway | 22 | 0.1 | 1.2 x 10-2 | 1.4 x 10-1 |
| Chemokine signaling pathway | 21 | 0.1 | 1.2 x 10-2 | 1.3 x 10-1 |
| Cancer | ||||
| Pathways in cancer | 42 | 0.3 | 4.1 x 10-5 | 3.4 x 10-3 |
| Glioma | 15 | 0.1 | 4.4 x 10-5 | 2.4 x 10-3 |
| Non-small cell lung cancer | 10 | 0.1 | 7.9 x 10-3 | 1.1 x 10-1 |
| Colorectal cancer | 13 | 0.1 | 8.4 x 10-3 | 1.1 x 10-1 |
| Chronic myeloid leukemia | 12 | 0.1 | 1.2 x 10-2 | 1.3 x 10-1 |
Table 4: KEGG Pathway analysis of DMRs identified from the rat Methyl-Seq.
Discussion
In this study, we designed and implemented the Methyl-Seq platform for the rat genome. By demonstrating its utility with a rat model of stress, we demonstrated that the experimental and analytical pipeline can provide differentially methylated regions between two comparison groups.
To ensure a successful implementation of the platform, several critical steps need to be observed. First, initial DNA quality and quantity has a significant impact on the quality and quantity of the final Methyl-Seq library. We used a fluorometer, rather than a spectrophotometer, to ensure that our DNA measurement reflected the quantity of double-stranded DNA present. The bioanalyzer was used to measure the molecular size and quantity of DNA following shearing and after adapter ligation. Verifying the molecular size "shift" between these steps is crucial to confirm the presence of adapters at the ends of each DNA fragment that will undergo adapter-mediated PCR in the subsequent steps. The quantity of DNA remaining at the end of the adapter ligation step is also important, since at least 100 ng of the library product is needed at this step to ensure sufficient quantity is available after the target enrichment and bisulfite conversion steps. A final high-sensitivity measurement was performed on the constructed Methyl-Seq library so that the library can be properly diluted for subsequent clustering on the next-generation sequencer. Finally, bisulfite pyrosequencing was employed as a highly-quantitative, independent method to assess the accuracy of the analytical pipeline. The final validation using the original samples and replication using additional animals are crucial steps to ensure that the experiment can detect biologically significant changes in DNA methylation.
We also include several guidelines in the event of deviation from the protocol or if problems are encountered. First, it is possible to lose too much DNA during end-repair, adapter ligation, or magnetic bead purification steps. Alternatively, starting amounts of DNA could be small (<200 ng) due to limited tissue/DNA availability or implementation of various enrichment methods such as fluorescence activated cell sorting. Increasing the cycle number during the two library amplification steps may be able to compensate for the excessive loss of DNA or low starting DNA amount throughout the library construction protocol. However, no more than an additional 2–3 cycles are recommended, as excessive template amplification is likely to lead to an increase in the number of duplicate reads being sequenced. These duplicates are excluded during the alignment step to prevent bias in percent methylation calculations. Second, if the average DNA size does not increase by more than 30 bps, check to ensure that the reagents are new, as T4 DNA polymerase, Klenow, and/or T4 ligase may be old. Commercially available replacement reagents can be used.
Additionally, it is possible that the predicted DMRs might not validate by pyrosequencing, where DNA methylation differences do not exist or are significantly less than those predicted by analysis. Poor validation of candidate regions is a problem too common for many genome-wide analyses, such as when pyrosequencing results do not confirm differential methylation or the effect size is much smaller than that predicted by the analysis. BSmooth is one analytical package that "smoothes" the methylation levels across a window of multiple CpGs. For the current experiment, BSmooth implicated a DMR whose methylation levels were validated by bisulfite pyrosequencing. However, there will likely be discrepancies between methylation levels predicted by BSmooth and those verified by pyrosequencing. The discrepancies arise from the smoothing function that estimates the average methylation values across all of the CpGs within a DMR, including consecutive CpGs that may differ in DNA methylation by more than 50% or CpGs whose methylation values were excluded due to sub-threshold read depth. R-packages such as MethylKit24 can be used to identify smaller windows of CpGs or even single CpGs whose methylation levels correlate strongly with those validated by pyrosequencing. Implementing different packages and testing their predicted regions or CpGs of differential methylation by pyrosequencing will ensure robustness of data. Alternatively, original Methyl-Seq libraries can be resequenced and added to the read files to increase read depth. Since determination of methylation levels are semi-quantitative and dictated by the number of reads [(# of CpGs)/(# of TpGs+CpGs)], increasing the read depth for a given CpG will increase the accuracy of its percent methylation value. In this study, we only considered CpGs whose methylation values were determined by at least ten reads and achieved an overall read coverage of 19x for each CpG.
The rat Methyl-Seq platform is not without its limitations. While it is more cost effective than whole-genome bisulfite sequencing, it is considerably more expensive than other methods. Nevertheless, most of the cost was for purchasing lanes on the sequencer and not for the capture system. Depending on the read depth necessary, with cross-tissue comparisons requiring less due to large (25–70%) differences12 in DNA methylation, the cost can be reduced by multiplexing more samples per lane and using a higher-capacity platform. Also, the sample preparation is more time-consuming than other methods. While similar to other pulldown approaches that incorporate next-generation sequencing, the added bisulfite conversion and purification steps add to the work load. Overall, the Methyl-Seq platform is a cost-effective alternative to whole-genome sequencing and provides base-pair resolution at more than 2.3 million CpGs, which is considerably more than those assayed by microarray-based platforms. To date, the commercially-available human and mouse Methyl-Seq platforms have been used to document alcohol-dependent changes in the macaque brain25,26, neurodevelopmental genes in the mouse brain9, and blood-brain targets of glucocorticoids10. Further, the ability to target specific regions regardless of sequence recognition by restriction enzymes makes it an ideal platform for cross-species comparisons. For this study, we designed the Methyl-Seq platform for the rat, for which many pharmacological, metabolic, and behavioral experiments are performed without the benefit of a genome-wide methylomic tool. Our data show that it can be used to detect DMRs in a rat model of stress and correlated other physiological parameters such as overall plasma CORT levels.
The Methyl-Seq platform is ideal for epigenetic experiments in animals with sequenced genomes that may not have enough experimental evidence documenting regulatory regions. When such regions are made available, additional regions may be custom-designed and attached to the current version. Further, the platform is ideal for comparative genomics, since the target enrichment is not constrained by restriction enzyme recognition. For instance, the promoter region of any gene of interest can be captured regardless of whether it harbors a specific restriction site. Similarly, any regulatory regions, such as those identified in mouse or humans, which are conserved in the genome of interest can be captured.
Disclosures
The manuscript is part of a contest prize from Agilent Technologies.
Acknowledgements
This study was funded by NIH grant MH101392 (RSL) and support from the following awards and foundations: a NARSAD Young Investigator Award, Margaret Ann Price Investigator Fund, the James Wah Mood Disorders Scholar Fund via the Charles T. Bauer Foundation, Baker Foundation, and the Project Match Foundation (RSL).
Materials
| Name | Company | Catalog Number | Comments |
| Radioimmuno assay (RIA) | MP Biomedicals | 7120126 | Corticosterone, 125I labeled |
| Master Pure DNA Purification Kit | Epicentre/Illumina | MC85200 | |
| Thermal-LOK 2-Position Dry Heat Bath | USA Scientific | 2510-1102 | Used with 1.5 mL tubes |
| Vortex Genie 2 | Fisher | 12-812 | Vortex Mixer |
| Ethyl alcohol, Pure | Sigma-Aldrich | E7023 | 100% Ethanol, molecular grade |
| Centrifuge 5424 R | Eppendorf | - | Must be capable of 20,000 x g |
| Qubit 2.0 | ThermoFisher Scientific | Q32866 | Fluorometer |
| Qubit dsDNA BR Assay Kit | ThermoFisher Scientific | Q32850 | |
| Qubit dsDNA HS Assay Kit | ThermoFisher Scientific | Q32851 | High sensitivity DNA detection reagents |
| Qubit Assay Tubes | ThermoFisher Scientific | Q32856 | |
| SureSelectXT Rat Methyl-Seq Reagent Kit | Agilent Technologies | G9651A | Reagents for preparing the Methyl-Seq library |
| SureSelect Rat Methyl-Seq Capture Library | Agilent Technologies | 931143 | RNA baits for enrichment of rat targets |
| IDTE, pH 8.0 | IDT DNA | 11-05-01-09 | 10 mM TE, 0.1 mM EDTA |
| DNA LoBind Tube 1.5 mL | Eppendorf | 22431021 | |
| Covaris E-series or S-series | Covaris | - | Isothermal sonicator |
| microTUBE AFA Fiber Pre-Slit Snap-Cap 6x16mm (25) | Covaris | 520045 | |
| Water, Ultra Pure (Molecular Biology Grade) | Quality Biological | 351-029-721 | |
| Veriti 96 Well-Thermal Cycler | Applied Biosystems | 4375786 | |
| AMPure XP Beads | Beckman Coulter | A63880 | DNA-Binding magnetic beads |
| 96S Super Magnet | ALPAQUA | A001322 | Magnetic plate for purification steps |
| 2200 TapeStation | Agilent Technologies | G2965AA | Electrophresis-based bioanalyzer |
| D1000 ScreenTape | Agilent Technologies | 5067-5582 | |
| D1000 ScreenTape High Sensitivity | Agilent Technologies | 5067-5584 | |
| D1000 Reagents | Agilent Technologies | 5067-5583 | |
| D1000 Reagents High Sensitivity | Agilent Technologies | 5067-5585 | |
| DNA110 SpeedVac | ThermoFisher Scientific | - | Vacuum Concentrator |
| Dynabeads MyOne Streptavidin T1 magnetic beads | Invitrogen | 65601 | Streptavidin magnetic beads |
| Labquake Tube Rotator | ThermoFisher Scientific | 415110Q | Nutator Mixer is also acceptable |
| EZ DNA Methylation-Gold Kit | Zymo Research | D5006 | Bisulfite conversion kit. Contains Binding, Wash, Desulphonation, and Elution buffers |
| Illumina Hi-Seq 2500 | Illumina | - | Next-generation sequencing machine |
| PCR and Pyrosequencing Primers | IDT DNA | Variable | |
| Taq DNA Polymerase with ThermoPol Buffer - 2,000 units | New England BioLabs | M0267L | |
| Deoxynucleotide (dNTP) Solution Set | New England BioLabs | N0446S | |
| Pyromark MD96 | QIAGEN | - | Pyrosequencing machine |
| Ethyl Alcohol 200 Proof | Pharmco-Aaper | 111000200 | 70% Ethanol solution |
| Sodium Hydroxide Pellets | Sigma-Aldrich | 221465 | 0.2 M NaOH denature buffer solution |
| Tris (Base) from J.T. Baker | Fisher Scientific | 02-004-508 | 10 mM Tris Acetate Buffer wash buffer solution |
| PyroMark Gold Q96 Reagents (50x96) | QIAGEN | 972807 | Reagents required for pyrosequencing |
| PyroMark Annealing Buffer | QIAGEN | 979009 | |
| PyroMark Binding Buffer (200 mL) | QIAGEN | 979006 | |
| Streptavidin Sepharose High Performance Beads | GE Healthcare | 17-5113-01 | Streptavidin-coated sepharose beads |
| PyroMark Q96 HS Plate | QIAGEN | 979101 | Pyrosequencing assay plate |
| Eppendorf Thermomixer R | Fisher Scientific | 05-400-205 | Plate mixer. 96-well block sold separately (cat. No 05-400-207) |
| SureDesign Website | Agilent Technologies | - | Target capture design software (https://earray.chem.agilent.com/suredesign/) |
| UCSC Genome Browser | University of California Santa Cruz | - | rat Nov 2004 rn4 assembly |
| Agilent Methyl-Seq Protocol | Agilent Technologies | - | https://www.agilent.com/cs/library/usermanuals/public/G7530-90002.pdf |
References
- Barski, A., et al. High-resolution profiling of histone methylations in the human genome. Cell. 129 (4), 823-837 (2007).
- Meissner, A., et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 454 (7205), 766-770 (2008).
- Bibikova, M., et al. High density DNA methylation array with single CpG site resolution. Genomics. 98 (4), 288-295 (2011).
- Naumov, V. A., et al. Genome-scale analysis of DNA methylation in colorectal cancer using Infinium HumanMethylation450 BeadChips. Epigenetics. 8 (9), 921-934 (2013).
- Wockner, L. F., et al. Genome-wide DNA methylation analysis of human brain tissue from schizophrenia patients. Translational Psychiatry. 4, e339 (2014).
- Meissner, A., et al. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Research. 33 (18), 5868-5877 (2005).
- Smith, Z. D., Gu, H., Bock, C., Gnirke, A., Meissner, A. High-throughput bisulfite sequencing in mammalian genomes. Methods. 48 (3), 226-232 (2009).
- Slieker, R. C., et al. Identification and systematic annotation of tissue-specific differentially methylated regions using the Illumina 450k array. Epigenetics Chromatin. 6 (1), 26 (2013).
- Hing, B., et al. Adaptation of the targeted capture Methyl-Seq platform for the mouse genome identifies novel tissue-specific DNA methylation patterns of genes involved in neurodevelopment. Epigenetics. 10 (7), 581-596 (2015).
- Seifuddin, F., et al. Genome-wide Methyl-Seq analysis of blood-brain targets of glucocorticoid exposure. Epigenetics. 12 (8), 637-652 (2017).
- Irizarry, R. A., et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nature Genetics. 41 (2), 178-186 (2009).
- Lee, R. S., et al. Adaptation of the CHARM DNA methylation platform for the rat genome reveals novel brain region-specific differences. Epigenetics. 6 (11), 1378-1390 (2011).
- Jankord, R., et al. Stress vulnerability during adolescent development in rats. Endocrinology. 152 (2), 629-638 (2011).
- Pellow, S., Chopin, P., File, S. E., Briley, M. Validation of open:closed arm entries in an elevated plus-maze as a measure of anxiety in the rat. Journal of Neuroscience Methods. 14 (3), 149-167 (1985).
- Krueger, F., Andrews, S. R. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 27 (11), 1571-1572 (2011).
- Langmead, B., Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nature Methods. 9 (4), 357-359 (2012).
- Langmead, B., Trapnell, C., Pop, M., Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biology. 10 (3), R25 (2009).
- Hansen, K. D., Langmead, B., Irizarry, R. A. BSmooth: from whole genome bisulfite sequencing reads to differentially methylated regions. Genome Biology. 13 (10), R83 (2012).
- Lee, R. S., et al. Chronic corticosterone exposure increases expression and decreases deoxyribonucleic acid methylation of Fkbp5 in mice. Endocrinology. 151 (9), 4332-4343 (2010).
- Lee, R. S., et al. A measure of glucocorticoid load provided by DNA methylation of Fkbp5 in mice. Psychopharmacology. , (2011).
- Bose, M., Olivan, B., Laferrere, B. Stress and obesity: the role of the hypothalamic-pituitary-adrenal axis in metabolic disease. Current Opinion in Endocrinology, Diabetes, and Obesity. 16 (5), 340-346 (2009).
- Brydon, L., Magid, K., Steptoe, A. Platelets, coronary heart disease, and stress. Brain, Behavior, and Immunity. 20 (2), 113-119 (2006).
- McKlveen, J. M., et al. Chronic Stress Increases Prefrontal Inhibition: A Mechanism for Stress-Induced Prefrontal Dysfunction. Biological Psychiatry. 80 (10), 754-764 (2016).
- Akalin, A., et al. methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biology. 13 (10), R87 (2012).
- Cervera-Juanes, R., Wilhelm, L. J., Park, B., Grant, K. A., Ferguson, B. Alcohol-dose-dependent DNA methylation and expression in the nucleus accumbens identifies coordinated regulation of synaptic genes. Translational Psychiatry. 7 (1), e994 (2017).
- Cervera-Juanes, R., Wilhelm, L. J., Park, B., Grant, K. A., Ferguson, B. Genome-wide analysis of the nucleus accumbens identifies DNA methylation signals differentiating low/binge from heavy alcohol drinking. Alcohol. 60, 103-113 (2017).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved