
Method Article
Eine Ratte Methyl-Seq-Plattform um epigenetische Veränderungen im Zusammenhang mit Stress Exposition zu identifizieren
In diesem Artikel
Zusammenfassung
Hier beschreiben wir das Protokoll und die Umsetzung von Methyl-Seq, eine epigenomischen-Plattform mit einem Rattenmodell epigenetische Veränderungen im Zusammenhang mit chronischem Stress ausgesetzt zu identifizieren. Ergebnisse zeigen, dass die Ratte-Methyl-Seq-Plattform ist in der Lage, die Methylierung Unterschiede, die durch Stress Exposition bei Ratten entstehen.
Zusammenfassung
Wie Genome eine breitere Palette von Tieren zur Verfügung stehen, gibt es ein zunehmender Bedarf an Tools, die dynamische epigenetische Veränderungen in diesen Tiermodellen erfassen können. Die Ratte ist ein bestimmtes Modell Tier wo eine epigenetische Tool viele pharmakologische und Verhaltensstörungen Studien aufschlussreiche mechanistischen Informationen ergänzen können. Zu diesem Zweck haben wir die SureSelect Capture Zielsystem (bezeichnet als Methyl-Seq) für die Ratte, die DNA-Methylierung Ebenen über die Ratte Genom einschätzen kann angepasst. Das Ratte-Design gezielt Promoter, CpG Inseln, Küsten und GC-reichen Regionen aus allen RefSeq Genen.
Zur Umsetzung der Plattform auf eine Ratte Experiment waren männliche Sprague-Dawley Ratten Variable Dauerstress für 3 Wochen ausgesetzt nach denen Blutproben für genomische DNA-Extraktion gesammelt wurden. Methyl-Seq-Bibliotheken wurden von der Ratte DNA-Proben von Scheren, Adapter Ligatur Ziel Bereicherung, Bisulfit-Konvertierung und Multiplexen gebaut. Bibliotheken wurden auf einer Plattform Next Generation Sequencing sequenziert und die sequenzielle Lesevorgänge wurden analysiert, um DMRs zwischen DNA von betonten und unbetonten Ratten zu identifizieren. Top-Kandidat DMRs wurden unabhängig von Bisulfit Pyrosequenzierung bestätigt die Robustheit der Plattform validiert.
Ergebnisse zeigen, dass die Ratte Methyl-Seq Plattform epigenetische nützlich ist, die Methylierung Veränderungen, die durch die Einwirkung auf stress erfassen können.
Einleitung
Fortschritte im Hochdurchsatz-Sequenzierung haben zu einer Fülle von Genomsequenzen für Modell und Modellorganismen geführt. Die Verfügbarkeit solcher Sequenzen hat Forschung in der Genetik, vergleichende Genomik und Transkriptom erleichtert. Zum Beispiel zur Verfügung Genomsequenzen sind sehr nützlich für die Ausrichtung von Sequenzierungsdaten von ChIP-Seq-Experimente, die basierend auf seine Assoziation mit Histon Änderungen1oder Bisulfit-Sequenzierung, DNA zu bereichern, der DNA-Methylierung von misst Erkennung von Uracil aus Bisulfit Umwandlung von unmethylated Cytosines2gebildet. Allerdings gab es Verzögerungen bei der Umsetzung des epigenomischen-Plattformen, die verfügbaren Genom-Sequenzierungsdaten in ihrem Design aufgrund mangelnder kommentierten Daten artspezifische regulatorischen Sequenzen zu integrieren, die Genfunktion beeinflussen können.
Insbesondere ist DNA-Methylierung eine der am häufigsten untersuchten epigenetischen Änderungen an der DNA, die verfügbare genomische Daten für den Aufbau einer Methylomic-Plattform nutzen können. Ein solches Beispiel ist eine Array-basierte Plattform für die menschlichen Methylome3, die in verschiedenen Disziplinen von Onkologie Psychiatrie4,5verbreitet. Leider sind ähnliche Plattformen für nichtmenschliche Tiermodellen knapp, gibt es praktisch keine weit verbreiteten Plattformen, die in ihrer ursprünglichen Gestaltung der genomischen Sequenz genutzt haben.
Eine gängige Methode, die Methylomic Landschaft der nichtmenschlichen Tiermodellen zu beurteilen ist verkleinerte Darstellung Bisulfit Sequenzierung (RRBS)6. Dieser Ansatz überwindet die Kosten des gesamten Genoms Bisulfit-Sequenzierung, die gleichzeitig eine umfassende Methylomic-Landschaft niedrigere lesen-Tiefe Abdeckung aufgrund von Kosten und begrenzten funktionelle Informationen in großen gen-armen Gebieten des Genoms2 bietet . RRBS beinhaltet Beschränkungsauswahl und Größenauswahl von genomischer DNA, für höchst GC-reichen Sequenzen wie CpG Inseln zu bereichern, die häufig gefunden in der Nähe von gen Promotoren und vermutlich eine Rolle bei gen Verordnung7spielen. Während die RRBS-Methode in einer Reihe von wichtigen Studien verwendet worden ist, ist seine Abhängigkeit von Restriktionsenzymen nicht ohne bemerkenswerte Herausforderungen und Einschränkungen. Zum Beispiel ist die Anreicherung des GC-reichen Sequenzen in RRBS völlig abhängig von das Vorhandensein von spezifischen Sequenzen vom Restriktionsenzym und anschließende Auswahl von Elektrophorese anerkannt. Dies bedeutet, dass alle genomischen Bereiche, die nicht diese Restriktionsschnittstellen enthalten während Größenauswahl ausgeschlossen sind. Cross-Arten Vergleiche sind auch anspruchsvoll, wenn die gleichen Restriktionsschnittstellen in den gleichen Loci unter den verschiedenen Arten vorhanden sind.
Ein Ansatz zur Überwindung der Einschränkungen von RRBS ist eine Bereicherung-Methode verwenden, die den veröffentlichten genomischen Sequenz in das Design der Plattform nutzt. Die Array-basierte menschliche Plattform nutzt Grundierung Sonden gegen bestimmte Verbrauchsgüter für Allel-spezifische (CG vs. TG nach Bisulfit-Konvertierung) Glühen und Primer Zielerweiterung entworfen. Das Design spiegelt nicht nur die verfügbaren menschlichen genomischen Sequenz, sondern experimentell verifiziert regulatorischen Regionen von mehreren Linien der Untersuchung, wie ENCODE und ENSEMBL8erworben. Trotz seiner breite Verwendung in menschlichen Methylomic Untersuchungen gibt es eine ähnliche Plattform für Modell Tiere nicht. Zusätzlich stellt die Array-basierte Format erhebliche Einschränkung für die Fläche für die Platzierung der Sonde zur Verfügung. In den letzten Jahren wurden Anstrengungen unternommen, die Ziel-Spezifität durch Aufnahme Sonde Design und Hochdurchsatz-Feature von Next Generation Sequencing gewährte zu kombinieren. Ein solches Unternehmen führte Sequenzierung-basierte Anreicherung Zielsystem für die Maus Genom (Maus Methyl-Seq), die zur Identifikation von Gehirn-spezifische oder Glukokortikoid-induzierte Unterschiede in der Methylierung9,10verwendet wurde. Ähnliche Plattformen für andere Modell und nicht-Modell Tiere sind erforderlich, um epigenomischen Forschung in diesen Tieren zu erleichtern.
Hier zeigen wir die Umsetzung dieser neuen Plattform Methylomic Analyse auf die Ratte leiten. Die Ratte diente als wichtige Tiermodell in der Pharmakologie, Metabolismus, Neuroendokrinologie und Verhalten. Zum Beispiel gibt es ein zunehmender Bedarf an die zugrunde liegenden Mechanismen zu verstehen, die Anlass zu Medikamententoxizität, Übergewicht, Stress-Reaktion oder Drogenabhängigkeit. Eine Hochdurchsatz-Plattform für den Fang von Methylomic Veränderungen im Zusammenhang mit diesen Bedingungen würde unser Verständnis der Mechanismen erhöhen. Da die Ratte Genom Anmerkung für regulatorischen Regionen immer noch fehlt, wir aufgenommen, nicht-redundanten Promotoren, CpG Inseln, Insel Ufer11, und zuvor identifizierten GC-reichen Sequenzen in der Ratte Methyl-Seq-Plattform12.
Um erfolgreiche Gestaltung und Umsetzung der SureSelect Ziel Bereicherung (allgemein bezeichnet als Methyl-Seq) Plattform für die Ratte Genom zu beurteilen, beschäftigten wir einem Rattenmodell Variable Dauerstress (CVS)13 , differentiell methylierte zu identifizieren Regionen zwischen unbetonten und gestressten Tieren. Unsere Plattformentwurf, Protokoll und Implementierung können nützlich für Ermittler, die wünschen können, um eine umfassende und objektive epigenetische Untersuchung auf den Organismus durchzuführen, deren genomischen Sequenz steht bereits aber bleibt schlecht kommentierte.
Protokoll
Alle Experimente wurden in Übereinstimmung und die Einhaltung aller relevanten rechtlichen und institutionellen Richtlinien, einschließlich der institutionellen Animal Care und Use Committee an der Johns Hopkins School of Medicine abgeschlossen.
(1) Tiere
- Erhalten Sie männliche Jugendliche Sprague-Dawley Ratten im Alter von 4 Wochen. Die Tiere in Polycarbonat Ratte Käfigen in einer Temperatur-Feuchte-kontrollierten Raum auf einer 12 h, 12 h dunkel Lichtzyklus mit leichten Beginn um 0600 h zu den Tieren mit Ad Libitum Zugang zu Wasser.
- Lassen Sie Ratten für 1 Woche zur Verringerung der Belastung, die mit der Beförderung verbundenen akklimatisieren. Paar-Haus unterstreichen die Tiere (N = 16) Isolierung Stress entgegen, und im Alter von 5 Wochen, beginnen die chronische Variable (CVS) Therapie für 3 Wochen.
2. Variable Dauerstress
- Verwalten Sie die CVS-Therapie einmal morgens (9-11:00) und einmal am Nachmittag (1 – 15:00) zu unregelmäßigen Zeiten, die Routine unberechenbar zu halten. Über Nacht milde Stressoren zu integrieren. Die CVS-Therapie umfasst: (1) 3 h in einem Zylinder Zurückhaltung; (2) 10 min. schwimmen; (3) 3 h Käfig kippen 4) 1 h langsam schütteln Plattform; und 5) 1 h in den 4 ° C kalten Raum.
Hinweis: Über Nacht Stressoren gehören soziale Verdrängung (5 pro Käfig), soziale Isolation, feuchte Einstreu, Essen Einschränkung und Lichter auf. Ein typischer Wochenplan der Stress-Therapie ist in Tabelle 1zur Verfügung gestellt.
3. endokrine Assays
-
Bestimmen, Ebenen von Corticosteron (CORT) mit Schweif Blut (~ 50 mL) Verkostungen gesammelt zur gleichen Zeit (09:00) zweimal pro Woche in das Experiment vor dem CVS-Schema zur Grundlinie Hormonspiegel (Tag 0), einmal in der Mitte der wöchentlichen Lebensläufe zu etablieren (Tage 4.11 und 18), alle 7 Tage von CVS (Tage 7 und 14), und beim Abschluss von CVS (Tag 21). Blutproben vor Stress-Tagesablauf zu sammeln.
- Sammeln Sie eine letzte Stamm Blutprobe während der Euthanasie (Tag 25) für RIA und genomische DNA-Extraktion.
- Zentrifugieren Sie alle Blutproben (600 X g, 4 ° C, 10 min), um das Plasma von den Blutzellen zu trennen. Pipettieren aus dem Plasma (überstand) und speichern die Proben bei-80 ° C.
- Tauen Sie auf und verwenden Sie das Plasma, um CORT Ebenen durch Radioimmunoassay (RIA) zu bestimmen. Stellen Sie sicher, dass die 3-Wochen-Plasmaspiegel CORT in den gestressten Tieren zu überprüfen, die Robustheit der Stress-Therapie erhöht sind.
4. Verhalten
- Nach der CVS-Therapie (Tage 23 – 24), bewerten jedes Tier für Angst-ähnliches Verhalten auf die erhöhte plus Labyrinth (EPM)14.
- Mit einer Videokamera, Aufzeichnung der Tiere auf der EPM-Apparat für 300 s und Gäste die Zeit in der Mitte verbrachten, Arme geschlossen und offenen Armen.
5. Gestaltung der Ratte Methyl-Seq
- Mithilfe der UCSC-Genom-Browsers erhalten Sie nicht-redundanten genomische Koordinaten (Ratte Nov 2004 rn4 Montage) für CpG Inseln und Küsten (± 1 kb flankierende CpG Inseln), Förderer (± 1 kb pro TSS) jedes RefSeq-gen und andere Sequenzen, die möglicherweise ab einschlägige Literatur.
Hinweis: Für die Ratte Methyl-Seq, zusätzliche GC-reichen Sequenzen von einer vorherigen Methylierung Array-basierte Plattform war hinzugefügt12. Für Regionen größer als 5 Kbit/s wurden wechselnde Regionen von 500 Basispunkten gesampelt, gefolgt von 1 KBit/s, die übersprungen wurden. Die letzte Ratte-Methyl-Seq-Design besteht aus 111 Mbit/s, 2,3 Millionen Verbrauchsgüter; und eine durchschnittliche Regionsgröße 594 BPS. Es richtet sich an 228.800 einzigartige Loci. - Geben Sie eine kompilierte Liste von genomischen Koordinaten in einem handelsüblichen Ziel erfassen-Design-Software für geeignete Sonde Design.
6. Aufbau der Ratte-Methyl-Seq-Bibliothek aus genomischer DNA
Hinweis: Um Batch-Effekte zu vermeiden, mehrere Proben gleichzeitig zu verarbeiten, sondern die master-Mixes entsprechend skalieren. Extrahieren Sie DNA mit einem im Handel erhältlichen DNA Extraktion Kit. Spalte oder Niederschlag-basierte Methoden beide ergeben qualitativ hochwertige genomischen DNA (260/280 Verhältnis ~ 1,8). Phenol-basierte Methoden sind nicht empfohlen. Eluieren Sie oder Aufschwemmen Sie DNA in Low-TE-Puffer (10 mM TE, 0, 1 mM EDTA, pH 8,0).
- Vorbereitung der Probe
Hinweis: Für jeden Schritt mit magnetischen Beads DNA-Bindung, sicherzustellen Sie, dass die Perlen sind gewöhnt, mindestens 30 min bei Raumtemperatur und vor Gebrauch gut vermischt.- Scher DNA
- Verwenden Sie ein Fluorometer um doppelsträngige DNA Ausgangskonzentration von jeder Probe zu bestimmen. Verdünnen Sie > 1 µg gDNA zu 50 µL mit Low-TE-Puffer (10 mM TE, 0, 1 mM EDTA, pH 8,0) in niedrigen DNA-bindende Mikrozentrifugenröhrchen.
- Proben mit einem isothermen Sonikator Scheren (10 % Duty Cycle, 5 Intensität, 200 Zyklen pro Burst, 6 Zyklen von 60 s, Frequenz fegen, 4 ° C).
- Beurteilen Sie die Qualität der DNA mit Hilfe einer Elektrophorese-basiertes System, das DNA-Größe und Menge misst.
Hinweis: Die DNA-Menge empfohlen ist 1 µg oder 3 µg. Es gibt begrenzte Ausgangsmaterial, sollte die niedrigste Eingangsbetrag > 500 ng, als niedrigere Beträge beeinträchtigt die Quantität und Qualität der Bibliotheken generiert.
- Enden der DNA zu reparieren.
- Verwenden Sie die Ratte Methyl-Seq-Kit um die Ende-Reparatur Master-Mix auf dem Eis vorzubereiten. Jede Probe 52 µL der Mischung hinzu und brüten in einem Thermocycler ohne beheizte Deckel (20 ° C für 30 min, 4 ° C halten).
Ende-Reparatur Master Mix (je Probe):
35,2 µL Wasser
10 µL Ende Reparatur Puffer (10 X)
1.6 µL dNTP-Mix
1 µL T4-DNA-Polymerase
2 µL Klenow-DNA-Polymerase
2.2 µL T4-Polynukleotid-Kinase - Proben mit 180 µL DNA-Bindung magnetische Beads und 400 µL frisch zubereitete 70 % Ethanol pro Probe zu reinigen. Jede Probe 180 µL Perlen hinzufügen und 5 min bei Raumtemperatur inkubieren. Pellet-Perlen, überstand zu entfernen und Aufschwemmen Pellet in 200 µL 70 % Ethanol. Ethanol zu entfernen und Waschen einmal wiederholen.
- Verwenden Sie eine Magnetplatte, pellet-Perlen und so viel Ethanol als möglich zu entfernen. In einem 37 ° C trocken Heatblock für 3 – 5 min bis die Perle Pellet vollständig trocknen. In 44 µL Nuklease-freies Wasser Aufschwemmen und ca. 42 µL des Überstandes zu sammeln.
Haltepunkt: Nach Ablauf der Reparatur von DNA Proben können versiegelt und bei-20 ° c gelagert
- Verwenden Sie die Ratte Methyl-Seq-Kit um die Ende-Reparatur Master-Mix auf dem Eis vorzubereiten. Jede Probe 52 µL der Mischung hinzu und brüten in einem Thermocycler ohne beheizte Deckel (20 ° C für 30 min, 4 ° C halten).
- Adenylat 3' endet.
- Adenylation Master-Mix auf dem Eis zubereiten. Jede Probe 9 µL Mischung hinzu und brüten in einem Thermocycler ohne beheizte Deckel (37 ° C für 30 min, 4 ° C halten).
Adenylation Master Mix (je Probe):
5 µL Klenow Puffer
1 µL dATP
3 µL Klenow-DNA-Polymerase - Proben mit 90 µL DNA-Bindung magnetische Beads und 400 µL frisch zubereitete 70 % Ethanol pro Probe zu reinigen. Jede Probe 90 µL Perlen hinzufügen und 5 min bei Raumtemperatur inkubieren. Pellet-Perlen, überstand zu entfernen und Aufschwemmen Pellet in 200 µL 70 % Ethanol. Ethanol zu entfernen und Waschen einmal wiederholen.
- Verwenden Sie eine Magnetplatte, pellet-Perlen und so viel Ethanol als möglich zu entfernen. In einem 37 ° C trocken Heatblock für 3 – 5 min bis die Perle Pellet vollständig trocknen. In 35 µL Nuklease-freies Wasser Aufschwemmen und ca. 33,5 µL des Überstandes zu sammeln.
- Adenylation Master-Mix auf dem Eis zubereiten. Jede Probe 9 µL Mischung hinzu und brüten in einem Thermocycler ohne beheizte Deckel (37 ° C für 30 min, 4 ° C halten).
- Verbinden Sie die methylierten Adapter.
- Bereiten Sie Ligation Master Mix auf Eis und jede Probe fügen Sie 16,5 µL der Mischung hinzu. Brüten Sie in einem Thermocycler ohne beheizte Deckel (20 ° C für 15 min, 4 ° C halten).
Ligatur Master Mix (je Probe):
2.5 µL Wasser
2.5 µL Methyl-Seq methyliert Adapter
10 µL T4 DNA-Ligase Puffer (5 X)
1,5 µL T4 DNA-Ligase - Proben mit 90 µL DNA-Bindung magnetische Beads und 400 µL frisch zubereitete 70 % Ethanol pro Probe zu reinigen. Jede Probe 90 µL Perlen hinzufügen und 5 min bei Raumtemperatur inkubieren. Pellet-Perlen, überstand zu entfernen und Aufschwemmen Pellet in 200 µL 70 % Ethanol. Ethanol zu entfernen und Waschen einmal wiederholen.
- Verwenden Sie eine Magnetplatte, pellet-Perlen und so viel Ethanol als möglich zu entfernen. In einem 37 ° C trocken Heatblock für 3 – 5 min bis die Perle Pellet vollständig trocknen. In 22 µL Nuklease-freies Wasser Aufschwemmen und ca. 22 µL des Überstandes zu sammeln. Bewerten Sie Qualität mit einem Bioanalyzer.
Hinweis: Wenn der Gesamtbetrag der DNA weniger als 500 ng, scher- und Prozess zusätzliche DNA bevor Sie die nachfolgenden Schritte ausführen ist. Wenn die durchschnittliche Größe der DNA nicht von mehr als 30 bps erhöht, prüfen Sie, um sicherzustellen, dass die Reagenzien sind neu, als T4-DNA-Polymerase, Klenow und/oder T4 Ligase kann alt sein.
Haltepunkt: Nach Ligation methylierte Adapter, Proben können versiegelt und bei-20 ° c gelagert
- Bereiten Sie Ligation Master Mix auf Eis und jede Probe fügen Sie 16,5 µL der Mischung hinzu. Brüten Sie in einem Thermocycler ohne beheizte Deckel (20 ° C für 15 min, 4 ° C halten).
- Scher DNA
- Hybridisierung
- Übertragen Sie Proben auf niedrigen DNA-bindende Mikrozentrifugenröhrchen und verwenden Sie einen beheizte Vakuum Konzentrator auf weniger als 3,4 µL. Stellen Proben zu 3,4 µL Probenvolumen zu reduzieren.
Hinweis: Konzentrat verdampft Proben etwa ~ 3 µL um sicherzustellen, dass Proben aus Vakuum Konzentrator vor alle Flüssigkeit entfernt werden. - Hybridisierung Puffer bei Raumtemperatur und Methyl-Seq-Block-Mix auf dem Eis zubereiten. Jede Probe fügen Sie 5,6 µL Methyl-Seq-Block-Mischung hinzu und inkubieren Sie im Thermocycler (95 ° C für 5 min, 65 ° C für 2 min, 65 ° C halten).
Hybridisierung-Puffer (je Probe):
6,63 µL Methyl-Seq Hyb 1
0,27 µL Methyl-Seq Hyb 2
2,65 µL Methyl-Seq Hyb 3
3.45 µL Methyl-Seq Hyb 4
Methyl-Seq-Block-Mix (je Probe):
2.5 µL Methyl-Seq Indizierung Block 1
2.5 µL Methyl-Seq-Block 2
0,6 µL Methyl-Seq-Block 3 - RNase-Block-Mix und erfassen Bibliothek Hybridisierung Mischung vorbereiten. Jede Probe 20 µL erfassen Bibliothek Hybridisierung Mix hinzu und bei 65 ° C für mindestens 16 h inkubieren.
RNase-Block-Mix (je Probe):
0,5 µL RNase-Block
1,5 µL Wasser
Bibliothek-Hybridisierung-Mix (je Probe) zu erfassen:
13 µL Hybridisierung Puffer
2 µL RNase-Block-Mix
5 µL der Ratte Methyl-Seq erfassen Bibliothek
Hinweis: Beim Hinzufügen von Hybridisierung Mix um unspezifische Schwergängigkeit zu verhindern Reaktionen bei 65 ° C zu halten . - Aliquoten 50 µL Streptavidin magnetische Beads pro Probe in einen neuen Schlauch 8-Well Streifen. Perlen mit 200 µL Methyl-Seq Binding Buffer zu waschen. Mit Magnetplatte pellet-Perlen und nehmen überstand zwischen jedem Waschgang für insgesamt 3 Wäschen. Aufschwemmen Sie nach der letzten Wäsche Streptavidin Perlen in 200 µL Methyl-Seq Binding Buffer.
- 200 µL der gewaschenen Streptavidin magnetische Beads Proben hinzu und Inkubation bei Raumtemperatur für 30 min mit einem rotierenden Mixer. Beim Mischen, aliquoten 200 µL von Methyl-Seq Wash Buffer 2 in dreifacher Ausfertigung Vertiefungen einer 96-Well-Platte pro Sample und Ort in einem Thermocycler bis zu 65 ° c Vorwärmen
- Nach der Inkubation pellet Streptavidin magnetische Beads mit Magnetplatte und die Perlen in 200 µL Aufschwemmen Methyl-Seq Wash Buffer 1. 15 min bei Raumtemperatur inkubieren. Verwenden Sie eine Magnetplatte pellet und überstand verwerfen.
- Waschen Sie Perlen 3 Mal mit Methyl-Seq waschen Puffer 2: Aufschwemmen Wulst Pellet in 200 µL waschen Puffer 2 (vorgewärmt in Schritt 6.2.5.) und inkubieren Sie Perlen im Thermocycler (65 ° C, 10 min) pellet-Perlen. Entsorgen Sie überstand nach jedem Waschen mit einer magnetischen Platte.
Hinweis: Pflegen Sie Hybridisierung Reaktionen bei 65 ° C, beim Hinzufügen von Wash Buffer 2 um unspezifische Schwergängigkeit zu verhindern. - Die gewaschenen Perlen 20 µL Methyl-Seq Elution Buffer hinzu und Inkubation bei Raumtemperatur für 20 min. Einsatz eine Magnetplatte, Pellet-Perlen und Übertragung auf einen neuen Streifen Schlauch überstand. Entsorgen Sie die Perlen.
Hinweis: Während der Inkubation, bereiten Sie Bisulfit-Konvertierung-Reagenz.
- Übertragen Sie Proben auf niedrigen DNA-bindende Mikrozentrifugenröhrchen und verwenden Sie einen beheizte Vakuum Konzentrator auf weniger als 3,4 µL. Stellen Proben zu 3,4 µL Probenvolumen zu reduzieren.
- Bisulfit-Konvertierung
Hinweis: Führen Sie Bisulfit Konvertierung von der eluierten SsDNA mit entsprechenden Reagenzien und Anweisungen von einem handelsüblichen Bisulfit-Umbausatz.- Überstand 130 µL bereit Bisulfit Konvertierung Reagenz aus vorherigen Schritt hinzufügen. Teilen Sie jede der 150 µL Reaktionen gleich zwei Brunnen. Brüten Sie in einem Thermocycler (64 ° C für 2,5 h, 4 ° C halten).
Hinweis: Die 150 µL Reaktion gliedert sich gleich in zwei eigene Brunnen, homogene Temperatur zu gewährleisten. Nach Inkubation für 2,5 h, sofort mit dem nächsten Schritt fortfahren. - Proben zu Spalten durch Zugabe von 600 µL Binding Buffer zu drehen und einmal mit 100 µL Waschpuffer waschen zu binden. Spalten (15.000 X g, 1 min) zwischen allen Bisulfit Konvertierungsschritte Zentrifugieren und Durchströmung zu verwerfen.
- Desulphonate Proben durch Zugabe von 200 µL Desulphonation-Puffer zu Spalten. Inkubation bei Raumtemperatur für 15-20 min. wiederholen Zentrifugation und Durchströmung zu verwerfen.
- Waschen Sie Spalten zweimal mit 200 µL Puffer waschen. Eluieren Sie jede Probe durch Zugabe von 10 µL der Elution Buffer auf die Spalte, Inkubation für 3 min bei Raumtemperatur und Zentrifugieren (15.000 X g, 1 min). Wiederholen Sie Schritt, Elution für eine Gesamtmenge von 20 µL.
- PCR-Reaktion Master Mix 1 auf Eis zubereiten. Jede Probe 82 µL der Mischung hinzufügen. Brüten Sie in einem Thermocycler mit dem folgenden Programm.
PCR-Reaktion Master Mix 1 (pro Probe):
30 µL Wasser
50 µL Methyl-Seq-PCR-Master-Mix
1 µL Methyl-Seq PCR1 Grundierung F
1 µL der Methyl-Seq PCR1 Primer R
Thermocycler-Programm:
Stufe 1, 1 Zyklus: 95 ° C 2 min
Stufe 2, 8 Zyklen: 95 ° C 30 s, 60 ° C 30 s, 72 ° C 30 s
Stufe 3, 1 Zyklus: 72 ° C 7 min
Stufe 4, 1 Zyklus: 4 ° C halten - Proben mit 180 µL DNA-Bindung magnetische Beads und 400 µL frisch zubereitete 70 % Ethanol pro Probe zu reinigen. Jede Probe 180 µL Perlen hinzufügen und 5 min bei Raumtemperatur inkubieren. Pellet-Perlen, überstand zu entfernen und Aufschwemmen Pellet in 200 µL 70 % Ethanol. Ethanol zu entfernen und Waschen einmal wiederholen.
- Verwenden Sie eine Magnetplatte, pellet-Perlen und so viel Ethanol als möglich zu entfernen. In einem 37 ° C trocken Heatblock für 3 – 5 min bis die Perle Pellet vollständig trocknen. In 21 µL Nuklease-freies Wasser Aufschwemmen und ca. 19,5 µL des Überstandes zu sammeln.
- Überstand 130 µL bereit Bisulfit Konvertierung Reagenz aus vorherigen Schritt hinzufügen. Teilen Sie jede der 150 µL Reaktionen gleich zwei Brunnen. Brüten Sie in einem Thermocycler (64 ° C für 2,5 h, 4 ° C halten).
- Indizierung
- PCR-Reaktion Master Mix 2 auf Eis zubereiten. Hinzufügen von 25,5 µL Master Mix 2 zu jeder Probe. Fügen Sie 5 µL kommerzielle Indizierung Primer an einzelne Samples und brüten in einem Thermocycler.
PCR-Reaktion Master Mix 2 (je Probe):
25 µL Methyl-Seq PCR Master Mix
0,5 µL Methyl-Seq gemeinsame Indizierung Primer
Thermocycler-Programm:
Stufe 1, 1 Zyklus: 95 ° C 2 min
Stufe 2, 6 Zyklen: 95 ° C 30 s, 60 ° C 30 s, 72 ° C 30 s
Stufe 3, 1 Zyklus: 72 ° C 7 min
Stufe 4, 1 Zyklus: 4 ° C halten
Hinweis: Zusätzliche Zyklen (2-3) möglicherweise erforderlich, wenn der Start DNA-Konzentration unterhalb der empfohlenen Werte. - Proben mit 90 µL DNA-Bindung magnetische Beads und 400 µL frisch zubereitete 70 % Ethanol pro Probe zu reinigen. Jede Probe 90 µL Perlen hinzufügen und 5 min bei Raumtemperatur inkubieren. Pellet-Perlen, überstand zu entfernen und Aufschwemmen Pellet in 200 µL 70 % Ethanol. Ethanol zu entfernen und Waschen einmal wiederholen.
- Verwenden Sie eine Magnetplatte, pellet-Perlen und so viel Ethanol als möglich zu entfernen. In einem 37 ° C trocken Heatblock für 3 – 5 min bis die Perle Pellet vollständig trocknen. In 24 µL Nuklease-freies Wasser Aufschwemmen und ca. 24 µL des Überstandes zu sammeln.
- Konzentration und bp-Größe mit hoher Empfindlichkeit DNA-Detektion Reagenzien auf einem Bioanalyzer zu beurteilen.
Hinweis: Wenn die Bioanalyzer nicht das Vorhandensein der Bibliothek DNA erkennt, wiederholen Sie die Vorbereitungsschritte mit zusätzlichen DNA.
Haltepunkt: nach der Reinigung, indizierte Proben versiegelt sein und bei-20 ° c gelagert - Bündelung von Proben für die entsprechenden Next Generation Sequencing-Plattform verwendet.
- Mit der Konzentrationsdaten aus Bioanalyzer, welche DNA Molarity basierend auf Bibliotheksgröße und Menge in einem bestimmten Volumen bestimmt, mit Low-TE-Puffer (6.1.1.1) verdünnen und kombinieren alle Proben, eine Endkonzentration von 15 Uhr.
Hinweis: Eine empfindlichere Methode zur Quantifizierung der Bibliothek ist durch quantitative Real-Time PCR Zündkapseln, die auf die aufgespaltenen Adapter verwenden. - Die Anzahl der Fahrspuren, die ausreichend für 4 Proben pro Bahn auf einer nächsten Generation Sequenzer weiterlaufen Sie Mischproben.
Hinweis: zum Beispiel wenn 16 Bibliothek Proben eindeutig indiziert und kombiniert haben, führen Sie die Bibliotheken über 4 Bahnen, 4 Proben pro Bahn entspricht.
- Mit der Konzentrationsdaten aus Bioanalyzer, welche DNA Molarity basierend auf Bibliotheksgröße und Menge in einem bestimmten Volumen bestimmt, mit Low-TE-Puffer (6.1.1.1) verdünnen und kombinieren alle Proben, eine Endkonzentration von 15 Uhr.
- PCR-Reaktion Master Mix 2 auf Eis zubereiten. Hinzufügen von 25,5 µL Master Mix 2 zu jeder Probe. Fügen Sie 5 µL kommerzielle Indizierung Primer an einzelne Samples und brüten in einem Thermocycler.
(7) die Sequenzierung auf einem Next-Generation-Sequenzer
- Senden Sie die Proben bis zum institutionellen Sequenzierung Kern für das clustering der Methyl-Seq-Bibliothek, gefolgt von Sequenzierung auf einem Computer Sequenzierung der nächsten Generation.
8. Analyse zur Ermittlung von DMRs
- Implementieren Sie Bismark15, die Fliege 2.0 als eine interne Reihenfolge Aligner16,17, aufruft, um raw input Lesezugriffe auf Bisulfit umgewandelt, Plus-Strang Genom auszurichten. Verwenden Sie nach Ausrichtung die Bismark_methylation_extractor Qualitätskontrolle durchführen und jedes CpG einen geschätzte Methylierung-Wert zuweisen.
- Erzeugt eine Liste von DMRs mit dem BS-Seq-Paket18 Bioconductor. Filter der DMRs basierend auf mit mehr als 3 aufeinanderfolgenden Verbrauchsgüter und P-Wert < 0,05.
Hinweis: Erzeugen eine DMR Liste mit genomischen Koordinaten, Entfernung zum nächstgelegenen RefSeq gen, Anzahl der Verbrauchsgüter innerhalb jedes DMR, durchschnittliche % CpG Methylierung Wert über den DMR für die zwei Vergleichsgruppen (z. B. vs. unbetont betont), der P-Wert, und der FDR (false Discovery Rate) Wert. Verwenden Sie die DMR-Liste, dh., genomische Koordinaten Pyrosequenzierung Primer für die Validierung zu entwerfen.
(9) Validierung von Bisulfit Pyrosequenzierung
-
Besser gekleideteres Design
- Primer für Bisulfit PCR und Pyrosequenzierung Design. Entwerfen Sie zwei Sätze von PCR-Primer (außen und verschachtelte), sodass die verschachtelte PCR 150 – 400 bps ein DMR verstärken wird.
Hinweis: im allgemeinen gestaltete Primer sind mindestens 24 Basen lang mit mindestens 4 – 5 nichtkonsekutiver G (C für die rückwärts-Primer) auf Konto für Ausglühen Temperatur aus dem Verlust von Sequenz Komplexität reduziert. Der geschachtelte Primer werden Biotin beschriftet und HPLC gereinigt. Jedoch sollte standard Primer den PCR Schritt zu optimieren, indem Sie die Reaktionen auf ein Agarosegel zu lösen als erster bestellt werden.- Die Pyrosequenzierung assay Grundierung, so dass es richtet sich an die komplementäre biotinylierte Design strand nur 1 – 2 Basen stromaufwärts von der Verbrauchsgüter, untersucht werden. Gestalten Sie mehrere Pyrosequenzierung Primer bei Bedarf, wie jede Pyrosequenzierung Grundierung zuverlässig 30 bps flussabwärts assay kann.
- Verwenden Sie für die Rt1-m4 Folgendes:
rRT1M4 draußen – F TGTAYGATTTTGGTTATYGTAAAT
rRT1M4 draußen – R AACTTACAAATTTCACCAACTCA
rRT1M4 verschachtelt – F GTGGGTTAYGTGGATAATATATAG
rRT1M4 Nested-R AATCACTTACCATTCTCTCTCTAACTA
rRT1M4 Pyro1 TAYGTGGATAATATATAGAT
rRT1M4 Pyro2 GATAGTTATTTGGYGAGTTAG
rRT1M4 Pyro3 GAGTATTTGGAGGAGTTGAT
rRT1M4 Pyro4 GGATTTTAATATTTGGT
- Primer für Bisulfit PCR und Pyrosequenzierung Design. Entwerfen Sie zwei Sätze von PCR-Primer (außen und verschachtelte), sodass die verschachtelte PCR 150 – 400 bps ein DMR verstärken wird.
-
Verwenden Sie eine handelsübliches Kit für Bisulfit Umwandlung der Ratte Blut gDNA.
Hinweis: Die Bisulfit Umwandlungsschritte angepasst wurden aus dem kommerziell erhältlichen Kit mit folgenden Änderungen: In Schritt 1, 50 – 100 ng von Blut gDNA hinzufügen und bis 20 µL mit Wasser verdünnen. Eluieren Sie in Schritt 9 20 µL pro Probe.- Bisulfit-Konvertierung-Reagenz laut Protokoll des Herstellers vorbereiten und mit verdünnten gDNA kombinieren. Brüten Sie im Thermocycler (64 ° C für 2,5 h, 4 ° C halten).
- Binding Buffer zu konvertierten gDNA in Spin Spalten und Zentrifuge (15.000 X g, 1 min) hinzufügen. Waschen Spalten einmal dann Desulphonation Puffer zu Spalten hinzufügen und 15 Minuten bei Raumtemperatur inkubieren. Zentrifuge (15.000 X g, 1 min).
- Waschen Sie die Spalte mit waschen-Puffer und Zentrifuge (15.000 X g, 1 min). Wiederholen Sie Waschschritt mit Zentrifugation (15.000 X g, 2 min). Fügen Sie 20 µL Elution Buffer und Zentrifuge (15.000 X g, 1 min) eluieren.
-
PCR-Amplifikation
- Bereiten Sie außen PCR Master Mix. 3,5 µL Bisulfit-konvertiert gDNA 21,5 µL Master Mix hinzu und führen Sie Thermocycler-Programm aus.
Äußeren PCR-Master-Mix:
16,25 µL Wasser
2.5 µL Polymerase-Puffer [10 X]
0,5 µL dNTP [10 mM]
1 µL Forward Primer [0,1 µM]
1 µL der rückwärts-Primer [0,1 µM]
0,25 µL Taq-DNA-Polymerase [5000 U/mL].
Thermocycler-Programm:
Stufe 1, 1 Zyklus: 94 ° C 4 min.
Stufe 2, 47 Zyklen: 94 ° C 1 min, 53 ° C 30 s, 72 ° C 1 min.
Stufe 3, 1 Zyklus: 72 ° C 8 min, 4 ° C halten - Bereiten Sie Nested PCR Master Mix. 2 µL der Probe vom äußeren PCR fügen Sie 23 µL Master Mix hinzu und wiederholen Sie das äußere PCR-Thermocycler-Programm. PCR-Produkt-Qualität durch Gelelektrophorese (1 X TAE Puffer, 1 % Agarosegel) zu beurteilen.
Nested PCR Master Mix:
17.75 µL Wasser
2.5 µL Polymerase-Puffer [10 X]
0,5 µL dNTP [10 mM]
1 µL Forward Primer [0,1 µM]
1 µL der rückwärts-Primer [0,1 µM]
0,25 µL Taq-DNA-Polymerase [5000 U/mL]
Hinweis: Für die verschachtelten PCR, muss entweder die vorwärts- oder rückwärts-Primer biotinylierte sein.
- Bereiten Sie außen PCR Master Mix. 3,5 µL Bisulfit-konvertiert gDNA 21,5 µL Master Mix hinzu und führen Sie Thermocycler-Programm aus.
-
Pyrosequenzierung
- Machen Sie einen master-Mix mit 38 µL Binding Buffer, 35 µL Wasser und 2 µL Streptavidin beschichteten Sepharose-Kügelchen pro Probe. Fügen Sie in einer 96-Well-Platte 75 µL des master-Mix und 5 µL des verschachtelten PCR-Produkt hinzu. Auf einer Platte Shaker für 15-60 min schütteln.
- Fügen Sie unter Schütteln 12 µL des Primers (0,5 µM, in verdünnter Glühen Puffer hinzu) in die Vertiefungen der Pyrosequenzierung Assay Platte.
- Nach dem Schütteln, Schritte waschen verbindliche Reaktion waschen Puffer verwenden. Vakuum Werkzeug im Trog mit Wasser gefüllt, dann Sammle Proben von Platte. Tauchen Sie ein Vakuum Werkzeug in halb gefüllte Tröge mit 70 % Ethanol, NaOH (0,2 M) und Tris-Acetat-Puffer (10 mM, pH 7.4). Trennen Sie vom Ort und Vakuum Vakuum Werkzeug in HS-Assay-Platte, Perlen zu übertragen.
- Heizblock legen Sie Platte auf und inkubieren Sie bei 80 ° C für 2 min. zulassen Platte für 5 min abkühlen lassen anschließend Pyro-Programm zu beginnen.
Ergebnisse
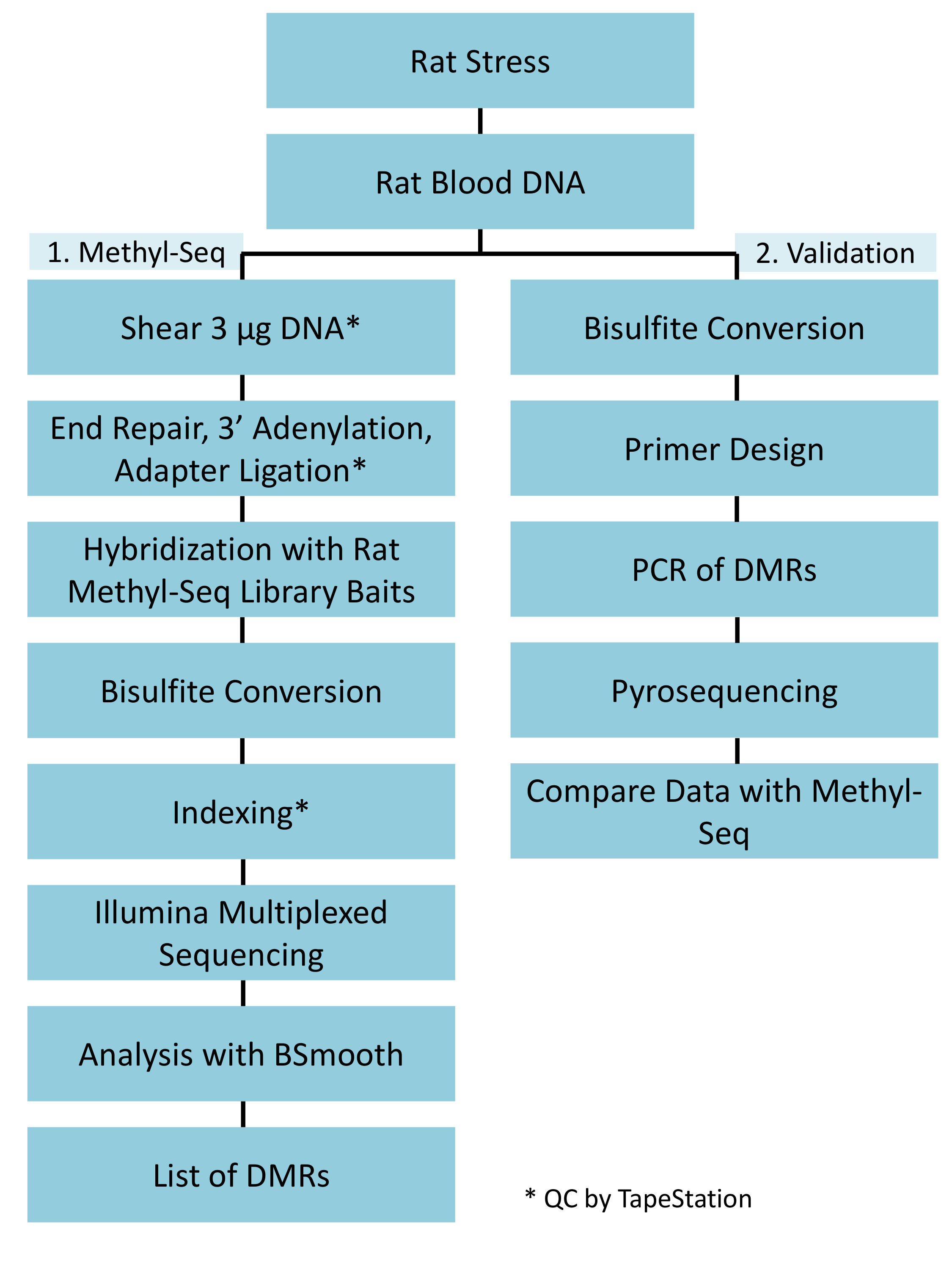
Eine erfolgreiche Umsetzung der Ratte-Methyl-Seq-Plattform richtet sich nach verschiedenen Kriterien. Abbildung 1 zeigt den gesamten Arbeitsablauf der Studie und hebt bestimmte Qualitätskontrolle (QC) Schritte, die erforderlich sind, bevor voran. Eines der ersten Faktoren zu berücksichtigen, ist die Robustheit des Tiermodell und die Stress-Therapie, die das Ausmaß der epigenetischen Veränderungen bestimmen, die über die Methylome auftreten. Da unsere tierische Arbeit auf unsere früheren Beobachtung das Corticosteron (CORT) Exposition zu Veränderungen der DNA-Methylierung19,20führen ausgesagt wird kann, mussten unsere Variable Dauerstress (CVS) Therapie der ausreichend strenge zu produzieren Ratten mit erhöhter Plasmaspiegel CORT betont. Eine typische wöchentliche CVS-Therapie ist in Tabelle 1 gezeigt und bestand aus täglichen Stressoren am Morgen, Nachmittag, und über Nacht, die werden ständig geändert, um zu verhindern, dass die Gewöhnung und vermindert Stress-Reaktion. Die 3-Wochen-Therapie, die betonten Tiere ausgestellt erheblich erhöhte Niveaus der mittlere Plasma CORT [4 – 21 Tage kontrollieren: 32,7 3,7 ng/mL, Stress: 103,0 11,9 ng/mL (Mittelwert SEM), P = 2,2 x 10-4, Abbildung 2A] über derjenigen unbetont, Kontrolltieren. Konsequent, zeigte diese Tiere auch größere Angst-ähnliches Verhalten auf die erhöhte plus Labyrinth (EPM), wie durch die deutlich mehr Zeit in den geschlossenen Armen EPM und weniger Zeit in die offenen Arme (Abbildung 2 b). Diese Ergebnisse zeigen, dass die CVS-Exposition zu bedeutenden endokrinen und Verhaltensänderungen führten, führt uns zu untersuchen, ob diese Änderungen bestimmter DNA-Methylierung Signaturen zugeordnet waren.
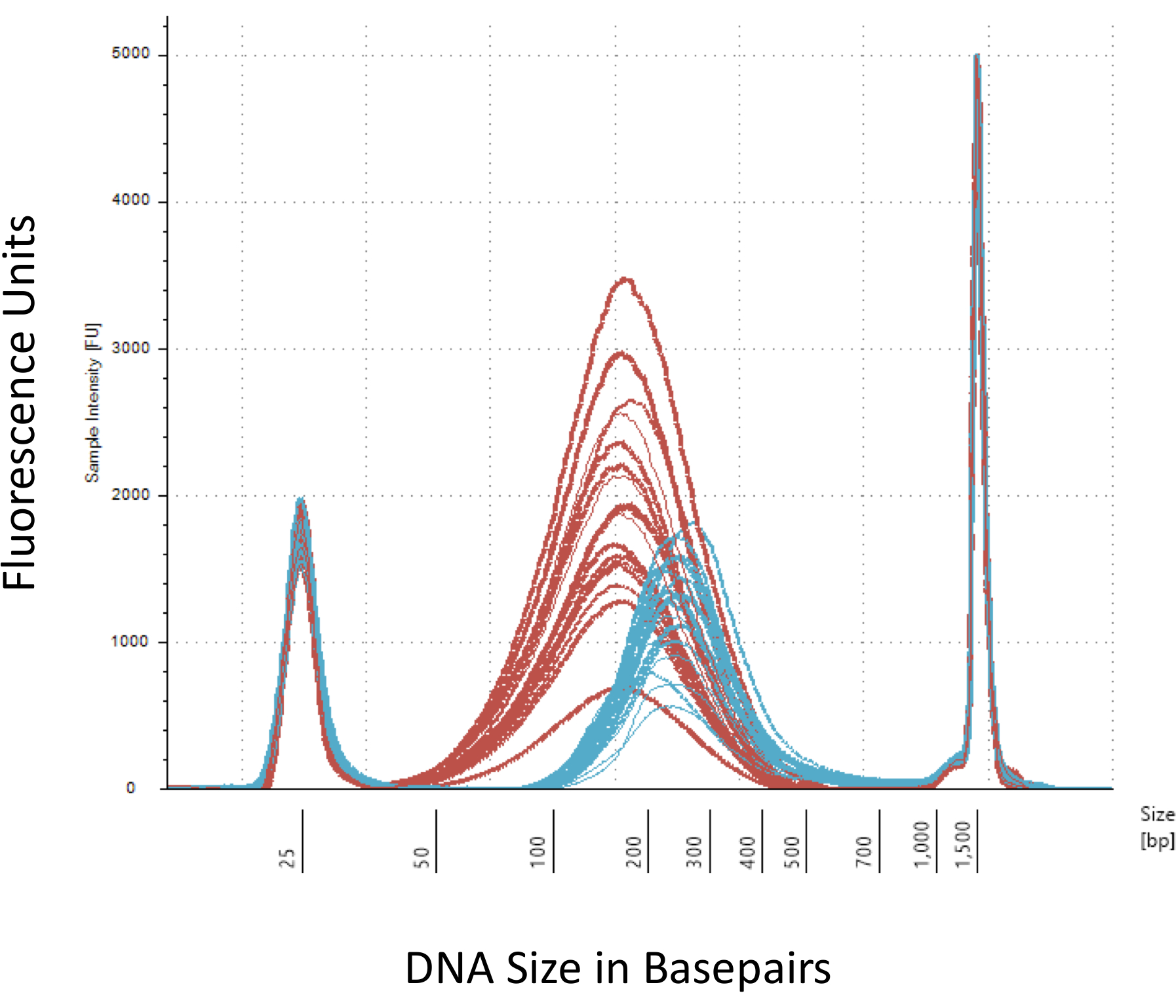
Wir legen Wert auf mehreren Checkpoints, die entscheidend für den erfolgreichen Aufbau der Methyl-Seq-Bibliothek. Beginnend mit einer ausreichenden Menge an DNA ist notwendig, als Beschallung, mehrere Wäsche/Reinigung, Ziel Bereicherung und Bisulfit Konvertierungsschritte sukzessive reduzieren die Menge der DNA in der fertigen Bibliothek. Obwohl mehrere Schritte der PCR-Amplifikation den Verlust der DNA Schablone lindern, können übermäßige PCR Zyklus zahlen höhere doppelte liest einzuführen. Für die aktuelle Studie an Ratten Methyl-Seq wurde 2 g Blut gDNA pro Ratte verwendet. Wir beachten, dass Methyl-Seq Bibliotheken gemacht werden können, mit der Startnummer DNA-Menge so niedrig wie 500 ng. Kleinere Ausgangsmaterial erlaubt Benutzern, Bibliotheken aus DNA isoliert durch FACS (Fluoreszenz-aktivierte Zellsortierung) generieren oder Nadel-Schläge, obwohl es besteht erhöhtes Risiko eine unzureichende Menge an Bibliotheken für die anschließende Sequenzierung zu produzieren. QC erfolgt durch Elektrophorese von 1 L der Probe auf einem Bioanalyzer DNA Molekulargewicht, Menge und Molarity vorsieht. Drei wichtige Schritte, die die Bioanalyzer benötigen sind: 1) nach Beschallung Schritt dafür ausreichend Scheren von DNA (~ 170 bp, rot, Abbildung 3); (2) folgende Adapter Ligatur Schritt angegeben durch eine Verschiebung in der durchschnittlichen Größe der DNA geschoren (~ 200 bp, blau, Abbildung 3) darauf ihre spätere Verstärkung durch PCR; und 3) nach der endgültigen Bibliothek Reinigungsstufe zur Gewährleistung der Menge und Größe der Bibliothek für die Sequenzierung.
Die R-Pakete BSSeq und BSmooth in Bioconductor wurden für die Analyse der Daten18-Sequenzierung Bisulfit verwendet. Dazu gehören Werkzeuge und Methoden für die Ausrichtung der Sequenz liest, Qualitätskontrolle, Durchführung und Identifizierung differentiell methyliert Regionen (DMRs). BSmooth Software ruft Bowtie 2.016,17 als eine interne Reihenfolge Aligner, CpG-Ebene Messung Zusammenfassungen durch Ausrichtung der raw input Lesezugriffe auf Bisulfit-konvertiert Genomsequenzen zu erhalten. Die ausgerichteten liest sind dann durch strenge Qualitätskontrollen gefiltert, die sich bemühen, systematische Sequenzierung und Basis-Aufruf Fehler, die die nachgeschaltete Analysen verzerren können. Eine Reihe von Parzellen werden generiert, um visuell in diesem Prozess der Filterung zu unterstützen. Sequenzierung Metriken entstehen auch auf Dokument relevante Informationen wie Anzahl der ausgerichteten liest, %-Ziel und pro CpG Abdeckung, unter anderem (Tabelle 2). Sobald die Daten gefiltert werden, erfolgt ein Glättung/Normalisierung Algorithmus, wo jeder CpG einen geschätzten Methylierung Wert basierend auf alle QC zugewiesen wird von jeder Probe liest und schätzt aus den benachbarten Verbrauchsgüter um genauere Berufung der Methylierung zu gewährleisten Stand auch in Fällen, in denen die Sequenz Abdeckung niedrig ist. Dieser Wert stellt eine geglättete Schätzung der Wahrscheinlichkeit der Methylierung an jedem Standort CpG. Vergleicht man den Mittelwert der geglätteten Methylierung Schätzungen jeder Probe zwischen den beiden Behandlungsgruppen und Ranking genomische Regionen aus den meisten deutlich anders aus als zuletzt, wird eine Liste der DMRs generiert (Tabelle 3).
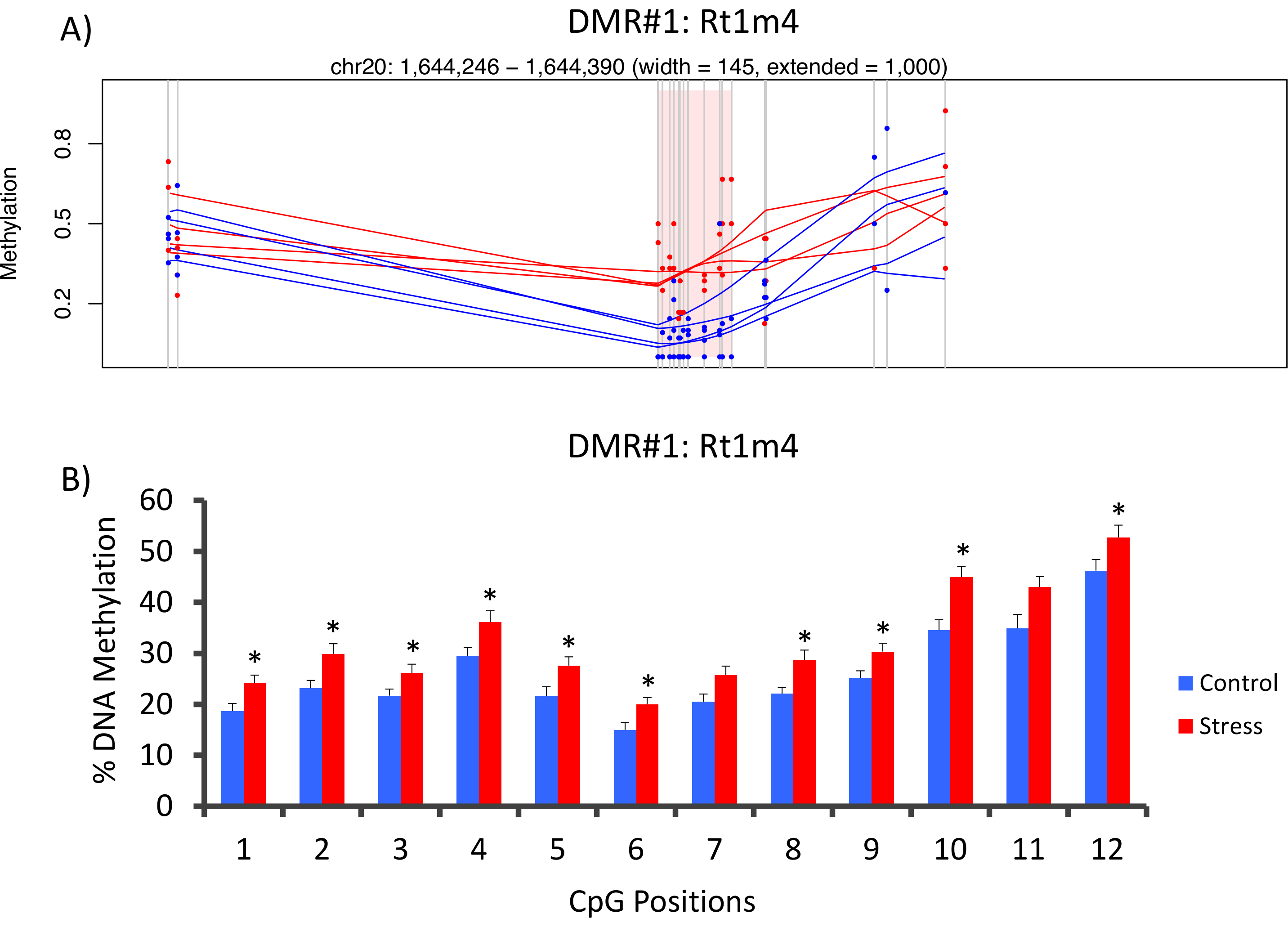
Die Top-DMR zwischen betonten und unbetonten Gruppen in der Promoter des Gens Ratte große Histocompatibility Rt1-m4, mit befand betonte Tiere ausstellen höhere Methylierung über alle Verbrauchsgüter als unbetont Tiere (Abb. 4A). Um die erfolgreiche Umsetzung der Methyl-Seq-Plattform und Analyse der Daten zu bestätigen, wurden Primer gegen die DMR und DNA-Methylierung Blutspiegel in die gesamte Kohorte von betonten und unbetonten Tiere (8 von Methyl-Seq sequenziert und 8 nicht sequenziert) entwickelt. wurden von Bisulfit Pyrosequenzierung bewertet. Ergebnisse zeigen deutliche Steigerung in der DNA-Methylierung in 10 aus den 12 Verbrauchsgüter untersucht (5.1-10.4 Änderung in % Methylierung, P < 0.037, Abbildung 4 b). KEGG Pathway Analyse erfolgte auf allen nominell erhebliche DMRs, Wege, die mit Stress verbunden zu identifizieren. Konsequent, verwickelt Wege DMR-assoziierten Krankheiten mit chronischem Stress ausgesetzt, wie Diabetes, Herz-Kreislauf-Erkrankungen und Krebs (Tabelle 4). 21 , 22 , 23 um einen Zusammenhang zwischen der epigenetischen Daten und den Grad der Exposition zu betonen zu demonstrieren, wurden Methylierung Ebenen am CpG-10 auf das mittlere 3-wöchigen CORT Niveau für jedes Tier verglichen. Die Ergebnisse zeigten eine bescheidene Korrelation zwischen den endokrinen und Methylierung Daten (R2= 0,54, P = 0,001, Abbildung 5).

Abbildung 1: insgesamt schematische Workflow für die Ratte Methyl-Seq-Plattform. 1 g der genomische DNA aus dem Blut extrahiert betont und Kontrolle Ratten wird zuerst verarbeitet, für den Bau der Methyl-Seq-Bibliotheken für Zielkennzeichnung, Sequenzierung und Analyse. Weitere 100 ng DNA wird für unabhängige Validierung der identifizierten epigenetische Ziele von Bisulfit Pyrosequenzierung verwendet. Bitte klicken Sie hier für eine größere Version dieser Figur.
{kind=link}

Abbildung 2: Belastung durch Variable Dauerstress (CVS) führt zu endokrinen und Verhaltensstörungen Veränderungen bei Ratten. (A) mehrere Kostproben von Corticosteron (CORT) zeigen die Robustheit der 3 Woche CVS-Therapie. Blutproben wurden am Morgen vor der täglichen Stress Therapie gesammelt. (B) Stressed Tiere mehr Zeit in den geschlossenen Armen und weniger Zeit in die offenen Arme der die erhöhte plus Labyrinth (EPM). Boxplots mit Datenpunkt für jedes Tier werden angezeigt. Studenten T-Test wurde die statistische Signifikanz uraufgeführt. * P < 0,05, ** P < 0,01, und *** P < 0,001. Bitte klicken Sie hier für eine größere Version dieser Figur.
{kind=link}

Abbildung 3: Quantifizierung der geschert und Adapter ligiert Ratte DNA auf einem Bioanalyzer. Die roten und blauen Kurven zeigen die Menge und Größe der genomischen DNA (rot) nach Scheren in einer isothermen Sonikator und Adapter Ligation, beziehungsweise. Jede Zeile repräsentiert eine Probe und die rote und blaue Kurven reflektieren sowohl Verlust der DNA während der mehrere Schritte (Ende-Reparatur, 3'-Adenylation und Probe Bereinigung) und Zunahme der Größe der bp wegen der Ligatur der Adapter. Scharfe Peaks bei 25 bp und 1500 bp sind standard-Marker, die in den Puffer laden hinzugefügt wurden. Bitte klicken Sie hier für eine größere Version dieser Figur.
{kind=link}

Abbildung 4: CVS-induzierte epigenetische Veränderungen sind röntgenologisch Ratte Methyl-f (A) Analyse der Ratte Methyl-Seq Daten zwischen gestresst (rot) und Kontrolle (blau) Ratten der Promoter des Gens Rt1m4 als ein differentiell methylierte Region (DMR) verwickelt. Die grafische Ausgabe für Rt1m4 DMR (rosa schattigen Bereich) zeigt jedes CpG (vertikale graue Linie), die vier Proben in jeder Gruppe (rote oder blaue Linien) und die % Methylierung Ebenen für jedes Tier (roter oder blauer Punkt). (B) zwölf Verbrauchsgüter innerhalb der DMR wurden von Bisulfit Pyrosequenzierung validiert. Die Balkendiagramme sind vertreten, da SEM bedeuten, und ein Student T-Test wurde durchgeführt, für die statistische Signifikanz. * P < 0,05. Bitte klicken Sie hier für eine größere Version dieser Figur.
{kind=link}

Abbildung 5: lineare Regressionsanalyse zeigte einer geringeren Korrelation zwischen % DNA Methylierung bei CpG-10 Rt1m4 und 3 Wochen meine Plasma CORT Ebenen beider betont und Tiere (N = 16) zu kontrollieren. Daten von gestressten Tieren werden durch rote Kreise dargestellt. Bitte klicken Sie hier für eine größere Version dieser Figur.
{kind=link}
| Woche | Tag1 | Tag2 | Tag3 | Tag 4 | Tag 5 | Tag 6 | Tag 7 |
| AM | Zurückhaltung | Schwimmen | Kühlraum | Schwimmen | Zurückhaltung | Shaker | Schwimmen |
| PM | Shaker | Käfig-Tilt | Zurückhaltung | Shaker | Kühlraum | Zurückhaltung | Kühlraum |
| Über Nacht | Essen zu beschränken | Nasse Einstreu | Isolierung | Licht auf | Verdrängung | Licht auf | Nasse Einstreu |
Tabelle 1: Eine typische Wochenplan der Variable Dauerstress Therapie (CVS).
| Sequenzierung Metriken | Stress-1 | Kontrolle1 |
| (n = 4) | (n = 4) | |
| Gepaart Ende liest (pro) | 89,290,397 | 80,165,674 |
| Eindeutig zugeordneten gekoppelten Ende liest (PARKTEN) | 39,200,255 | 35,013,406 |
| Ausrichtung-Rate/Mapping-Effizienz (PARKTEN / pro) | 44 % | 44 % |
| Duplikat liest (% der PARKTEN) | 73 % | 65 % |
| Deduplizierte PARKTEN | 10,481,031 | 12,306,018 |
| Im Durchschnitt lesen Sie Tiefe Abdeckung (x) (ARDC) | 6 x | 6 x |
| Verbrauchsgüter (N) | 12,056,878 | 12,056,878 |
| ARDC (X) der Verbrauchsgüter | 2 x | 2 x |
| Verbrauchsgüter mit mindestens 10 mal gelesen (N) | 481.383 | 595.850 |
| ARDC (X) der Verbrauchsgüter mit mindestens 10 mal gelesen | 19 | 19 |
| Am Ziel Verbrauchsgüter (vollständige Überschneidung mit Sonde Zielregionen) | 1.923.872 | 2.007.638 |
| Am Ziel ARDC (X) der Verbrauchsgüter | 7 x | 8 x |
| Am Ziel Verbrauchsgüter mit mindestens 10 mal gelesen (N) | 428.249 | 531.419 |
| Am Ziel ARDC (X) der Verbrauchsgüter mit mindestens 10 mal gelesen | 18 x | 18 x |
| Auf Ziel (PER mit 1 oder mehreren Basenpaaren Überlappung mit Sonde Zielregionen) (PARKTEN) | 8.277.715 | 9.369.523 |
| % (Auf deduplizierte PARKTEN) | 78 % | 77 % |
| Am Ziel (Gesamtunterseiten abgebildet) Mb | 125 mb | 128 mb |
| Auf durchschnittliche Lese-Tiefe Zielabdeckung (x) (ARDC) | 9 x | 10 x |
| 1 Sequenzierung Metriken basiert auf Durchschnittswerten über Themen in den einzelnen Gruppen |
Tabelle 2: Sequenzierung Metriken aus der Ratte-Methyl-Seq-Plattform gewonnen.
| Chr | Start | Ende | gen | Entfernung | areaStat | meanDiff | Stress | Kontrolle | Richtung |
| chr20 | 1.644.246 | 1.644.390 | RT1-M4 | in_gene | 93.03 | 0,22 | 0,33 | 0,11 | zu gewinnen |
| chr5 | 160,361,352 | 160,361,564 | LOC690911 | in_gene | -70.75 | -0.19 | 0,72 | 0.91 | Verlust |
| chr3 | 61,138,281 | 61,138,330 | RGD1564319 | 265569 | 61.79 | 0.21 | 0,94 | 0,72 | zu gewinnen |
| ChR2 | 143,064,811 | 143,065,010 | Ufm1 | 8569 | -59.48 | -0.11 | 0.13 | 0,24 | Verlust |
| chr7 | 30,764,111 | 30,764,284 | Ntn4 | in_gene | 57,04 | 0.21 | 0,94 | 0,73 | zu gewinnen |
| chr17 | 12,469,112 | 12,469,218 | Idnk | 41996 | -50.91 | -0.13 | 0,74 | 0,88 | Verlust |
| chr7 | 47,101,725 | 47,101,930 | Pawr | in_gene | -50.54 | -0,12 | 0.64 | 0,76 | Verlust |
| chr5 | 76,111,248 | 76,111,822 | Txndc8 | 151703 | -50.38 | -0.11 | 0.85 | 0,96 | Verlust |
| chr11 | 80,640,132 | 80,640,356 | Dgkg | in_gene | -50.07 | -0.16 | 0,73 | 0,89 | Verlust |
| chr8 | 71,759,248 | 71,759,411 | Mir190 | 210226 | -47.84 | -0.17 | 0,58 | 0,75 | Verlust |
Tabelle 3: Top 10 differentiell methyliert Regionen. Für jede DMR die Ausgabetabelle zeigt von Links zur rechten Spalte: chromosomalen Position (chr), koordiniert (Beginn/Ende), gen Namen, Entfernung der Transkription Startseite, differenzierte Statistiken zwischen betont und Steuern Gruppen (AreaStat), meine differenzielle Methylierung (MeanDiff), mittlere Methylierung Ebenen über jede DMR für betont und Kontrollgruppen (Stressbewältigung) und Richtung der Methylierung von Steuerelementen ändern.
| KEGG Pathway Begriffe | Gen zählen | % | P-Wert | Benjamini |
| Diabetes | ||||
| Diabetes Mellitus Typ II | 12 | 0.1 | 3.6 x 10-4 | 9,8 x 10-3 |
| Herz-Kreislauf-Krankheit | ||||
| Vaskulären glatten Muskel-Kontraktion | 18 | 0.1 | 1.6 x 10-3 | 3.6 x 10-2 |
| Arrhythmogenic rechtsventrikuläre Kardiomyopathie (ARVC) | 13 | 0.1 | 4.0 x 10-3 | 7.1 x 10-2 |
| Dilatative Kardiomyopathie | 14 | 0.1 | 7.6 x 10-3 | 1.2 x 10-1 |
| Neuron-Funktion | ||||
| Langzeit-Potenzierung | 11 | 0.1 | 1,5 x 10-2 | 1.4 x 10-1 |
| Signalisierung | ||||
| MAPK Signalweg | 35 | 0,2 | 2.4 x 10-4 | 9.9 x 10-3 |
| Kalzium-Signalweg | 22 | 0.1 | 1.2 x 10-2 | 1.4 x 10-1 |
| Chemokin-Signalweg | 21 | 0.1 | 1.2 x 10-2 | 1,3 x 10-1 |
| Krebs | ||||
| Wege in der Krebstherapie | 42 | 0,3 | 4.1 x 10-5 | 3.4 x 10-3 |
| Gliom | 15 | 0.1 | 4.4 x 10-5 | 2.4 x 10-3 |
| Nicht-kleinzelliger Lungenkrebs | 10 | 0.1 | 7,9 x 10-3 | 1.1 x 10-1 |
| Kolorektales Karzinom | 13 | 0.1 | 8,4 x 10-3 | 1.1 x 10-1 |
| Chronisch-myeloischer Leukämie | 12 | 0.1 | 1.2 x 10-2 | 1,3 x 10-1 |
Tabelle 4: KEGG Pathway Analyse der DMRs anhand der Ratte Methyl-seq.
Diskussion
In dieser Studie haben wir konzipiert und umgesetzt die Methyl-Seq-Plattform für die Ratte Genom. Durch den Nachweis seiner Dienstprogramm mit einem Rattenmodell von Stress, haben wir bewiesen, dass die experimentelle und analytische Pipeline differentiell methylierte Regionen zwischen zwei Vergleichsgruppen bieten kann.
Um eine erfolgreiche Umsetzung der Plattform zu gewährleisten, müssen einige wichtige Schritte zu beachten. Ersten, ursprünglichen DNA-Qualität und Quantität hat erhebliche Auswirkungen auf die Qualität und Quantität der endgültigen Methyl-Seq-Bibliothek. Wir verwendet ein Fluorometer, anstatt einem Spektrophotometer, um sicherzustellen, dass unsere DNA-Messung die Menge der doppelsträngigen DNA vorhanden reflektiert. Die Bioanalyzer wurde verwendet, um die molekularen Größe und Menge der DNA nach Scheren und nach Adapter Ligatur zu messen. Überprüfung der Molekülgröße "Schicht" zwischen diesen Schritten ist entscheidend für das Vorhandensein von Adaptern an den Enden der einzelnen DNA-Fragment zu bestätigen, die Adapter-mediated PCR in den nachfolgenden Schritten durchlaufen werden. Die Menge der DNA, die am Ende des Adapters Ligatur Schrittes ist auch wichtig, seit mindestens 100 ng des Messguts Bibliothek bei diesem Schritt erforderlich, um sicherzustellen, ausreichender Menge steht nach dem Ziel Bereicherung und Bisulfit Konvertierungsschritte. Eine Nachmessung hochempfindlichen wurde auf die konstruierten Methyl-Seq-Bibliothek durchgeführt, so dass die Bibliothek richtig verdünnt werden kann, für das anschließende clustering auf der nächsten Generation-Sequencer. Zu guter Letzt wurde Bisulfit Pyrosequenzierung als hoch-quantitative, unabhängige Methode eingesetzt, um die Genauigkeit der analytischen Pipeline zu bewerten. Die endgültige Validierung mithilfe der ursprünglichen Proben und Replikation über zusätzliche Tiere sind entscheidende Schritte um sicherzustellen, dass das Experiment biologisch signifikante Veränderungen in der DNA-Methylierung erkennen kann.
Wir gehören auch mehrere Richtlinien im Falle einer Abweichung von dem Protokoll oder wenn Probleme auftreten. Erstens ist es möglich, zu viel DNA während der Ende-Reparatur, Adapter Ligatur oder magnetischer Wulst Reinigungsschritte zu verlieren. Alternativ ab Mengen DNA könnte sein, klein (< 200 ng) aufgrund von begrenzten Gewebe/DNA-Verfügbarkeit oder Umsetzung von verschiedenen Anreicherungsverfahren wie Fluoreszenz aktiviert Zellsortierung. Zahl der Zyklus während der zwei Bibliothek Verstärkung Schritte möglicherweise kompensieren die übermäßigen Verlust von DNA oder niedrige DNA-Menge im gesamten Bibliothek Bau Protokoll ab. Jedoch sind nicht mehr als eine zusätzliche 2-3 Zyklen empfohlen, da übermäßige Vorlage Verstärkung führen zu einer Erhöhung der Anzahl der doppelten Lesevorgänge sequenziert werden. Diese Duplikate sind ausgeschlossen, bei der Ausrichtung, Neigung in Prozent Methylierung Berechnungen zu verhindern. Zweitens, wenn die durchschnittliche Größe der DNA nicht von mehr als 30 bps erhöht, überprüfen Sie um sicherzustellen, dass die Reagenzien sind neu, als T4-DNA-Polymerase, Klenow und/oder T4 Ligase kann alt sein. Im Handel erhältlichen Ersatz Reagenzien können verwendet werden.
Darüber hinaus ist es möglich, dass die vorhergesagten DMRs nicht durch Pyrosequenzierung, bestätigen könnte, wo DNA-Methylierung Unterschiede existieren nicht oder sind deutlich geringer als die von Analyse vorhergesagt. Armen Validierung der Kandidat Regionen ein Problem allzu häufig für viele genomweite Analysen, z. B. wenn Pyrosequenzierung Ergebnisse nicht differenzielle Methylierung bestätigen oder die Effektgröße ist viel kleiner als die von der Analyse vorhergesagt. BSmooth ist ein analytischer Paket, dass "glättet" die Methylierung Ebenen über ein Zeitfenster von mehreren Verbrauchsgüter. Für das aktuelle Experiment verwickelt BSmooth ein DMR deren Methylierung Ebenen von Bisulfit Pyrosequenzierung validiert wurden. Allerdings werden wahrscheinlich Diskrepanzen zwischen Methylierung von BSmooth vorhergesagt und diejenigen von Pyrosequenzierung überprüft. Die Unterschiede ergeben sich aus der Glättungsfunktion, der schätzt die durchschnittliche Methylierung-Werte aller Verbrauchsgüter innerhalb eines DMR, einschließlich aufeinanderfolgenden Verbrauchsgüter, die in der DNA-Methylierung von mehr als 50 % abweichen oder Verbrauchsgüter, deren Methylierung Werte wegen ausgeschlossen wurden Sub-Schwelle lesen Tiefe. R-Pakete wie MethylKit24 können verwendet werden, um kleinere Fenster Verbrauchsgüter oder sogar einzelne Verbrauchsgüter, deren Methylierung-Spiegel stark mit denen von Pyrosequenzierung validiert korrelieren, zu identifizieren. Verschiedene Pakete implementieren und testen ihre prognostizierten Regionen oder Verbrauchsgüter der differenziellen Methylierung von Pyrosequenzierung werden die Robustheit der Daten sichergestellt. Alternativ können original Methyl-Seq-Bibliotheken Endbiegung und hinzu kommt die lesen Sie Dateien lesen Sie Tiefe zu erhöhen. Da die Bestimmung der Methylierung sind semi-quantitativen und diktiert durch die Anzahl der Lesevorgänge [(# of CpGs) / (Anzahl der TpGs + Therapieoptimierungs-)], Erhöhung erfahren Sie Tiefe für einen bestimmten CpG die Genauigkeit seines Prozent Methylierung-Wertes zu erhöhen. Nur als wir in dieser Studie Verbrauchsgüter, deren Methylierung-Werte wurden bestimmt von mindestens zehn Mal gelesen und eine Insgesamte lesen Sie Abdeckung von 19 X für jedes CpG erreicht.
Die Ratte-Methyl-Seq-Plattform ist nicht ohne seine Grenzen. Während es kostengünstiger als Vollständiggenom Bisulfit-Sequenzierung ist, ist es erheblich teurer als andere Methoden. Dennoch war den größten Teil der Kosten für den Kauf von Fahrspuren auf der Sequencer und nicht für die Capture-System. Je nach Bedarf mit Kreuz-Gewebe Vergleiche erfordern weniger durch große (25-70 %) Unterschiede12 in der DNA-Methylierung lesen Sie Tiefe kann die Kosten durch multiplexing mehr Proben pro Bahn und mit Hilfe einer höheren Kapazität Plattform reduziert werden. Darüber hinaus ist die Probenvorbereitung mehr Zeit in Anspruch als andere Methoden. Während ähnlich wie bei anderen Pulldown-Ansätze, die Sequenzierung der nächsten Generation zu integrieren, hinzufügen die zusätzlichen Bisulfit-Umwandlung und Reinigung Schritte die Arbeitsbelastung. Insgesamt die Methyl-Seq-Plattform ist eine kostengünstige Alternative zur Sequenzierung des gesamten Genoms und bietet Basenpaar Auflösung auf mehr als 2,3 Millionen Verbrauchsgüter, das ist deutlich mehr als die von Microarray-basierte Plattformen getestet. Bis heute wurden die handelsüblichen Mensch und Maus wurden Methyl-Seq-Plattformen zur dokumentieren alkoholabhängig Änderungen in der Makaken Gehirn25,26, Entwicklungsstörungen Gene in der Maus Gehirn9und die Blut-Hirn- Ziele von Glukokortikoiden10. Darüber hinaus macht es die Fähigkeit, bestimmte Regionen unabhängig von der Reihenfolge Anerkennung durch Restriktionsenzyme Ziel eine ideale Plattform für Cross-Arten Vergleiche. Für diese Studie haben wir entwickelt die Methyl-Seq-Plattform für die Ratte, wofür viele pharmakologische, metabolische und Verhaltensstörungen Experimente ohne den Vorteil eines Werkzeugs genomweite Methylomic durchgeführt werden. Unsere Daten zeigen, dass es verwendet werden, kann um DMRs in einem Rattenmodell von Stress zu erkennen und andere physiologische Parameter wie allgemeine CORT-Plasmaspiegel korreliert.
Die Methyl-Seq-Plattform ist ideal für epigenetische Experimente an Tieren mit sequenzierten Genome, die möglicherweise nicht über genügend experimentelle Beweise regulatorische Regionen zu dokumentieren. Wenn solche Regionen zur Verfügung gestellt werden, können zusätzliche Regionen speziell angefertigte und angebracht auf die aktuelle Version. Darüber hinaus ist die Plattform ideal für vergleichende Genomik, da die Ziel-Anreicherung von Restriktionsenzym Anerkennung nicht eingeschränkt wird. Zum Beispiel kann der Promotorregion jedes Gen des Interesses erfasst werden, unabhängig davon, ob es eine bestimmte Einschränkung Website birgt. Ebenso können regulatorischen Regionen, wie im Maus oder Menschen, die in das Genom von Interesse konserviert sind genannten erfasst werden.
Offenlegungen
Das Manuskript ist Teil eines Wettbewerbs-Preises von Agilent Technologies.
Danksagungen
Diese Studie wurde von NIH Grant MH101392 (RSL) und Unterstützung durch die folgenden Auszeichnungen und Stiftungen finanziert: eine NARSAD Young Investigator Award, Margaret Ann Preis Ermittler Fonds, dem James Wah Stimmung Störungen Gelehrter Fonds über Charles T. Bauer Foundation, Baker Stiftung und der Projekt-Match-Stiftung (RSL).
Materialien
| Name | Company | Catalog Number | Comments |
| Radioimmuno assay (RIA) | MP Biomedicals | 7120126 | Corticosterone, 125I labeled |
| Master Pure DNA Purification Kit | Epicentre/Illumina | MC85200 | |
| Thermal-LOK 2-Position Dry Heat Bath | USA Scientific | 2510-1102 | Used with 1.5 mL tubes |
| Vortex Genie 2 | Fisher | 12-812 | Vortex Mixer |
| Ethyl alcohol, Pure | Sigma-Aldrich | E7023 | 100% Ethanol, molecular grade |
| Centrifuge 5424 R | Eppendorf | - | Must be capable of 20,000 x g |
| Qubit 2.0 | ThermoFisher Scientific | Q32866 | Fluorometer |
| Qubit dsDNA BR Assay Kit | ThermoFisher Scientific | Q32850 | |
| Qubit dsDNA HS Assay Kit | ThermoFisher Scientific | Q32851 | High sensitivity DNA detection reagents |
| Qubit Assay Tubes | ThermoFisher Scientific | Q32856 | |
| SureSelectXT Rat Methyl-Seq Reagent Kit | Agilent Technologies | G9651A | Reagents for preparing the Methyl-Seq library |
| SureSelect Rat Methyl-Seq Capture Library | Agilent Technologies | 931143 | RNA baits for enrichment of rat targets |
| IDTE, pH 8.0 | IDT DNA | 11-05-01-09 | 10 mM TE, 0.1 mM EDTA |
| DNA LoBind Tube 1.5 mL | Eppendorf | 22431021 | |
| Covaris E-series or S-series | Covaris | - | Isothermal sonicator |
| microTUBE AFA Fiber Pre-Slit Snap-Cap 6x16mm (25) | Covaris | 520045 | |
| Water, Ultra Pure (Molecular Biology Grade) | Quality Biological | 351-029-721 | |
| Veriti 96 Well-Thermal Cycler | Applied Biosystems | 4375786 | |
| AMPure XP Beads | Beckman Coulter | A63880 | DNA-Binding magnetic beads |
| 96S Super Magnet | ALPAQUA | A001322 | Magnetic plate for purification steps |
| 2200 TapeStation | Agilent Technologies | G2965AA | Electrophresis-based bioanalyzer |
| D1000 ScreenTape | Agilent Technologies | 5067-5582 | |
| D1000 ScreenTape High Sensitivity | Agilent Technologies | 5067-5584 | |
| D1000 Reagents | Agilent Technologies | 5067-5583 | |
| D1000 Reagents High Sensitivity | Agilent Technologies | 5067-5585 | |
| DNA110 SpeedVac | ThermoFisher Scientific | - | Vacuum Concentrator |
| Dynabeads MyOne Streptavidin T1 magnetic beads | Invitrogen | 65601 | Streptavidin magnetic beads |
| Labquake Tube Rotator | ThermoFisher Scientific | 415110Q | Nutator Mixer is also acceptable |
| EZ DNA Methylation-Gold Kit | Zymo Research | D5006 | Bisulfite conversion kit. Contains Binding, Wash, Desulphonation, and Elution buffers |
| Illumina Hi-Seq 2500 | Illumina | - | Next-generation sequencing machine |
| PCR and Pyrosequencing Primers | IDT DNA | Variable | |
| Taq DNA Polymerase with ThermoPol Buffer - 2,000 units | New England BioLabs | M0267L | |
| Deoxynucleotide (dNTP) Solution Set | New England BioLabs | N0446S | |
| Pyromark MD96 | QIAGEN | - | Pyrosequencing machine |
| Ethyl Alcohol 200 Proof | Pharmco-Aaper | 111000200 | 70% Ethanol solution |
| Sodium Hydroxide Pellets | Sigma-Aldrich | 221465 | 0.2 M NaOH denature buffer solution |
| Tris (Base) from J.T. Baker | Fisher Scientific | 02-004-508 | 10 mM Tris Acetate Buffer wash buffer solution |
| PyroMark Gold Q96 Reagents (50x96) | QIAGEN | 972807 | Reagents required for pyrosequencing |
| PyroMark Annealing Buffer | QIAGEN | 979009 | |
| PyroMark Binding Buffer (200 mL) | QIAGEN | 979006 | |
| Streptavidin Sepharose High Performance Beads | GE Healthcare | 17-5113-01 | Streptavidin-coated sepharose beads |
| PyroMark Q96 HS Plate | QIAGEN | 979101 | Pyrosequencing assay plate |
| Eppendorf Thermomixer R | Fisher Scientific | 05-400-205 | Plate mixer. 96-well block sold separately (cat. No 05-400-207) |
| SureDesign Website | Agilent Technologies | - | Target capture design software (https://earray.chem.agilent.com/suredesign/) |
| UCSC Genome Browser | University of California Santa Cruz | - | rat Nov 2004 rn4 assembly |
| Agilent Methyl-Seq Protocol | Agilent Technologies | - | https://www.agilent.com/cs/library/usermanuals/public/G7530-90002.pdf |
Referenzen
- Barski, A., et al. High-resolution profiling of histone methylations in the human genome. Cell. 129 (4), 823-837 (2007).
- Meissner, A., et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 454 (7205), 766-770 (2008).
- Bibikova, M., et al. High density DNA methylation array with single CpG site resolution. Genomics. 98 (4), 288-295 (2011).
- Naumov, V. A., et al. Genome-scale analysis of DNA methylation in colorectal cancer using Infinium HumanMethylation450 BeadChips. Epigenetics. 8 (9), 921-934 (2013).
- Wockner, L. F., et al. Genome-wide DNA methylation analysis of human brain tissue from schizophrenia patients. Translational Psychiatry. 4, e339 (2014).
- Meissner, A., et al. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Research. 33 (18), 5868-5877 (2005).
- Smith, Z. D., Gu, H., Bock, C., Gnirke, A., Meissner, A. High-throughput bisulfite sequencing in mammalian genomes. Methods. 48 (3), 226-232 (2009).
- Slieker, R. C., et al. Identification and systematic annotation of tissue-specific differentially methylated regions using the Illumina 450k array. Epigenetics Chromatin. 6 (1), 26 (2013).
- Hing, B., et al. Adaptation of the targeted capture Methyl-Seq platform for the mouse genome identifies novel tissue-specific DNA methylation patterns of genes involved in neurodevelopment. Epigenetics. 10 (7), 581-596 (2015).
- Seifuddin, F., et al. Genome-wide Methyl-Seq analysis of blood-brain targets of glucocorticoid exposure. Epigenetics. 12 (8), 637-652 (2017).
- Irizarry, R. A., et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nature Genetics. 41 (2), 178-186 (2009).
- Lee, R. S., et al. Adaptation of the CHARM DNA methylation platform for the rat genome reveals novel brain region-specific differences. Epigenetics. 6 (11), 1378-1390 (2011).
- Jankord, R., et al. Stress vulnerability during adolescent development in rats. Endocrinology. 152 (2), 629-638 (2011).
- Pellow, S., Chopin, P., File, S. E., Briley, M. Validation of open:closed arm entries in an elevated plus-maze as a measure of anxiety in the rat. Journal of Neuroscience Methods. 14 (3), 149-167 (1985).
- Krueger, F., Andrews, S. R. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 27 (11), 1571-1572 (2011).
- Langmead, B., Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nature Methods. 9 (4), 357-359 (2012).
- Langmead, B., Trapnell, C., Pop, M., Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biology. 10 (3), R25 (2009).
- Hansen, K. D., Langmead, B., Irizarry, R. A. BSmooth: from whole genome bisulfite sequencing reads to differentially methylated regions. Genome Biology. 13 (10), R83 (2012).
- Lee, R. S., et al. Chronic corticosterone exposure increases expression and decreases deoxyribonucleic acid methylation of Fkbp5 in mice. Endocrinology. 151 (9), 4332-4343 (2010).
- Lee, R. S., et al. A measure of glucocorticoid load provided by DNA methylation of Fkbp5 in mice. Psychopharmacology. , (2011).
- Bose, M., Olivan, B., Laferrere, B. Stress and obesity: the role of the hypothalamic-pituitary-adrenal axis in metabolic disease. Current Opinion in Endocrinology, Diabetes, and Obesity. 16 (5), 340-346 (2009).
- Brydon, L., Magid, K., Steptoe, A. Platelets, coronary heart disease, and stress. Brain, Behavior, and Immunity. 20 (2), 113-119 (2006).
- McKlveen, J. M., et al. Chronic Stress Increases Prefrontal Inhibition: A Mechanism for Stress-Induced Prefrontal Dysfunction. Biological Psychiatry. 80 (10), 754-764 (2016).
- Akalin, A., et al. methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biology. 13 (10), R87 (2012).
- Cervera-Juanes, R., Wilhelm, L. J., Park, B., Grant, K. A., Ferguson, B. Alcohol-dose-dependent DNA methylation and expression in the nucleus accumbens identifies coordinated regulation of synaptic genes. Translational Psychiatry. 7 (1), e994 (2017).
- Cervera-Juanes, R., Wilhelm, L. J., Park, B., Grant, K. A., Ferguson, B. Genome-wide analysis of the nucleus accumbens identifies DNA methylation signals differentiating low/binge from heavy alcohol drinking. Alcohol. 60, 103-113 (2017).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenWeitere Artikel entdecken
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten