
Method Article
쥐 메 틸-Seq 플랫폼 관련 스트레스에 노출 된 후 변경 내용을 확인 하
요약
여기, 우리 프로토콜 및 관련 만성 스트레스에 노출 된 후 변경 내용을 확인을 쥐 모델을 사용 하 여 메 틸 Seq, epigenomic 플랫폼의 구현을 설명 합니다. 결과 쥐 메 틸-Seq 플랫폼은 쥐에서 스트레스 노출에서 발생 하는 메 틸 화 차이 감지 수를 보여 줍니다.
초록
다양 한 동물의 게놈을 사용할 수 있게,이 동물 모델에 동적 epigenetic 변화를 캡처할 수 있는 도구에 대 한 증가 필요가 있다. 쥐 epigenetic 도구 통찰력 기계 정보를 제공 하는 많은 약리학 및 행동 연구를 보완 수 있는 하나의 특정 모델 동물입니다. 이 위해, 우리는 쥐 쥐 게놈에서 DNA 메 틸 화 수준을 평가할 수 있는 SureSelect 캡처 시스템 (메 틸-Seq 라고 함)를 적응. 쥐 디자인 대상 발기인, CpG 섬, 섬 해안, 및 모든 RefSeq 유전자에서 GC 풍부한 지역.
쥐 실험에는 플랫폼을 구현 하기 위해 남성 Sprague Dawley 쥐 혈액 샘플 게놈 DNA 추출에 대 한 수집 된 후 3 주 동안 만성 변수 스트레스에 노출 되었다. 메 틸-Seq 라이브러리는 전단, 어댑터 결 찰, 대상 농축, 황산 변환 및 멀티플렉싱 하 여 쥐 DNA 견본에서 건설 되었다. 라이브러리는 다음-세대 시퀀싱 플랫폼에서 시퀀싱 했다 고 시퀀스 읽기 강조 하 고 강세가 없는 쥐의 DNA 사이 DMRs를 식별 하기 위해 분석 했다. 최고 후보자 DMRs 했다 독립적으로 유효성을 검사 하지 황산 pyrosequencing 플랫폼의 견고성을 확인 하 여.
결과 쥐 메 틸-Seq 플랫폼은 스트레스에 노출에 의해 유도 된 메 틸 화 변화를 캡처할 수 있는 유용한 epigenetic 도구를 보여 줍니다.
서문
높 처리량 연속에 발전은 다양 한 모델 및 비 모형 유기 체 게놈 시퀀스에 이르렀다. 같은 시퀀스의 여부 유전학, 비교 유전체학, 및 transcriptomics 연구를 촉진 했다. 예를 들어, 사용 가능한 게놈 시퀀스는 매우 풍요롭게 히스톤 수정1또는 황산 시퀀싱와 연결에 따라 DNA 칩 Seq 실험에서 시퀀싱 데이터를 정렬 하는 데 유용는 DNA 메 틸 화에 의해 측정 unmethylated cytosines2의 황산 변환에서 형성 uracil 감지. 그러나, 유전자 기능에 영향을 미칠 수 있는 종의 규제 시퀀스의 주석된 데이터의 부족 때문에 그들의 디자인에서 사용 가능한 게놈 시퀀싱 데이터를 통합 하는 epigenomic 플랫폼의 구현에서 지연 되었습니다.
특히, DNA 메 틸 화 methylomic 플랫폼을 구축 하기 위한 사용 가능한 게놈 데이터를 활용할 수 있는 DNA에 가장 널리 공부 후 성적인 수정 중 하나입니다. 1 개의 그런 보기는 인간 methylome3, 정신과4,5종양학에서 다양 한 분야에서 널리 사용 되었습니다 배열 기반 플랫폼입니다. 불행 하 게도, 비인간적인 동물 모델에 대 한 비슷한 플랫폼은 거의 아무 널리 사용 플랫폼을 그들의 초기 디자인에서 게놈 시퀀스의 장점을 받아들으로 부족, 있습니다.
비 인간 동물 모델의 methylomic 프리를 평가 하는 일반적인 방법은 이다 감소 표현 황산 시퀀싱 (RRBS)6. 이 접근 방식을 제공 하는 포괄적인 methylomic 풍경, 낮은 읽기-심층 취재 비용 및 게놈2의 큰 유전자 가난한 지역에서 제한 된 기능 정보를 제공 하는 전체 게놈 황산 시퀀싱 비용 극복 . RRBS 제한 다이제스트 및 일반적으로 유전자 발기인 근처 발견 하 고 유전자 규칙7에 역할을 생각 하는 CpG 섬 등 매우 다양 한 GC 시퀀스에 대 한 풍부 하 게 genomic DNA의 크기 선택 포함 한다. 중요 한 연구의 숫자에 RRBS 메서드를 사용 하는 동안 제한 효소에 의존이 아니다 주목할 만한 도전과 한계 없이. 예를 들어, GC 풍부한 시퀀스 RRBS의 농축은 전기 이동 법에 의해 제한 효소와 후속 크기 선택에 의해 인식 하는 특정 순서의 존재에 전적으로 의존. 즉, 이러한 금지 사이트를 포함 하지 않는 모든 게놈 영역 크기 선택 시 제외 됩니다. 또한, 크로스-종 비교 동일한 제한 사이트는 다른 종 중 동일한 loci에 존재 하지 않는 도전 하.
RRBS의 한계를 극복 하는 한 가지 방법은 사용 하는 플랫폼의 설계에 게시 된 게놈 시퀀스를 활용 하는 농축 방법. 어레이 기반 인간 플랫폼 뇌관 프로브 대립 유전자 특정 (황산 변환 후 TG 대 CG) 대상 어 닐 링 및 뇌관 확장을 위한 특정 cpgs 내 겠에 대 한 설계를 사용 합니다. 그것의 디자인 뿐만 아니라 사용할 수 있는 인간 게놈 시퀀스, 하지만 실험적으로 확인 된 규제 영역 문의 인코딩 및 합8등의 여러 줄에서 인수를 반영 한다. 인간 methylomic 조사에 그것의 광범위 한 사용에도 불구 하 고 비슷한 플랫폼 모델 동물에 대 한 존재 하지 않습니다. 또한, 배열 기반 형식 프로브 배치에 사용할 수 있는 영역에 중요 한 제약 조건을 배치합니다. 지난 몇 년 동안, 노력 캡처 프로브 설계 및 차세대 시퀀싱의 높은 처리량 기능 제공 대상-특이성 결합 되었습니다. 이러한 노력 결과 마우스 게놈 (마우스 메 틸-Seq), 메 틸 화9,10에 두뇌 또는 glucocorticoid 유발 차이 식별 하는 데 사용 했다의 시퀀싱 기반 대상 농축 시스템입니다. 다른 모델과 아닌 모델 동물에 대 한 비슷한 플랫폼이이 동물에 epigenomic 연구를 촉진 하기 위하여 필요 합니다.
여기, 우리는 쥐에 methylomic 분석을 소설이 플랫폼의 구현 방법을 보여 줍니다. 쥐는 약리학, 신진 대사, 내분비학, 동작에 중요 한 동물 모델을 역임 했다. 예를 들어 약물 독성, 비만, 스트레스 반응, 또는 마약 중독을 기본 메커니즘을 이해 하는 증가 필요가 있다. 이러한 조건과 관련 된 methylomic 변경 캡처 수 있는 높은 처리량 플랫폼 메커니즘에 대 한 우리의 이해를 증가할 것입니다. 쥐 게놈은 아직 규제 지역에 대 한 주석 부족, 이후 비 중복 발기인, CpG 섬, 섬 해안11, 통합 하 고 이전 쥐 메 틸-Seq 플랫폼12GC 풍부한 시퀀스를 식별 합니다.
쥐 게놈에 대 한 SureSelect 대상 농축 (일반적으로 메 틸-Seq 라고도 함) 플랫폼의 성공적인 설계 및 구현 평가, 우리는 만성 변수 스트레스 (CVS)13 차동 methylated 식별 하의 쥐 모델 고용 강세와 스트레스 동물 사이의 영역입니다. 우리의 플랫폼, 프로토콜, 디자인과 구현 체 누구의 게놈 시퀀스는 이미 사용할 수 있지만 제대로 주석이 남아에 포괄적이 고 편견 후 조사를 실시 하 고 싶은 수 있습니다 수사에 대 한 유용할 수 있습니다.
프로토콜
모든 실험은 따라와 모든 관련 규제 및 제도 지침 준수, 기관 동물 관리 및 사용 위원회 의학의 존스 홉킨스 학교에서를 포함 하 여 완료 되었습니다.
1입니다. 동물
- 나이의 4 주에 남성 사춘기 Sprague-Dawley 쥐를 얻을. 온도에서 폴 리 카보 네이트 쥐 감 금 소에 있는 동물을 집-0600 h.에서 가벼운 증상과 12 h, 12 h 어두운 주기에 습도 제어 룸 제공 물 광고 libitum 액세스와 동물.
- 운송과 관련 된 스트레스를 줄이기 위해 1 주 두었던 쥐를 수 있습니다. 쌍 집 격리 스트레스를 배제 하 고 나이의 5 주, 만성 변수 시작 동물 (N = 16) 3 주 (CVS) 처방 스트레스.
2. 만성 변수 스트레스
- 루틴을 예측할 수 없는 계속 불규칙 한 시간에 CVS 처방 한번 아침 (오전 9-11)에 한번 오후 (오후 1-3)에서 관리 합니다. 밤새 가벼운 스트레스를 통합 합니다. CVS 처방 포함: 구속 실린더; 1) 3 h 2) 10 분 수영; 3) 3 h 케이지 기울기 4) 1 시간 느린 떨고 플랫폼; 그리고 5) 4 ° C의 차가운 룸에서 1 시간.
참고: 하룻밤 스트레스는 사회 군집 (5 새 장 당), 사회적 고립, 젖은 침구, 음식 제한, 그리고 조명. 스트레스 처방의 전형적인 주간 일정은 표 1에 제공 됩니다.
3. 내 분 비 분석 실험
-
주간 CVS의 중앙 동안에 한 번 초기 호르몬 수준 (0 일) 설정에 CVS 처방 사전 실험을 통해 주당 두 번 같은 시간 (오전 9)에서 수집 된 corticosterone (CORT) 꼬리 혈액 (50 mL) samplings를 사용 하 여의 수준 결정 (일 4,11, 그리고 18), CVS의 모든 7 일 후 (일 7 및 14), CVS (21 일)의 결론에서. 매일 스트레스 처방 전에 혈액 샘플을 수집 합니다.
- RIA 및 게놈 DNA 추출을 위한 안락사 (25 일) 동안 한 마지막 트렁크 혈액 샘플을 수집 합니다.
- 모든 혈액 샘플 (600 x g, 4 ° C, 10 분)는 혈에서 플라스마를 분리 원심 피펫으로 플라즈마 (상쾌한) 고-80 ° c.에 샘플 저장
- 해 동 고 방사 면역 검정 법 (RIA)에 의해 CORT 수준을 결정 하는 플라즈마를 사용 합니다. 3 주 플라즈마 CORT 수준 스트레스 처방의 견고성을 확인 하기 위해 스트레스 동물에서 승격 되 있는지 확인 합니다.
4입니다. 동작
- CVS 처방 후 (23-24 일) 평가 상승에 불안 같은 동작에 대 한 각 동물 플러스 미로 (EPM)14.
- 기록, 비디오 카메라를 사용 하 여 300 s와 점수는 센터에서 보낸 시간에 대 한 EPM 기구에 동물 팔, 폐쇄 하 고 무기를 엽니다.
5. 쥐 메 틸-Seq의 디자인
- UCSC 게놈 브라우저를 사용 하 여 얻을 CpG 섬 및 섬 해안 (± 1 kb CpG 섬 측면)에서 사용할 수 있는 다른 시퀀스와 각 RefSeq 유전자의 발기인 (± 각 TSS의 1 킬로바이트)에 대 한 비중복 게놈 좌표 (2004 년 11 월 rn4 어셈블리 쥐) 관련 문학입니다.
참고: 쥐 메 틸-Seq, 이전 배열 기반 메 틸 화 플랫폼에서 추가 GC 풍부한 순서12를 추가 되었습니다. 5 kbps 보다 큰 지역에 대 한 500 bps의 교류 영역 1 kbps 건너뛴 다음 샘플링 했다. 마지막 쥐 메 틸-Seq 디자인 구성 111 Mbps, 2.3 백만 cpgs 내 겠; 그리고 594 bps의 평균 지역 크기입니다. 228,800 고유 loci 대상. - 적절 한 프로브 디자인에 대 한 상용 대상 캡처 디자인 소프트웨어에 게놈 좌표의 컴파일된 목록을 입력 합니다.
6. 쥐 메 틸-Seq 라이브러리 Genomic DNA에서의 건설
참고: 일괄 처리 효과 제거 하려면, 동시에 여러 개의 샘플을 처리 하 고 따라 마스터 믹스 최대 규모. 상업적으로 이용 가능한 DNA 추출 키트를 사용 하 여 DNA를 추출 합니다. 열 또는 강 수 기반 방법의 얻을 높은 품질 genomic DNA (260/280 비율 ~ 1.8). 페 놀 기반 방법의 사용을 권장 하지 않습니다. Elute 또는 낮은 테 버퍼 (10 m m 테, 0.1 m m EDTA, pH 8.0)에서 DNA를 resuspend.
- 샘플 준비
참고: DNA 바인딩을 사용 하 여 모든 단계를 위한 자석 구슬, 구슬 적어도 30 분 동안 실내 온도에 풍토에 순화와 잘 혼합 사용 하기 전에 있는지 확인 합니다.- DNA를 전단
- 각 샘플의 초기 이중 가닥 DNA 농도 결정 하는 fluorometer를 사용 합니다. 희석 > 1 µ g의 낮은 테 버퍼 (10 m m 테, 0.1 m m EDTA, pH 8.0) 낮은 DNA 바인딩 microcentrifuge 튜브에서 50 µ L gDNA.
- 등온선 sonicator를 사용 하 여 샘플 전단 (10% 듀티 사이클, 5 강도, 버스트, 60 s, 주파수 연소, 4 ° C의 6 주기 당 200 사이클).
- DNA 크기와 수량을 측정 하는 전기 영동 기반 시스템을 사용 하 여 DNA의 품질을 평가 합니다.
참고: 권장 DNA 금액은 1 µ g, 또는 3 µ g. 경우에 제한 된 시작 물자, 최저 입력된 금액 이어야 한다 > 500 ng, 낮은 금액으로 부정적인 영향을 미칠 생성 라이브러리의 품질과 수량.
- 복구 DNA 끝납니다.
- 쥐 메 틸-Seq 키트를 사용 하 여 얼음에 최종 복구 마스터 믹스를 준비 하. 각 샘플에 혼합의 52 µ L을 추가 하 고 온수 뚜껑 (20 ° C 30 분, 4 ° C 대기) 없이 열 cycler에서 품 어.
(샘플) 당 끝 복구 마스터 믹스:
35.2 µ L의 물
최종 수리 버퍼의 10 µ L (10 배)
DNTP 믹스의 1.6 µ L
T4 DNA 중 합 효소의 1 µ L
Klenow DNA 중 합 효소의 2 µ L
T4 Polynucleotide 키의 2.2 µ L - DNA 묶는 자석 구슬의 180 µ L 400 µ L의 샘플 당 갓된 70% 에탄올을 사용 하 여 샘플을 정화. 각 샘플에 구슬의 180 µ L을 추가 하 고 실 온에서 5 분 동안 품 어. 작은 구슬, 상쾌한 제거 하 고 70% 에탄올의 200 µ L에 펠 릿을 resuspend. 에탄올을 제거 하 고 세척을 한 번 반복.
- 자석 접시를 사용 하 여 작은 구슬 하 고 가능한 많은 에탄올을 제거. 37 ° C에서 드라이 비드 펠 렛은 완전히 때까지 3-5 분 heatblock 건조. Nuclease 무료 물 44 µ L에서 resuspend와 상쾌한의 약 42 µ L를 수집 합니다.
중지 포인트: 수리 DNA 끝나면 샘플 봉인 되 고-20 ° c.에 저장
- 쥐 메 틸-Seq 키트를 사용 하 여 얼음에 최종 복구 마스터 믹스를 준비 하. 각 샘플에 혼합의 52 µ L을 추가 하 고 온수 뚜껑 (20 ° C 30 분, 4 ° C 대기) 없이 열 cycler에서 품 어.
- Adenylate 3' 끝.
- Adenylation 마스터 믹스에 얼음을 준비 합니다. 각 샘플에 9 µ L 믹스를 추가 하 고 온수 뚜껑 (37 ° C 30 분, 4 ° C 대기) 없이 열 cycler에서 품 어.
(샘플) 당 Adenylation 마스터 믹스:
Klenow 버퍼의 5 µ L
DATP의 1 µ L
Klenow DNA 중 합 효소의 3 µ L - DNA 묶는 자석 구슬의 90 µ L 400 µ L의 샘플 당 갓된 70% 에탄올을 사용 하 여 샘플을 정화. 각 샘플에 구슬의 90 µ L을 추가 하 고 실 온에서 5 분 동안 품 어. 작은 구슬, 상쾌한 제거 하 고 70% 에탄올의 200 µ L에 펠 릿을 resuspend. 에탄올을 제거 하 고 세척을 한 번 반복.
- 자석 접시를 사용 하 여 작은 구슬 하 고 가능한 많은 에탄올을 제거. 37 ° C에서 드라이 비드 펠 렛은 완전히 때까지 3-5 분 heatblock 건조. Nuclease 무료 물 35 µ L에서 resuspend와 상쾌한의 약 33.5 µ L를 수집 합니다.
- Adenylation 마스터 믹스에 얼음을 준비 합니다. 각 샘플에 9 µ L 믹스를 추가 하 고 온수 뚜껑 (37 ° C 30 분, 4 ° C 대기) 없이 열 cycler에서 품 어.
- Methylated 어댑터 선
- 얼음 결 찰 마스터 믹스를 준비 하 고 각 샘플에 혼합의 16.5 µ L를 추가. 열띤된 뚜껑 (20 ° C 15 분, 4 ° C 대기) 없이 열 cycler에서 품 어.
(샘플) 당 결 찰 마스터 믹스:
물 2.5 µ L
메 틸-Seq의 2.5 µ L 갠 어댑터
T4 DNA 리가 버퍼의 10 µ L (5 배)
T4 DNA 리가의 1.5 µ L - DNA 묶는 자석 구슬의 90 µ L 400 µ L의 샘플 당 갓된 70% 에탄올을 사용 하 여 샘플을 정화. 각 샘플에 구슬의 90 µ L을 추가 하 고 실 온에서 5 분 동안 품 어. 작은 구슬, 상쾌한, 제거 하 고 70% 에탄올의 200 µ L에 펠 릿을 resuspend. 에탄올을 제거 하 고 세척을 한 번 반복.
- 자석 접시를 사용 하 여 작은 구슬 하 고 가능한 많은 에탄올을 제거. 37 ° C에서 드라이 비드 펠 렛은 완전히 때까지 3-5 분 heatblock 건조. Nuclease 무료 물 22 µ L에서 resuspend와 상쾌한의 약 22 µ L를 수집 합니다. bioanalyzer을 사용 하 여 품질을 평가 합니다.
참고: 경우 DNA의 총 금액은 500 ng, 전단 및 프로세스 추가 DNA 후속 단계를 진행 하기 전에. 평균 DNA 크기 30 bps 이상으로 증가 하지 않는다는 시 약은 새로운 T4 DNA 중 합 효소, Klenow, 또는 T4로 리가 나이가 있을 수 있습니다 있는지 확인 합니다.
중지 포인트: 갠 어댑터 ligating, 후 샘플 수 봉인 있으며-20 ° c.에 저장
- 얼음 결 찰 마스터 믹스를 준비 하 고 각 샘플에 혼합의 16.5 µ L를 추가. 열띤된 뚜껑 (20 ° C 15 분, 4 ° C 대기) 없이 열 cycler에서 품 어.
- DNA를 전단
- 교 잡
- 낮은 DNA 바인딩 microcentrifuge 관에 샘플을 전송 하 고 온수 진공 집중 장치를 사용 하 여 샘플 볼륨 미만 3.4 µ L. 다시 구성 견본 3.4 µ L 줄일.
참고: 집중 샘플 샘플 모든 액체 전에 진공 집중 장치에서 제거 되도록 약 3 µ L 증발. - 실내 온도 및 얼음에 메 틸-Seq 블록 믹스에서 교 잡 버퍼를 준비 합니다. 각 샘플을 메 틸-Seq 블록 혼합의 5.6 µ L을 추가 하 고 열 cycler 5 분, 2 분, 65 ° C 보류 65 ° C (95 ° C)에서 품 어.
(샘플) 당 교 잡 버퍼:
메 틸-Seq Hyb 1의 6.63 µ L
메 틸-Seq Hyb 2의 0.27 µ L
메 틸-Seq Hyb 3의 2.65 µ L
메 틸-Seq Hyb 4의 3.45 µ L
(샘플당) 메 틸-Seq 블록 믹스:
메 틸-Seq 색인 블록 1의 2.5 µ L
메 틸-Seq 블록 2의 2.5 µ L
메 틸-Seq 블록 3의 0.6 µ L - RNase 블록 혼합 및 캡처 라이브러리 교 잡 믹스를 준비 합니다. 각 샘플 캡처 라이브러리 교 잡 믹스 20 µ L을 추가 하 고 적어도 16 h 65 ° C에서 품 어.
(샘플) 당 RNase 블록 믹스:
RNase 블록의 0.5 µ L
물 1.5 µ L
(샘플) 당 도서관 교 잡 믹스를 캡처:
교 잡 버퍼의 13 µ L
RNase 블록 혼합의 2 µ L
5 µ L 쥐 메 틸-Seq의 캡처 라이브러리
참고: 65 ° C에서 반응 유지 비 특정 바인딩 방지 하기 위해 교 잡 믹스를 추가할 때. - 새로운 8 잘 스트립 관으로 샘플 당 streptavidin 자석 구슬의 aliquot 50 µ L. 비즈 메 틸-Seq 바인딩 버퍼의 200 µ L로 씻는 다. 자석 접시를 사용 하 여 작은 공의 구슬 3 세척의 총 각 씻어 사이의 상쾌한 제거 하. 최종 세척 후 메 틸-Seq 바인딩 버퍼의 200 µ L에서 streptavidin 구슬 resuspend.
- 씻어 streptavidin 자석 구슬의 200 µ L에 샘플을 추가 하 고 회전 믹서를 사용 하 여 30 분 동안 실 온에서 품 어. 샘플 및 65 ° c.에 미리 따뜻한 열 cycler에서 당 96 잘 접시의 triplicate 우물에 메 틸-Seq 워시 버퍼 2의 aliquot 200 µ L를 혼합 하는 동안
- 부 화, 후 작은 streptavidin 자석 구슬 자석 접시를 사용 하 여 그리고 200 µ L에 구슬 resuspend 메 틸-Seq 워시 버퍼 1. 실 온에서 15 분 동안 품 어. 자석 접시를 사용 하 여 작은 상쾌한 삭제 하.
- 메 틸-Seq 워시 버퍼 2와 3 번 구슬을 세척: 세척 버퍼 2 (사전 단계 6.2.5에서에서 따뜻하게 합니다.)의 200 µ L에서 비드 펠 렛 resuspend, (65 ° C, 10 분), 열 cycler에 구슬을 품 어 및 작은 구슬. 자석 접시를 사용 하 여 각 세척 후 상쾌한을 버리십시오.
참고: 때 비 특정 바인딩 방지 하기 위해 세척 버퍼 2 추가 65 ° C에서 교 잡 반응을 유지 한다. - 씻어 구슬에 메 틸-Seq 차입 버퍼의 20 µ L을 추가 하 고 20 분 사용 펠 릿 구슬 및 전송 표면에 뜨는 새로운 스트립 튜브를 자기 접시에 대 한 실 온에서 품 어. 비즈를 삭제 합니다.
참고: 잠복기, 동안 황산 변환 시 약 준비.
- 낮은 DNA 바인딩 microcentrifuge 관에 샘플을 전송 하 고 온수 진공 집중 장치를 사용 하 여 샘플 볼륨 미만 3.4 µ L. 다시 구성 견본 3.4 µ L 줄일.
- 황산 변환
참고: 적절 한 시 약 및 상용 황산 변환 키트에서 지침을 사용 하 여 eluted ssDNA의 황산 변환을 수행 합니다.- 이전 단계에서 상쾌한을 130 µ L 준비 황산 변환 시 약을 추가 합니다. 2 개의 우물에 동등 하 게 150 µ L 반응의 각각 분할 한다. 열 cycler (2.5 h, 4 ° C 보류에 대 한 64 ° C)에서 품 어.
참고: 150 µ L 반응 균일 온도 보장 하기 위해 두 개의 별도 웰 스도 동등 하 게 분할 된다. 2.5 h에 대 한 잠복기 후 즉시 다음 단계를 진행 합니다. - 샘플 바인딩 버퍼의 600 µ L을 추가 하 여 그리고 세척 버퍼의 100 µ L로 한 번 씻어 열을 바인딩하십시오. 모든 황산 변환 단계 사이 열 (15000 x g, 1 분)을 원심 및 삭제를 통해 흐름.
- 열을 Desulphonation 버퍼의 200 µ L을 추가 하 여 Desulphonate 샘플. 15-20 분 반복 원심 분리에 대 한 실 온에서 incubate 그리고 흐름을 통해 삭제.
- 워시 워시 버퍼의 200 µ L로 두 번 열. 열, 실내 온도에 3 분 동안 잠복기 centrifuging (15000 x g, 1 분)을 차입 버퍼의 10 µ L을 추가 하 여 각 샘플을 elute. 20 µ L의 총 차입 단계를 반복 합니다.
- PCR 반응 마스터 믹스 1 얼음을 준비 합니다. 각 샘플에 혼합의 82 µ L를 추가 합니다. 다음 프로그램으로 열 cycler에서 품 어.
PCR 반응 마스터 믹스 1 (샘플):
물 30 µ L
메 틸-Seq PCR 주인 혼합의 50 µ L
메 틸-Seq PCR1 뇌관 F의 1 µ L
메 틸-Seq PCR1 뇌관 R의 1 µ L
열 Cycler 프로그램:
단계 1, 1 주기: 95 ° C 2 분
단계 2, 8 주기: 95 ° C 30 초, 60 ° C 30 s, 72 ° C 30 s
단계 3, 1 주기: 72 ° C 7 분
단계 4, 1 주기: 4 ° C 보류 - DNA 묶는 자석 구슬의 180 µ L 400 µ L의 샘플 당 갓된 70% 에탄올을 사용 하 여 샘플을 정화. 각 샘플에 구슬의 180 µ L을 추가 하 고 실 온에서 5 분 동안 품 어. 작은 구슬, 상쾌한 제거 하 고 70% 에탄올의 200 µ L에 펠 릿을 resuspend. 에탄올을 제거 하 고 세척을 한 번 반복.
- 자석 접시를 사용 하 여 작은 구슬 하 고 가능한 많은 에탄올을 제거. 37 ° C에서 드라이 비드 펠 렛은 완전히 때까지 3-5 분 heatblock 건조. 21 µ L nuclease 무료 물에서 resuspend와 상쾌한의 약 19.5 µ L를 수집 합니다.
- 이전 단계에서 상쾌한을 130 µ L 준비 황산 변환 시 약을 추가 합니다. 2 개의 우물에 동등 하 게 150 µ L 반응의 각각 분할 한다. 열 cycler (2.5 h, 4 ° C 보류에 대 한 64 ° C)에서 품 어.
- 인덱싱
- PCR 반응 마스터 믹스 2 얼음을 준비 합니다. 추가 25.5 µ L 각 샘플 마스터 믹스 2. 개별 샘플을 뇌관을 인덱싱 상업 5 µ L을 추가 하 고 열 cycler에서 품 어.
PCR 반응 마스터 믹스 2 (샘플):
25 µ L 메 틸-Seq PCR 마스터 믹스
0.5 µ L 메 틸-Seq 일반적인 인덱싱 뇌관
열 Cycler 프로그램:
단계 1, 1 주기: 95 ° C 2 분
단계 2, 6 주기: 95 ° C 30 초, 60 ° C 30 s, 72 ° C 30 s
단계 3, 1 주기: 72 ° C 7 분
단계 4, 1 주기: 4 ° C 보류
참고: 추가 사이클 (2-3) 시작 DNA 농도 권장된 값 보다 경우 필요할 수 있습니다. - DNA 묶는 자석 구슬의 90 µ L 400 µ L의 샘플 당 갓된 70% 에탄올을 사용 하 여 샘플을 정화. 각 샘플에 구슬의 90 µ L을 추가 하 고 실 온에서 5 분 동안 품 어. 작은 구슬, 상쾌한 제거 하 고 70% 에탄올의 200 µ L에 펠 릿을 resuspend. 에탄올을 제거 하 고 세척을 한 번 반복.
- 자석 접시를 사용 하 여 작은 구슬 하 고 가능한 많은 에탄올을 제거. 37 ° C에서 드라이 비드 펠 렛은 완전히 때까지 3-5 분 heatblock 건조. 24 µ L nuclease 무료 물에서 resuspend 고 상쾌한의 약 24 µ L를 수집 합니다.
- 농도 혈압의 크기는 bioanalyzer에 고감도 DNA 검출 시 약을 사용 하 여 평가 합니다.
참고: 라이브러리 DNA의 존재를 감지 하는 bioanalyzer 못하면 추가 dna 준비 단계를 반복 합니다.
중지 포인트: 정화, 후 인덱싱된 샘플 봉인 되 고-20 ° c.에 저장 - 사용 하는 적절 한 다음-세대 시퀀싱 플랫폼에 대 한 샘플을 풀링.
- 낮은 테 버퍼 (6.1.1.1)로 희석 하 고 15의 최종 농도에 모든 샘플을 결합 하 여 DNA 몰 주어진된 볼륨에 라이브러리 크기에 따라 결정, bioanalyzer에서 농도 데이터를 사용 하 여 오후.
참고: 정량 실시간 PCR 뇌관 대상으로 합자 어댑터를 사용 하 여 라이브러리를 측정의 더 중요 한 방법이입니다. - 다음-세대 시퀀서에 레인 당 4 샘플에 대 한 충분 한 차선 수에 풀링된 샘플을 실행 합니다.
참고: 예를 들어, 경우 16 라이브러리 샘플 고유 색인 되었고 결합, 실행 라이브러리 이상 4 차선, 차선 당 4 샘플에 해당 합니다.
- 낮은 테 버퍼 (6.1.1.1)로 희석 하 고 15의 최종 농도에 모든 샘플을 결합 하 여 DNA 몰 주어진된 볼륨에 라이브러리 크기에 따라 결정, bioanalyzer에서 농도 데이터를 사용 하 여 오후.
- PCR 반응 마스터 믹스 2 얼음을 준비 합니다. 추가 25.5 µ L 각 샘플 마스터 믹스 2. 개별 샘플을 뇌관을 인덱싱 상업 5 µ L을 추가 하 고 열 cycler에서 품 어.
7. 다음-세대 Sequencer에서 시퀀싱
- 다음-세대 시퀀싱 컴퓨터에서 시퀀싱 다음 메 틸-Seq 라이브러리의 클러스터링에 대 한 제도적 시퀀싱 코어 샘플을 보냅니다.
8. DMRs를 식별 하는 분석
- 비스마르크15, 황산 변환, 플러스 물가 게놈 원시 입력된 읽기를 정렬 하는 내부 시퀀스 aligner16,17, Bowtie 2.0을 호출 하를 구현 합니다. 맞춤에 따라 품질 관리를 수행 하 고 각 포장 소비재를 예상된 메 틸 화 값을 할당 하는 Bismark_methylation_extractor를 사용 합니다.
- Bioconductor에서 학사-Seq 패키지18 함께 DMRs의 목록을 생성 합니다. 필터는 DMRs 3 연속 cpgs 내 겠 고 P-값 보다 큰 데에 따라 < 0.05.
참고: 게놈 좌표, 거리 가까운 RefSeq 유전자, 각 DMR, 평균 %CpG 메 틸 화 값 (예: 강세 대 압박), P-값, 두 개의 비교 그룹에 대 한 DMR 내 cpgs 내 겠 수를 포함 하는 DMR 목록을 생성 하 고 루즈벨트 (틀린 발견 비율) 값입니다. DMR 목록, 즉사용., 유효성 검사를 위한 pyrosequencing 뇌관 디자인 게놈 좌표.
9입니다. 황산 Pyrosequencing에 의해 유효성 검사
-
뇌관 디자인
- 황산 PCR 위한 뇌관 및 pyrosequencing 디자인. 중첩 된 PCR은 증폭 한 DMR의 150-400 bps (외부와 중첩) PCR 뇌관의 2 세트를 디자인.
참고: 일반적으로 설계 된 뇌관은 최소 24 기지 어 닐 링 온도 시퀀스 복잡도의 손실에서 감소에 대 한 계정에 적어도 4-5 비 연속 G의 (C의 반전 뇌관을 위한)와 긴. 중첩 된 뇌관 중 하나 biotin 분류 및 HPLC 정화 될 것입니다. 그러나, 표준 뇌관 PCR 단계는 agarose 젤에 반응을 확인 하 여 최적화를 먼저를 주문 한다.- 디자인은 pyrosequencing 시험 뇌관을 보완 biotinylated 대상 상류 수 분석을 cpgs 내 겠 단지 1-2 기초 물가. 각 pyrosequencing 뇌관 30 bps 다운스트림 분석 결과 안정적으로 수로 필요에 따라 여러 pyrosequencing 뇌관 디자인.
- Rt1 m 4에 대 한 다음을 사용 합니다.
외부-F TGTAYGATTTTGGTTATYGTAAAT rRT1M4
외부-R AACTTACAAATTTCACCAACTCA rRT1M4
rRT1M4 중첩-F GTGGGTTAYGTGGATAATATATAG
rRT1M4 중첩-R AATCACTTACCATTCTCTCTCTAACTA
rRT1M4 Pyro1 TAYGTGGATAATATATAGAT
rRT1M4 Pyro2 GATAGTTATTTGGYGAGTTAG
rRT1M4 Pyro3 GAGTATTTGGAGGAGTTGAT
rRT1M4 Pyro4 GGATTTTAATATTTGGT
- 황산 PCR 위한 뇌관 및 pyrosequencing 디자인. 중첩 된 PCR은 증폭 한 DMR의 150-400 bps (외부와 중첩) PCR 뇌관의 2 세트를 디자인.
-
쥐 피 gDNA의 황산 변환을 위한 상용 키트를 사용 합니다.
참고: 황산 변환 단계는 다음과 같은 수정 상용 키트에서 적응 되었습니다: 1 단계에서 혈액 gDNA의 50-100 ng을 추가 하 고 20 µ L 물으로 희석. 9 단계에서 샘플 당 20 µ L을 elute.- 제조 업체의 프로토콜에 따라 황산 변환 시 약을 준비 하 고 희석된 gDNA를 결합. 열 cycler (2.5 h, 4 ° C 보류에 대 한 64 ° C)에서 품 어.
- 스핀 열과 원심 분리기 (15000 x g, 1 분)에서 변환된 gDNA를 바인딩 버퍼를 추가 합니다. 워시 열 한 다음 열에 Desulphonation 버퍼를 추가 하 고 실 온에서 15 분 동안 품 어. 원심 분리기 (15000 x g, 1 분)
- 워시 워시 버퍼와 원심 분리기 (15000 x g, 1 분) 열. 원심 분리 (15000 x g, 2 분)와 세척 단계를 반복 합니다. 20 µ L을 추가 차입 버퍼 및 원심 분리기 (15000 x g, 1 분) elute.
-
PCR 증폭
- 외부 PCR 주인 혼합을 준비 합니다. 3.5 µ L 황산 변환 gDNA를 마스터 믹스의 21.5 µ L을 추가 하 고 열 cycler 프로그램 실행.
외부 PCR 마스터 믹스:
물의 16.25 µ L
중 합 효소 버퍼의 2.5 µ L [10 배]
DNTP [10mm]의 0.5 µ L
앞으로 뇌관의 1 µ L [0.1 µ M]
반전 뇌관의 1 µ L [0.1 µ M]
Taq DNA 중 합 효소의 0.25 µ L [5000 U/mL].
열 cycler 프로그램:
단계 1, 1 주기: 94 ° C 4 분
단계 2, 47 주기: 94 ° C 1 분, 53 ° C 30 초, 72 ° C 1 분
단계 3, 1 주기: 72 ° C 8 분, 4 ° C 보류 - 중첩된 한 PCR 주인 혼합을 준비 합니다. 외부 PCR에서 2 µ L의 샘플을 마스터 믹스의 23 µ L을 추가 하 고 반복 외부 PCR 열 cycler 프로그램. 젤 전기 이동 법 (1 x TAE 버퍼, 1 %agarose 젤) 통해 PCR 제품의 품질을 평가 합니다.
중첩된 한 PCR 마스터 믹스:
17.75 µ L의 물
중 합 효소 버퍼의 2.5 µ L [10 배]
DNTP [10mm]의 0.5 µ L
앞으로 뇌관의 1 µ L [0.1 µ M]
반전 뇌관의 1 µ L [0.1 µ M]
Taq DNA 중 합 효소의 0.25 µ L [5000 U/mL]
참고: 중첩 된 PCR를 위해 앞으로 또는 반전 뇌관 되어야 합니다 biotinylated.
- 외부 PCR 주인 혼합을 준비 합니다. 3.5 µ L 황산 변환 gDNA를 마스터 믹스의 21.5 µ L을 추가 하 고 열 cycler 프로그램 실행.
-
Pyrosequencing
- 바인딩 버퍼의 38 µ L, 35 µ L의 물, 그리고 샘플 당 sepharose streptavidin 입히는 구슬의 2 µ L 마스터 믹스를 확인 합니다. 96 잘 접시에서 75 µ L 마스터 믹스와 중첩 된 PCR 제품의 5 µ L를 추가 합니다. 15-60 분 판 통에 흔들.
- 떨고, 하는 동안 pyrosequencing 분석 결과 접시의 우물에 뇌관 (0.5 µ M, 어 닐 링 버퍼에 희석된)의 12 µ L를 추가 합니다.
- 떨고, 후 바인딩 반응 워시 버퍼를 사용 하 여 세척 단계를 수행 합니다. 장소 진공 도구 물마루에서 물으로 채워진 다음 접시에서 샘플을 수집 합니다. 반쯤 채워진 골짜기 70% 에탄올, NaOH (0.2 M), 그리고 트리 아세테이트 버퍼 (10 m m, pH 7.4)에 진공 도구 잠수함 진공 및 장소 진공 도구 전송 구슬 HS 분석 결과 플레이트에서에서 분리 하십시오.
- 열 블록에 접시를 놓고 2 분 5 분 기다린 다음 파이 프로그램 시작 허용 플레이트 80 ° C에서 품 어.
결과
쥐 메 틸-Seq 플랫폼을 성공적으로 구현 하는 몇 가지 기준에 따라 달라 집니다. 그림 1 연구의 전체 워크플로 보여 줍니다 및 앞으로 이동 하기 전에 필요한 특정 품질 관리 (QC) 단계를 하이라이트. 고려해 야 할 첫 번째 요소 중 하나는 methylome에 걸쳐 발생 하는 epigenetic 변화의 크기를 결정 하는 동물 모델 및 스트레스 처방의 견고성입니다. 이후 우리의 동물 작품 corticosterone (CORT) 노출 변화 DNA 메 틸 화19,20에 이어질 수 있는 우리의 이전의 관찰에 입각 한, 우리의 만성 변수 스트레스 (CVS) 처방 필요한 생산 충분 한 사후 경직 높은 플라즈마 CORT 수준으로 쥐를 강조 했다. 전형적인 주간 CVS 처방은 표 1 에 나와 아침, 오후, 매일 스트레스의 구성 그리고 하룻밤 habituation를 방지 하기 위해 지속적으로 변경 되 고 스트레스 반응 감소. 3 주 처방을 통해 스트레스 동물 의미 플라즈마 CORT의 상당히 높은 수준의 전시 [일 4-21, 제어: 32.7 3.7 ng/mL, 스트레스: 103.0 11.9 ng/mL (말은 sem의), P = 10-4, 그림 2Ax 2.2]의 강세, 그 이상의 동물을 제어 합니다. 일관 되 게,이 동물 또한 보였다는 상승에 더 큰 불안 같은 행동 플러스 미로 (EPM), EPM 및 더 적은 시간 (그림 2B) 두 팔을 벌려에서 닫힌된 팔에는 훨씬 더 많은 시간으로 표시. 이러한 결과 CVS 노출 중요 일 분 비 및 행동 변화, 이러한 변화 했다 특정 DNA 메 틸 화 서명 관련 여부 조사 선도 이끌어 보여줍니다.
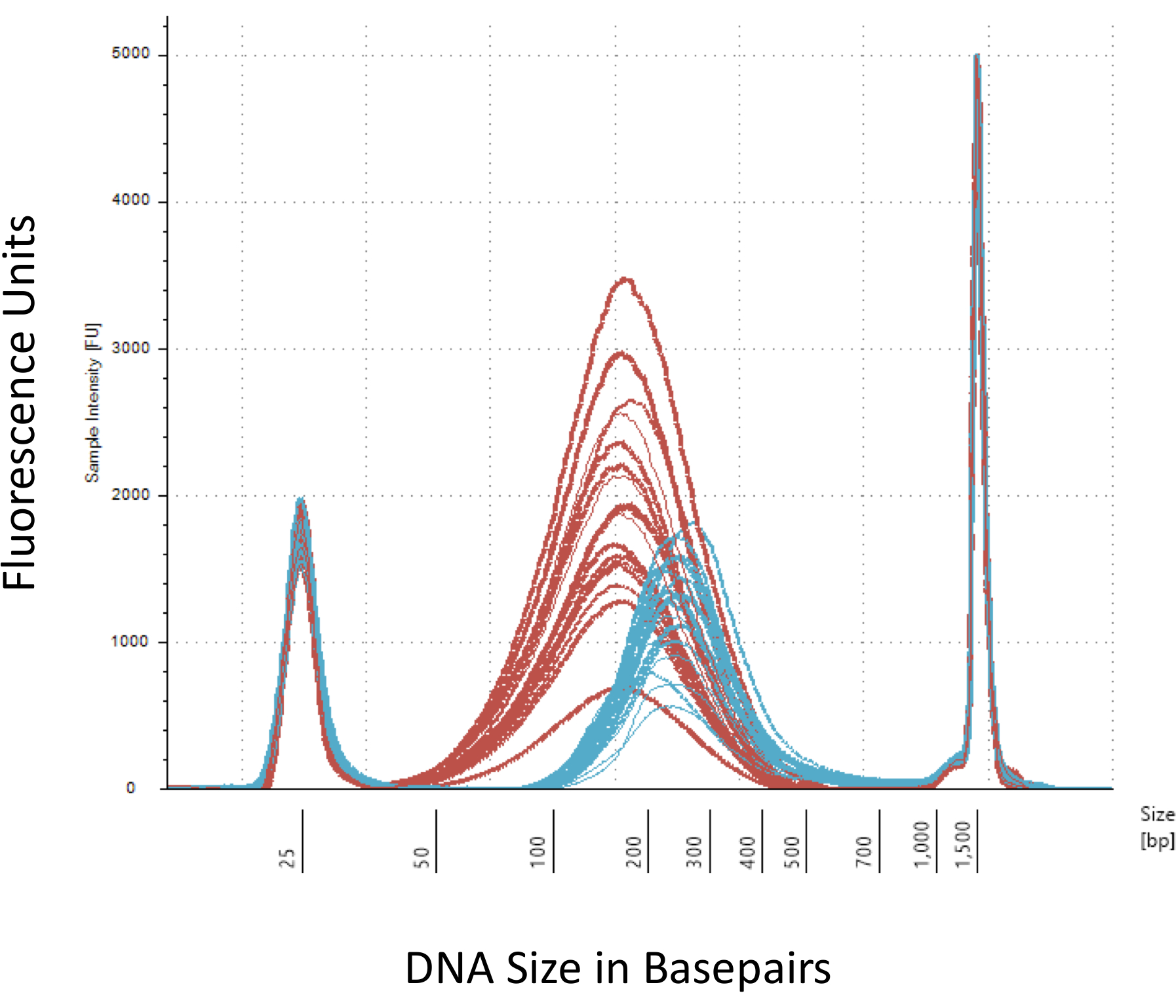
우리는 메 틸-Seq 라이브러리의 성공적인 건설에 대 한 중요 한 여러 검사점을 강조. 충분 한 양의 DNA로 시작 필요, 쥡니다, 여러 세척/정화, 대상 농축 이며 황산 변환 단계 연속적으로 완성 된 라이브러리에서 DNA의 양을 감소. 비록 몇 가지 PCR 증폭 단계 완화 DNA 템플렛의 손실, 과도 한 PCR 주기 숫자 높은 중복 읽기 발생할 수 있습니다. 현재 쥐 메 틸-Seq 연구를 위해 쥐 당 혈액 gDNA의 2 세대 사용 되었다. 우리 주의 메 틸-Seq 라이브러리 시작 DNA 금액 500으로 만들어질 수 있다 니. 작은 시작 물자 후속 시퀀싱에 대 한 라이브러리의 부족 한 금액을 생산의 위험이 증가 하지만 FACS (형광 활성화 된 세포 분류)에 의해 고립 된 DNA에서 라이브러리를 생성 하거나 바늘 펀치, 사용자 수 있습니다. QC는 DNA 분자 무게, 양, 및 몰 제공 하는 bioanalyzer에 샘플의 1 L의 전기 이동 법에 의해 수행 됩니다. bioanalyzer의 사용을 필요로 하는 3 개의 중요 한 단계는: 1) (~ 170 혈압, 레드, 그림 3); DNA의 충분 한 전단 되도록 쥡니다 단계를 다음 (~ 200 bp, 블루, 그림 3) PCR;에 의해 그들의 연속적인 증폭 되도록 전단된 DNA의 평균 크기에 변화에 의해 표시 2) 다음 어댑터 결 찰 단계 그리고 3) 수량 및 시퀀싱에 대 한 라이브러리의 크기를 되도록 최종 라이브러리 정화 단계를 다음과 같은.
R 패키지 BSSeq와 Bioconductor에 BSmooth 시퀀싱 데이터18황산을 분석 하는 데 사용 되었다. 그들은 도구와 정렬 시퀀스 읽습니다, 품질 관리를 수행 하기 위한 메서드를 포함 하 고 지역 (DMRs) 갠 차동 식별. BSmooth 소프트웨어는 황산 변환 게놈 시퀀스를 원시 입력된 읽기의 정렬에 의해 CpG 수준 측정 요약을 얻기 위해 내부 시퀀스 aligner로 Bowtie 2.016,17 를 호출 합니다. 정렬 된 읽기 다음 체계적인 시퀀싱 및 다운스트림 분석을 왜곡 수 있습니다 기본 호출 오류를 식별 하는 엄격한 품질 관리 절차를 통해 필터링 됩니다. 플롯의 시리즈는 시각적으로 필터링이 과정에 도움 생성 됩니다. 시퀀싱 통계와 같은 정렬된 읽기, % 대상의 포장 소비재 범위, 다른 (표 2) 중 당 문서 관련 정보에도 생성 됩니다. 일단 데이터 필터링, 스무 딩/정규화 알고리즘 수행, 모든 소비재 모든 QC에 따라 예상된 메 틸 화 값 할당 각 샘플에서 읽습니다 메 틸 화의 더 정확한 호출 되도록 cpgs 내 겠 이웃에서 추정 시퀀스 범위 낮은 경우에도 상태. 이 값에는 메 틸 화 CpG 사이트 각에서 확률의 매끄러운된 견적을 제공합니다. 두 치료 그룹 및 순위 이상에 가장 크게 다른 게놈 영역 사이 각 샘플의 부드러운된 메 틸 견적의 평균을 비교 하 여 DMRs의 목록이 생성 됩니다 (표 3).
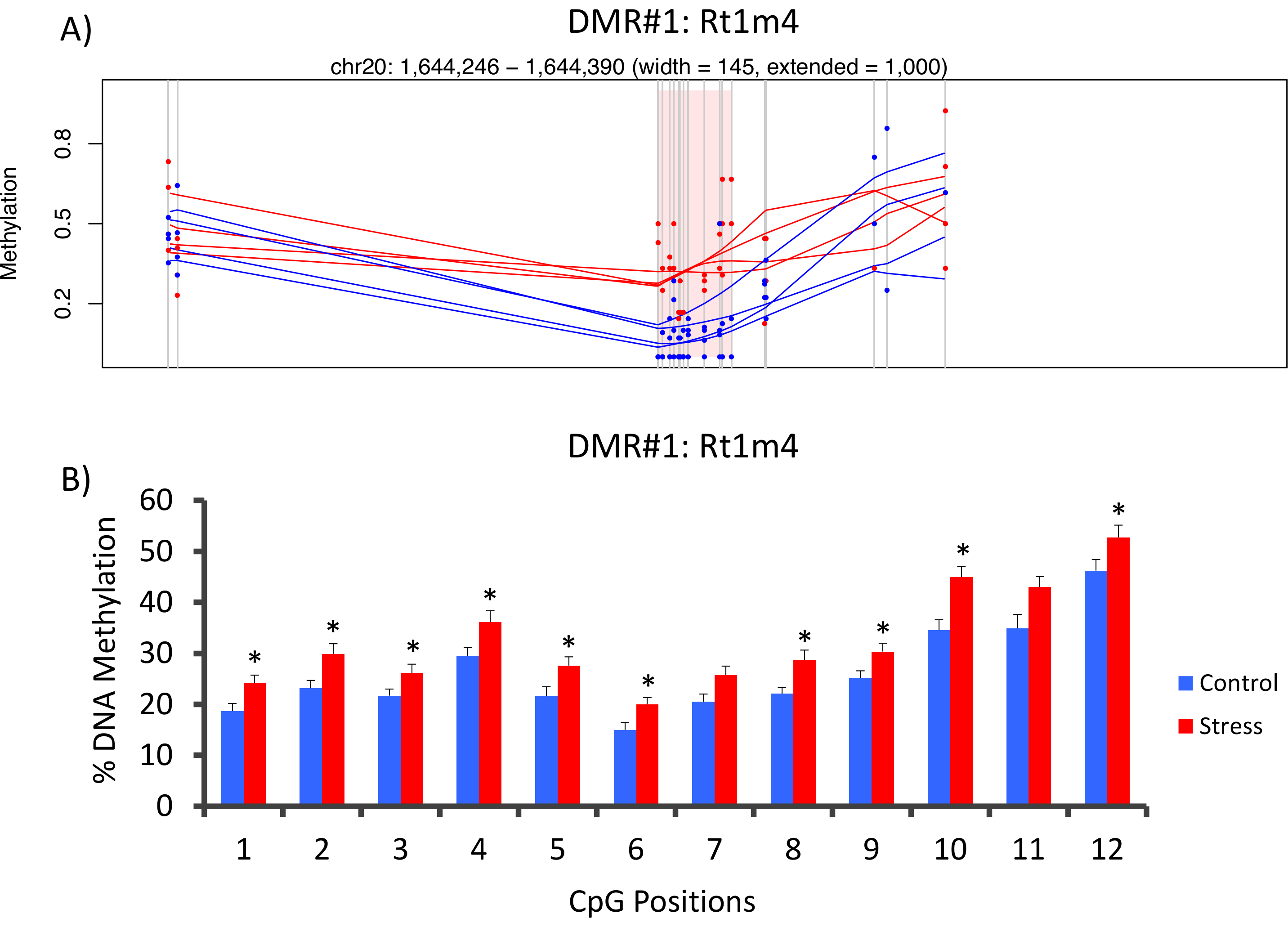
스트레스와 강세 그룹 사이 DMR와 Rt1 m4, 쥐 중요 한 조직 적합성 유전자의 발기인에 있는 최고 강세 동물 (그림 4A) 보다 모든 cpgs 내 겠 전체 메 틸 화 상부를 전시 하는 동물을 강조 했다. 메 틸-Seq 플랫폼 및 데이터 분석의 성공적인 구현을 확인, 뇌관 DMR, 및 압박 하 고 강세가 없는 동물의 (메 틸-Seq에 의해 시퀀스 8 하지 시퀀스 8) 전체 코 호트에서 혈액 DNA 메 틸 화 수준에 대 한 설계 되었습니다. 황산 pyrosequencing에 의해 평가 됐다. 결과 분석 12 cpgs 내 겠 밖으로 10에 걸쳐 DNA 메 틸 화에 상당한 증가 입증 (5.1-10.4% 메 틸 화, P에에서 변화 < 0.037, 그림 4B). KEGG 통로 분석은 모든 스트레스와 관련 된 경로 식별 하기 위해 명목상 상당한 DMRs에서 수행 되었다. 일관 되 게, DMR 관련 경로 질병 당뇨병, 심혈 관 질환 및 암 (표 4)와 같은 만성 스트레스 노출과 관련 된 연루. 21 , 22 , 23 스트레스 후 데이터와 노출의 정도 사이 연결을 보여주는, 메 틸 화 CpG-10 레벨 각 동물에 대 한 평균 3 주 CORT 수준 비교 되었다. 결과 내 분 비 및 메 틸 화 데이터 사이 겸손 한 상관 관계를 보였다 (R2= 0.54, P = 0.001, 그림 5).

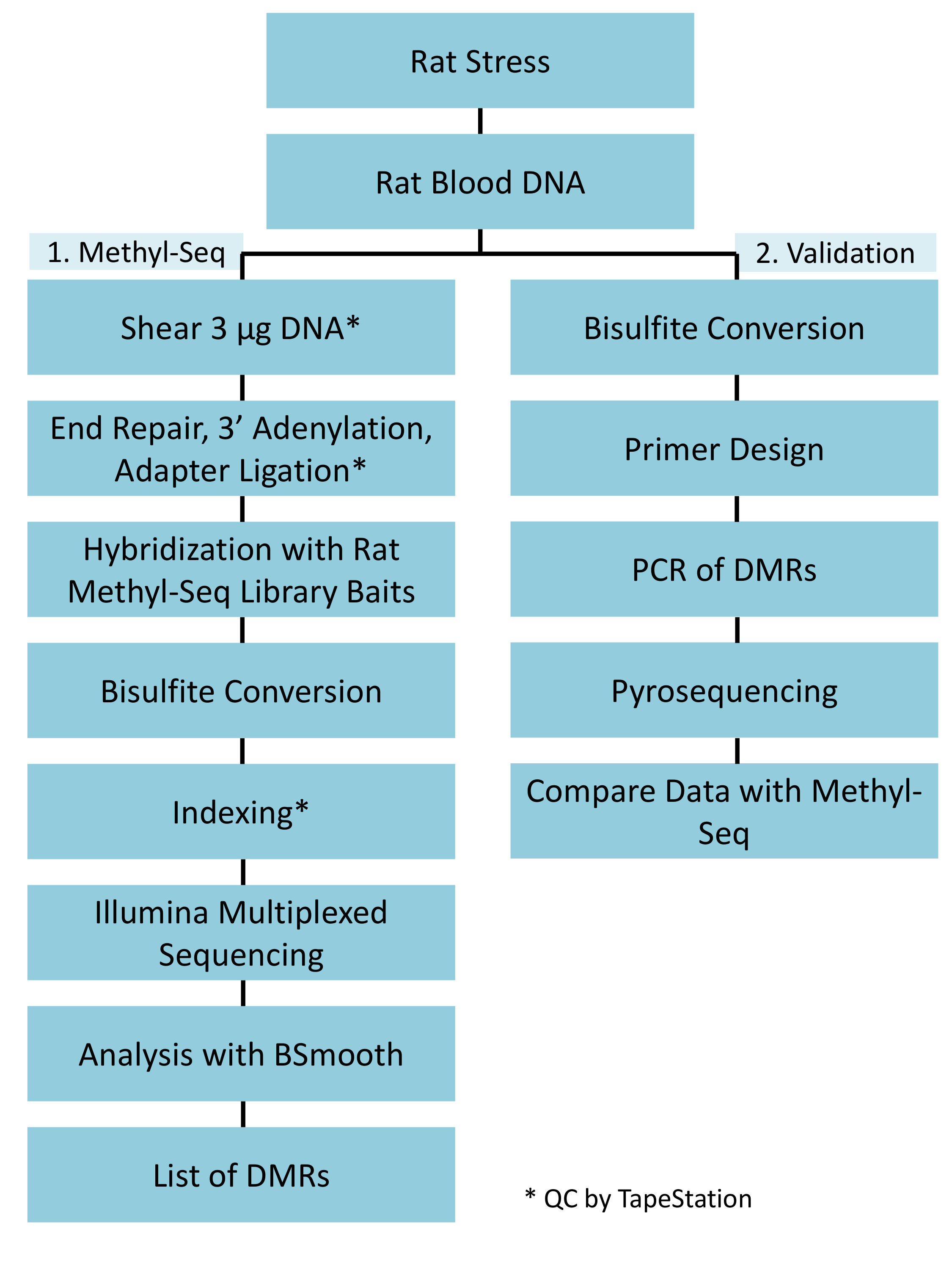
그림 1: 쥐 메 틸-Seq 플랫폼에 대 한 전반적인 회로도 워크플로. 1 g의 혈액에서 추출한 게놈 DNA의 스트레스 및 제어 쥐 먼저 시퀀싱, 분석 및 대상 식별에 대 한 메 틸-Seq 라이브러리 구성에 대 한 처리. 또 다른 100 DNA의 ng 황산 pyrosequencing에 의해 확인 된 후 대상의 독립적인 유효성 검사에 사용 됩니다. 이 그림의 더 큰 버전을 보려면 여기를 클릭 하십시오.
{kind=link}

그림 2: 만성 변수 스트레스 (CVS)에 노출 쥐에 내 분 비 및 행동 변화를 리드. (A) 여러 samplings corticosterone (CORT)의 설명의 견고성 CVS 처방 3 주. 혈액 샘플은 매일 스트레스 처방 전에 아침에 수집 되었다. (B) 스트레스 동물 미로 (EPM) 플러스 시간에는 상승의 두 팔을 벌려 덜 닫힌된 무기에 더 많은 시간을 보냈다. Boxplots 각 동물에 대 한 데이터 요소와 함께 표시 됩니다. 스튜던트 T-검정은 통계적 의미에 대 한 수행 했습니다. * P < 0.05, * * P < 0.01, 그리고 * * * P < 0.001. 이 그림의 더 큰 버전을 보려면 여기를 클릭 하십시오.
{kind=link}

그림 3:는 bioanalyzer에 전단 및 어댑터 출혈 쥐 DNA의 정량. 빨간색과 파란색 곡선 수량과 각각 등온 sonicator 및 어댑터 결 찰, 전단 뒤 genomic DNA (레드)의 크기를 표시 합니다. 각 줄 한 샘플 및 빨간색 나타내고 파란색 곡선 여러 단계 (최종 수리, 3'-adenylation, 및 샘플 정리) DNA의 두 손실 반영 및 bp 크기는 어댑터의 결 찰 때문에 증가. 날카로운 봉우리 25 bp와 1500 bp는 표준 마커를 로딩 버퍼에 추가 되었습니다. 이 그림의 더 큰 버전을 보려면 여기를 클릭 하십시오.
{kind=link}

그림 4: CVS 유도 후 성적인 변화는 쥐 메 틸이 검색 쥐 메 틸-Seq 데이터의 (A) 분석 스트레스 (빨강) 및 컨트롤 (블루) 쥐 사이 차동 methylated 지역 (DMR)로 서 Rt1m4 유전자의 발기인 연루. Rt1m4 DMR (분홍색 음영된 지역)에 대 한 그래픽 출력 각 그룹 (빨간색 이나 파란색 라인), 그리고 각 동물 (빨간색 또는 파란색 점)에 대 한 % 메 틸 화 수준에 각 포장 소비재 (회색 세로선), 4 개의 샘플을 표시합니다. (B) 12 cpgs 내 겠은 DMR 내 황산 pyrosequencing에 의해 확인 했다. 막대 그래프는 SEM, 의미 표시 되 고 통계적 의미에 대 한 스튜던트 T-검정 수행 했다. * P < 0.05. 이 그림의 더 큰 버전을 보려면 여기를 클릭 하십시오.
{kind=link}

그림 5: 선형 회귀 분석 보여 겸손 상관관계 % DNA CpG-10 Rt1m4 의 메 틸 화와 3 주 의미 플라즈마 CORT 수준을 모두 스트레스의 제어 동물 (N = 16). 데이터 스트레스 동물에서 빨간색 원으로 표시 됩니다. 이 그림의 더 큰 버전을 보려면 여기를 클릭 하십시오.
{kind=link}
| 주 | 주 1 | 주 2 | 3 일 | 4 일 | 5 일째 | 주 6 | 7 일 |
| 오전 | 구속 | 수영 | 콜드 룸 | 수영 | 구속 | 쉐이 커 | 수영 |
| 오후 | 쉐이 커 | 케이지 기울기 | 구속 | 쉐이 커 | 콜드 룸 | 구속 | 콜드 룸 |
| 하룻밤 | 음식 제한 | 젖은 침구 | 절연 | 빛에 | 크롤 링 | 빛에 | 젖은 침구 |
표 1: 만성 변수 스트레스 처방 (CVS)의 전형적인 주간 일정.
| 시퀀싱 통계 | 스트레스1 | 제어1 |
| (n = 4) | (n = 4) | |
| 끝 읽기 (당) 한 쌍 | 89,290,397 | 80,165,674 |
| 고유 하 게 매핑된 짝된 끝 (UMPER)를 읽으십시오 | 39,200,255 | 35,013,406 |
| 정렬 속도/매핑 효율 (UMPER / 1) | 44% | 44% |
| 중복 읽기 (UMPER %) | 73% | 65% |
| Deduplicated UMPER | 10,481,031 | 12,306,018 |
| 평균 읽기 깊이 범위 (x) (ARDC) | 6 x | 6 x |
| Cpgs 내 겠 (N) | 12,056,878 | 12,056,878 |
| Cpgs 내 겠 ARDC (x) | 2 x | 2 x |
| 10 읽기 (N)으로 cpgs 내 겠 | 481,383 | 595,850 |
| 10 읽기로 cpgs 내 겠 ARDC (X) | 19 | 19 |
| 대상 cpgs 내 겠 (완전 한 조사 대상 지역 중복에) | 1,923,872 | 2,007,638 |
| 대상의 cpgs 내 겠 ARDC (x) | 7 x | 8 배속 |
| 10 읽기 (N)와 대상 cpgs 내 겠에 | 428,249 | 531,419 |
| Cpgs 내 겠 적어도 10와 대상 ARDC (x)에 읽습니다. | 18 x | 18 x |
| 대상 (대상 지역 조사와 1 이상의 기본적인 쌍 중복 당) (UMPER) | 8,277,715 | 9,369,523 |
| % (Deduplicated UMPER)의 대상에 | 78% | 77% |
| 대상 (총 기지 매핑)에 Mb | 125 mb | 128 mb |
| 에 대상 평균 읽기 깊이 범위 (x) (ARDC) | 9 x | 10 배 |
| 1 각 그룹에 과목 전체 평균에 따라 시퀀싱 통계 |
표 2: 시퀀싱 쥐 메 틸-Seq 플랫폼에서 얻은 통계.
| chr | 시작 | 끝 | 유전자 | 거리 | areaStat | meanDiff | 스트레스 | 제어 | 방향 |
| chr20 | 1,644,246 | 1,644,390 | RT1-M4 | in_gene | 93.03 | 0.22 | 0.33 | 0.11 | 이득 |
| chr5 | 160,361,352 | 160,361,564 | LOC690911 | in_gene | -70.75 | -0.19 | 0.72 | 0.91 | 손실 |
| chr3 | 61,138,281 | 61,138,330 | RGD1564319 | 265569 | 61.79 | 0.21 | 0.94 | 0.72 | 이득 |
| chr2 | 143,064,811 | 143,065,010 | Ufm1 | 8569 | -59.48 | -0.11 | 0.13 | 0.24 | 손실 |
| chr7 | 30,764,111 | 30,764,284 | Ntn4 | in_gene | 57.04 | 0.21 | 0.94 | 0.73 | 이득 |
| chr17 | 12,469,112 | 12,469,218 | Idnk | 41996 | -50.91 | -0.13 | 0.74 | 0.88 | 손실 |
| chr7 | 47,101,725 | 47,101,930 | Pawr | in_gene | -50.54 | -0.12 | 0.64 | 0.76 | 손실 |
| chr5 | 76,111,248 | 76,111,822 | Txndc8 | 151703 | -50.38 | -0.11 | 0.85 | 0.96 | 손실 |
| chr11 | 80,640,132 | 80,640,356 | Dgkg | in_gene | -50.07 | -0.16 | 0.73 | 0.89 | 손실 |
| chr8 | 71,759,248 | 71,759,411 | Mir190 | 210226 | -47.84 | -0.17 | 0.58 | 0.75 | 손실 |
표 3: 최고 10 차동 갠 지역. 각 DMR에 대 한 출력 테이블 오른쪽 열에 왼쪽에서 보여줍니다: 의미, 염색체 위치 (chr), 좌표 (시작/종료), 유전자 이름, 거리는 전사에서 사이트를 시작, 차동 지역 통계 사이 강조 하 고 제어 그룹 (areaStat) 차동 메 틸 화 (meanDiff)에 대 한 각 DMR에서 말은 메 틸 화 수준의 스트레스와 제어 그룹 (스트레스/제어), 및 메 틸 화의 방향 컨트롤에서 변경.
| KEGG 통로 용어 | 유전자 수 | % | P-값 | Benjamini |
| 당뇨병 | ||||
| 타입 II 당뇨병 mellitus | 12 | 0.1 | 3.6 x 10-4 | 9.8 x 10-3 |
| 심혈 관 질환 | ||||
| 혈관 평활 근 수축 | 18 | 0.1 | 1.6 x 10-3 | 3.6 x 10-2 |
| Arrhythmogenic 오른쪽 심 실 심근 (ARVC) | 13 | 0.1 | 10-3 x 4.0 | 7.1 x 10-2 |
| 동 공이 확장 되어 심장 근육 병 증 | 14 | 0.1 | 7.6 x 10-3 | 1.2 x 10-1 |
| 신경 기능 | ||||
| 장기 potentiation | 11 | 0.1 | 1.5 x 10-2 | 1.4 x 10-1 |
| 신호 | ||||
| MAPK 신호 통로 | 35 | 0.2 | 2.4 x 10-4 | 9.9 x 10-3 |
| 칼슘 신호 전달 경로 | 22 | 0.1 | 1.2 x 10-2 | 1.4 x 10-1 |
| Chemokine 신호 통로 | 21 | 0.1 | 1.2 x 10-2 | 1.3 x 10-1 |
| 암 | ||||
| 암에 경로 | 42 | 0.3 | 4.1 x 10-5 | 3.4 x 10-3 |
| Glioma | 15 | 0.1 | 4.4 x 10-5 | 2.4 x 10-3 |
| 비 작은 세포 폐 암 | 10 | 0.1 | 7.9 x 10-3 | 10-1 x 1.1 |
| 대 장 암 | 13 | 0.1 | 8.4 x 10-3 | 10-1 x 1.1 |
| 만성 골수성 백혈병 | 12 | 0.1 | 1.2 x 10-2 | 1.3 x 10-1 |
표 4: KEGG 통로 분석 DMRs 메 틸이 쥐에서 확인
토론
이 연구에서 우리는 설계 및 쥐 게놈에 대 한 메 틸-Seq 플랫폼 구현. 스트레스의 쥐 모델의 유틸리티를 시연 함으로써 우리는 실험 및 분석 파이프라인 두 비교 그룹 사이 차동 갠 영역을 제공할 수 있습니다 설명 했다.
플랫폼의 성공적인 구현을 보장 하기 위해, 몇 가지 중요 한 단계를 관찰 해야 합니다. 첫 번째, 초기 DNA 품질 및 수량 최종 메 틸-Seq 라이브러리의 양과 품질에 큰 영향을 있다. 우리는 fluorometer 보다는 분 광 광도 계, 우리의 DNA 측정 현재 이중 가닥 DNA의 수량을 반영 되도록 사용. 분자 크기와 어댑터 결 찰 후 전단에 따라 DNA의 수량을 측정 하는 bioanalyzer 사용 되었다. 분자 크기 다음이 단계 사이 "변화"를 확인 하는 것은 후속 단계에서 PCR 어댑터 중재를 받게 될 것 이다 각 DNA 파편의 끝에 어댑터의 존재를 확인 중요 합니다. DNA는 어댑터 결 찰 단계의 끝에 남은 수량 또한 중요 하다, 적어도 100부터 ng 라이브러리 제품의 충분 한 수량 대상 농축 황산 변환 단계 후 사용할 수 있도록 하려면이 단계에서 필요 하다. 최종 높은 감도 측정 라이브러리는 제대로 다음-세대 시퀀서에 후속 클러스터링에 대 한 희석 수 있도록 생성 된 메 틸-Seq 라이브러리에서 수행 되었다. 마지막으로, 황산 pyrosequencing 높은 양적, 독립적인 방법으로 분석 파이프라인의 정확도 평가 하기 위해 고용 되었다. 원래 샘플 및 추가적인 동물을 사용 하 여 복제를 사용 하 여 최종 검증은 실험 DNA 메 틸 화에 생물학적으로 상당한 변화를 감지할 수 있도록 중요 한 단계입니다.
우리는 또한 프로토콜에서 편차 또는 경우 문제 발생 시 몇 가지 지침을 포함 합니다. 첫째, 그것은 최종 수리, 어댑터 결 찰, 또는 자석 구슬 정화 단계 동안 너무 많은 DNA를 잃을 수입니다. 시작 하는 양의 DNA 작은 수 또는 (< 200 ng) 제한 된 조직/DNA 가용성 또는 형광 활성화 된 세포 분류 등 다양 한 농축 방법의 구현. DNA의 과도 한 손실에 대 한 보상을 수 또는 도서관 건설 프로토콜에 걸쳐 DNA 양을 시작 낮은 사이클 수를 증가 하는 두 개의 라이브러리 증폭 단계 수 있습니다. 그러나, 더 이상 추가 2-3 주기는 권장, 과도 한 템플릿 증폭 시퀀싱 되 고 중복 읽기의 증가를 끌 것입니다. 이러한 중복 백분율 메 틸 화 계산에 바이어스를 방지 하기 위해 맞춤 단계에서 제외 됩니다. 둘째, 평균 DNA 크기 30 bps 이상으로 증가 하지 않습니다, 경우는 시 약은 새로운 T4 DNA 중 합 효소, Klenow, 또는 T4로 리가 나이가 있을 수 있습니다 있는지 확인 합니다. 상업적으로 이용 가능한 대체 시 약을 사용할 수 있습니다.
또한, 예측된 DMRs pyrosequencing, DNA 메 틸 화 차이 존재 하지 않는 또는 분석에 의해 예측 보다 훨씬 적은 있다 의해 확인 하지 수도 가능 하다. 후보 지역 가난한 정품 확인은 너무 때 pyrosequencing 결과 차동 메 틸 화를 확인 하지 않습니다 같은 많은 게놈 넓은 분석에 대 한 일반적인 문제 또는 효과 크기는 분석에 의해 예측 보다 훨씬 작습니다. BSmooth 여러 cpgs 내 겠 창 한 분석 패키지는 "부드럽게" 메 틸 화 수준입니다. 현재 실험에 대 한 BSmooth 연루는 DMR의 메 틸 화 수준의 황산 pyrosequencing에 의해 확인 되었다. 그러나, 거기 있을 것 이다 메 틸 화 수준 예측 BSmooth와 pyrosequencing에 의해 확인 사이의 불일치. 모든 연속 DNA 메 틸 화에 50% 이상 차이가 날 수 있습니다 cpgs 내 겠 또는 메 틸 화 값으로 인해 제외 된 cpgs 내 겠은 DMR 내 cpgs 내 겠 평균 메 틸 화 값을 추정 다듬기 기능에서 발생 하는 불일치 하위 임계값 깊이 읽기. R-패키지 MethylKit24 cpgs 내 겠 또는 심지어 단일 cpgs 내 겠 그 메 틸 화 수준 연관 강하게 pyrosequencing의 유효성을 검사 하는 그의 작은 윈도우를 식별 하기 위해 사용할 수 있습니다. 다른 패키지를 구현 하 고 pyrosequencing에 의해 그들의 예측된 영역 또는 차동 메 틸 화의 cpgs 내 겠 테스트 데이터의 안정성을 보장 합니다. 또는, 원래 메 틸-Seq 라이브러리 resequenced 하 고 읽기 깊이 증가 읽기 파일에 추가 될 수 있습니다. 때문에 메 틸 화 수준의 결정 반 양적 및 구술 읽기 수로 [(# of CpGs) / (TpGs + cpgs 내 겠 #)], 주어진된 포장 소비재의 백분율 메 틸 화 값의 정확도 높일 것에 대 한 읽기 깊이 증가. 이 연구에서 우리 메 틸 화 값 적어도 10 읽기에 의해 결정 되었다 고 각 포장 소비재에 대 한 19 x의 전체 읽기 범위 달성 cpgs 내 겠 고려.
쥐 메 틸-Seq 플랫폼의 제한 없이 아니다. 전체 게놈 황산 시퀀싱 보다 비용 효과적인 동안, 그것은 상당히 다른 방법 보다 더 비싸다입니다. 그럼에도 불구 하 고, 비용의 대부분을 캡처 시스템 그리고 시퀀서에 차선을 구매 했다. 크로스-조직 비교 덜 큰 (25-70%) 차이12 DNA 메 틸 화에 따른 요구와 필요한 읽기 깊이 따라 레인 당 더 많은 샘플을 멀티플렉싱 높은 용량 플랫폼을 사용 하 여 비용을 줄일 수 있습니다. 또한, 샘플 준비는 다른 방법 보다 더 많은 시간이 걸리는. 차세대 시퀀싱을 통합 하는 다른 풀 다운 방식와 마찬가지로, 하는 동안 추가 황산 변환 및 정화 단계 작업 부하를 추가 합니다. 전반적으로, 메 틸-Seq 플랫폼 전체 게놈 시퀀싱에 대 한 비용 효율적인 대안 이며 상당히 microarray 기반 플랫폼 분석 보다 더 이상의 2.3 백만 cpgs 내 겠에서 기본적인 쌍 해상도 제공. 날짜 하려면, 상용 인간과 마우스 메 틸-Seq 플랫폼 알코올 의존 변화 원숭이 뇌25,26, 마우스 뇌9, 및 혈액 neurodevelopmental 유전자를 사용 되었습니다. 스테로이드 제제10대상 또한, 제한 효소에 의해 시퀀스 인식에 특정 영역을 대상으로 기능 그것은 종 간 비교에 대 한 이상적인 플랫폼을 통해 있습니다. 이 연구를 위해 우리는 많은 약리학, 대사, 행동 실험 게놈 넓은 methylomic 도구의 이점 없이 수행 되는 쥐에 대 한 메 틸-Seq 플랫폼을 설계 되었습니다. 우리의 데이터는 스트레스의 쥐 모델에서 DMRs를 검색 하기 위해 사용할 수 있습니다을 전체 플라즈마 CORT 수준 같은 다른 생리 적인 매개 변수 상관을 보여준다.
메 틸-Seq 플랫폼 규제 영역을 문서화 하는 충분 한 실험적 증거를 없을 수 있습니다 시퀀스 유전자와 동물에 있는 후 성적인 실험에 이상적입니다. 이러한 영역은 사용할 수 있습니다 때 추가 지역 맞춤형 고 현재 버전에 연결 된 수 있습니다. 또한, 플랫폼 대상 농축 제한 효소 인식에 의해 제약 되지 않습니다 때문에, 비교 유전체학에 이상적입니다. 예를 들어, 그것은 특정 제한 사이트를 비호 하는 여부에 관심사의 어떤 유전자의 발기인 지구를 캡처할 수 있습니다. 마찬가지로, 마우스 또는 관심의 게놈에 보존은 인간에서 확인 된 그들과 같은 어떤 규제 영역을 캡처할 수 있다.
공개
원고는 Agilent 기술에서 경연 대회 상금의 일부입니다.
감사의 말
이 연구는 NIH 그랜트 MH101392 (RSL) 및 다음 수상 및 재단에서 지원에 의해 투자 되었다: NARSAD 젊은 수 사관 상, 마가렛 앤 가격 조사 기금, 찰스 T. 바 우 어 재단, 베이커를 통해 제임스 Wah 기분 장애 학자 기금 재단, 그리고 프로젝트 일치 재단 (RSL).
자료
| Name | Company | Catalog Number | Comments |
| Radioimmuno assay (RIA) | MP Biomedicals | 7120126 | Corticosterone, 125I labeled |
| Master Pure DNA Purification Kit | Epicentre/Illumina | MC85200 | |
| Thermal-LOK 2-Position Dry Heat Bath | USA Scientific | 2510-1102 | Used with 1.5 mL tubes |
| Vortex Genie 2 | Fisher | 12-812 | Vortex Mixer |
| Ethyl alcohol, Pure | Sigma-Aldrich | E7023 | 100% Ethanol, molecular grade |
| Centrifuge 5424 R | Eppendorf | - | Must be capable of 20,000 x g |
| Qubit 2.0 | ThermoFisher Scientific | Q32866 | Fluorometer |
| Qubit dsDNA BR Assay Kit | ThermoFisher Scientific | Q32850 | |
| Qubit dsDNA HS Assay Kit | ThermoFisher Scientific | Q32851 | High sensitivity DNA detection reagents |
| Qubit Assay Tubes | ThermoFisher Scientific | Q32856 | |
| SureSelectXT Rat Methyl-Seq Reagent Kit | Agilent Technologies | G9651A | Reagents for preparing the Methyl-Seq library |
| SureSelect Rat Methyl-Seq Capture Library | Agilent Technologies | 931143 | RNA baits for enrichment of rat targets |
| IDTE, pH 8.0 | IDT DNA | 11-05-01-09 | 10 mM TE, 0.1 mM EDTA |
| DNA LoBind Tube 1.5 mL | Eppendorf | 22431021 | |
| Covaris E-series or S-series | Covaris | - | Isothermal sonicator |
| microTUBE AFA Fiber Pre-Slit Snap-Cap 6x16mm (25) | Covaris | 520045 | |
| Water, Ultra Pure (Molecular Biology Grade) | Quality Biological | 351-029-721 | |
| Veriti 96 Well-Thermal Cycler | Applied Biosystems | 4375786 | |
| AMPure XP Beads | Beckman Coulter | A63880 | DNA-Binding magnetic beads |
| 96S Super Magnet | ALPAQUA | A001322 | Magnetic plate for purification steps |
| 2200 TapeStation | Agilent Technologies | G2965AA | Electrophresis-based bioanalyzer |
| D1000 ScreenTape | Agilent Technologies | 5067-5582 | |
| D1000 ScreenTape High Sensitivity | Agilent Technologies | 5067-5584 | |
| D1000 Reagents | Agilent Technologies | 5067-5583 | |
| D1000 Reagents High Sensitivity | Agilent Technologies | 5067-5585 | |
| DNA110 SpeedVac | ThermoFisher Scientific | - | Vacuum Concentrator |
| Dynabeads MyOne Streptavidin T1 magnetic beads | Invitrogen | 65601 | Streptavidin magnetic beads |
| Labquake Tube Rotator | ThermoFisher Scientific | 415110Q | Nutator Mixer is also acceptable |
| EZ DNA Methylation-Gold Kit | Zymo Research | D5006 | Bisulfite conversion kit. Contains Binding, Wash, Desulphonation, and Elution buffers |
| Illumina Hi-Seq 2500 | Illumina | - | Next-generation sequencing machine |
| PCR and Pyrosequencing Primers | IDT DNA | Variable | |
| Taq DNA Polymerase with ThermoPol Buffer - 2,000 units | New England BioLabs | M0267L | |
| Deoxynucleotide (dNTP) Solution Set | New England BioLabs | N0446S | |
| Pyromark MD96 | QIAGEN | - | Pyrosequencing machine |
| Ethyl Alcohol 200 Proof | Pharmco-Aaper | 111000200 | 70% Ethanol solution |
| Sodium Hydroxide Pellets | Sigma-Aldrich | 221465 | 0.2 M NaOH denature buffer solution |
| Tris (Base) from J.T. Baker | Fisher Scientific | 02-004-508 | 10 mM Tris Acetate Buffer wash buffer solution |
| PyroMark Gold Q96 Reagents (50x96) | QIAGEN | 972807 | Reagents required for pyrosequencing |
| PyroMark Annealing Buffer | QIAGEN | 979009 | |
| PyroMark Binding Buffer (200 mL) | QIAGEN | 979006 | |
| Streptavidin Sepharose High Performance Beads | GE Healthcare | 17-5113-01 | Streptavidin-coated sepharose beads |
| PyroMark Q96 HS Plate | QIAGEN | 979101 | Pyrosequencing assay plate |
| Eppendorf Thermomixer R | Fisher Scientific | 05-400-205 | Plate mixer. 96-well block sold separately (cat. No 05-400-207) |
| SureDesign Website | Agilent Technologies | - | Target capture design software (https://earray.chem.agilent.com/suredesign/) |
| UCSC Genome Browser | University of California Santa Cruz | - | rat Nov 2004 rn4 assembly |
| Agilent Methyl-Seq Protocol | Agilent Technologies | - | https://www.agilent.com/cs/library/usermanuals/public/G7530-90002.pdf |
참고문헌
- Barski, A., et al. High-resolution profiling of histone methylations in the human genome. Cell. 129 (4), 823-837 (2007).
- Meissner, A., et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 454 (7205), 766-770 (2008).
- Bibikova, M., et al. High density DNA methylation array with single CpG site resolution. Genomics. 98 (4), 288-295 (2011).
- Naumov, V. A., et al. Genome-scale analysis of DNA methylation in colorectal cancer using Infinium HumanMethylation450 BeadChips. Epigenetics. 8 (9), 921-934 (2013).
- Wockner, L. F., et al. Genome-wide DNA methylation analysis of human brain tissue from schizophrenia patients. Translational Psychiatry. 4, e339 (2014).
- Meissner, A., et al. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Research. 33 (18), 5868-5877 (2005).
- Smith, Z. D., Gu, H., Bock, C., Gnirke, A., Meissner, A. High-throughput bisulfite sequencing in mammalian genomes. Methods. 48 (3), 226-232 (2009).
- Slieker, R. C., et al. Identification and systematic annotation of tissue-specific differentially methylated regions using the Illumina 450k array. Epigenetics Chromatin. 6 (1), 26 (2013).
- Hing, B., et al. Adaptation of the targeted capture Methyl-Seq platform for the mouse genome identifies novel tissue-specific DNA methylation patterns of genes involved in neurodevelopment. Epigenetics. 10 (7), 581-596 (2015).
- Seifuddin, F., et al. Genome-wide Methyl-Seq analysis of blood-brain targets of glucocorticoid exposure. Epigenetics. 12 (8), 637-652 (2017).
- Irizarry, R. A., et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nature Genetics. 41 (2), 178-186 (2009).
- Lee, R. S., et al. Adaptation of the CHARM DNA methylation platform for the rat genome reveals novel brain region-specific differences. Epigenetics. 6 (11), 1378-1390 (2011).
- Jankord, R., et al. Stress vulnerability during adolescent development in rats. Endocrinology. 152 (2), 629-638 (2011).
- Pellow, S., Chopin, P., File, S. E., Briley, M. Validation of open:closed arm entries in an elevated plus-maze as a measure of anxiety in the rat. Journal of Neuroscience Methods. 14 (3), 149-167 (1985).
- Krueger, F., Andrews, S. R. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 27 (11), 1571-1572 (2011).
- Langmead, B., Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nature Methods. 9 (4), 357-359 (2012).
- Langmead, B., Trapnell, C., Pop, M., Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biology. 10 (3), R25 (2009).
- Hansen, K. D., Langmead, B., Irizarry, R. A. BSmooth: from whole genome bisulfite sequencing reads to differentially methylated regions. Genome Biology. 13 (10), R83 (2012).
- Lee, R. S., et al. Chronic corticosterone exposure increases expression and decreases deoxyribonucleic acid methylation of Fkbp5 in mice. Endocrinology. 151 (9), 4332-4343 (2010).
- Lee, R. S., et al. A measure of glucocorticoid load provided by DNA methylation of Fkbp5 in mice. Psychopharmacology. , (2011).
- Bose, M., Olivan, B., Laferrere, B. Stress and obesity: the role of the hypothalamic-pituitary-adrenal axis in metabolic disease. Current Opinion in Endocrinology, Diabetes, and Obesity. 16 (5), 340-346 (2009).
- Brydon, L., Magid, K., Steptoe, A. Platelets, coronary heart disease, and stress. Brain, Behavior, and Immunity. 20 (2), 113-119 (2006).
- McKlveen, J. M., et al. Chronic Stress Increases Prefrontal Inhibition: A Mechanism for Stress-Induced Prefrontal Dysfunction. Biological Psychiatry. 80 (10), 754-764 (2016).
- Akalin, A., et al. methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biology. 13 (10), R87 (2012).
- Cervera-Juanes, R., Wilhelm, L. J., Park, B., Grant, K. A., Ferguson, B. Alcohol-dose-dependent DNA methylation and expression in the nucleus accumbens identifies coordinated regulation of synaptic genes. Translational Psychiatry. 7 (1), e994 (2017).
- Cervera-Juanes, R., Wilhelm, L. J., Park, B., Grant, K. A., Ferguson, B. Genome-wide analysis of the nucleus accumbens identifies DNA methylation signals differentiating low/binge from heavy alcohol drinking. Alcohol. 60, 103-113 (2017).
재인쇄 및 허가
JoVE'article의 텍스트 или 그림을 다시 사용하시려면 허가 살펴보기
허가 살펴보기더 많은 기사 탐색
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. 판권 소유