
Method Article
Una pantalla de supresor asistida por secuenciación profunda, espontánea en la levadura de fisión Schizosaccharomyces pombe
En este artículo
Resumen
Presentamos un protocolo de pantalla simple supresor en fisión levaduras. Este método es eficiente, libre de mutágeno y selectivo para las mutaciones que ocurren a menudo en un locus genómico. El protocolo es conveniente para aislar los supresores que paliar defectos de crecimiento en cultivo líquido que son causados por una mutación o una droga.
Resumen
Una pantalla genética de alelos del mutante que suprimir defectos fenotípicos causados por una mutación es un poderoso enfoque para identificar los genes que pertenecen a las vías bioquímicas relacionadas estrechamente. Los métodos anteriores como el análisis de la matriz genética sintética (SGA) y técnicas de mutagénesis al azar mediante radiación ultravioleta (UV) o químicos como Metanosulfonato de etilo (EMS) o N-etil-N-nitrosourea (ENU), han sido ampliamente utilizados pero a menudo son costosos y laborioso. Además, estos métodos basado en el mutágeno son con frecuencia asociados con graves efectos secundarios sobre el organismo, induciendo mutaciones múltiples que se suman a la complejidad de aislar a los supresores. Aquí, presentamos un protocolo sencillo y eficaz para identificar las mutaciones supresoras en mutantes que confieren un defecto de crecimiento en Schizosaccharomyces pombe. La aptitud de las células con deficiencia de crecimiento en medios líquidos enriquecidos estándar o medios líquidos sintéticos puede controlarse usando un lector de placa de 96 pocillos automatizado durante un período prolongado de recuperación. Una vez que una célula adquiere una mutación supresora en la cultura, sus descendientes vencen los de las células parentales. Las células recuperadas que tienen una ventaja competitiva crecimiento sobre las células parentales se pueden aisladas y retrocruzas con las células parentales. Las mutaciones supresoras entonces se identifican con la secuenciación del genoma completo. Usando este acercamiento, hemos aislado con éxito varios supresores que paliar los defectos de crecimiento severo causados por la pérdida de Elf1, una familia de AAA + ATPasa que es importante en el transporte de ARNm nuclear y mantenimiento de la estabilidad genómica. Actualmente hay más de 400 genes en S. pombe con mutantes que confieren un defecto de crecimiento. Como muchos de estos genes no caracterizados, proponemos que nuestro método será acelerar la identificación de nuevas interacciones funcionales con este método fácil de usar, alto rendimiento.
Introducción
La base de la comprensión de los vínculos funcionales entre los genes se basa en la capacidad para identificar los mecanismos moleculares que divergen rasgos genéticos complejos para producir fenotipos diversos1. En la levadura de fisión Schizosaccharomyces pombe (S. pombe), la mayoría de los genes de la proteína-codificación es prescindible para viabilidad2. Este resultado no habla a la insignificancia de estos genes, sino a los intrincados mecanismos compensatorios subyace a las vías bioquímicas a las que pertenecen dichos genes. Estos mecanismos compensatorios de disección ha generado mapas de epistasis, que han descubierto interacciones genéticas integrales y ampliado nuestra comprensión de las vías bioquímicas funcionales3,4.
Métodos de alto rendimiento (por ejemplo, análisis de la matriz genética sintética o SGA) se han desarrollado para identificar las interacciones genéticas genoma en levadura de florecimiento y se han ampliado para su uso en la levadura de fisión5,6. Estos enfoques a menudo dependen de una biblioteca de las cepas que contienen todas viables solo proteína-codificación canceladuras del gene (alrededor 3.300 mutantes de la canceladura haploide cubriendo más del 92% del genoma de la levadura de fisión) y requieren de un brazo robótico para realizar las cruzas genéticas entre las cepa de interés y todas las tensiones posibles en la library6. Además, técnicas SGA dependen de la capacidad de las cepas de la biblioteca para tener acoplamiento adecuado y eficiente, un fenotipo que es anormal a 444 actualmente caracterizado genes en S. pombe2.
A pesar de la complejidad de las interacciones genéticas, comparando el fenotipo de una cepa con mutaciones en dos genes para el fenotipo de dos cepas con mutaciones individuales de cada gen puede tener uno de dos resultados notables: 1) el fenotipo mutante doble es peor que los fenotipos parentales multiplicativos esperados en forma de enfermedad o, en el caso más extremo, mortalidad. Esto se conoce como una interacción genética negativo y es generalmente una señal de que los dos genes actúan en las vías biológicas paralelas. 2) el fenotipo mutante doble es mejor que la esperada combinación de fenotipos de los padres, también conocido como una interacción genética positiva. Una positiva interacción genética es particularmente interesante porque indica que estos genes funcionan en el mismo proceso. Dos genes interactuando positivamente tienen tres relaciones posibles: un gen mutante puede para arriba-regular la expresión de otro gen en una vía paralela, los dos genes pueden trabajar en conjunto dentro de la misma vía aguas abajo de uno con el otro o los dos genes codifican proteínas que interactúan directamente entre sí. Por lo tanto, las interacciones genéticas positivas pueden utilizarse para clasificar genes desacostumbrados en las vías bioquímicas7,8y mapa nodos reguladoras de genes.
Un supresor es una mutación que puede aliviar el fenotipo de la enfermedad de la mutación de otro gen, que normalmente representa una positiva interacción genética entre dos genes9,10. Mutaciones supresoras en un locus diferente de la de la mutación que se suprimen son conocidas como supresores extragenic. Son especialmente valiosos en el estudio de mutaciones genéticas no viables por rescatar sintéticamente el fenotipo letal (también conocido como el efecto Lázaro)11. También tienen potenciales aplicaciones terapéuticas en el tratamiento de enfermedades hereditarias12,13.
Por todas estas razones, la identificación de las mutaciones supresoras en diversos organismos modelo ha sido utilizada ampliamente para facilitar la comprensión de varias vías bioquímicas14,15,16. Proyección para supresores generalmente se basa en el fenotipo de la mutación en cuestión y requiere realizar mutagénesis al azar para aislar las mutaciones que aliviarían el fenotipo. Casi todos los organismos modelo han establecido métodos de mutagénesis al azar. Por ejemplo, N-ethyl-N-nitrosourea (ENU) y ethylmethanesulfonate (EMS), dos mutágenos que son capaces de inducir mutaciones puntuales en el ADN, se emplean extensamente en varios modelos de las bacterias a ratones17,18,19 . Además, cloruro de manganeso se ha utilizado mucho en las levaduras para que la capacidad del catión manganeso para inhibir la ADN reparación vías20. Otro método común es mutagénesis UV-inducido, que genera dímeros de pirimidina mutagénicos genoma21,22.
Aunque la utilización de la mutagénesis química para identificar mutaciones supresoras ha sido popular, el método tiene muchas desventajas, incluyendo el uso de productos químicos peligrosos, las tasas de éxito muy variable y la introducción de variables confusoras extra presentado por los efectos secundarios negativos del mutágeno en múltiples procesos celulares23,24. Además, mutagénesis química a menudo induce múltiples mutaciones en el genoma que se suma a la complejidad de la utilización de genética y técnicas de secuenciación para identificar la mutación exacta que el fenotipo supresor en el organismo25.
Para abordar las deficiencias de los enfoques actuales de la mutagénesis, presentamos un método a la pantalla para las mutaciones espontáneas supresor en la levadura de fisión que no depende de ningún mutágenos o una biblioteca de eliminación. El método aísla a supresores a través de un análisis de selección positiva. El principio de este método se basa en la ventaja del crecimiento de la subpoblación de supresor mutado en la cultura líquida, que puede ser monitoreada por un lector automatizado. Apareamiento y la meiosis se utilizan solamente si uno quisiera limpiar el fondo genético o confirmar la presencia de alelos monogénicas de supresores antes de secuenciación de genoma completo. Si el fenotipo de supresión es causado por una mutación, el fenotipo supresor separe 2:2 después de retrocruzamiento con las cepas parentales. Las mutaciones supresoras pueden identificarse entonces con la secuenciación del genoma completo. Proponemos que este método es aplicable para la detección de supresores en todos los microorganismos que pueden crecer a una gran población en cultivo líquido.
Protocolo
1. colar la preparación y construcción
- Generar una mutación o una canceladura del gene (yfm, su mutación favorito) mediante mutagénesis sitio-dirigida estándar (SDM) como se describe anteriormente26.
- Antes de comenzar la pantalla, retrocruzamiento (óptimo) las cepas mutantes con una cepa de tipo salvaje para limpiar el fondo genético y generar células mutantes recién nacido como las cepas parentales. Estría la cepa parental a colonias individuales en placas de medios enriquecidos estándar. Selección al azar ocho a dieciséis colonias independientes (Replica biológica) con las mutaciones deseadas para el ensayo de placa lector (véase 3.1).
Nota: Este protocolo es eficaz sólo cuando las cepas parentales tienen un crecimiento del defecto en medios líquidos (mínimo o ricos, con o sin drogas o con cambios de temperatura que causan el defecto de crecimiento). Todas las cepas parentales deben ser haploides y por lo tanto capaces de cruzarse genéticamente con otras cepas haploides con un tipo de apareamiento complementario.
2. Análisis de lector de placa
- Con un aplicador estéril, tomar una pequeña cantidad de cada una de las colonias preparadas en el paso 1.1 (no cantidad exacta necesaria para inocular la puesta en marcha de la cultura) y colocar en una microplaca de poliestireno de 96 pocillos. Suspender cada una de las colonias en 200 μL de medio líquido apropiado (ricos o mínima, con o sin drogas). Incluyen un pozo en blanco para cada fila en la placa que contiene 200 μL de los mismos medios (sin células).
- Ejecutar el siguiente protocolo en un software de detección de placa lector conectado a un lector automatizado de microplacas: establecer un programa de cinético para 24 h y la temperatura a 30 ° C, con agitación orbital rápido continuo (cpm 425, amplitud de 3 mm). Establecer que las lecturas ópticas para medir la dispersión de la luz en una longitud de onda de 600 nm para densidad óptica y la luz para leer por debajo de la placa a una frecuencia de lectura de 2 minutos (721 lecturas total durante un período de 24 horas por pozo).
- Después de 24 horas, grabar las lecturas de densidad óptica blanco final (OD600 cubiertas) y utilice la siguiente fórmula para determinar el volumen necesario para diluir cada muestra a O.D. = 0.1:

Nota: Exportar los datos desde el software de lector de la placa y utilizar un software de hoja de cálculo para insertar la fórmula anterior como una función al lote de proceso el volumen de la dilución a utilizar de cada pozo experimental. - Cada 24 h, diluir cada una de las muestras utilizando los mismos medios como día 0 a O.D. = 0.1 (1.5 x 106 células/mL) utilizando la fórmula indicada en el paso 2.3. Guardar todo crecimiento curvas generan diariamente y observe cualquier Colonia individual que muestra una tasa de crecimiento mayor, juzgada por un diámetro externo final que es significativamente mayor que el resto de la cohorte con el mismo fondo genético o por una curva de crecimiento similar a la de colonias de tipo salvaje.
Nota: Este ensayo toma generalmente cerca de 7-14 días. Realizar todos los pasos bajo condiciones estériles.
3. selección de colonias supresor y confirmación del fenotipo.
- Desde el último día de la prueba del lector de placa (paso 2.4), guardar el líquido culturas que tienen una tasa de crecimiento notablemente recuperado, probablemente adquiriendo una mutación supresora que puede aliviar el fenotipo de la mutación de los padres. Transferencia y cultivo líquido de 250 μl a un criotubo que contiene 250 μl de glicerol al 50% de la mezcla. Flash congelar las células en nitrógeno líquido y guardar las cepas en - 80oC indefinidamente.
- Para confirmar que la mutación supresora es un elemento genético heredable, utilizar métodos de cruce genético estándar para cruzar yfm P (para los padres, la cepa utilizada al principio del ensayo la placa lector) con yfm S (para el supresor, la cepa guardada en el extremo de lo análisis de los lectores de la placa). Si la mutación supresora es de hecho un elemento genético heredable, yfm yfm de P × S debe generar tétradas en la que dos colonias tienen el fenotipo de la enfermedad de la cepa parental y dos colonias tienen el crecimiento recuperado del supresor de cepa.
- De la Cruz del paso 3.2, escoger tres colonias con el fenotipo supresor (cepa S) y tres colonias con el fenotipo parental (cepa P) de la misma genética cruzan (3 repeticiones biológicos para cada uno) y proceden con la extracción de ADN genómica y secuencia pasos.
Nota: Pasos 3.2 y 3.3 son muy recomendables, pero no son necesarios. Alternativamente, uno recuperado cultura líquida recogida de 3.1 en medio rico en colonias solo, entonces escoge al azar tres colonias como triplicado biológica para la secuenciación del genoma completo sin más confirmación genética. En este caso, tres triplicado biológica de la cepa parental debe utilizarse para la comparación de la secuencia genomic.
4. genomic DNA extracción, producción de biblioteca y secuenciación.
- Para extracción de ADN, preparación de biblioteca y secuenciación, escoger al azar tres réplicas biológicas por cepa yfm P , y biológico tres réplicas de cada individual presentado yfm S cepas de las cruzas genéticas (paso 3.2) o de la placas que han sido difundidas para obtener solo colonias de la cepa S (paso 3.3 Nota).
- Crecen las cepas en las culturas de 10 mL en medios enriquecidos a fase logarítmica media (D.E. = 0,5 – 0,8, de 0.75-1.2 x 107 células/mL) y utilizar una incubadora de agitación para crecer culturas líquidas a 30 ° C con agitación continua a 250 rpm. Recoger las células por centrifugación a 4 ° C durante 5 minutos a 1000 x g.
- Suspender las células sedimentadas en 400 μL de tampón de extracción de ADN (2% Tritón X-100, 1% SDS, 100 mM NaCl, 10 mM Tris-Cl (pH 8.0), 1 mM2de Na-EDTA), añadir 400 μL de bolitas de cristal y 400 μL de fenol: cloroformo: isoamílico alcohol de 25:24:1. Vortex vigorosamente durante 2 min a 4 ° C.
- Agregar un adicional 200 μL de tampón de extracción de ADN y mezcle por inversión varias veces. Centrifugar 5 min a 4 ° C a 20.000 x g.
- Transferir la fase acuosa a un tubo limpio Añadir 20 μg de mezcla de Rnasa A/T1 e incubar a 37 ° C durante 15 minutos.
- Añadir un volumen igual de alcohol de 25:24:1 fenol: cloroformo: isoamílico, centrifugado durante 5 minutos a 4 ° C a 20.000 x g, luego transferir la fase acuosa a un tubo limpio.
- Añadir un volumen igual de cloroformo, mezclar por inversión varias veces, gira durante 5 min a 4 ° C a 20.000 x g, luego transferir la fase acuosa a un tubo limpio.
- ADN precipitado con dos volúmenes de etanol al 100% y 10% del volumen de 3 M de NaOAC (pH 4.3) a-20 ° C durante al menos 2 h, luego girar durante 5 minutos a 4 ° C a 20.000 x g y recoger el diábolo.
- Lavar el pellet (ADN precipitado) dos veces con etanol al 70% frío (centrifugar a 20.000 x g, 5 min, 4 ° C) y suspender el sedimento en 50 μl de 10 mM de tampón Tris (pH 7.4).
- Usar una preparación de biblioteca kit (véase Tabla de materiales) según las recomendaciones del fabricante para preparar la biblioteca de la secuenciación del genoma completo.
Nota: recomendamos el kit figuran en la Tabla de materiales ya que permite la construcción de la biblioteca genómica sin amplificación por PCR, que minimiza las mutaciones errores generadas durante la amplificación por PCR. Además, durante la preparación de la biblioteca genómica, no permita que los granos a completamente seco al acortar el cordón a 1-2 min el tiempo de secado. - Para los parámetros de corte durante la preparación de la biblioteca, utilizar un sonicador centrada (véase Tabla de materiales) y el factor trabajo al 20%, pico de potencia de 175 W, con 200 ciclos por explosión, y modo barrido de frecuencia a 5.5oC a 6 ° C durante 45 s. alternativamente, utilizar un ADN y chomatin sistema de corte (véase Tabla de materiales) con los siguientes parámetros: amplitud del 50% a 4 ° C con el modo de pulso, 15 s s on y 15 off por 10 min, con un tiempo total de procesamiento de 20 minutos.
- Es esencial para manejar los materiales peligrosos utilizados en este paso con cuidado. Consulte la apropiadas hojas de datos de seguridad Material y de institución salud ambiental y oficina de seguridad para el manejo de NaOAC, etanol, alcohol de fenol: cloroformo: isoamílico 25:24:1 y cloroformo.
- Las bibliotecas genómicas resultantes de la secuencia. La Lee de la secuencia entera debe cubrir por lo menos tres veces de todo el genoma con la resolución en un rango de un solo nucleótido. Se recomienda la secuencia terminó emparejado (o tecnología).
5. bioinformática análisis para la identificación de las mutaciones supresoras
- Realizar el análisis de bioinformática para centrarse en los cambios genómicos que constantemente se identifican entre los padres y suprimido yfm cepas en biológico todas réplicas.
Nota: El proceso de canalización completa se describe a continuación, pero, además, dos-BASH-script archivos de texto plano, fastq_to_vcf.sh y vcfprocess.sh, se incluyen como materiales complementarios para mostrar ejemplos del flujo de trabajo de procesamiento de Lee a la variante VCF archivos y procesamiento y cruce de la VCF archivos, respectivamente. - Trim corta-Lee con cizalla (https://github.com/jbpease/shear) siguiendo las líneas de comandos (todos otra opciones por defecto):
Shear.py--fq1 $FASTQ1--fq2 $FASTQ2 - out1 $OUTFQ1 - out2 $OUTFQ2 \
--barcodes1 $BARCODE--plataforma TruSeq--trimqual 20:20 \
trimpolyat--0--trimambig--filterlength 50--filterunpaired - Mapa lee la referencia de S. pombe genoma v2.30 obtenida de PomBase (ftp://ftp.ebi.ac.uk/pub/databases/pombase/pombe/Chromosome_Dumps/fasta/) usando BWA v0.7.1527. Utilice la siguiente línea de comandos (todos otra opciones por defecto):
BWA mem -t 8 $GENOME $OUTFQ1 $OUTFQ2 > $SAM1 - Poner los archivos de alineación SAM por GATK mejores prácticas tuberías28 para llamar variante usando GATK v3.629, PicardTools v2.5.0 (http://broadinstitute.github.io/picard) y SAMtools v1.3.130. Utilice las siguientes líneas de comandos y parámetros (todos otra opciones por defecto):
Java-Xmx30g-picard.jar AddOrReplaceReadGroups entrada del tarro = $SAM1 \
SALIDA = RGID $BAMMARKED = 1 RGLB = lib01 RGPL = illumina \
RGPU = RGSM $BARCODE = $SAMPLENUMBER
samtools fixmate bam - O $BAMMARKED $BAMFIXED
samtools tipo - O bam -o $BAMSORTED -T /home/peasejb/tmp $BAMFIXED
Índice de samtools $BAMSORTED
Java-Xmx30g-jar GenomeAnalysisTK.jar -T HaplotypeCaller \
-R $GENOME-I $BAMSORTED--genotyping_mode descubrimiento \
-stand_emit_conf 10 - stand_call_conf 30 -o $VCFRAW - Comprimir y ficheros VCF con tabix INDICE:
bgzip $VCFRAW.vcf
Tabix $VCFRAW.vcf.gz - Comparar archivos VCF entre los padres y supresor de cepas repeticiones de la secuencia usando BCFtools v1.3.127. Utilice las siguientes líneas de comandos y parámetros (todos otros opciones izquierda por defecto):
bcftools isec - n + 1 $VCFPARENTAL1.gz $VCFPARENTAL2.gz $VCFPARENTAL3.gz \
$VCFMUTANT1.gz $VCFMUTANT2.gz $VCFMUTANT3.gz > common_variants.list
Nota: Este comando rindió un archivo codificado con patrones binarios en variantes de la secuencia que aparece en el primer mutante sólo sería binario codificado como "000100", el segundo mutante solamente como "000010", todas tres mutantes como "000111," etcetera. Estos archivos fueron generados para cada conjunto de padres y mutante replicar archivos VCF. - Compilar los archivos de lista de intersección variante junto con el nombre de archivo que se anexa a cada línea con el comando grep de UNIX:
grep "." *.list > all.list - Referencias cruzadas de la variante lista completa con el archivo de anotación actual de GFF3 (ftp://ftp.ebi.ac.uk/pub/databases/pombase/pombe/Chromosome_Dumps/gff3/schizosacchar omyces_pombe.chr.gff3) usando un script en Python personalizado (variant_characterize.py) identificar sitios SNP consistente en proteína-codificación regiones (sinónimas y no sinónimas), UTRs 5′ y 3′ y ncRNA.
python3 variant_characterize.py--lista common_variants.list \
schizosaccharomyces_pombe.chr.gff3--gff \
--fasta Schizosaccharomyces_pombe. ASM294v2.30.DNA.Genome.fa \
-000100 - hacia fuera all.list.filter.000100 del patrón
Repita esta secuencia de comandos modifica el patrón y el sufijo del archivo de salida (--a) utilizando el binario
patrones: 000010 000001, 000110, 000011, 000101 y 000111 - Combinar la salida de todos estos funcionamientos de secuencia de comandos en un archivo separados por tabulador, para ser visto como una hoja de cálculo. La tabla anotada de variantes incluye a aquellos que aparecen en uno o los dos cepas mutantes de positivo en relación con el fondo. El campo binario bandera denota cualquier aspecto de una sola cepa mutante (000100, 000010, 000001), dos cepas mutantes (000011, 000101, 000110), o las tres cepas mutantes (000111).
- Analizar las listas de salida anotada de variantes no se encuentran en las muestras de los padres, pero encuentra en uno, dos o las tres muestras mutantes. La anotación denota la localización genómica y clase de variante (sinónimo/no-sinónimas en una región de la codificación, 3' / 5' UTR, no codificante, etcetera.). De la lista de mutaciones de candidato, un ejemplo de un candidatos fuertemente relevante podría ser una variante de codificación no sinónimo que aparecen constantemente en las tres cepas. Otro tipo de candidato fuerte sería una acumulación de varias mutaciones reglamentación no sinónimo o supuestas en las cepas mutantes que aparecen cerca o dentro del mismo gene.
Resultados
Crecimiento lento mutantes muestran recuperación fenotípica en cultivo líquido
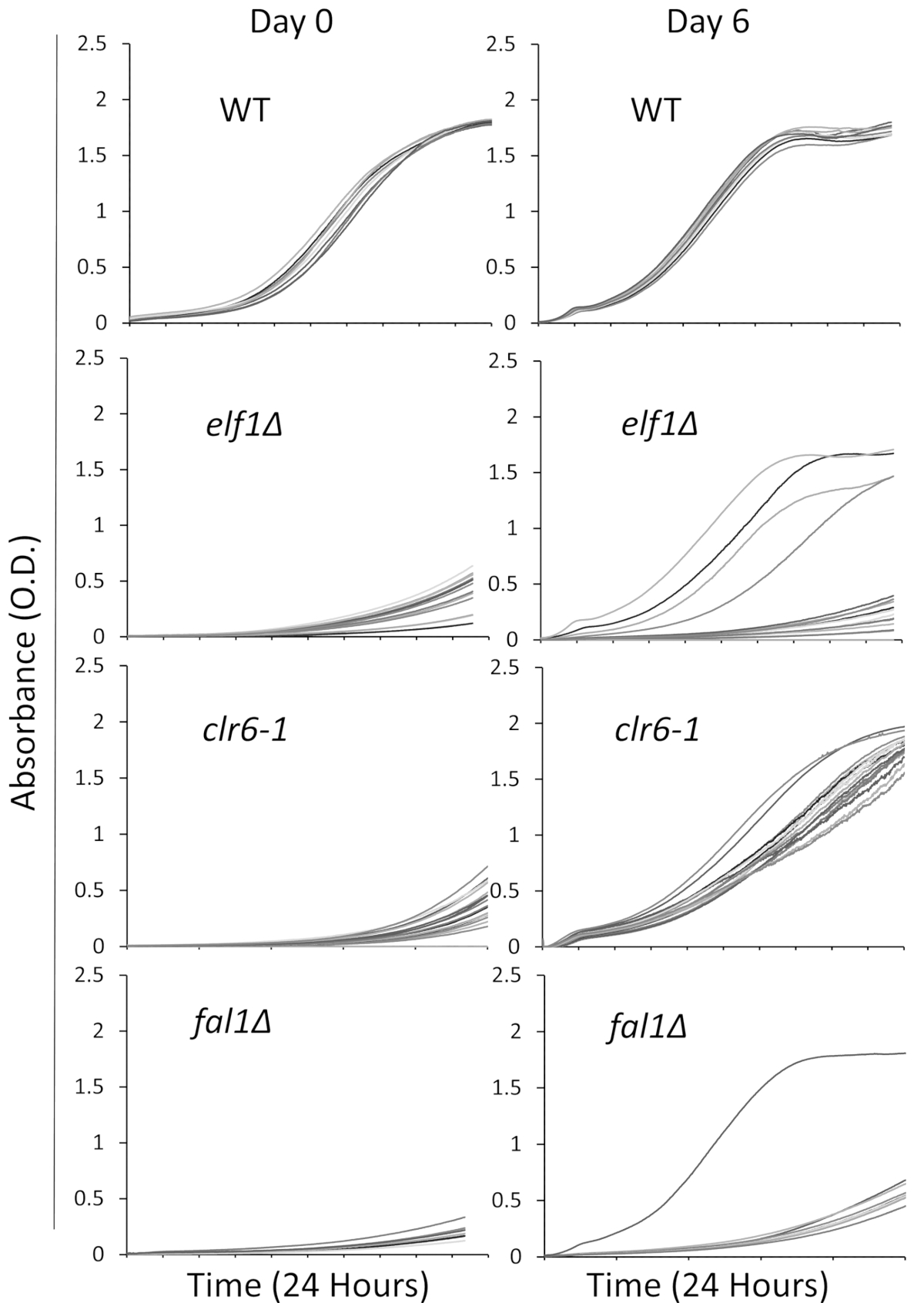
Se seleccionaron tres mutantes implicados en una variedad de vías biológicas con un fenotipo enfermo, crecimiento lento,: AAA familia Elf1 de ATPasa, histona deacetilasa Clr6 y exón cruce complejo componente Fal1. Cepas con mutaciones de estos tres genes que habían sido retrocruzas con cepas de tipo salvaje y una cepa de tipo salvaje se trata a colonias individuales y 16 colonias individuales fueron seleccionadas al azar para ser cultivadas en medios líquidos enriquecidos usando la placa de 96 pocillos como descrito anteriormente. Las curvas de crecimiento de colonias individuales fueron registradas en el momento inicial (día 0) y 6 días con monitorización continua con el lector. Como se esperaba, colonias de tipo no muestran cambios notables en sus curvas de crecimiento a lo largo del experimento31 (figura 1). En particular, cuatro colonias con el fondo de elf1∆ y una colonia de fal1∆ muestran un cambio dramático en el crecimiento de crecimiento lento para algunos niveles variables de crecimiento similar o cercano de las colonias de tipo salvaje. Dramáticamente, todos mutantes clr6-1 muestran una consistente recuperación fenotípica, creciendo a un ritmo más rápido al final del ensayo31 (figura 1). Para caracterizar los fenotipos diferentes, nos referimos a las cepas originales que son de crecimiento lento como "Cepas de P" (o las cepas parentales) y a las cepas mostrando recuperación fenotípica como "Cepas de S" (o suprimida de las cepas). Tenga en cuenta que figura 1 es un ejemplo de una ronda del experimento de la proyección y no representan los supresores no complementarios total identificado y secuenciado en los siguientes resultados representativos.
Recuperación fenotípica se atribuye a los rasgos hereditarios
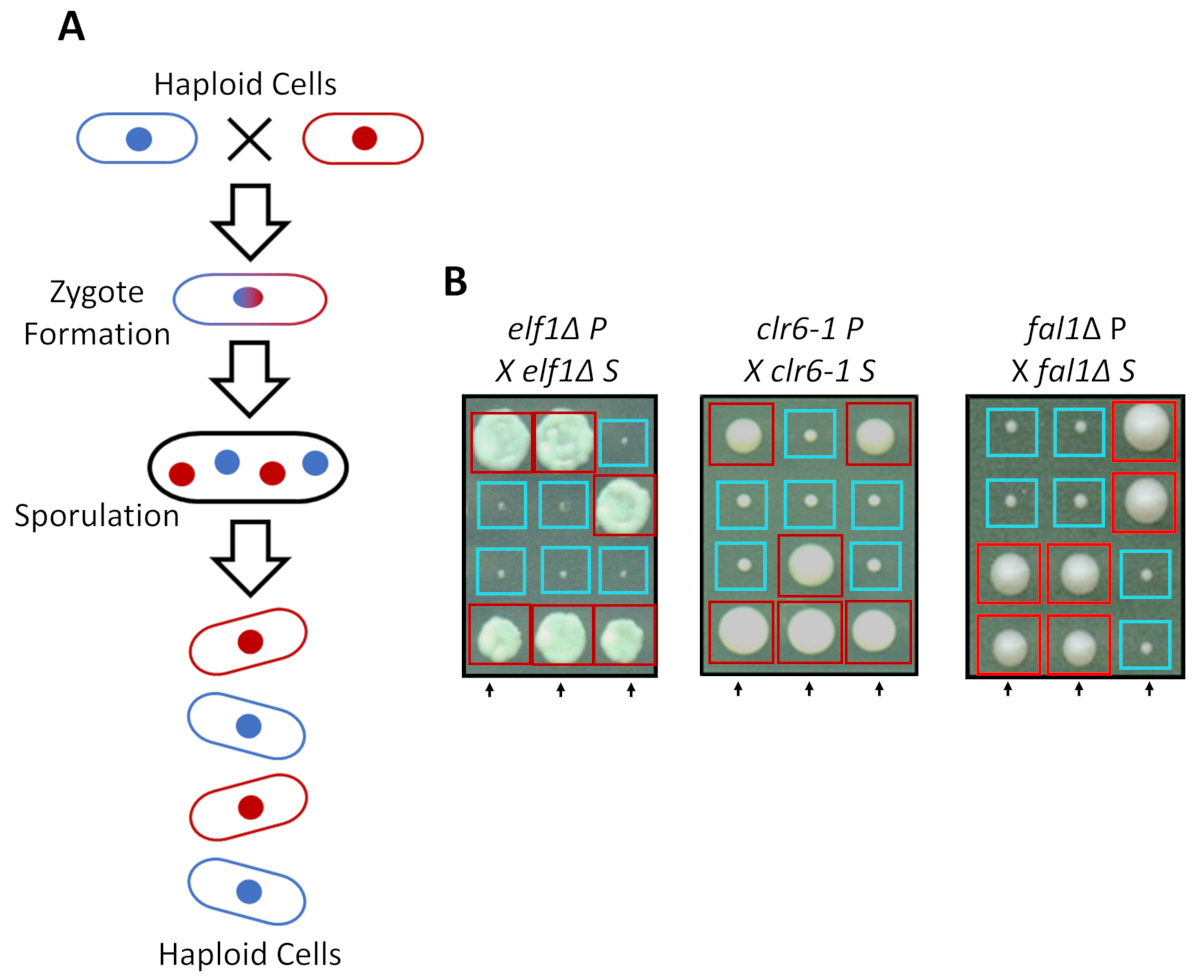
S. pombe puede crecer como un haploid en medios enriquecidos, pero dos cepas haploides complementario apareamiento mate de tipos con hambre de nitrógeno. Meiosis en la levadura de fisión consiste en una ronda de duplicación seguida por dos rondas de división celular. El ciclo sexual se traduce en la formación de cuatro esporas haploides que lleva el material genético de la cepa parental con 2:2 segregación de rasgos genéticos siguiendo las reglas de la genética mendeliana clásica (figura 2A). Cuando se cultiva en el mismo plato por la misma cantidad de tiempo, confirmamos la segregación 2:2 cuando todos cruzamientos suprimieron las cepas (cepas S) con sus cepas parentales (cepas de P), que dio lugar a 2 pequeños (defecto de crecimiento) y 2 grandes (fenotipo supresor) colonias. Ejemplos individuales para suprimidos elf1∆, clr6-1 y fal1∆ las células se muestran en la figura 2B. Hemos confirmado que todas las cepas aisladas de S llevan un elemento genético monogénica que suprime el fenotipo de crecimiento lento de sus cepas de P (datos no mostrados).
Secuenciación del genoma completo con éxito identifica mutaciones supresoras
Por ejemplo, utilizamos secuenciación del genoma entero de extremo apareado para identificar los elementos genéticos responsables de la recuperación fenotípica de cepas de S elf1∆ . Una descripción más completa de análisis de datos está disponible en línea31. Brevemente, se emplean triplicados biológicas de dos cepas de elf1∆ P independientemente generados y duplicados biológicas de cinco grupos no complementarios de elf1∆ S cepas, cada una de ellas contiene supresores de la distintos. Después de que obtuvimos una lista de las variantes anotadas de análisis bioinformáticos (6.1-10), priorizamos ciertas clases de variantes que fueron relevantes para nuestro análisis. Nos hemos centrado en la identificación de constantes cambios genomic que eran idénticos en todas las repeticiones biológicas de cepas de elf1∆ S individuales en comparación con sus cepas parental de elf1∆ P (figura 3 y suplementario tablas 1-4 ). Se identificaron cinco cambios nonsynonymous en regiones CDS en los cinco elf1∆ S cepas diferentes, incluyendo rli1 +, SPBPJ4664.02, cue2 + y rpl2702 +. S-A1 y A2 S contienen transformado SPBPJ4664.02, aunque las mutaciones se producen en diferentes aminoácidos. Porque SPBPJ4664.02 es un gen largo (11.916 nucleótidos) con cientos de repeticiones, las mutaciones no pudieron confirmarse mediante la realización de PCR seguida por la secuencia. S-A3 contiene a un mutante de eliminación en rli1 que es constante en ambos duplicados biológicas. Sin embargo, el mutante no segregar conjuntamente con el fenotipo de S en elf1∆ fondo. Identificamos a un mutante cue2 (cue2-1) en S-B1, con los aminoácidos que faltan 396-400. S-B2 contiene a un mutante rpl2702 (rpl2702-1), que cambia el aminoácido en la posición 45 de glicina aspartato31. Cue2-1 y rpl2702-1 fueron confirmados como supresores de elf1∆ como se muestra a continuación.
Confirmación genética de mutaciones identificadas supresor verifica la heredabilidad del fenotipo de recuperación
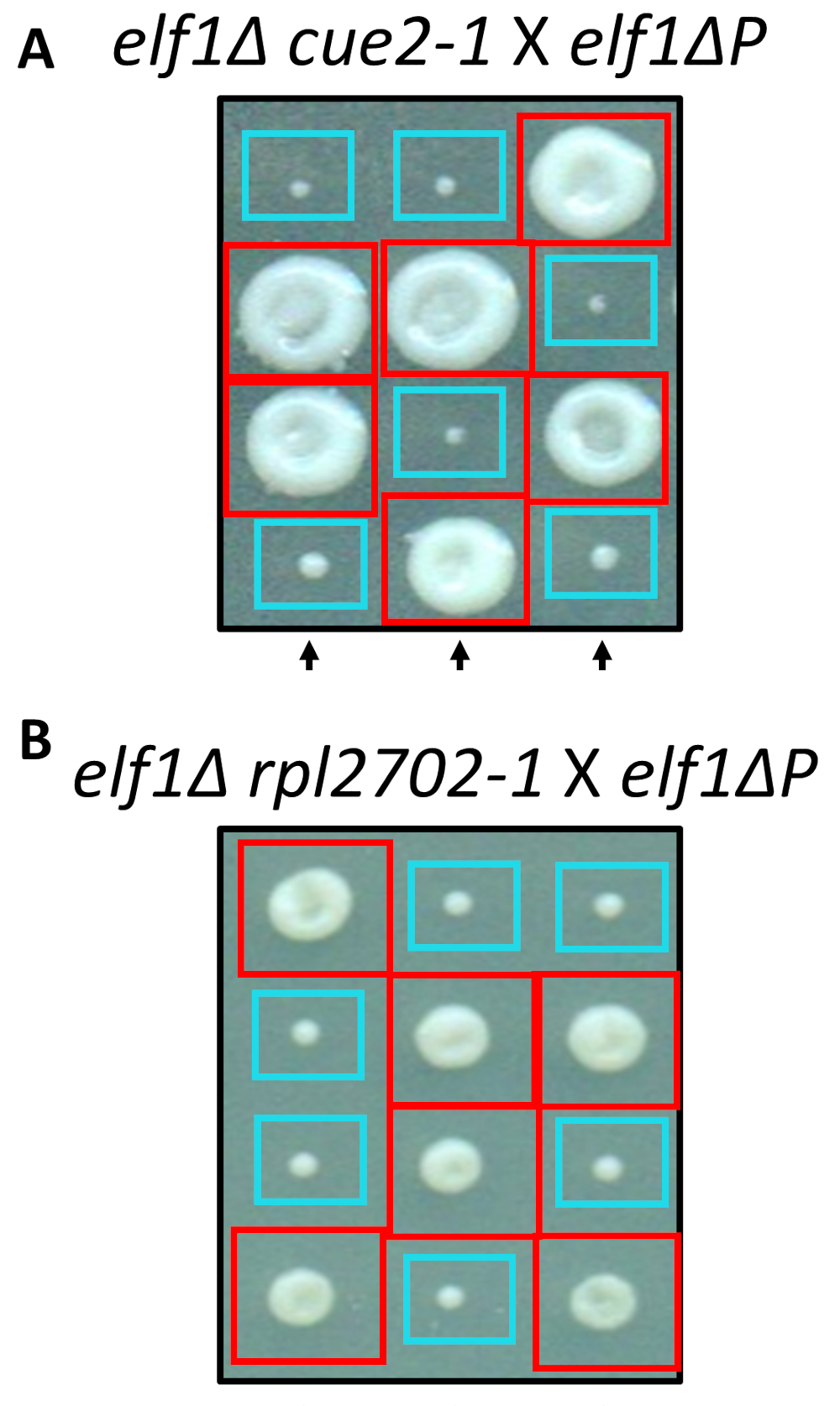
Dos de los identificados cambios nonsynonymous, cue2-1 y rpl2702-1, fueron reconstruidas en el laboratorio utilizando protocolos estándar de mutagénesis sitio-dirigida. Doble mutante cepas cue2 1 P elf1∆ y rpl2702-1 elf1∆ P fueron cruzados con la conexión elf1∆ P cepa31 (figura 4). Si las mutaciones nonsynonymous, identificadas a través de esta pantalla, fueron suficientes para suprimir la P elf1∆, entonces las tétradas resultantes mostraría 2:2 pequeñas y gran relación en las colonias resultantes de las 4 esporas en cada tétrada. De hecho, cruce genético demostró que las mutaciones supresoras identificados son exitosas en la supresión del fenotipo de crecimiento lento de P elf1∆ y son heredables.

Figura 1: recuperación fenotípica puede controlarse mediante el registro de las curvas de crecimiento en un lector de placas. Dieciséis colonias individuales de tipo salvaje (WT), elf1∆, clr6-1y fal1∆ se colocaron en una placa de 96 pocillos. Las curvas de crecimiento se registraron durante un período de 24 horas y las colonias fueron nuevamente diluidas en medios enriquecidos. El defecto de crecimiento es evidente por la absorbencia baja (O.D.) al final del período de 24 horas en el día 0. Cepas fenotípicamente recuperadas son los que muestran una curva de crecimiento similar, o cerca, la de tipo salvaje durante el período de 24 horas el día 6. Cuatro colonias de elf1∆, una colonia de fal1∆y todas las colonias de clr6-1 mostraron varios niveles de recuperación fenotípica después de 6 días. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 2: cruce genético puede confirmar que la recuperación fenotípica se atribuye a un solo alelo heredable. (A) cuando las células sufren hambre de nitrógeno, dos células haploides con un tipo de apareamiento complementario de la levadura de fisión puede generar un cigoto que sporulates para generar una tétrada de 4 esporas. El material genético paterno se segregan durante la meiosis siguiendo las reglas de la genética mendeliana. (B) recuperado fenotípicamente colonias (S etiquetados, para suprimido) con indican genotipos parentales fueron cruzado con su Colonia los padres gratuito (que no demuestra ninguna recuperación fenotípica, con la etiqueta P, para parental). Genéticas cruces mostrando pequeñas 2:2 (pobre aptitud) a colonias grandes (fitness recuperados) demuestran que la recuperación fenotípica es heredable y puede atribuirse a un solo elemento genético. Cajas rojas son colonias llevan el alelo de supresor, y cajas azules son colonias llevan el alelo paterno. Esta figura se ha modificado desde Marayati et al., 201831. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 3: Análisis de los datos de secuenciación del genoma para identificar elementos genéticos responsables de la recuperación fenotípica. Biológico de tres réplicas de dos cepas parentales de la "P" (P A y P-B), y dos réplicas biológicas de cinco recuperado fenotípicamente las cepas "S" (S-A1 y A2 S S - A3 recuperaron de P-A; S-B1 y S-B2 de P-B), fueron ordenados y las mutaciones fueron organizadas como una lista de cada mutación en la cepa recuperada en comparación con el genoma de la cepa parental fue derivado (por ejemplo, P-A VS S-A1, etcetera). El número total de mutaciones detectadas a través de todo el genoma de todas tales comparaciones pares fue 660. Se identificó un total de 44 mutaciones cuando se seleccionaron solo las mutaciones que se producen en ambas réplicas biológicas de la misma cepa de la "S". De 44 mutaciones, 12 mutaciones eran inserción/deleción (INDEL) o no mutaciones sinónimas. Fuera de las 12 mutaciones no sinónimas o INDEL, cinco ocurrieron en la secuencia de codificación de la proteína. Las cinco mutaciones potencialmente se correlacionan con el elemento genético responsable de las cepas fenotípicamente recuperadas: una mutación no sinónima en SPBPJ4664.02 encuentra en S-A1 y S-A2, INDEL en rli1 en S-A3, INDEL en cue2 en S-B1 y las mutaciones no sinónimas en rpl2702 en S-B2. Secuencia detallada información sobre las mutaciones y el fondo filtrado está incluido en el suplemento tablas 1-4. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 4: confirmación de los supresores identificados a través de la secuenciación del genoma completo. Resultados de la secuenciación del genoma completo fueron confirmados independientemente generando las mutaciones y realizar cruces genéticos para confirmar la recuperación fenotípica al cruzar una variedad elf1∆ cue2-1 con una cepa de P elf1∆ y elf1∆ rpl2702-1 elf1∆ P la tensión. Se muestran tres tétradas vertical representativas. Cajas rojas son colonias doble mutante (elf1 cue2-1o elf1 rpl2702-1); cajas azules son colonias de elf1∆ . Esta figura se ha modificado desde Marayati et al., 201831. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Cuadro suplementario 1. Haga clic aquí para descargar esta tabla.
Tabla 2 suplementaria . Haga clic aquí para descargar esta tabla.
3 tabla suplementaria . Haga clic aquí para descargar esta tabla.
4 tabla suplementaria . Haga clic aquí para descargar esta tabla.
Codificación de archivos suplementarios . Haga clic aquí para descargar los archivos.
Discusión
El protocolo descrito aquí representa un novedoso y simple pantalla para las mutaciones espontáneas supresor detectables mediante recuperación fenotípica de mutaciones que confieren un crecimiento lento en la levadura de la fisión, un fenotipo característico de los más de 400 genes en S. pombe, la función de muchos de los cuales permanece desconocido2,32. Los métodos anteriores han adoptado otros métodos para detectar las mutaciones supresoras en microorganismos, incluyendo el uso de mutágenos21, o la aplicación de un cambio de temperatura en entornos mutantes sensibles a la temperatura33. Por el contrario, este protocolo muestra que fenotípica recuperación ocurra sin interferencia química ambiental adicional y destaca la ventaja de la aptitud de la subida de las mutaciones de supresión eventualmente hacerse cargo de los recursos disponibles en líquido cultura. Esta pantalla permite el aislamiento de supresores de bypass o supresores de interacción ya es efectiva para ambas mutaciones de pérdida de función como elf1∆ o fal1∆ y las mutaciones de punto como clr6-1, mientras que los mutantes demostrar aptitud defectos en cultivo líquido.
Hasta ahora, las cepas recuperadas de S que hemos investigado han demostrado diversos grados de recuperación fenotípica. Según lo detectado por cruce genético, el fenotipo recuperado es atribuible a un solo elemento genético y es heredables (los ejemplos que se muestra en la figura 2). Esta es una de las ventajas más significativas de este método en comparación con pantallas supresor químico o UV, que a menudo múltiples loci genómicos. Es común observar uno o dos colonias recuperadas de 16 colonias, cepa (alrededor del 10%) dentro de una semana. Sin embargo, notar que algunos mutantes, tales como la pérdida de función de Rrp6, la subunidad de exosomas nucleares específicos, nunca se recuperó a la tasa de crecimiento de tipo casi salvaje observaron en células de elf1Δ 31. Es probable que la función de Rrp6 sólo puede ser compensada parcialmente por supresores, a diferencia de la función de los otros mutantes probado, incluyendo fal1∆, que fue demostrada para causar un severo defecto meiótico a través de su importante función en la regulación empalme de34. Creemos que supresor alternativo métodos de cribado sería sujeto al mismo problema cuando yfg tiene funciones únicas, no se puede reemplazar en el crecimiento de la célula.
Antes de realizar la secuencia genomic, es óptimo a la espalda-Cruz las colonias fenotípicamente recuperadas, identificadas desde el lector de placas, con las cepas parentales para aclarar el fondo genético y para obtener réplicas biológicas. Además, la secuenciación de genoma completo profundo identifica cientos de cambios de un solo nucleótido, la mayoría de los cuales no es idéntica entre las réplicas biológicas que son de poco interés para la investigación. Por ejemplo, encontramos un total de 660 alteraciones genómicas en todos los tres cromosomas entre las dos elf1Δ P y las cinco cepas diferentes de S (figura 3). No comúnmente observamos las mutaciones idénticas entre biológicos secuenciados réplicas de cada cepa, sugiriendo que durante el cultivo de células de elf1Δ antes de la construcción de la biblioteca genómica pueden aparecer nuevas mutaciones o errores al azar pueden ser introducidos durante la construcción de la biblioteca y la secuenciación. Por lo tanto, aislando mutaciones que son consistentes a través de repeticiones biológicas es un aspecto importante en la identificación exitosa de supresores mediante secuenciación de genoma completo.
Se identificaron y confirmaron dos supresores en las regiones CDS en cinco cepas secuenciadas de S. Aunque se detectaron mutaciones en SPBPJ4664.02 en S-A1 y A2 S cepas, es improbable que SPBPJ4664.02 sea un supresor válido ya que S-A1 y A2 S no contienen un supresor en el mismo gen como no son complementarias entre sí ( datos no mostrados). Nosotros también no confirmaron rli1 en S-A3, que no segregan conjuntamente con el fenotipo de S cuando retrocruzas con elf1Δ. Por otra parte, se encontraron mutaciones específicas en regiones no codificantes en S-A1, S-A2 y A3 S. Es posible que estas regiones genómicas no codificante alteradas aliviar el fenotipo de elf1Δ , que será tratado en nuestros estudios futuros. En comparación con los métodos tradicionales como un ensayo de acoplamiento, que puede tomar años para una mutación genética, se identificaron dos supresores dentro de dos meses después de confirmar que un elemento monogénica había causada el fenotipo de S. Con el rápido desarrollo de la tecnología de secuenciación de genoma completo, somos optimistas que este método será más eficiente para identificar mutaciones genéticas consistentes en un futuro previsible.
En Resumen, este protocolo proporciona instrucciones paso a paso para identificar con éxito las mutaciones supresoras para cualquier gen de interés con un defecto de crecimiento lento en cultivo líquido. La simplicidad de este ensayo permite la proyección a gran escala de múltiples fondos genéticos de interés con poco entrenamiento práctico. Hay espacio para más automatizar el proceso usando un robot de manejo de líquidos para realizar las diluciones diario. Manipulación de laboratorio de microorganismos requiere inevitablemente el crecimiento en cultivo líquido, un proceso que es inherentemente selectivo para fitness, proponemos que este protocolo se puede aplicar ampliamente a otros organismos de gran población modelo tales como bacterias y otras especies de levadura.
Divulgaciones
Los autores no declaran aprobaciones por los fabricantes de los instrumentos utilizados en este método y no intereses financieros en competencia.
Agradecimientos
Este trabajo fue financiado por el Instituto Nacional del General médica Ciencias, becas 1R15GM119105-01 a K.Z. Agradecemos a todos los revisores de perspicaces observaciones. También agradecemos a James Tucker, Alicia Anderson, Elizabeth negro y Glen Marrs para la discusión y comentarios sobre este manuscrito.
Materiales
| Name | Company | Catalog Number | Comments |
| Adenine, Powder | Acros Organics | 147441000 | Use at 75 mg/L to make liquid and solid rich media (YEA) |
| Bacteriology Petri Dish | Corning, Falcon | C351029 | 100 ×15 mm, use to grow strains to single colonies on solid rich media |
| D-Glucose Anhydrous, Powder | Fisher Chemical | D16-1 | Use at 30 g/L to make liquid and solid rich media (YEA) |
| Difco Agar, Granuated | Becton, Dickinson and Co. | 214530 | Use at 20 g/L to make solid rich media (YEA) |
| DNA extraction buffer | 2% Triton X-100, 1% SDS, 100 mM NaCl, 10 mM Tris-Cl (pH 8.0), 1mM Na2-EDTA | ||
| Focused-ultrasonicator | Covaris Inc. | S220 | Alternatively, use QSonica Q800R sonicator/DNA and chromatin shearing system |
| Gen5 Data Collection and Analysis Software | Biotek, Inc. | GEN5SECURE | Or equivalent, must be compatible with the micro-plate reader, use to export data readings from the micro-plate reader |
| Hydrochloric Acid 1N, Liquid | Fisher Chemical | SA48-4 | Use to adjust pH to 5.5 in liquid and solid rich media |
| Liquid Rich Media (liquid YEA) | 30 g/L D-Glucose, 5 g/L Yeast Extract, 75 mg/L Adenine, pH adjusted to 5.5 with 1 M HCl | ||
| Microplate Reader, Synergy H1 Hybrid Multi-Mode Reader | Biotek, Inc. | BTH1MG | Or equivalent, must read visible light at 600 nm wavelength range |
| Rich Media agar plates (YEA plates) | 30 g/L D-Glucose, 5 g/L Yeast Extract, 75 mg/L Adenine, 20 g/L Agar, pH adjusted to 5.5 with 1 M HCl. | ||
| RNase A/T1 mix | Thermo Fisher Scientific | EN0551 | Use according to manufacturer recommendation |
| Sterile Polystyrene Inoculating Loop | Corning, Inc. | OS101 | Or equivalent, use to transfer colonies from agar plates to 96-well plate |
| Sterile workspace and burners | |||
| Tissue Culture Plate, 96-well Optical Flat Bottom with Low Evaporation Lid | Corning, Falcon | C353072 | Or equivalent, must have optical flat bottom for micro-plate ready |
| TruSeq DNA PCR-Free LT/HT Library Prep Kit | Illumina, Inc. | 20015962 | Use to prepare the whole-genome sequencing library |
| Yeast Extract, Powder | Fisher Chemical | BP1422-500 | Use at 5 g/L to make liquid and solid rich media (YEA) |
Referencias
- McKay, J. K., Latta, R. G. Adaptive population divergence: Markers, QTL and traits. Trends in Ecology and Evolution. 17 (6), 285-291 (2002).
- Wood, V., Harris, M. A., et al. PomBase: A comprehensive online resource for fission yeast. Nucleic Acids Research. 40 (D1), (2012).
- de Visser, J. A. G. M., Cooper, T. F., Elena, S. F. The causes of epistasis. Proceedings of the Royal Society B: Biological Sciences. 278 (1725), 3617-3624 (2011).
- Sailer, Z. R., Harms, M. J. Detecting high-order epistasis in nonlinear genotype-phenotype maps. Genetics. 205 (3), 107911088(2017).
- Kuzmin, E., Costanzo, M., Andrews, B., Boone, C. Synthetic genetic arrays: Automation of yeast genetics. Cold Spring Harbor Protocols. 2016 (4), 326-332 (2016).
- Tong, A. H. Y., Boone, C. Synthetic genetic array analysis in Saccharomyces cerevisiae. Methods in Molecular Biology. 313 (1), 171-192 (2006).
- Dixon, S. J., Costanzo, M., Baryshnikova, A., Andrews, B., Boone, C. Systematic Mapping of Genetic Interaction Networks. Annual Review of Genetics. 43 (1), 601-625 (2009).
- Boone, C., Bussey, H., Andrews, B. J. Exploring genetic interactions and networks with yeast. Nature Reviews Genetics. 8 (6), 437-449 (2007).
- Bai, X., Yang, Z., Jiang, H., Lin, S., Zon, L. I. Genetic suppressor screens in haploids. Methods in Cell Biology. , 129-136 (2011).
- Manson, M. D. Allele-specific suppression as a tool to study protein-protein interactions in bacteria. Methods. 20 (1), 18-34 (2000).
- Motter, A. E., Gulbahce, N., Almaas, E., Barabási, A. L. Predicting synthetic rescues in metabolic networks. Molecular Systems Biology. 4, 168(2008).
- Peterson, R. T., Shaw, S. Y., et al. Chemical suppression of a genetic mutation in a zebrafish model of aortic coarctation. Nature Biotechnology. 22 (5), 595-599 (2004).
- Giorgini, F., Guidetti, P., Nguyen, Q., Bennett, S. C., Muchowski, P. J. A genomic screen in yeast implicates kynurenine 3-monooxygenase as a therapeutic target for Huntington disease. Nature Genetics. 37 (5), 526-531 (2005).
- Forsburg, S. L., Patton, E., et al. The art and design of genetic screens. Nature reviews. Genetics. 2 (9), 659-668 (2001).
- Johnston, D. S. The art and design of genetic screens. Genetics. 3 (March), 176-188 (2002).
- Jorgensen, E. M., Mango, S. E. The art and design of genetic screens: Caenorhabditis elegans. Nature Reviews Genetics. 3 (5), 356-369 (2002).
- Gocke, E., Müller, L. In vivo studies in the mouse to define a threshold for the genotoxicity of EMS and ENU. Mutation Research - Genetic Toxicology and Environmental Mutagenesis. 678 (2), 101-107 (2009).
- Suzuki, T., Hayashi, M., et al. A comparison of the genotoxicity of ethylnitrosourea and ethyl methanesulfonate in lacZ transgenic mice (Muta(TM)Mouse). Mutation Research - Genetic Toxicology and Environmental Mutagenesis. 395 (1), 75-82 (1997).
- Uttam, J., Alberico, C., De Stasio, E. ENU Mutagenesis. International C. elegans Meeting. , (1995).
- Putrament, A., Baranowska, H., Ejchart, A., Prazmo, W. Manganese Mutagenesis in Yeast. Methods in Cell Biology. 20, 25-34 (1978).
- Bose, J. L. Chemical and UV mutagenesis. Methods in Molecular Biology. 1373, 111-115 (2016).
- Ikehata, H., Ono, T. The Mechanisms of UV Mutagenesis. Journal of Radiation Research. 52 (2), 115-125 (2011).
- Shrivastav, N., Li, D., Essigmann, J. M. Chemical biology of mutagenesis and DNA repair: cellular responses to DNA alkylation. Carcinogenesis. 31 (1), 59-70 (2010).
- De Stasio, E. A., Dorman, S. Optimization of ENU mutagenesis of Caenorhabditis elegans. Mutation Research - Genetic Toxicology and Environmental Mutagenesis. 495 (1-2), 81-88 (2001).
- Probst, F. J., Justice, M. J. Mouse mutagenesis with the chemical supermutagen ENU. Methods in Enzymology. 477 (C), 297-312 (2010).
- Bähler, J., Wu, J. Q., et al. Heterologous modules for efficient and versatile PCR-based gene targeting in Schizosaccharomyces pombe. Yeast. 14 (10), 943-951 (1998).
- Li, H., Durbin, R. Fast and accurate short read alignment with Burrows – Wheeler transform. Bioinformatics. 25 (14), 1754-1760 (2009).
- Van der Auwera, G. A., Carneiro, M. O., et al. From fastQ data to high-confidence variant calls: The genome analysis toolkit best practices pipeline. Current Protocols in Bioinformatics. 43, 11.10.1-11.10.33 (2013).
- Mckenna, A., Hanna, M., et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Research. 20, 1297-1303 (2010).
- Li, H., Handsaker, B., et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 25 (16), 2078-2079 (2009).
- Marayati, B. F., Drayton, A. L., et al. Loss of Elongation-Like Factor 1 Spontaneously Induces Diverse, RNase H-Related Suppressor Mutations in Schizosaccharomyces pombe. Genetics. 209 (4), 967-981 (2018).
- Harris, M. A., Lock, A., Bähler, J., Oliver, S. G., Wood, V. FYPO: The fission yeast phenotype ontology. Bioinformatics. 29 (13), 1671-1678 (2013).
- Xu, X., Wang, L., Yanagida, M. Whole-Genome Sequencing of Suppressor DNA Mixtures Identifies Pathways That Compensate for Chromosome Segregation Defects in Schizosaccharomyces pombe. G3: Genes|Genomes|Genetics. 8 (3), 1031-1038 (2018).
- Marayati, B. F., Hoskins, V., et al. The fission yeast MTREC and EJC orthologs ensure the maturation of meiotic transcripts during meiosis. RNA. 22 (9), 1349-1359 (2016).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados