
Method Article
Uma tela de supressor de Deep-sequenciamento-assistida, espontânea na fissão levedura Schizosaccharomyces pombe
Neste Artigo
Resumo
Apresentamos um protocolo de tela simples supressor em fissão fermento. Este método é eficiente, livre de mutagênico e seletiva para as mutações que ocorrem frequentemente em um único locus genômico. O protocolo é apropriado para isolar supressores que aliviar defeitos de crescimento em cultura líquida que são causados por uma mutação ou uma droga.
Resumo
Uma tela de genética para alelos mutantes que suprimir defeitos fenotípicos causados por uma mutação é uma poderosa abordagem para identificar genes que pertencem a caminhos bioquímicos estreitamente relacionados. Métodos anteriores, como a matriz genética sintético (SGA) análise e técnicas de mutagênese aleatória usando radiação ultravioleta (UV) ou produtos químicos como Metanossulfonato de etila (EMS) ou N-etil-N-Nitrosoureia (ENU), têm sido amplamente utilizados, mas muitas vezes são caros e laborioso. Além disso, estes métodos de triagem baseada em agente mutagénico são frequentemente associados a efeitos colaterais graves no organismo, induzindo várias mutações que aumentar a complexidade de isolar os supressores. Aqui, apresentamos um protocolo simples e eficaz para identificar as mutações supressor em mutantes que conferem um defeito de crescimento em Schizosaccharomyces pombe. A aptidão de células com uma deficiência de crescimento em meio líquido rico padrão ou meios líquidos sintéticos pode ser monitorada para recuperação utilizando um leitor de placa de 96 poços automatizados durante um período prolongado. Uma vez que uma célula adquire uma mutação supressor na cultura, seus descendentes outcompete aqueles das células parentais. As células recuperadas que têm uma vantagem competitiva crescimento sobre as células dos pais podem ser isoladas e retrocruzadas com as células parentais. As mutações do supressor então são identificadas usando o sequenciamento do genoma de todo. Usando essa abordagem, isolamos vários supressores que atenuar os defeitos de crescimento grave causados pela perda de Elf1, uma família AAA + ATPase que é importante no transporte do RNAm nuclear e a manutenção da estabilidade genômica com êxito. Existem atualmente mais de 400 genes em S. pombe com mutantes, conferindo um defeito de crescimento. Como muitos destes genes são descaracterizados, propomos que o nosso método apressarei a identificação do romance interações funcionais com esta abordagem amigável, alta produtividade.
Introdução
A base da compreensão de relações funcionais entre genes depende a capacidade de identificar o mecanismo (s) molecular pelo qual complexos traços genéticos divergem para produzir fenótipos diversos1. No fermento de fissão, Schizosaccharomyces pombe (S. pombe), a maioria dos genes codificantes de proteínas é dispensável para a viabilidade de2. Este resultado não fala para a insignificância destes genes, mas sim para os intrincados mecanismos compensatórios subjacentes os caminhos bioquímicos ao qual pertencem tais genes. Dissecar esses mecanismos compensatórios gerou mapas de epistasia, que tem descoberto abrangentes interações genéticas e ampliou a nossa compreensão das vias bioquímicas funcional3,4.
Métodos de alta produtividade (por exemplo, análise sintética matriz genética, ou SGA) foram desenvolvidos para identificar todo o genoma interações genéticas em leveduras brotamento e expandiram-se para uso em fissão fermento5,6. Tais abordagens muitas vezes dependem de uma biblioteca de cepas que contém todos os viável único-codificação da proteína do gene exclusões (cerca de 3.300 mutantes de exclusão haploides cobrindo mais de 92% do genoma de leveduras de fissão) e exigem um braço robótico para realizar os cruzamentos genéticos entre os estirpe de interesse e todas as cepas possíveis no library6. Além disso, técnicas SGA dependem a capacidade de estirpes de biblioteca de ter adequada e eficiente de acasalamento, um fenótipo que é anormal para 444 atualmente caracterizada genes em S. pombe2.
Apesar da complexidade das interações genéticas, comparar o fenótipo de uma estirpe carregando mutações em dois genes para o fenótipo de duas linhagens carregando mutações individuais de cada gene pode ter um dos dois resultados notáveis: 1) é o fenótipo mutante duplo pior do que o esperado fenótipos parentais multiplicativos em forma de doença ou, no caso mais extremo, letalidade. Isto é referido como uma interação genética negativa e é geralmente um sinal de que os dois genes agem em paralelas vias biológicas. 2) o fenótipo mutante duplo é melhor do que a esperada combinação de fenótipos parentais, também conhecido como uma interação positiva e genética. Uma interação positiva e genética é particularmente interessante porque indica que estes genes funcionam no mesmo processo. Dois genes interagindo positivamente têm três relações possíveis: um gene mutante pode acima-regula a expressão do outro gene em um caminho paralelo, dois genes podem trabalhar em conjunto dentro da mesma via a jusante de um outro, ou os dois genes que codificam proteínas que interagem diretamente com o outro. Portanto, interações genéticas positivas podem ser usadas para mapear nós regulamentar de gene e classificar descaracterizados genes em vias bioquímicas de7,8.
Um supressor é uma mutação que pode aliviar o fenótipo da doença da mutação de um gene, geralmente representando uma interação genética positiva entre os dois genes9,10. Mutações de supressor em um locus diferente da mutação, suprime são conhecidas como supressores extragenic. Eles são especialmente valiosos no estudo de mutações genéticas não viáveis salvando sinteticamente o fenótipo letal (também conhecido como o efeito de Lazarus)11. Eles também têm potenciais aplicações terapêuticas no tratamento de doenças hereditárias12,13.
Por todas estas razões, a identificação de mutações de supressor em vários organismos modelo tem sido amplamente utilizada para facilitar a nossa compreensão de várias vias bioquímicas14,15,16. Triagem para supressores baseia-se geralmente o fenótipo da mutação em questão e requer a realização de mutagênese aleatória para isolar as mutações que aliviaria o fenótipo. Quase todos os organismos modelo estabeleceram métodos de mutagênese aleatória. Por exemplo, N-etil-N-Nitrosoureia (ENU) e ethylmethanesulfonate (EMS), dois agentes mutagénicos que são capazes de induzir mutações de ponto no DNA, são amplamente utilizados em vários modelos de bactérias para ratos17,18,19 . Além disso, cloreto de manganês tem sido muito utilizado em leveduras para a capacidade do cátion manganês para inibir o DNA reparação caminhos20. Outra abordagem comum é mutagênese induzida por UV, o que gera todo o genoma de pirimidina mutagénicas dímeros21,22.
Embora a utilização de mutagênese química para identificar mutações supressor tem sido popular, o método tem muitas desvantagens, incluindo a utilização de produtos químicos perigosos, as taxas de sucesso muito variável e a introdução de variáveis de confundimento extra apresentado pelos efeitos colaterais negativos do mutagênico em vários processos celulares23,24. Além disso, mutagênese química induz frequentemente várias mutações no genoma que aumenta a complexidade do uso genética e técnicas de sequenciamento para identificar a mutação exata que confere o fenótipo supressor no organismo25.
Para resolver as deficiências dos métodos atuais de mutagénese, apresentamos um método de tela para as mutações espontâneas supressor no fermento de fissão que não dependem de qualquer mutagénicas ou uma biblioteca de exclusão. O método isola supressores através de um ensaio de selecção positiva. O princípio deste método baseia-se a vantagem de crescimento da subpopulação de supressor mutado na cultura do líquido, que pode ser monitorizada por um leitor automatizado. Acasalamento e meiose são usados somente se uma gostaria de limpar o fundo genético ou confirmar a presença de alelos monogénicas de supressores antes de sequenciamento do genoma de todo. Se o fenótipo de supressão é causado por uma mutação única, o fenótipo do supressor vai segregar 2:2 após retrocruzamento com as linhagens parentais. As mutações do supressor então podem ser identificadas usando o sequenciamento do genoma de todo. Propomos que este método é aplicável para a seleção de supressores em todos os microorganismos que podem crescer para uma grande população em cultura líquida.
Protocolo
1. tensão de construção e preparação
- Gere uma mutação ou uma deleção do gene (yfm, sua mutação favorita), usando padrão mutagenesis local-dirigido (SDM), conforme descrito anteriormente,26.
- Antes de iniciar a tela, backcross (idealmente) as cepas mutantes de uma estirpe de tipo selvagem para limpar o fundo genético e gerar as células mutantes recém-nascido como as linhagens parentais. Raia da estirpe parental para colônias individuais em placas padrão de mídia avançada. Aleatoriamente escolhe oito ou 16 colônias independentes (Replica biológica) com as mutações desejadas para o ensaio de leitor de placa (ver ponto 3.1).
Nota: Este protocolo é eficaz somente quando as linhagens parentais têm um crescimento de defeitos em meio líquido (mínimo ou rico, com ou sem drogas ou com mudanças de temperatura que causam o defeito do crescimento). Todas as cepas parentais devem ser haploides e assim poder ser geneticamente cruzado com outras estirpes haploides com um tipo de acasalamento complementar.
2. ensaio de leitor de placa
- Com um aplicador estéril, pegue uma pequena quantidade de cada uma das colónias preparadas no passo 1.1 (não cultura nenhuma quantidade exata necessária para inocular o ponto de partida) e coloque em uma microplaca de poliestireno de 96 poços. Suspenda a cada uma das colónias em 200 µ l de meio líquido adequado (rico ou mínimo, com ou sem drogas). Incluem um poço em branco para cada linha na placa contendo 200 µ l da mesma mídia (sem pilhas).
- Executar o seguinte protocolo em um software de detecção de leitor de placa conectado a um leitor de microplacas automatizado: definir um programa cinético para 24h e a temperatura em 30 ° C, com contínua agitação orbital rápido (425 cpm, amplitude de 3 mm). Definir o lê ótico para medir a dispersão de luz no comprimento de onda de 600nm de densidade óptica e definir a luz para ler abaixo a placa em uma frequência de leitura de 2 min (721 leituras totais durante um período de 24 horas por alvéolo).
- Depois de 24 h, gravar as leituras da densidade óptica branco final (OD600 branco) e use a seguinte fórmula para determinar o volume necessário para diluir cada uma das amostras até O.D. = 0.1:

Nota: Os dados de exportação a partir do software de leitor de placa e usar um software de planilha para inserir a fórmula acima como uma função de lote processar o volume de diluição deve ser usado em cada poço experimental. - Cada 24 h, diluir cada uma das amostras usando a mesma mídia como dia 0 para OD = 0.1 (cerca de 1,5 x 106 células/mL) usando a fórmula indicada na etapa 2.3. Salve todo crescimento curvas geradas diariamente e anote qualquer colônia individual que mostra uma taxa de aumento do crescimento, julgada por uma overdose de final que é significativamente maior do que o resto da coorte com o mesmo fundo genético ou por uma curva de crescimento que é semelhante da colônias do tipo selvagem.
Nota: Este ensaio geralmente leva cerca de 7-14 dias. Execute todas as etapas em condições estéreis.
3. seleção de colônias supressor e confirmação do fenótipo.
- Desde o último dia de ensaio de leitor de placa (passo 2.4), salve as culturas de líquido que tem uma taxa de crescimento visivelmente recuperado, presumivelmente, ganhando uma mutação de supressor que pode aliviar o fenótipo da mutação parental. Transferência e misturar cultura líquida de 250 µ l de uma criotubo contendo 250 µ l de glicerol a 50%. Flash congelar as células em nitrogênio líquido e salvar as cepas - 80oC indefinidamente.
- Para confirmar que a mutação de supressor é um elemento geneticamente hereditário, usar métodos padrão de cruzamento genético para cruzar yfm P (para parental, a tensão usada no início do ensaio do leitor de placa) com yfm S (para o supressor, a tensão que salvou no final placa leitor do ensaio de). Se a mutação do supressor é certamente um elemento geneticamente hereditário, yfm P × yfm S devem produzir tétrades em que duas colónias têm o fenótipo da doença da estirpe parental e duas colónias têm a taxa de crescimento recuperados do supressor estirpe.
- Da Cruz do passo 3.2, escolher três colônias com o fenótipo supressor (estirpe S) e três colônias com o fenótipo parental (estirpe P) da mesma genética cruzam (3 réplicas biológicas para cada um) e proceda com a extração de DNA genômica e passos de sequenciamento abaixo.
Nota: As etapas 3.2 e 3.3 são altamente recomendadas, mas não são necessárias. Alternativamente, um pode espalhar cultura líquida recuperada coletada dos 3.1 em meio rico em colônias única e, em seguida, escolher aleatoriamente três colônias como triplica biológico para o sequenciamento do genoma inteiro sem confirmação genética. Neste caso, três triplica biológica da estirpe parental deve ser usado para comparação de sequenciamento genômico.
4. genomic DNA extração, produção de biblioteca e sequenciamento.
- Para DNA extração, preparação de biblioteca e sequenciamento, escolhem aleatoriamente três réplicas biológicas por yfm P estirpe, e três biológicos Replica de cada cepas individualmente levantado yfm S dos cruzamentos genéticos (passo 3.2) ou do placas que foram distribuídas para obter colônias única da cepa S (nota passo 3.3).
- Crescer estirpes em culturas de 10ml em mídia rica para log de meio da fase (O.D. = 0.5-0.8, sobre 0,75 – 1.2 x 107 células/mL) e usar uma tremendo incubadora para crescer culturas líquidas a 30 ° C, agitando continuamente a 250 rpm. Colete as células por centrifugação a 4 ° C por 5 min a 1000 x g.
- Suspender as células peletizadas em 400 µ l de tampão de extração de DNA (2% Triton X-100, 1% SDS, 100 mM de NaCl, 10 mM Tris-Cl (pH 8.0), 1 mM Na2-EDTA), em seguida, adicione 400 µ l de grânulos de vidro e 400 µ l de 25:24:1 fenol: clorofórmio: isoamílico álcool. Vórtice vigorosamente por 2 min a 4 ° C.
- Adicione um adicionais 200 µ l de tampão de extração de DNA e misture por inversão várias vezes. Centrifugue por 5 min a 4 ° C a 20.000 x g.
- Transferir a fase aquosa para um tubo limpo, adicionar 20 µ g de mistura de RNase A/T1 e incubar a 37 ° C por 15 min.
- Adicionar um volume igual de 25:24:1 fenol: clorofórmio: isoamílico álcool, rotação por 5 min a 4 ° C a 20.000 x g, em seguida, transferir a fase aquosa para um tubo limpo.
- Adicionar um volume igual de clorofórmio, misture por inversão várias vezes, em seguida, girar por 5 min a 4 ° C a 20.000 x ge, em seguida, transferir a fase aquosa para um tubo limpo.
- Precipitado de DNA com dois volumes de etanol 100%, mais 10% do volume de NaOAC de 3 M (pH 4,3) a-20 ° C durante pelo menos 2 h, em seguida girar por 5 min a 4 ° C, a 20.000 x g e coletar o sedimento.
- Lave o pellet (DNA precipitado) duas vezes com etanol a 70% refrigerados (centrifugar 20.000 x g, 5 min, 4 ° C) e suspender o sedimento em 50 µ l de tampão Tris (pH 7,4) de 10 mM.
- Uso um prep biblioteca kit (veja a Tabela de materiais) por recomendações do fabricante para preparar a biblioteca de sequenciamento do genoma de todo.
Nota: Recomendamos o kit listado na Tabela de materiais , porque permite a construção de biblioteca genômica sem amplificação por PCR, que minimiza as mutações de erro geradas durante a amplificação por PCR. Além disso, durante a preparação da biblioteca genômica, não permita que os grânulos de totalmente seco, encurtando o talão para 1 – 2 min o tempo de secagem. - Para a tosquia parâmetros durante a preparação da biblioteca, use um sonicador concentrado (veja a Tabela de materiais) e definir o factor de serviço de 20%, potência de pico de 175 W, com 200 ciclos por explosão, e frequência modo varrendo a 5.5oC a 6 ° C durante 45 s. Alternativamente, usar uma DNA e chomatin sistema de corte (ver Tabela de materiais) com as seguintes configurações: amplitude de 50% em 4 ° C, com modo de pulso, 15 s s on e 15 off durante 10 minutos, com um tempo total de processamento de 20 min.
- É essencial para lidar com os materiais perigosos utilizados nesta etapa com cuidado. Consulte o apropriado Material Safety Data Sheets e de instituição saúde ambiental e escritório de segurança para a manipulação de NaOAC, etanol, 25:24:1 fenol: clorofórmio: isoamílico álcool e clorofórmio.
- Sequencie as bibliotecas genômicas resultantes. O sequenciamento inteiro lê deve cobrir pelo menos três vezes do genoma inteiro com a resolução em uma gama de nucleotídeo único. Recomenda-se a sequenciação emparelhado-terminou (ou tecnologia).
5. Bioinformática análise para a identificação das mutações supressor
- Realizar a análise de Bioinformática para focar as alterações genômicas que são consistentemente identificadas entre parentais e reprimida yfm estirpes em todos biológico Replica.
Nota: O processo completo de gasoduto é descrito abaixo, mas, além disso, dois arquivos de texto simples BASH-script, o fastq_to_vcf.sh e vcfprocess.sh, são incluídos como materiais complementares para mostrar exemplos de fluxo de trabalho de processamento de leituras a variante de FCR interseção entre o FCR e arquivos e processamento de arquivos, respectivamente. - Corte curto-leituras usando tesoura (https://github.com/jbpease/shear) seguindo as linhas de comando (todas as outra opções padrão):
Shear.py - fq1 $FASTQ1 - fq2 $FASTQ2 - out1 $OUTFQ1 - out2 $OUTFQ2 \
..--barcodes1 $BARCODE - TruSeq - trimqual de plataforma 20:20 \
..--trimpolyat 0..--trimambig..--filterlength 50..--filterunpaired - Mapa lê para o S. pombe referência genoma 2.30 obtidos de PomBase (ftp://ftp.ebi.ac.uk/pub/databases/pombase/pombe/Chromosome_Dumps/fasta/) usando o BWA v0.7.1527. Use a seguinte linha de comando (todas as outra opções padrão):
BWA mem -t 8 $GENOME $OUTFQ1 $OUTFQ2 > $SAM1 - Colocar arquivos de alinhamento SAM através de GATK melhores práticas oleoduto28 por variante ligar usando o GATK v 3.629, PicardTools v2.5.0 (http://broadinstitute.github.io/picard) e SAMtools v 1.3.130. Use as seguintes linhas de comando e parâmetros (todas as outra opções padrão):
Java-Xmx30g-jar picard.jar entrada AddOrReplaceReadGroups = $SAM1 \
SAÍDA = $BAMMARKED Especificadosrgid = 1 RGLB = lib01 RGPL = illumina \
RGPU = $BARCODE RGSM = $SAMPLENUMBER
samtools fixmate - O bam $BAMMARKED $BAMFIXED
samtools classificar - O bam -o $BAMSORTED -T /home/peasejb/tmp $BAMFIXED
índice de samtools $BAMSORTED
Java-Xmx30g-jar GenomeAnalysisTK.jar -T HaplotypeCaller \
-R $GENOME-eu $BAMSORTED..--genotyping_mode descoberta \
-stand_emit_conf 10 - stand_call_conf 30 -o $VCFRAW - Comprimir e indexar arquivos VCF usando tabix:
bgzip $VCFRAW.vcf
tabix $VCFRAW.vcf.gz - Comparar arquivos VCF entre parentais e supressor de cepas de sequenciamento é replicada usando o BCFtools v 1.3.127. Use as seguintes linhas de comando e parâmetros (todas as outra opções esquerda padrão):
isec bcftools - n + 1 $VCFPARENTAL1.gz $VCFPARENTAL2.gz $VCFPARENTAL3.gz \
$VCFMUTANT1.gz $VCFMUTANT2.gz $VCFMUTANT3.gz > common_variants.list
Nota: Este comando rendeu um arquivo codificado com os padrões binários onde variantes de sequência, aparecendo no primeiro mutante só seria binário-codificado como "000100", o segundo mutante apenas como "000010", todos três mutantes como "000111", etc. Esses arquivos foram gerados para cada conjunto de parental e mutante replicar arquivos VCF. - Compile os arquivos de lista de interseção variante juntamente com o nome do arquivo anexado a cada linha usando o comando grep do UNIX:
grep "." *.list > all.list - Referência cruzada variante lista completa com o atual arquivo de anotação de GFF3 (ftp://ftp.ebi.ac.uk/pub/databases/pombase/pombe/Chromosome_Dumps/gff3/schizosacchar omyces_pombe.chr.gff3) usando um script Python personalizado (variant_characterize.py) para identificar sites de SNP consistentes em regiões (sinônimas e não-sinónimas), UTRs 5 ' e 3 ' e ncRNA codificantes de proteínas.
python3 variant_characterize.py - lista common_variants.list \
-gff schizosaccharomyces_pombe.chr.gff3 \
..--fasta Schizosaccharomyces_pombe. ASM294v2.30.DNA.Genome.FA \
-padrão 000100..--fora all.list.filter.000100
Repita este script modificando o padrão e o sufixo do arquivo de saída (..--fora) usando o binário
padrões: 000010, 000001, 000110, 000011, 000101 e 000111 - Combine a saída de todos estes executa script em um arquivo separado por tabulações, para ser visto como uma planilha. A tabela anotada de variantes inclui aqueles que aparecem em um ou ambos estirpe mutante em relação ao fundo. O campo sinalizador binário denota qualquer aparência de uma única estirpe mutante (000100, 000010, 000001), duas estirpes mutantes (000011, 000101, 000110), ou todas as três estirpes mutantes (000111).
- Analise as listas de saída anotada de variantes não encontrado nas amostras dos pais, mas encontrou em um, dois ou todos os três amostras mutantes. A anotação denota o genoma localização e classe de variante (sinônimo/não-sinônimo em uma região da codificação, 3' / 5' UTR, não-codificantes, etc.). Esta lista de mutações de candidato, um exemplo de um candidatos altamente relevantes pode ser uma variante não-sinônimo de codificação aparecendo consistentemente em todas as três estirpes. Um outro tipo de forte candidato seria um acúmulo de várias mutações reguladoras não-sinônimos ou putativos em cepas mutantes aparecem próximos um do outro, ou dentro do mesmo gene.
Resultados
Crescimento lento mutantes mostram recuperação fenotípica em cultura líquida
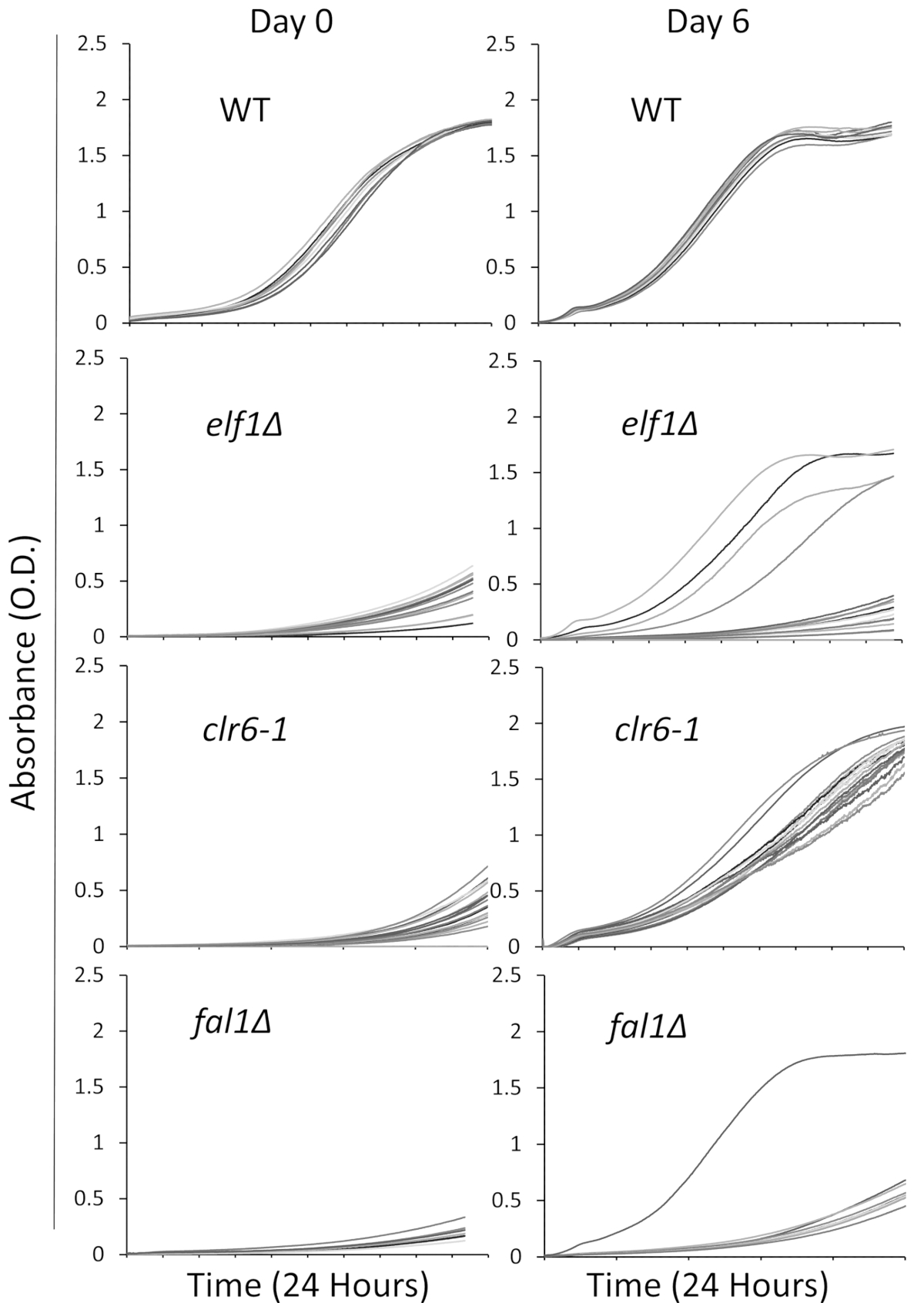
Nós selecionamos três mutantes envolvidos em uma variedade de caminhos biológicos com um fenótipo de doente, de crescimento lento,: AAA família ATPase Elf1, histona deacetilase Clr6 e Exon Junction complexo componente Fal1. Uma estirpe de tipo selvagem e estirpes carregando mutações destes três genes que tinha sido traçada com as estirpes de tipo selvagem foram listradas para colônias individuais e 16 única colônias foram selecionadas aleatoriamente para ser cultivadas em meio líquido rico usando a placa de 96 poços, como acima descrito. Curvas de crescimento de colônias individuais foram gravadas no ponto inicial de tempo (dia 0) e por 6 dias com monitoramento contínuo, usando o leitor de placa. Como esperado, selvagem-tipo colônias não mostram nenhuma alteração perceptível em suas curvas de crescimento em todo o experimento31 (Figura 1). Notavelmente, quatro colônias com o fundo de elf1∆ e uma colônia de fal1∆ mostram uma mudança dramática no crescimento de crescimento lento para alguns níveis variados de crescimento semelhante ou próximo do selvagem-tipo colônias. Dramaticamente, todos mutantes clr6-1 mostram uma recuperação consistente fenotípica, crescendo em um ritmo mais rápido no final do ensaio31 (Figura 1). Para caracterizar os diferentes fenótipos, referimo-nos para as cepas originais que são de crescimento lento como "Estirpes de P" (ou linhagens parentais) e as estirpes mostrando recuperação fenotípica como "Estirpes de S" (ou estirpes reprimidas). Por favor, note que a Figura 1 é um exemplo de uma rodada de experimento a triagem e não representa os totais não-complementares supressores identificados e sequenciados nos seguintes resultados representativos.
Recuperação fenotípica é atribuída a traços hereditários
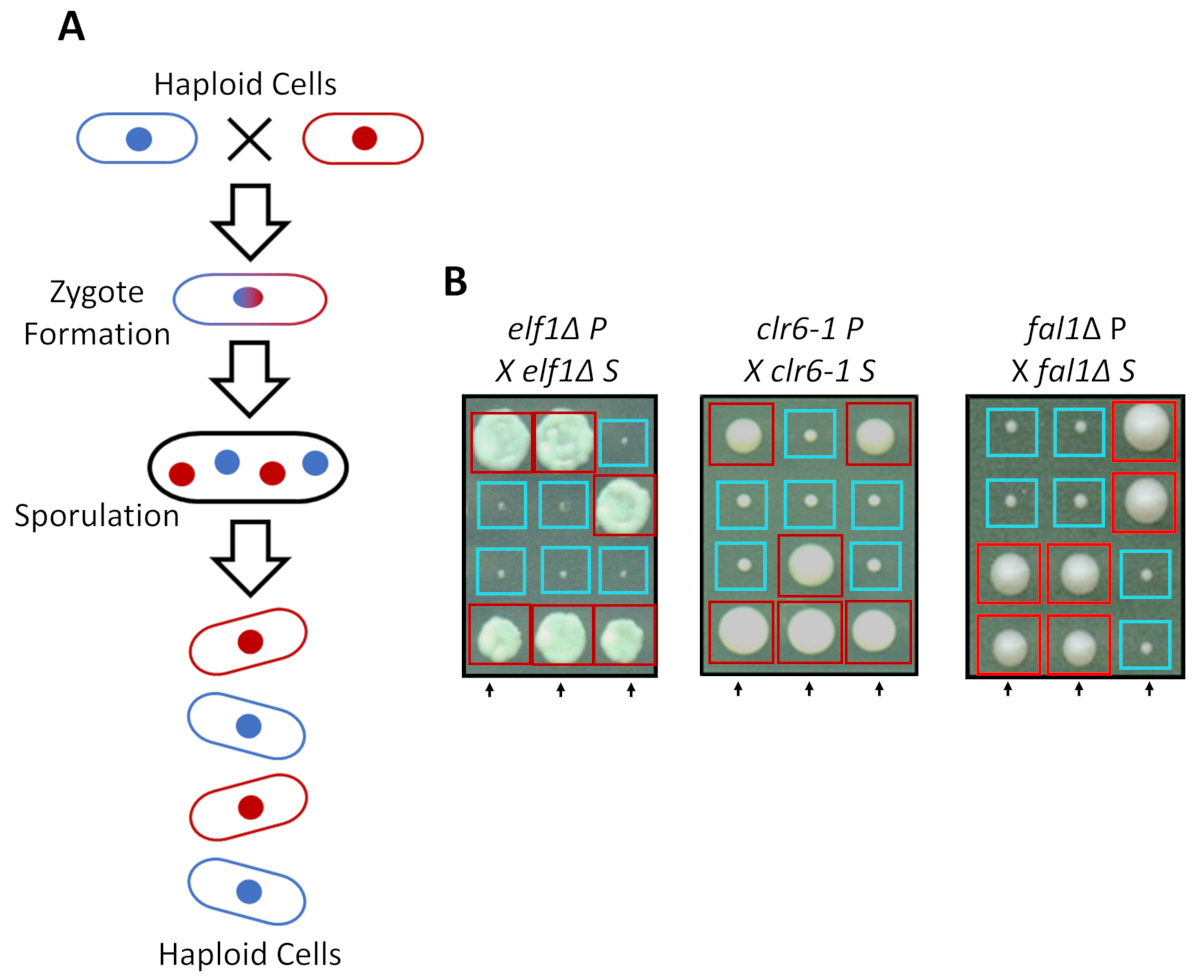
S. pombe pode crescer como um haploid em mídia avançada, mas duas linhagens haploides com o companheiro de tipos complementares do acasalamento sob inanição de nitrogênio. Meiosis em fermento de fissão implica uma rodada de duplicação, seguida por duas rodadas de divisão celular. O ciclo sexual resulta na formação de quatro esporos haploides, carregando o material genético da estirpe parental com 2:2 segregação de características genéticas, seguindo as regras da genética mendeliana clássica (Figura 2A). Quando crescido no mesmo prato para a mesma quantidade de tempo, nós confirmamos a segregação de 2:2 quando tudo volta-travessia suprimidas cepas (linhagens de S) com suas cepas parentais (estirpes de P), que resultou em 2 pequenas (defeito de crescimento) e 2 grandes (fenótipo supressor) colónias. Exemplos individuais para as células elf1∆, clr6-1 e fal1∆ suprimidos são mostrados na Figura 2B. Confirmamos que todas as cepas isoladas de S carregam um elemento genético monogénicas que suprime o fenótipo de crescimento lento de suas estirpes de P (dados não mostrados).
Sequenciamento do genoma inteiro identifica com êxito as mutações supressor
Como exemplo, usamos o sequenciamento do genoma inteiro emparelhado-final para identificar os responsável pela recuperação fenotípica em cepas de S de elf1∆ de elementos genéticos. Uma descrição mais completa de análise de dados está disponível on-line31. Brevemente, utilizamos triplica biológico de duas linhagens de elf1∆ P independentemente gerado e duplicatas biológicas de cinco grupos não-complementares de estirpes de elf1∆ S , cada um dos quais contém diferentes supressores. Depois conseguimos uma lista de variantes anotadas da análise bioinformática (6.1-10), priorizamos certas classes de variantes que foram relevantes para nossa análise. Focamos na identificação de alterações genômicas consistentes que eram idênticas em todas as repetições biológicas de cepas de S de elf1∆ individual em comparação com suas estirpes de P elf1∆ parental (Figura 3 e suplementar tabelas 1-4 ). Identificamos cinco alterações nonsynonymous em regiões CDS em todas cinco diferentes elf1∆ S as estirpes, incluindo rli1 +, SPBPJ4664.02, cue2 + e rpl2702 +. Ambos S-A1 e A2-S contêm mutante SPBPJ4664.02, embora as mutações ocorrem em diferentes aminoácidos. Porque o SPBPJ4664.02 é um gene longo (11.916 nucleótidos) com centenas de repetições, as mutações foram incapazes de ser confirmado através da realização de PCR seguido de sequenciação. S-A3 contém um mutante de exclusão no rli1 que é consistente em ambas as duplicatas biológicas. No entanto, o mutante não co segregar com o fenótipo de S em elf1∆ plano. Nós identificamos um mutante de cue2 (cue2-1) no S-B1, com os aminoácidos 396-400 desaparecidos. S-B2 contém um mutante de rpl2702 (rpl2702-1), que altera o aminoácido na posição 45 de glicina-aspartato31. Ambos os cue2-1 e rpl2702-1 foram confirmados como supressores de elf1∆ , como mostrado abaixo.
Confirmação genética de mutações identificadas supressor verifica a herdabilidade do fenótipo recuperação
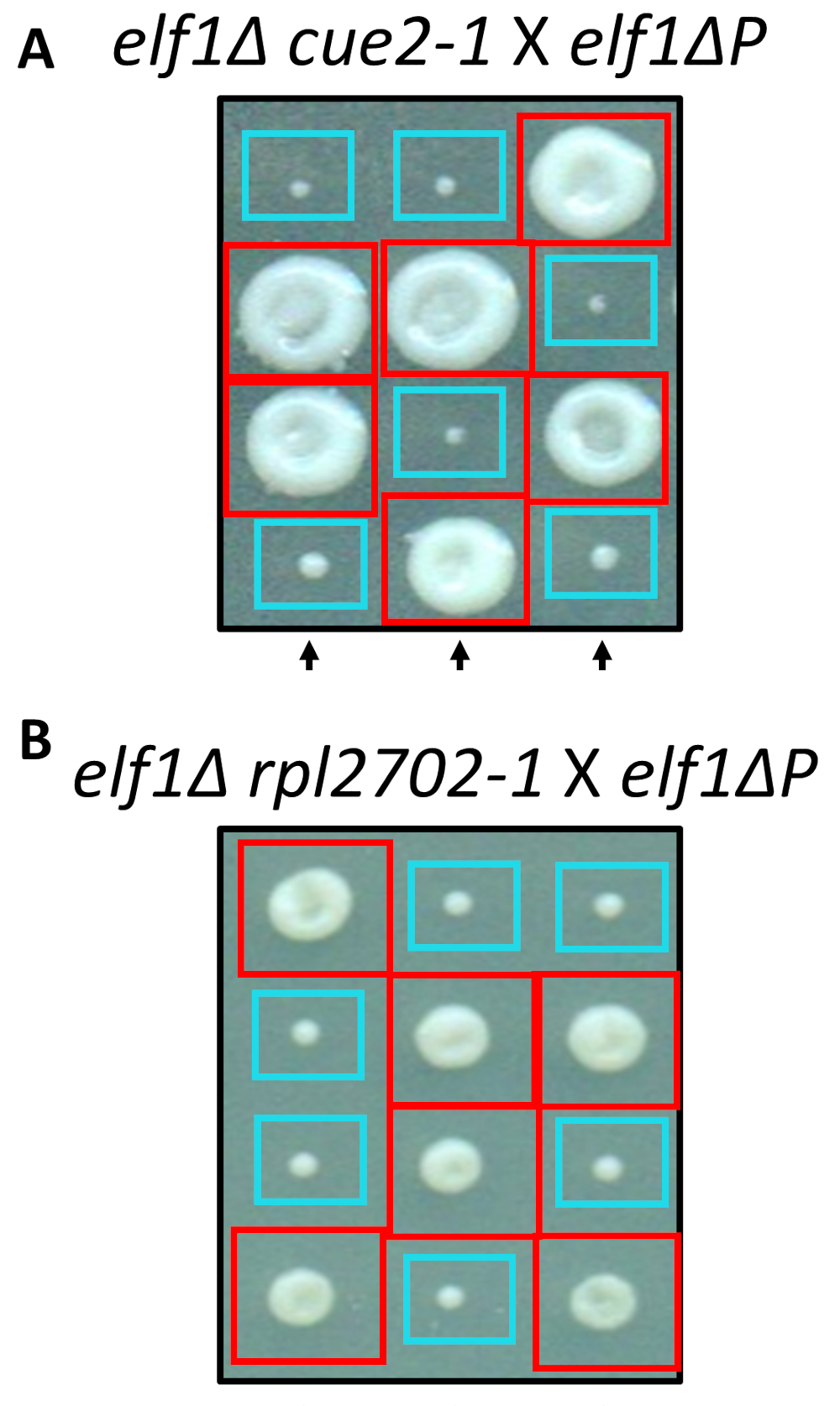
Duas das mudanças identificadas nonsynonymous, cue2-1 e rpl2702-1, foram reconstruídas no laboratório usando protocolos padrão para o mutagenesis local-dirigido. Cepas de mutante duplo cue2-1 elf1∆ P e rpl2702-1 elf1∆ P foram cruzados com a cortesia elf1∆ P tensão31 (Figura 4). Se as mutações nonsynonymous, identificadas por este ecrã, foram suficientes para suprimir elf1∆ P, então as resultantes tétrades mostrar 2:2 pequeno grande proporção nas colônias resultantes os 4 esporos em cada Tétrade. Com efeito, cruzamento genético mostrou que as mutações identificadas supressor são bem sucedidas em suprimir o fenótipo de crescimento lento de P elf1∆ e são hereditárias.

Figura 1: recuperação fenotípica pode ser monitorizada por gravação de curvas de crescimento em um leitor de placa. Dezesseis colônias única do selvagem-tipo (WT), elf1∆, clr6-1e fal1∆ foram colocadas em uma placa de 96 poços. Curvas de crescimento foram gravadas ao longo de um período de 24 horas e colônias foram re-diluídas diariamente na mídia. O defeito de crescimento é evidente pelo baixa absorvância (OD) no final do período de 24h no dia 0. Estirpes fenotipicamente recuperadas são aqueles que exibem uma curva de crescimento semelhante, ou próximo, do tipo selvagem ao longo do período de 24 horas no dia 6. Quatro colônias de elf1∆, uma colônia de fal1∆e todas as colônias de clr6-1 mostraram vários níveis de recuperação fenotípico após 6 dias. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 2: cruzamento genético pode confirmar que a recuperação fenotípica é atribuída a um único alelo hereditário. (A) quando o fermento de fissão células estão sujeitos à fome de nitrogênio, duas células haploides com um tipo de acasalamento complementar pode gerar um zigoto que sporulates para gerar uma Tétrade de 4 esporos. Os materiais genéticos dos pais vão segregar durante a meiose, seguindo as regras da genética mendeliana. (B) recuperado fenotipicamente colônias (etiquetados S, para suprimida) com indicavam a genótipos parentais retrocruzada com sua cortesia colônia parental (que mostra sem recuperação fenotípica, rotulada P, para parental). Genéticas cruzes mostrando 2:2 pequenas (aptidão pobre) para colônias grandes (aptidão recuperados) demonstram que a recuperação fenotípica é hereditária e pode ser atribuída a um único elemento genético. Caixas vermelhas são portadores do alelo de supressor de colônias, e caixas azuis são colônias carregando o alelo parental. Esta figura foi modificada de Marayati et al., 201831. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 3: análise de dados de sequenciamento de genoma-largo para identificar elementos genéticos responsáveis de recuperação fenotípica. Biológico de três repetições de duas linhagens parentais de "P" (P-A e P-B), e duas repetições biológicas de cinco fenotipicamente recuperado trocou as cepas de "S" (S-A1, A2-S e S - A3 recuperaram de P-A; S-B1 e S-B2 do P-B), foram sequenciados e as mutações foram organizadas como uma lista de cada mutação na estirpe recuperada em comparação com o genoma da estirpe parental é derivado (por exemplo, P-A vs S-A1, etc.). O número total de mutações detectadas através de todo o genoma de todos tais comparações emparelhadas foi 660. Um total de 44 mutações foram identificadas quando foram selecionadas apenas as mutações que ocorrem em ambas as réplicas biológicas da mesma estirpe "S". Dos 44 mutações, 12 mutações foram a inserção/exclusão (INDEL) ou não - mutações sinónimas. Fora a 12 INDEL ou mutações não-sinónimas, cinco ocorreram na proteína sequência de código. As cinco mutações potencialmente correlacionam com o único elemento genético responsável para as cepas fenotipicamente recuperadas: uma mutação não-sinónimas em SPBPJ4664.02 encontrada no S-A1 e A2-S, INDEL em rli1 em S-A3, INDEL em cue2 encontrados em S-B1 e mutações não-sinónimas em rpl2702 , encontrado em S-B2. Sequência detalhada informações sobre as mutações e o fundo filtrado são incluídas no suplementar tabelas 1-4. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 4: confirmação dos supressores identificados através do sequenciamento do genoma inteiro. Resultados do sequenciamento do genoma inteiro foram confirmados independentemente, gerando as mutações e realizando cruzamentos genéticos para confirmar a recuperação fenotípica do cruzamento de uma estirpe de elf1∆ cue2-1 com uma estirpe de P elf1∆ e elf1∆ rpl2702-1 com tensão de elf1∆ P . Três representante tétrades verticais são mostradas. Caixas vermelhas são colônias de duplo-mutante (elf1 cue2-1, ou elf1 rpl2702-1); caixas azuis são colônias elf1∆ . Esta figura foi modificada de Marayati et al., 201831. Clique aqui para ver uma versão maior desta figura.
{kind=link}
Quadro suplementar 1. Clique aqui para baixar esta tabela.
Suplementar tabela 2 . Clique aqui para baixar esta tabela.
Suplementar tabela 3 . Clique aqui para baixar esta tabela.
Quadro suplementar 4 . Clique aqui para baixar esta tabela.
Suplementar de codificação de arquivos . Clique aqui para baixar os arquivos.
Discussão
O protocolo descrito aqui representa uma novela e uma simples tela de mutações espontâneas supressor detectáveis através de recuperação fenotípica das mutações conferindo crescimento lento no fermento de fissão, um fenótipo característico de mais de 400 genes em S. pombe, o função de muitos dos que permanece desconhecido2,32. Métodos anteriores tomaram outras abordagens para a tela de mutações de supressor em microorganismos, incluindo o uso de agentes mutagênicos21, ou a aplicação de uma mudança de temperatura em contextos mutantes sensíveis à temperatura,33. Em contraste, este protocolo mostra que recuperação fenotípica ocorre sem interferência de química ambiental/adicional e destaca a vantagem de aptidão da ascensão de mutações de supressão eventualmente assumir os recursos disponíveis no líquido cultura. Esta tela permite que o isolamento de ambos os supressores de desvio ou supressores de interação porque é eficaz para ambas as mutações de perda-de-função como elf1∆ ou fal1∆ e mutações pontuais, tais como clr6-1, contanto que os mutantes demonstre aptidão defeitos em cultura líquida.
Até agora, todas as cepas S recuperadas que temos investigado têm demonstrado vários graus de recuperação fenotípica. Como detectado através de cruzamento genético, o fenótipo recuperado é atribuível a um único elemento genético e hereditários (exemplos mostrados na Figura 2). Esta é uma das mais significativas vantagens desse método em comparação com telas baseadas em química ou UV supressor, que muitas vezes alvo de múltiplos loci genômicos. É comum observar uma ou duas colônias recuperadas fora 16 colônias/tensão (cerca de 10%) dentro de uma semana. No entanto, percebemos que certos mutantes, tais como a perda da função de Rrp6, a subunidade exossomo nuclear específico, nunca se recuperou para a taxa de crescimento quase selvagem-tipo observaram em células de elf1Δ 31. É provável que a função de Rrp6 só pode ser parcialmente compensada por supressores, diferentemente da função dos outros mutantes testados, incluindo fal1∆, que foi mostrado para causar um grave defeito meiótico através de sua importante função na regulação emenda34. Acreditamos que essa alternativa supressor métodos de rastreio estaria sujeita o mesmo problema quando yfg possui funções únicas, não substituível em crescimento celular.
Antes de executar o sequenciamento genômico, é ideal para o cruzamento das colônias fenotipicamente recuperados, identificadas a partir do leitor de placa, com as linhagens parentais para limpar o fundo genético e obter réplicas biológicas. Além disso, sequenciamento de fundo do inteiro-genoma identifica centenas de alterações de nucleotídeo único, a maioria dos quais não é idêntica entre réplicas biológicas que são de pouco interesse para o rastreio. Por exemplo, encontramos um total de 660 alterações genômicas em todos os três cromossomos entre os dois elf1Δ P e as cinco diferentes estirpes de S (Figura 3). Nós não comumente observaram mutações idênticas entre biológico sequenciado Replica de cada estirpe, sugerindo que novas mutações podem ocorrer durante o cultivo de células de elf1Δ antes da construção de biblioteca genômica ou erros aleatórios podem ser introduzidas durante a construção da biblioteca e sequenciamento. Daí, isolar as mutações que são consistentes através de repetições biológicas é um aspecto importante na identificação bem sucedida de supressores usando o sequenciamento do genoma de todo.
Identificamos e confirmou dois supressores em regiões CDS em cinco estirpes sequenciadas de S. Embora as mutações em SPBPJ4664.02 foram detectadas em S-A1 e estirpes de S-A2, é improvável que o SPBPJ4664.02 é um supressor válido porque S-A1 e A2-S não contém um supressor sobre o mesmo gene, como eles não são complementares com uns aos outros ( dados não mostrados). Nós também não confirmou rli1 em S-A3, que não co segregar com o fenótipo de S quando retrocruzada com elf1Δ. Alternativamente, encontramos mutações específicas em regiões não-codificantes na S-A1, A2-S e S-A3. É possível que estas regiões genômicas alteradas não-codificantes aliviar o fenótipo elf1Δ , que será abordado em estudos futuros. Em comparação com métodos tradicionais como um ensaio de ligação, que pode levar anos para mapear uma mutação genética, identificamos dois supressores dentro de dois meses após a confirmação de que um elemento monogénicas causou o fenótipo de S. Com o rápido desenvolvimento da tecnologia de sequenciamento do genoma inteiro, estamos otimistas de que esse método será mais eficiente para identificar mutações genéticas consistentes num futuro previsível.
Em resumo, este protocolo fornece instruções passo a passo para identificar com sucesso as mutações supressor para qualquer gene de interesse com um defeito de crescimento lento em cultura líquida. A simplicidade deste teste permite o rastreio em larga escala de múltiplas origens genéticas de interesse com pouco treinamento hands-on. Há espaço para mais, automatizar o processo usando um robô de manipulação de líquidos para realizar as diluições diárias. Desde que a manipulação laboratorial de microrganismos requer inevitavelmente o crescimento em cultura líquida, um processo que é inerentemente seletivo para fitness, propomos que este protocolo pode ser amplamente aplicado a outros organismos modelo de grande população, tais como bactérias e outras espécies de leveduras.
Divulgações
Os autores declaram não menções pelos fabricantes dos instrumentos utilizados neste método, e sem interesses financeiros concorrentes.
Agradecimentos
Este trabalho foi financiado pelo Instituto Nacional de General Medical Ciências, bolsas 1R15GM119105-01 a K.Z. Agradecemos a todos os comentadores para observações perspicazes. Agradecemos também James Tucker, Alicia Anderson, Elizabeth Black e Glen Marrs para a discussão e comentários sobre este manuscrito.
Materiais
| Name | Company | Catalog Number | Comments |
| Adenine, Powder | Acros Organics | 147441000 | Use at 75 mg/L to make liquid and solid rich media (YEA) |
| Bacteriology Petri Dish | Corning, Falcon | C351029 | 100 ×15 mm, use to grow strains to single colonies on solid rich media |
| D-Glucose Anhydrous, Powder | Fisher Chemical | D16-1 | Use at 30 g/L to make liquid and solid rich media (YEA) |
| Difco Agar, Granuated | Becton, Dickinson and Co. | 214530 | Use at 20 g/L to make solid rich media (YEA) |
| DNA extraction buffer | 2% Triton X-100, 1% SDS, 100 mM NaCl, 10 mM Tris-Cl (pH 8.0), 1mM Na2-EDTA | ||
| Focused-ultrasonicator | Covaris Inc. | S220 | Alternatively, use QSonica Q800R sonicator/DNA and chromatin shearing system |
| Gen5 Data Collection and Analysis Software | Biotek, Inc. | GEN5SECURE | Or equivalent, must be compatible with the micro-plate reader, use to export data readings from the micro-plate reader |
| Hydrochloric Acid 1N, Liquid | Fisher Chemical | SA48-4 | Use to adjust pH to 5.5 in liquid and solid rich media |
| Liquid Rich Media (liquid YEA) | 30 g/L D-Glucose, 5 g/L Yeast Extract, 75 mg/L Adenine, pH adjusted to 5.5 with 1 M HCl | ||
| Microplate Reader, Synergy H1 Hybrid Multi-Mode Reader | Biotek, Inc. | BTH1MG | Or equivalent, must read visible light at 600 nm wavelength range |
| Rich Media agar plates (YEA plates) | 30 g/L D-Glucose, 5 g/L Yeast Extract, 75 mg/L Adenine, 20 g/L Agar, pH adjusted to 5.5 with 1 M HCl. | ||
| RNase A/T1 mix | Thermo Fisher Scientific | EN0551 | Use according to manufacturer recommendation |
| Sterile Polystyrene Inoculating Loop | Corning, Inc. | OS101 | Or equivalent, use to transfer colonies from agar plates to 96-well plate |
| Sterile workspace and burners | |||
| Tissue Culture Plate, 96-well Optical Flat Bottom with Low Evaporation Lid | Corning, Falcon | C353072 | Or equivalent, must have optical flat bottom for micro-plate ready |
| TruSeq DNA PCR-Free LT/HT Library Prep Kit | Illumina, Inc. | 20015962 | Use to prepare the whole-genome sequencing library |
| Yeast Extract, Powder | Fisher Chemical | BP1422-500 | Use at 5 g/L to make liquid and solid rich media (YEA) |
Referências
- McKay, J. K., Latta, R. G. Adaptive population divergence: Markers, QTL and traits. Trends in Ecology and Evolution. 17 (6), 285-291 (2002).
- Wood, V., Harris, M. A., et al. PomBase: A comprehensive online resource for fission yeast. Nucleic Acids Research. 40 (D1), (2012).
- de Visser, J. A. G. M., Cooper, T. F., Elena, S. F. The causes of epistasis. Proceedings of the Royal Society B: Biological Sciences. 278 (1725), 3617-3624 (2011).
- Sailer, Z. R., Harms, M. J. Detecting high-order epistasis in nonlinear genotype-phenotype maps. Genetics. 205 (3), 107911088(2017).
- Kuzmin, E., Costanzo, M., Andrews, B., Boone, C. Synthetic genetic arrays: Automation of yeast genetics. Cold Spring Harbor Protocols. 2016 (4), 326-332 (2016).
- Tong, A. H. Y., Boone, C. Synthetic genetic array analysis in Saccharomyces cerevisiae. Methods in Molecular Biology. 313 (1), 171-192 (2006).
- Dixon, S. J., Costanzo, M., Baryshnikova, A., Andrews, B., Boone, C. Systematic Mapping of Genetic Interaction Networks. Annual Review of Genetics. 43 (1), 601-625 (2009).
- Boone, C., Bussey, H., Andrews, B. J. Exploring genetic interactions and networks with yeast. Nature Reviews Genetics. 8 (6), 437-449 (2007).
- Bai, X., Yang, Z., Jiang, H., Lin, S., Zon, L. I. Genetic suppressor screens in haploids. Methods in Cell Biology. , 129-136 (2011).
- Manson, M. D. Allele-specific suppression as a tool to study protein-protein interactions in bacteria. Methods. 20 (1), 18-34 (2000).
- Motter, A. E., Gulbahce, N., Almaas, E., Barabási, A. L. Predicting synthetic rescues in metabolic networks. Molecular Systems Biology. 4, 168(2008).
- Peterson, R. T., Shaw, S. Y., et al. Chemical suppression of a genetic mutation in a zebrafish model of aortic coarctation. Nature Biotechnology. 22 (5), 595-599 (2004).
- Giorgini, F., Guidetti, P., Nguyen, Q., Bennett, S. C., Muchowski, P. J. A genomic screen in yeast implicates kynurenine 3-monooxygenase as a therapeutic target for Huntington disease. Nature Genetics. 37 (5), 526-531 (2005).
- Forsburg, S. L., Patton, E., et al. The art and design of genetic screens. Nature reviews. Genetics. 2 (9), 659-668 (2001).
- Johnston, D. S. The art and design of genetic screens. Genetics. 3 (March), 176-188 (2002).
- Jorgensen, E. M., Mango, S. E. The art and design of genetic screens: Caenorhabditis elegans. Nature Reviews Genetics. 3 (5), 356-369 (2002).
- Gocke, E., Müller, L. In vivo studies in the mouse to define a threshold for the genotoxicity of EMS and ENU. Mutation Research - Genetic Toxicology and Environmental Mutagenesis. 678 (2), 101-107 (2009).
- Suzuki, T., Hayashi, M., et al. A comparison of the genotoxicity of ethylnitrosourea and ethyl methanesulfonate in lacZ transgenic mice (Muta(TM)Mouse). Mutation Research - Genetic Toxicology and Environmental Mutagenesis. 395 (1), 75-82 (1997).
- Uttam, J., Alberico, C., De Stasio, E. ENU Mutagenesis. International C. elegans Meeting. , (1995).
- Putrament, A., Baranowska, H., Ejchart, A., Prazmo, W. Manganese Mutagenesis in Yeast. Methods in Cell Biology. 20, 25-34 (1978).
- Bose, J. L. Chemical and UV mutagenesis. Methods in Molecular Biology. 1373, 111-115 (2016).
- Ikehata, H., Ono, T. The Mechanisms of UV Mutagenesis. Journal of Radiation Research. 52 (2), 115-125 (2011).
- Shrivastav, N., Li, D., Essigmann, J. M. Chemical biology of mutagenesis and DNA repair: cellular responses to DNA alkylation. Carcinogenesis. 31 (1), 59-70 (2010).
- De Stasio, E. A., Dorman, S. Optimization of ENU mutagenesis of Caenorhabditis elegans. Mutation Research - Genetic Toxicology and Environmental Mutagenesis. 495 (1-2), 81-88 (2001).
- Probst, F. J., Justice, M. J. Mouse mutagenesis with the chemical supermutagen ENU. Methods in Enzymology. 477 (C), 297-312 (2010).
- Bähler, J., Wu, J. Q., et al. Heterologous modules for efficient and versatile PCR-based gene targeting in Schizosaccharomyces pombe. Yeast. 14 (10), 943-951 (1998).
- Li, H., Durbin, R. Fast and accurate short read alignment with Burrows – Wheeler transform. Bioinformatics. 25 (14), 1754-1760 (2009).
- Van der Auwera, G. A., Carneiro, M. O., et al. From fastQ data to high-confidence variant calls: The genome analysis toolkit best practices pipeline. Current Protocols in Bioinformatics. 43, 11.10.1-11.10.33 (2013).
- Mckenna, A., Hanna, M., et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Research. 20, 1297-1303 (2010).
- Li, H., Handsaker, B., et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 25 (16), 2078-2079 (2009).
- Marayati, B. F., Drayton, A. L., et al. Loss of Elongation-Like Factor 1 Spontaneously Induces Diverse, RNase H-Related Suppressor Mutations in Schizosaccharomyces pombe. Genetics. 209 (4), 967-981 (2018).
- Harris, M. A., Lock, A., Bähler, J., Oliver, S. G., Wood, V. FYPO: The fission yeast phenotype ontology. Bioinformatics. 29 (13), 1671-1678 (2013).
- Xu, X., Wang, L., Yanagida, M. Whole-Genome Sequencing of Suppressor DNA Mixtures Identifies Pathways That Compensate for Chromosome Segregation Defects in Schizosaccharomyces pombe. G3: Genes|Genomes|Genetics. 8 (3), 1031-1038 (2018).
- Marayati, B. F., Hoskins, V., et al. The fission yeast MTREC and EJC orthologs ensure the maturation of meiotic transcripts during meiosis. RNA. 22 (9), 1349-1359 (2016).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados