
Method Article
Ein Deep-Sequenzierung-gestützte, spontane Suppressor-Bildschirm in der Spalthefe Schizosaccharomyces pombe
In diesem Artikel
Zusammenfassung
Wir präsentieren eine einfache Suppressor Bildschirm Protokoll in Kernspaltung Hefe. Diese Methode ist effizient, Mutagen-frei und selektiv für Mutationen, die oft auf einen einzigen genomischen Locus auftreten. Das Protokoll ist geeignet für die Isolierung von Unterdrücker, die Wachstumsstörungen in Flüssigkultur lindern, die durch eine Mutation oder ein Medikament verursacht werden.
Zusammenfassung
Ein genetische Bildschirm für die mutierten Allele, die phänotypische Defekte, die durch eine Mutation zu unterdrücken ist ein leistungsfähiger Ansatz, Gene zu identifizieren, die zu eng verwandten biochemischen Bahnen gehören. Bisherige Methoden wie synthetische genetische Array (SGA) Analyse und zufällige Mutagenese Techniken mit Ultraviolett (UV) oder Chemikalien wie Ethyl Methanesulfonate (EMS) oder N-Ethyl-N-Nitrosourea (DEU), am meisten benutzt gewesen, aber sind oft teuer und mühsam. Außerdem sind diese Mutagen-basierten Screening-Methoden häufig assoziiert mit schweren Nebenwirkungen auf den Organismus, induzieren mehrere Mutationen, die die Komplexität der Unterdrücker zu isolieren. Hier präsentieren wir Ihnen ein einfaches und effektives Protokoll zur Suppressor Mutationen in Mutanten zu identifizieren, die einen Wachstum Defekt in Schizosaccharomyces Pombeverleihen. Die Fitness der Zellen mit einem Wachstum Mangel in standard reiche flüssige Medien oder synthetische flüssigen Medien kann für die Wiederherstellung mit einem automatisierten 96-Well-Platte-Reader über einen längeren Zeitraum überwacht werden. Sobald eine Zelle eine Suppressor-Mutation in der Kultur erwirbt, Esel-seine Nachkommen derjenigen die elterliche Zellen. Die wiederhergestellten Zellen, die ein wettbewerbsfähiges Wachstum gegenüber den elterlichen Zellen Vorteil können dann isoliert und backcrossed mit den elterlichen Zellen. Die Suppressor-Mutationen werden dann durch Sequenzierung des gesamten Genoms identifiziert. Diesen Ansatz haben wir erfolgreich mehrere Entstörer isoliert, die lindern die schwere Wachstumsstörungen, verursacht durch Verlust von Elf1, einer AAA + Familie ATPase, die in nuklearen mRNA Transport und Wartung der genomischen Stabilität wichtig ist. Es gibt derzeit über 400 Gene in S. Pombe mit Mutanten, die Verleihung eines Mangels Wachstum. Wie viele dieser Gene Fußgelenkes, schlagen wir vor, dass unsere Methode die Identifizierung von neuartigen funktionalen Wechselwirkungen mit diesem benutzerfreundlichen, Hochdurchsatz-Ansatz beschleunigen wird.
Einleitung
Die Grundlage des Verstehens funktionaler Verbindungen zwischen den Genen beruht auf der Fähigkeit, die molekularen Mechanismen zu identifizieren, durch die komplexen genetische Merkmalen abweichen, um unterschiedliche Phänotypen1zu produzieren. In der Spalthefe Schizosaccharomyces Pombe (S. Pombe), sind die meisten Protein-kodierenden Gene für Lebensfähigkeit2entbehrlich. Dieses Ergebnis spricht nicht auf die Unwichtigkeit dieser Gene, sondern um die komplizierten kompensatorischen Mechanismen, die die biochemischen Bahnen, zu denen solche Gene gehören. Diese Kompensationsmechanismen sezieren hat Epistasis Karten, erzeugt die umfassende genetische Interaktionen aufgedeckt und erweitert unser Verständnis der funktionalen biochemischen Bahnen3,4.
Hochdurchsatz-Methoden (z. B. synthetische genetische Array-Analysen oder SGA) wurden entwickelt, um genomweite genetische Interaktionen in der angehenden Hefe zu identifizieren, und wurden für den Einsatz in Kernspaltung Hefe5,6erweitert. Solche Ansätze verlassen sich oft auf eine Bibliothek von Stämmen, enthält alle lebensfähigen einzelne Protein-kodierenden gen Streichungen (rund 3.300 haploiden Löschung durch Mutation entstehende Variationen mit über 92 % des Hefegenoms Kernspaltung), und erfordern einen Roboterarm der genetischen Kreuzungen zwischen durchführen der Belastung von Interesse und alle möglichen Sorten in der library6. Darüber hinaus SGA Techniken richten sich auf die Fähigkeit der Bibliothek Stämme, korrekte und effiziente Paarung haben, ein Phänotyp, die abnorme, 444 derzeit gekennzeichnet Gene in S. Pombe2.
Trotz der Komplexität der genetische Interaktionen, vergleicht man den Phänotyp eines Stammes mit Mutationen in zwei Genen, der Phänotyp der beiden Stämme tragen einzelne Mutationen pro gen eines zwei bemerkenswerte Ergebnisse haben kann: 1) der doppelten mutierte Phänotyp ist schlimmer als die erwarteten multiplikativen elterlichen Phänotypen in Form von Krankheit oder im extremsten Fall Letalität. Dies wird als eine negative genetische Interaktion bezeichnet und ist in der Regel ein Zeichen, das die beiden Gene in parallele biologischer Signalwege zu handeln. (2) der doppelte mutierte Phänotyp ist besser als die erwartete Kombination der elterlichen Phänotypen, auch bekannt als eine positive genetische Interaktion. Eine positive genetische Interaktion ist besonders interessant, weil es zeigt, dass diese Gene im gleichen Prozess funktionieren. Zwei positiv interagierender Gene haben drei mögliche Beziehungen: ein mutiertes Gen kann bis-regulieren die Expression des Gens in einen parallelen Weg, die beiden Gene können in Konzert innerhalb der gleichen Weg flussabwärts voneinander arbeiten oder die beiden Gene kodieren Proteine, die direkt miteinander kommunizieren. Daher können positive genetische Interaktionen gen regulatorischen Knoten zuordnen und klassifizieren Fußgelenkes Gene in biochemischen Bahnen7,8verwendet werden.
Ein Suppressor ist eine Mutation, die die Krankheit Phänotyp ein weiteres Gen-Mutation lindern können in der Regel eine positive genetische Interaktion zwischen den zwei Gene9,10. Suppressor-Mutationen auf eine andere von der Mutation, die sie unterdrücken werden Stents Entstörer genannt. Sie sind besonders wertvoll bei der Untersuchung nicht lebensfähige Mutationen durch die synthetisch Rettung der tödliche Phänotyp (auch bekannt als der Lazarus-Effekt)11. Sie haben auch therapeutische Einsatzmöglichkeiten bei der Behandlung von Erbkrankheiten12,13.
Aus all diesen Gründen ist die Identifizierung von Suppressor-Mutationen in verschiedenen Modellorganismen weithin verwendet worden, um unser Verständnis von verschiedenen biochemischen Bahnen14,15,16zu erleichtern. Screening für Entstörer basiert normalerweise auf den Phänotyp der Mutation in Frage und erfordert die Durchführung von zufälligen Mutagenese um die Mutationen zu isolieren, die den Phänotyp lindern würde. Fast alle Modellorganismen haben zufällige Mutagenese Methoden etabliert. Zum Beispiel N-Ethyl-N-Nitrosourea (ENU) und Ethylmethanesulfonate (EMS), zwei mutagenen, die sind in der Lage, induzierende Punktmutationen in DNA, sind weit verbreitet in verschiedenen Modellen von Bakterien auf Mäuse17,18,19 eingesetzt . Darüber hinaus hat Mangan Chlorid lange in Hefen für die Fähigkeit der Mangan-kation, DNA-Reparatur Wege20hemmen eingesetzt. Eine andere Möglichkeit ist UV-induzierten Mutagenese, die genomweite mutagene Pyrimidine Dimere21,22erzeugt.
Obwohl die Nutzung der chemische Mutagenese, Suppressor-Mutationen zu identifizieren populär gewesen ist, hat die Methode viele Nachteile, einschließlich der Verwendung von gefährlichen Chemikalien, sehr variabel Erfolgsraten und extra konfundierenden Variablen eingeführt werden präsentiert durch die negativen Nebenwirkungen von Mutagen auf mehrere Zellprozesse23,24. Darüber hinaus induziert chemische Mutagenese oft mehrere Mutationen im Genom trägt zur Komplexität der Verwendung von genetischen und Sequenzierung Techniken, um die exakte Mutation zu identifizieren, die die Suppressor-Phänotyp in der Organismus25verliehen.
Um die Mängel des aktuellen Mutagenese Ansätze präsentieren wir eine Methode, um Bildschirm für spontane Suppressor Mutationen in Spalthefe, die nicht auf eine mutagene oder eine Löschung Bibliothek angewiesen ist. Die Methode isoliert Unterdrücker durch eine Positivauswahl Assay. Das Prinzip dieser Methode basiert auf den Vorteil, Wachstum der mutierten Suppressor Teilgesamtheit in Flüssigkultur, die durch einen automatisierten Platte Reader überwacht werden kann. Paarung und Meiose werden nur verwendet, wenn man möchte den genetischen Hintergrund bereinigen oder bestätigen das Vorhandensein von monogenen Allele der Unterdrücker vor der Sequenzierung des gesamten Genoms. Wenn die Unterdrückung Phänotyp durch eine einzige Mutation verursacht wird, wird die Suppressor-Phänotyp 2:2 nach Rückkreuzung mit den Eltern zu trennen. Die Suppressor-Mutationen können dann mit der Sequenzierung des gesamten Genoms identifiziert werden. Wir schlagen vor, dass diese Methode ist anwendbar für das screening von Unterdrücker in alle Mikroorganismen, die einen Großteil der Bevölkerung in Flüssigkultur wachsen können.
Protokoll
1. Aufbau und Vorbereitung belasten
- Generieren Sie eine Mutation oder eine Deletion des Gens (Yfm, Ihre Lieblings-Mutation) mit standard Site-verwiesene Mutagenese (SDM) wie zuvor26beschrieben.
- Vor dem Start des Bildschirms (optimal) backcross mutierte Stämme mit einem Wildtyp Stamm zu reinigen den genetischen Hintergrund und mutierte Zellen frisch geboren, als die elterliche Stämme zu generieren. Streifen der elterlichen Belastung einzelner Kolonien auf standard rich-Media-Platten. Nach dem Zufallsprinzip auswählen acht bis sechzehn unabhängigen Kolonien (biologische Wiederholungen) mit den gewünschten Mutationen für die Platte Leser Assay (siehe 3.1).
Hinweis: Dieses Protokoll ist effektiv, nur, wenn die elterliche Stämme haben eine Wachstum in flüssigen Medien (minimal oder reich, mit oder ohne Droge oder mit Temperaturverschiebungen, die den Wachstum Defekt verursachen) defekt. Alle elterlichen Stämme sollte haploiden und somit in der Lage, mit anderen haploiden Stämmen mit einem komplementären Paarung genetisch überschritten werden.
2. Platte Leser assay
- Nehmen Sie mit einem sterilen Applikator eine kleine Menge der einzelnen Kolonien in Schritt 1.1 (keine genaue Höhe notwendig, die ab impfen Kultur) zubereitet und in einer 96-Well Polystyrol Mikrotestplatte. Jeder der Kolonien in 200 µL des entsprechenden flüssigen Medien (reich oder minimal, mit oder ohne Droge) aussetzen. Schließen Sie einen leeren Brunnen für jede Zeile auf dem Teller mit 200 µL der gleichen Medien (keine Zellen).
- Lauf das folgende Protokoll auf eine Platte Leser Erkennungssoftware zu einem automatisierten Mikrotestplatte-Reader verbunden: eine kinetische Programm für 24 h und Temperatur einstellen, bei 30 ° C, mit kontinuierlichen schnell orbital schütteln (425 cpm, 3 mm Amplitude). Legen Sie der optischen liest, Lichtstreuung bei einer Wellenlänge von 600nm für optische Dichte zu messen, und das Licht von unten die Platte bei einer Lesung Frequenz von 2 min (721 Gesamt gelesen über einen Zeitraum von 24 h pro Well) zu lesen.
- Nach 24 h, die endgültige ausgeblendeten optische Dichte Lesungen (ausgeblendete OD600) aufzeichnen und verwenden Sie die folgende Formel um das musste jeder der Proben bis zu O.D verdünnen Volumen bestimmen = 0,1:

Hinweis: Exportieren Sie die Daten von der Platte-Reader-Software und verwenden Sie eine Tabellenkalkulations-Software, um die obige Formel einfügen, wie eine Funktion Batch verarbeiten die Verdünnung aus jeder experimentellen Brunnen verwendet werden. - Alle 24 h, verdünnen Sie jedes der Beispiele verwenden die gleichen Medien als Tag 0 zu O.D = 0,1 (ca. 1,5 x 106 Zellen/mL) mit Hilfe der Formel in Schritt 2.3 angegeben. Speichern Sie alle Wachstum Kurven täglich generiert und notieren Sie jede einzelne Kolonie, die zeigt eine erhöhte Wachstumsrate, beurteilt durch eine endgültige O.D, die deutlich höher ist als der Rest der Kohorte mit den gleichen genetischen Hintergrund oder einer Wachstumskurve, die derjenigen ähnlich ist Wildtyp Kolonien.
Hinweis: Dieser Test dauert in der Regel ca. 7-14 Tage. Führen Sie alle Schritte unter sterilen Bedingungen.
3. Auswahl der Suppressor-Kolonien und Bestätigung der Phänotyp.
- Ab dem letzten Tag des Platte Leser Assays (Schritt 2.4) speichern Sie die flüssige Kulturen, die haben eine merklich erholt Wachstumsrate, vermutlich durch die Gewinnung einer Suppressor-Mutation, die den Phänotyp der elterlichen Mutation lindern können. Transfer und 250 µL Flüssigkultur, eine Cryotube mit 250 µL 50 % Glycerin mischen. Blitz der Zellen in flüssigem Stickstoff eingefroren und die Stämme in - 80oC auf unbestimmte Zeit zu sparen.
- Um zu bestätigen, dass die Suppressor-Mutation ein genetisch vererbbar Element, standard genetische Kreuzung Methoden verwenden, um Yfm P überqueren (für Eltern, die Dehnung verwendet am Anfang des Platte-Leser-Tests) mit Yfm S (für Suppressor, der Belastung am Ende gespeichert von der Platte-Leser-Test). Wenn die Suppressor-Mutation in der Tat ein genetisch vererbbar Element ist, sollte Yfm P × Yfm S Rendite Tetraden in dem zwei Kolonien haben die Krankheit Phänotyp des elterlichen Stammes und zwei Kolonien haben die wiederhergestellten Wachstumsrate der die suppressor Zuchtlinie
- Vom Kreuz Schritt 3.2 Wählen Sie drei Kolonien mit der Suppressor-Phänotyp (S-Stamm) und drei Kolonien mit dem elterlichen Phänotyp (P-Stamm) aus dem gleichen genetischen überqueren (3 biologische Wiederholungen für jeden), und fahren Sie mit der genomischen DNA-Extraktion und Sequenzierung Schritte aus.
Hinweis: Die Schritte 3.2 und 3.3 sind sehr zu empfehlen, aber sind nicht erforderlich. Alternativ kann eine wiederhergestellte Flüssigkultur gesammelt von 3,1 auf reiche Medium in einzelnen Kolonien verbreiten, dann nach dem Zufallsprinzip wählen drei Kolonien als biologische Triplicates für ganze Genom-Sequenzierung ohne weitere genetische Bestätigung. In diesem Fall sollten drei biologischen Triplicates des elterlichen Belastung für den Vergleich der Genom-Sequenzierung verwendet werden.
4. genomische DNA-Extraktion, Bibliothek-Produktion und Sequenzierung.
- DNA-Extraktion, Bibliothek Vorbereitung und Sequenzierung, nach dem Zufallsprinzip drei biologische Wiederholungen pro Yfm P Stamm wählen und drei biologische repliziert jedes einzeln entstandene Yfm S -Stämme aus der genetischen Kreuzungen (Schritt 3.2) oder aus der Platten, die verbreitet wurde, um einzelne Kolonien des S-Stammes (Schritt 3.3 Hinweis) zu erhalten.
- Wachsen-Stämme in 10 mL Kulturen in rich-Media, Mid Log-Phase (O.D = 0,5 – 0,8, etwa 0,75 – 1,2 x 107 Zellen/mL), und verwenden Sie einen schütteln Inkubator, um flüssige Kulturen bei 30 ° C mit schütteln kontinuierlich bei 250 u/min zu wachsen. Sammeln Sie Zellen durch Zentrifugation bei 4 ° C für 5 min bei 1000 X g.
- Gebeizte Zellen in 400 µL DNA Extraktionspuffer auszusetzen (2 % Triton x-100, 1 % SDS, 100 mM NaCl, 10 mM Tris-Cl (pH 8,0), 1 mM Na2-EDTA), dann 400 µL von Glasperlen und 400 µL 25:24:1 Phenol: Chloroform: Isoamyl Alkohol dazugeben. Vortex energisch für 2 min bei 4 ° C.
- Fügen Sie eine zusätzliche 200 µL der DNA Extraktionspuffer und Mix durch invertieren mehrmals. Zentrifuge für 5 min bei 4 ° C bei 20.000 X g.
- Übertragen der wässrigen Phase in ein sauberes Röhrchen, 20 µg RNase A/T1-Mix hinzufügen und bei 37 ° C für 15 min inkubieren.
- Fügen Sie ein gleiches Volumen an 25:24:1 Phenol: Chloroform: Isoamyl Alkohol, Spin für 5 min bei 4 ° C bei 20.000 X g, dann übertragen Sie die wässrige Phase in ein sauberes Röhrchen zu.
- Fügen Sie ein gleiches Volumen von Chloroform, mischen, indem Sie mehrere Male das invertieren spin für 5 min bei 4 ° C bei 20.000 X gdann die wässrige Phase in ein sauberes Röhrchen zu übertragen.
- Voreilige DNA mit zwei Bänden von 100 % Ethanol plus 10 % Vol. 3 M NaOAC (pH 4,3) bei-20 ° C für mindestens 2 h, dann für 5 min bei 4 ° C bei 20.000 x g und sammeln das Pellet drehen.
- Pellet (gefällte DNA) zweimal mit gekühlten 70 % Ethanol (Zentrifugieren bei 20.000 X g, 5 min, 4 ° C) waschen und das Pellet in 50 µL 10 mM Tris-Puffer (pH 7,4) auszusetzen.
- Verwendung eine Bibliothek Prep kit (siehe Tabelle der Materialien), pro des Herstellers, die Sequenzierung des gesamten Genoms Bibliothek vorzubereiten.
Hinweis: Wir empfehlen das Kit in die Tabelle der Materialien aufgeführt, weil dadurch die Bauarbeiten für die genomische Bibliothek ohne PCR Verstärkung, wodurch Fehler Mutationen erzeugt während der PCR-Amplifikation minimiert wird. Darüber hinaus während der Vorbereitung genomische Bibliothek erlauben nicht die Perlen, durch die Verkürzung der Wulstes Trocknungszeit um 1 – 2 min. völlig trocken. - Für Scheren Parameter bei der Bibliothek-Vorbereitung, verwenden eine fokussierte Sonikator (siehe Tabelle der Materialien) und setzen Sie das Tastverhältnis auf 20 %, Spitzenleistung auf 175 W, mit 200 Zyklen pro platzen, und geschwungenen Frequenzmodus bei 5.5oC bis 6 ° C für 45 s. Alternativ, verwenden Sie eine DNA und Chomatin Scheren System (siehe Tabelle der Materialien) mit den folgenden Einstellungen: 50 % Amplitude bei 4 ° C mit Pulse-Modus, 15 s ein und 15 s aus 10 min. mit einer Gesamtbearbeitungszeit von 20 min lang.
- Es ist wichtig, gefährlichen Stoffen verwendet in diesem Schritt mit Sorgfalt zu behandeln. Konsultieren Sie die entsprechenden Sicherheitsdatenblätter und Institution Umwelt und Gesundheit und Arbeitssicherheit für den Umgang mit NaOAC, Ethanol, 25:24:1 Phenol: Chloroform: Isoamyl Alkohol und Chloroform.
- Die daraus resultierende genomischen Bibliotheken-Sequenz. Die komplette Sequenzierung liest sollte mindestens dreimal des gesamten Genoms mit der Auflösung in einem Einzel-Nukleotid-Bereich abdecken. Gepaart endete Sequenzierung (oder neueste Technologie) wird empfohlen.
(5) Bioinformatik-Analyse zur Identifizierung von Suppressor-Mutationen
- Führen Sie die Bioinformatik-Analyse auf der genomischen Veränderungen konzentrieren, die konsequent zwischen elterlichen identifiziert werden und unterdrückte Yfm Stämme in allen biologischen repliziert.
Hinweis: Die komplette Pipeline-Prozess wird weiter unten beschrieben, aber zusätzlich zwei Klartext-BASH-Skript-Dateien, fastq_to_vcf.sh und vcfprocess.sh, werden als ergänzende Materialien, Beispiele des Workflows von der Verarbeitung der Lesevorgänge auf VCF Variante zeigen bzw. Dateien und Verarbeitung und Kreuzung der VCF-Dateien. - Stutzen kurz liest mit Schere (https://github.com/jbpease/shear) nach die Befehlszeilen (alle anderen Optionen Standard):
Shear.py--fq1 $FASTQ1--fq2 $FASTQ2--out1 $OUTFQ1--out2 $OUTFQ2 \
--barcodes1 $BARCODE--Plattform TruSeq--Trimqual 20:20 \
--Trimpolyat 0--Trimambig--Filterlength 50 - filterunpaired - Karte liest die S. Pombe Bezug Genom v2.30 gewonnenen PomBase (ftp://ftp.ebi.ac.uk/pub/databases/pombase/pombe/Chromosome_Dumps/fasta/) mit BWA v0.7.1527. Verwenden Sie die folgende Befehlszeile (alle anderen Optionen Standard):
BWA Mem -t 8 $GENOME $OUTFQ1 $OUTFQ2 > $SAM1 - GATK Ausrichtung SAM Dateien durchgestellt pipeline bewährte28 für variant Berufung mit GATK 3.629, PicardTools v2.5.0 (http://broadinstitute.github.io/picard) und SAMtools v1.3.130. Verwenden Sie die folgenden Befehlszeilen und Parameter (alle anderen Optionen Standard):
Java-Xmx30g-jar picard.jar AddOrReplaceReadGroups INPUT$ SAM1 = \
Ausgang = $BAMMARKED RGID = 1 RGLB = lib01 RGPL = Illumina \
RGPU = $BARCODE RGSM = $SAMPLENUMBER
SamTools Fixmate - O Bam $BAMMARKED $BAMFIXED
SamTools sortieren - O Bam -o $BAMSORTED -T /home/peasejb/tmp $BAMFIXED
SamTools Index $BAMSORTED
Java-Xmx30g-jar GenomeAnalysisTK.jar -T HaplotypeCaller \
-R $GENOME-ich $BAMSORTED--Genotyping_mode Entdeckung \
-Stand_emit_conf 10 - Stand_call_conf 30 -o $VCFRAW - Komprimieren und index Tabix mit VCF-Dateien:
Bgzip $VCFRAW.vcf
TABIX $VCFRAW.vcf.gz - VCF-Dateien unter elterliche vergleichen und Suppressor Stämme Sequenzierung Wiederholungen mit BCFtools v1.3.127. Verwenden Sie die folgenden Befehlszeilen und Parameter (alle anderen Optionen links Standard):
Bcftools Isec - n + 1 $VCFPARENTAL1.gz $VCFPARENTAL2.gz $VCFPARENTAL3.gz \
$VCFMUTANT1.gz $VCFMUTANT2.gz $VCFMUTANT3.gz > common_variants.list
Hinweis: Dieser Befehl ergab eine Datei codiert mit binären Mustern wo wäre Sequenzvarianten, die in den ersten Mutanten nur binär codiert als "000100", den zweiten Mutanten nur als "000010", alle drei Mutanten als "000111", etc.. Diese Dateien wurden generiert für jede Gruppe von Eltern und Mutant replizieren VCF-Dateien. - Kompilieren Sie die Variante Kreuzung Listendateien zusammen mit den Namen der Datei an jede Zeile mit dem UNIX Grep-Befehl angehängt:
Grep "." *.list > all.list - Querverweis auf die vollständige Variante Liste mit der aktuellen GFF3 Annotation Datei (ftp://ftp.ebi.ac.uk/pub/databases/pombase/pombe/Chromosome_Dumps/gff3/schizosacchar omyces_pombe.chr.gff3) mit einem benutzerdefinierten Python-Skript (variant_characterize.py) konsequente SNP-Standorten in Protein-kodierenden Regionen (Synonym und nicht gleichbedeutend), 5 ' und 3 ' wo und NcRNA identifizieren.
python3 variant_characterize.py--Liste common_variants.list \
Gff - schizosaccharomyces_pombe.chr.gff3 \
--Fasta Schizosaccharomyces_pombe. ASM294v2.30.DNA.Genome.FA \
--Muster 000100--, all.list.filter.000100
Wiederholen Sie dieses Skript ändern--Muster und das Suffix der Ausgabedatei (--heraus) mit den binären
Muster: 000010 000001, 000110, 000011, 000101 und 000111 - Kombinieren Sie die Ausgabe von diesen Skript läuft in eine tabulatorgetrennte Datei als Tabelle angezeigt werden. Die kommentierte Tabelle der Varianten gehören diejenigen, die angezeigt werden in einem oder beiden mutierten Angabe bezogen auf den Hintergrund. Die binäre Attributfeld bezeichnet entweder aussehen in einem einzigen mutierten Stamm (000100, 000010, 000001), zwei mutierte Stämme (000011, 000101, 000110) oder alle drei mutierte Stämme (000111).
- Analysieren Sie die kommentierten Ausgabelisten Varianten nicht in die elterliche Proben, aber in einem, zwei oder alle drei mutierten Proben gefunden. Die Anmerkung kennzeichnet die genomische Lage und Klasse Variante (Synonym/nicht-Synonym in kodierenden Region, 3' / 5' UTR, nicht-kodierenden, etc..). Aus dieser Liste von Kandidaten Mutationen möglicherweise ein Beispiel für eine stark relevanten Kandidaten nicht gleichbedeutend Codierung Variante, die konsequent in alle drei Stämme. Eine andere Art von starker Kandidat wäre eine Ansammlung von verschiedenen nicht Synonym oder vermeintliche regulatorische Mutationen in die mutierte Stämme erscheinen dicht beieinander oder innerhalb des gleichen Gens.
Ergebnisse
Langsam wachsende Mutanten anzeigen phänotypischen Erholung in Flüssigkultur
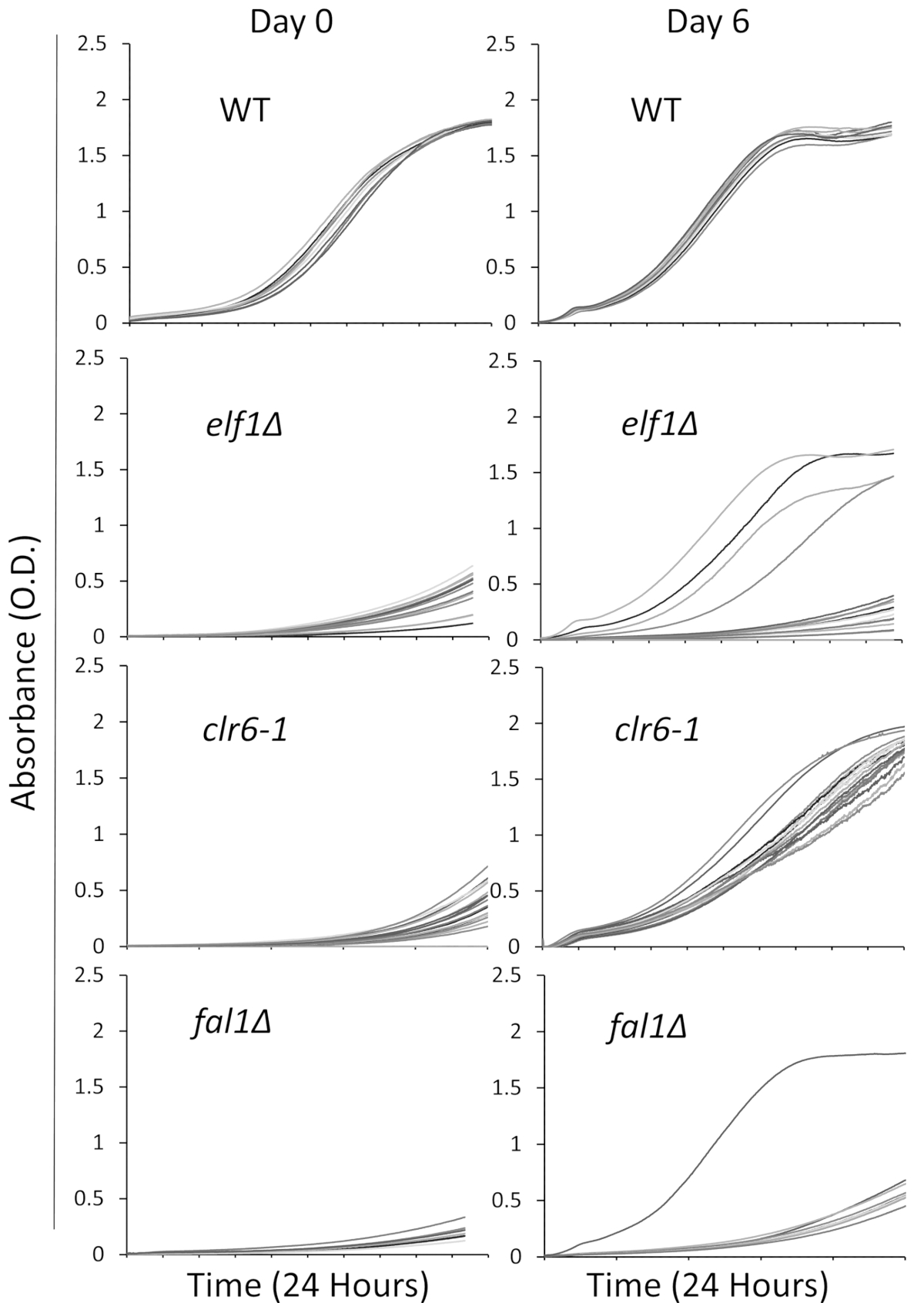
Wir drei Mutanten eingebunden in eine Vielzahl biologischer Signalwege mit einem Kranken, langsamwüchsig, Phänotyp ausgesucht: AAA Familie ATPase Elf1, Histon Deacetylase Clr6 und Exon-Junction-Komplex Komponente Fal1. Ein Wildtyp Stamm und Stämme tragen Mutationen dieser drei Gene, die mit Wildtyp Stämmen backcrossed worden waren wurden in einzelnen Kolonien durchzogen, und 16 einzelne Kolonien wurden zufällig ausgewählt, um im Reich Flüssigmedien mit 96-Well-Platte als kultiviert werden wie oben beschrieben. Wachstumskurven der einzelnen Kolonien wurden zum ersten Zeitpunkt (Tag 0) und für 6 Tage mit kontinuierliche Überwachung mit dem Reader Platte aufgezeichnet. Wie erwartet, zeigen Wildtyp Kolonien keine spürbaren Veränderungen in ihre Wachstumskurven im gesamten Experiment31 (Abbildung 1). Insbesondere zeigen vier Kolonien mit elf1∆ Hintergrund und eine fal1∆ Kolonie eine dramatische Verschiebung im Wachstum von langsamwüchsig bis einige unterschiedlicher Wachstum ähnlich oder in der Nähe der Wildtyp Kolonien. Dramatisch, zeigen alle clr6-1 -Mutanten eine konsequente phänotypische Erholung, wächst mit einer schnelleren Rate bis zum Jahresende die Assay-31 (Abbildung 1). Um die unterschiedlichen Phänotypen zu charakterisieren, verweisen wir auf die ursprünglichen Stämme, die als "P-Stämme" (oder elterliche Stämme) langsamwüchsig und zu den Klängen zeigt phänotypischen Erholung als "S-Stämme" (oder unterdrückte Stämme). Bitte beachten Sie, dass Abbildung 1 ein Beispiel für eine Runde des Experiments Screening ist und repräsentiert nicht die Gesamt-ergänzende Unterdrücker identifiziert und in den folgenden repräsentative Ergebnisse sequenziert.
Phänotypische Erholung wird vererbbare Eigenschaften zugeschrieben
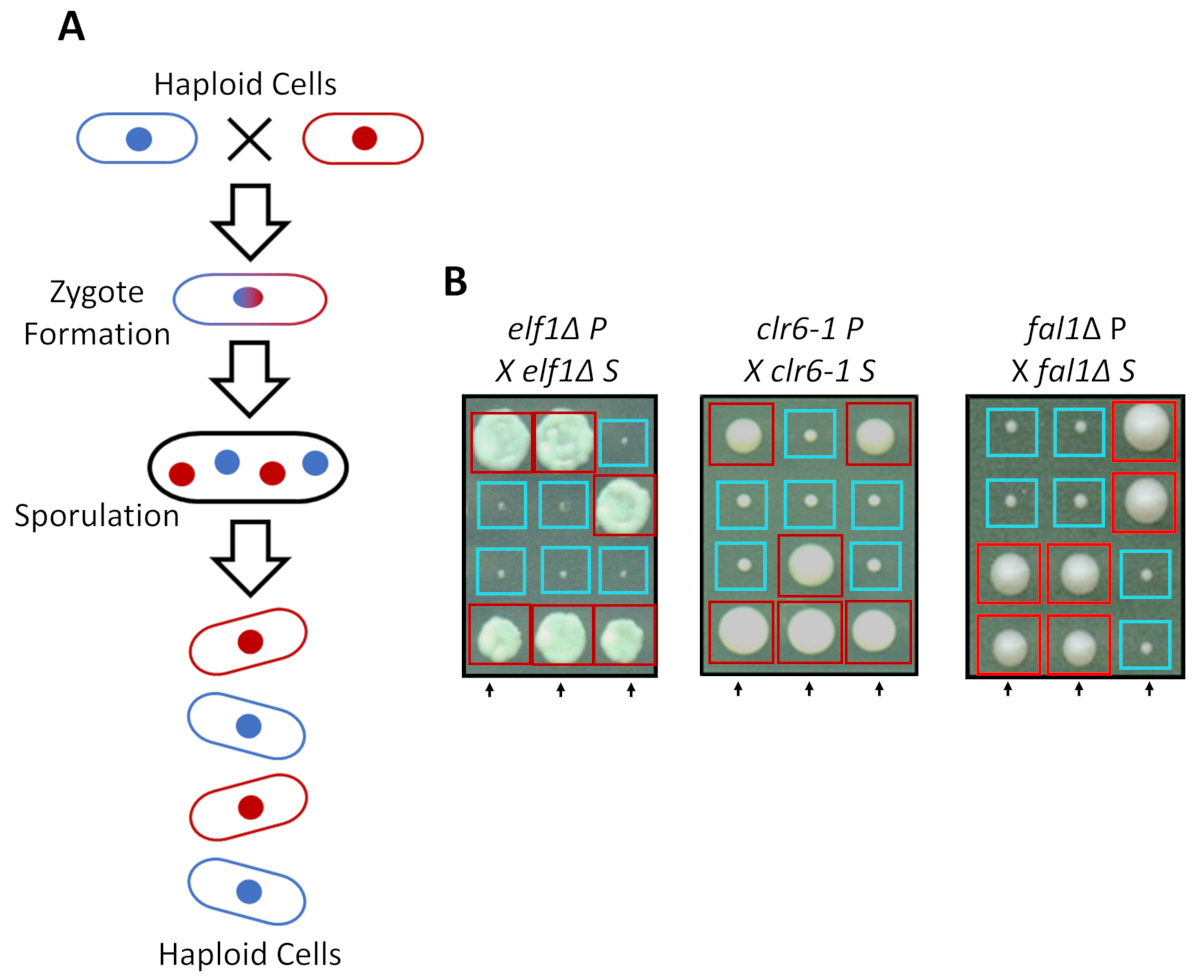
S. Pombe kann als ein Haploid in rich-Media, sondern zwei haploiden Stämme mit ergänzenden Paarung Arten Mate unter Stickstoff Hunger wachsen. Meiose in Spalthefe bringt eine Runde von Doppelarbeit, gefolgt von zwei Runden der Zellteilung. Die sexuelle Zyklus führt zur Bildung von vier haploide Sporen tragen das genetische Material des elterlichen Stammes mit 2:2 Trennung von genetischen Merkmalen, die nach den Regeln der klassischen Mendelschen Genetik (Abbildung 2A). Wenn für die gleiche Menge an Zeit auf dieselbe Platte gewachsen, bestätigt wir die 2:2-Trennung, wenn Rückkreuzung alle Sorten (S-Stämme) mit ihrer elterlichen Sorten (P-Stämme), unterdrückt die 2 kleinen (Wachstum defekt) und 2 große (Suppressor Phänotyp) geführt Kolonien. Einzelne Beispiele für unterdrückte elf1∆, clr6-1 und fal1∆ Zellen sind in Abbildung 2 bdargestellt. Wir haben bestätigt, dass alle isolierten S-Stämme ein monogene genetisches Element tragen, das den langsam wachsenden Phänotyp ihrer P-Sorten (Daten nicht gezeigt) unterdrückt.
Die Sequenzierung des gesamten Genoms identifiziert erfolgreich Suppressor-Mutationen
Als Beispiel zur wir gepaart-End kompletten Genoms genetische Elemente verantwortlich für die phänotypische Erholung in elf1∆ S -Stämme verwendet. Eine ausführlichere Beschreibung der Datenanalyse ist verfügbar online-31. Kurz, beschäftigten wir biologische Triplicates zweier unabhängig generierten elf1∆ P -Sorten und biologische Duplikate von fünf nicht-komplementären Gruppen elf1∆ S -Stämme, die jeweils unterschiedliche Entstörer enthalten. Nachdem wir eine Liste mit kommentierten Varianten von Bioinformatik Analyse (6,1-10), erhalten priorisiert wir bestimmte Klassen von Varianten, die für unsere Analyse relevant waren. Wir konzentrierten uns auf die Identifizierung von konsistenten genomischen Veränderungen, die in allen biologischen Wiederholungen von einzelnen elf1∆ S -Stämme, die im Vergleich zu ihrer elterlichen elf1∆ P -Sorten (Abbildung 3 und zusätzliche Tabellen 1-4 identisch sind ). Wir identifizierten fünf nonsynonymous Änderungen bei CDS Regionen in allen fünf verschiedene elf1∆ S Stämme, einschließlich rli1 +, SPBPJ4664.02, cue2 + und rpl2702 +. S-A1 und A2-S enthalten mutierte SPBPJ4664.02, obwohl die Mutationen an verschiedenen Aminosäuren. Da SPBPJ4664.02 eine lange gen (11.916 Nukleotide) mit Hunderten von Wiederholungen ist, konnten die Mutationen durch Durchführung von PCR gefolgt von Sequenzierung bestätigt werden. S-A3 enthält eine Löschung Mutante in rli1 , die in beiden biologischen Duplikate übereinstimmt. Jedoch das mutierte nicht mit der S-Phänotyp in elf1∆ Hintergrund Co zu trennen. Wir identifizierten einer cue2 Mutante (cue2-1) in S-B1, mit den Aminosäuren, 396-400 fehlt. S-B2 enthält eine rpl2702 Mutante (rpl2702-1), die Aminosäure an Position 45 aus Glycin und Aspartat-31ändert. Cue2-1 und rpl2702-1 wurden als elf1∆ Entstörer bestätigt, wie unten dargestellt.
Genetische Bestätigung der identifizierten Suppressor Mutationen überprüft die Erblichkeit des Phänotyps Erholung
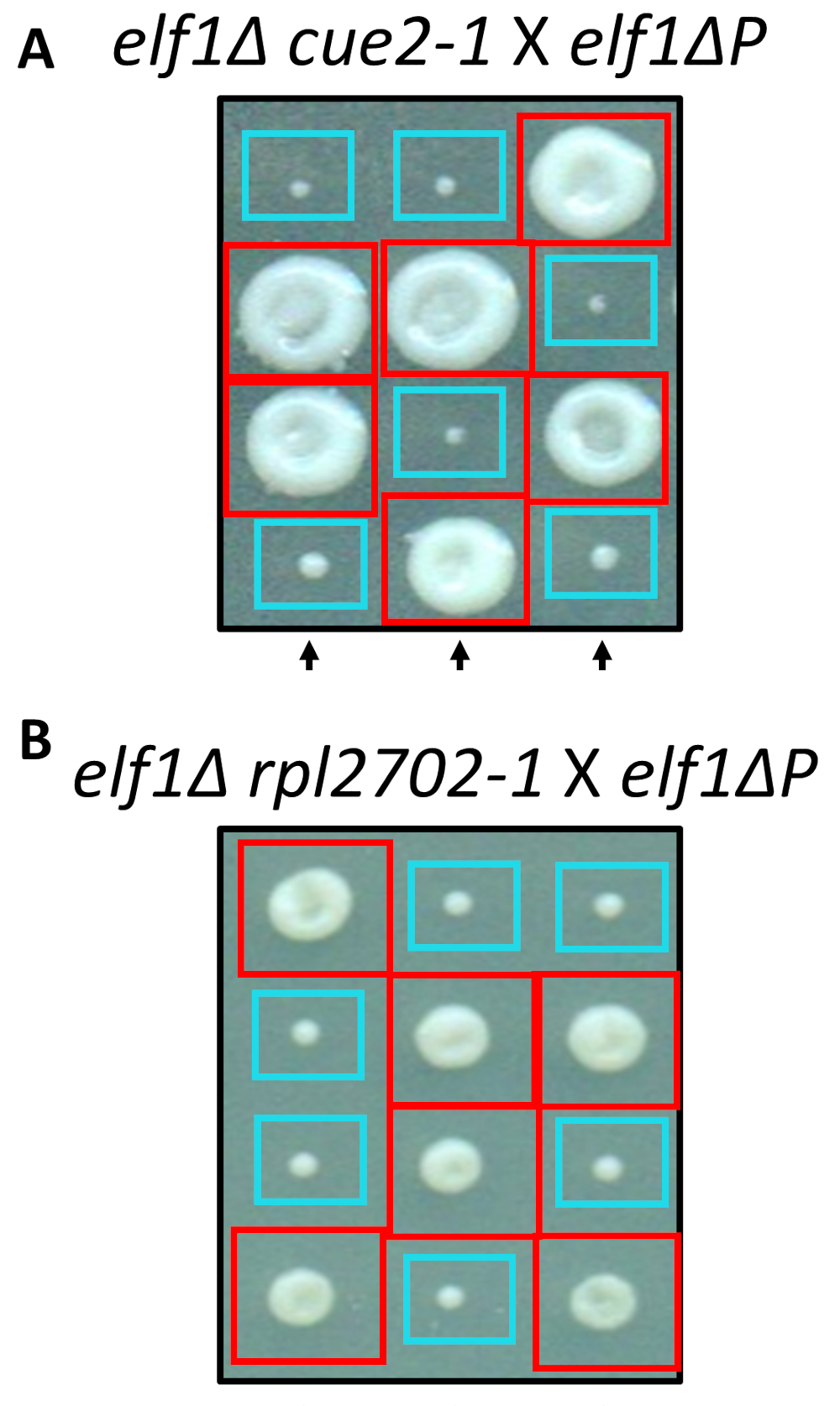
Zwei der identifizierten nonsynonymous Änderungen, cue2-1 und rpl2702-1, wurden im Labor mit Standardprotokolle für Site-verwiesene Mutagenese rekonstruiert. Doppelte mutierte Stämme cue2-1 elf1∆ P und rpl2702-1 elf1∆ P wurden mit der kostenlosen elf1∆ P Stamm31 (Abbildung 4) gekreuzt. Würden die nonsynonymous Mutationen identifiziert über diesen Bildschirm ausreicht, um elf1∆ Pzu unterdrücken, würde dann die resultierenden Tetraden ein 2:2 kleine bis große Verhältnis in den Kolonien aus den 4 Sporen in jeder Tetrade zeigen. Genetische Kreuzung zeigte, dass die identifizierten Suppressor-Mutationen erfolgreich sind bei der Unterdrückung des langsam wachsenden Phänotyps elf1∆ P und vererbbar.

Abbildung 1: phänotypische Erholung durch die Aufnahme von Wachstumskurven in einem Teller Reader überwacht werden kann. Sechzehn einzelne Kolonien der Wildtyp (WT), elf1∆, clr6-1und fal1∆ wurden in einer 96-Well-Platte gelegt. Wachstumskurven wurden über einen Zeitraum von 24 Stunden aufgezeichnet und Kolonien waren wieder verdünnt täglich in rich-Media. Der Wachstum defekt ist offensichtlich durch die geringe Absorption (O.D) am Ende der Periode 24 h am Tag 0. Phänotypisch wiederhergestellten Stämme sind diejenigen, die Anzeigen eine Wachstumskurve ähnlich, oder in der Nähe der Wildtyp-im Zeitraum von 24 h am Tag 6. Vier Kolonien von elf1∆, eine Kolonie von fal1∆und alle Kolonien von clr6-1 zeigte verschiedene Ebenen der phänotypischen Erholung nach 6 Tagen. Bitte klicken Sie hier für eine größere Version dieser Figur.
{kind=link}

Abbildung 2: genetische Kreuzung kann bestätigen, dass die phänotypische Erholung ein einzelnes vererbbar Allel zugeschrieben wird. (A) bei Spalthefe Zellen Stickstoff Hunger, zwei haploiden Zellen mit einer ergänzenden Paarung Art ausgesetzt sind eine Zygote, die sporulates generieren kann, um eine Tetrade von 4 Sporen zu generieren. Die elterlichen genetischen Materials werden während der Meiose, die nach den Regeln der Mendelian Genetik zu trennen. (B) erholt phänotypisch Kolonien (beschriftete S für unterdrückt) mit angegeben elterlichen Genotypen wurden rückgekreuzt mit ihrer kostenlosen elterliche Kolonie (zeigt keine phänotypischen Erholung, mit der Bezeichnung P, für Eltern). Genetische Kreuze zeigen 2:2 kleine (schlechte Fitness) (wiederhergestellte Fitnessraum) Kolonien zeigen, dass die phänotypische Erholung erblich ist und auf einem einzigen genetischen Element zugeschrieben werden kann. Rote Kästchen sind Kolonien tragen die Suppressor-Allel und blauen Kästen Kolonien tragen das elterliche Allel. Diese Zahl wurde von Marayati Et Al., 201831geändert. Bitte klicken Sie hier für eine größere Version dieser Figur.
{kind=link}

Abbildung 3: Analyse der genomweite Sequenzierungsdaten identifizieren genetische Elemente der phänotypischen Erholung verantwortlich. Drei biologische repliziert der beiden elterlichen "P" Stämme (P-A und P-B), und zwei biologische Wiederholungen von fünf phänotypisch erholt "S" Stämme (S-A1, S-A2 und A3 S - erholt von P-A; S-B1 und S-B2 von P-B), wurden sequenziert und die Mutationen wurden als eine Liste von jede Mutation in der wiederhergestellten Belastung im Vergleich zu den elterlichen Belastung Genom es abgeleitet wurde (z. B. PA vs. S-A1 usw.) organisiert. Die Gesamtzahl der gefundenen Mutationen über das gesamte Genom von solchen paarweisen Vergleichen war 660. Insgesamt 44 Mutationen wurden identifiziert als einzige Mutationen, die auftreten, in beiden biologischen Wiederholungen der gleichen Sorte "S" ausgewählt wurden. Aus 44 Mutationen wurden 12 Mutationen einfügen/löschen (INDEL) oder nicht - Synonym für Mutationen. Aus den 12 INDEL oder nicht gleichbedeutend Mutationen, fünf in das Protein kodieren Sequenz aufgetreten. Die fünf Mutationen möglicherweise korrelieren mit dem einzigen genetischen Element verantwortlich für die phänotypisch wiederhergestellten Stämme: eine nicht gleichbedeutend Mutation in SPBPJ4664.02 gefunden in S-A1 und S-A2, INDEL in rli1 in S-A3, INDEL in cue2 in S-B1 und nicht gleichbedeutend Mutationen im rpl2702 in S-B2 gefunden gefunden. Genaue Ablauf Informationen über die Mutationen und dem gefilterten Hintergrund beinhaltet ergänzende Tabellen 1-4. Bitte klicken Sie hier für eine größere Version dieser Figur.
{kind=link}

Abbildung 4: Bestätigung der Unterdrücker identifiziert durch Sequenzierung des gesamten Genoms. Ergebnisse vom gesamten Genoms wurden durch selbstständig erzeugen die Mutationen und Durchführung bestätigt, genetische Kreuze bestätigen die phänotypische Erholung durch die Kreuzung einer elf1∆ cue2-1 -Stamm mit einem elf1∆ P -Stamm und elf1∆ rpl2702-1 mit elf1∆ P -Stamm. Drei repräsentative vertikale Tetraden werden angezeigt. Rote Felder sind Doppel-Mutante Kolonien (elf1 cue2-1oder elf1 rpl2702-1); blaue Boxen sind elf1∆ Kolonien. Diese Zahl wurde von Marayati Et Al., 201831geändert. Bitte klicken Sie hier für eine größere Version dieser Figur.
{kind=link}
Ergänzende Tabelle1. Klicken Sie hier, um diese Tabelle herunterzuladen.
Zusätzliche Tabelle 2 . Klicken Sie hier, um diese Tabelle herunterzuladen.
Zusätzliche Tabelle 3 . Klicken Sie hier, um diese Tabelle herunterzuladen.
Zusätzliche Tabelle 4 . Klicken Sie hier, um diese Tabelle herunterzuladen.
Zusätzliche Dateien kodieren . Klicken Sie bitte hier, um die Dateien herunterzuladen.
Diskussion
Das hier beschriebene Protokoll stellt eine neuartige und einfache Bildschirm für spontane Suppressor Mutationen nachweisbar durch phänotypische Rückgewinnung von Mutationen verleihen langsames Wachstum in Spalthefe, einen Phänotyp charakteristisch für über 400 Gene in S. Pombe, das Funktion von vielen von denen bleibt unbekannt2,32. Bisherige Methoden wurden andere Ansätze für Suppressor Mutationen in Mikroorganismen, einschließlich der Verwendung von mutagenen21oder die Anwendung eine Temperaturverschiebung temperatursensible Mutanten Hintergründe33auf den Bildschirm. Im Gegensatz dazu zeigt dieses Protokolls phänotypischen Erholung erfolgt ohne zusätzliche Umwelt-/chemische Interferenzen und highlights den Fitness-Vorteil des Aufstiegs der Unterdrückung Mutationen schließlich übernahm die verfügbaren Ressourcen in Flüssigkeit Kultur. Auf diesem Bildschirm können die Isolation der Bypass Unterdrücker oder Interaktion Entstörer, weil es effektiv für beide Loss-of-Function-Mutationen wie elf1∆ oder fal1∆ und Punktmutationen z. B. clr6-1, solange die Mutanten Fitness-Mängel in der flüssigen Kultur zu demonstrieren.
Bisher haben alle wiederhergestellten S-Stämme, die wir untersucht haben unterschiedliche Grade der phänotypischen Erholung gezeigt. Durch genetische Kreuzung erkannt, der wiederhergestellten Phänotyp resultiert aus einem einzigen genetischen Element und ist vererbbar (Beispiele siehe Abbildung 2). Dies ist einer der wichtigsten Vorteile dieser Methode im Vergleich zu chemischen oder UV-basierten Suppressor-Bildschirme, die oft mehrere genomic Loci Ziel. Es ist üblich, ein oder zwei Kolonien erholt aus 16 Kolonien/Stamm (ca. 10 %) zu beobachten innerhalb einer Woche. Allerdings haben wir bemerkt, dass bestimmte Mutanten, wie z. B. die Verlustfunktion Rrp6, die Atom-spezifischen Exosom Untereinheit nie erholte sich die fast Wildtyp-Wachstumsrate in elf1Δ Zellen31beobachtet. Es ist wahrscheinlich, dass die Funktion des Rrp6 nur teilweise kann, durch Entstörer, im Gegensatz zur Funktion der anderen Mutanten getestet kompensiert werden, darunter fal1∆, die gezeigt wurde, um eine schwere meiotische defekt durch seine wichtige Funktion in regulatorischen verursachen Spleißen34. Wir glauben, dass das gleiche Problem, dass alternative Suppressor screening-Verfahren unterliegen würden, wenn Yfg einzigartige, nicht austauschbaren Rollen in Zelle Wachstum hat.
Bevor Sie Genom-Sequenzierung durchführen, ist es optimal zu Rücken-Kreuz phänotypisch wiederhergestellten Kolonien, identifiziert aus der Platte-Leser, den elterlichen Belastungen den genetischen Hintergrund und biologischer Replikate zu erhalten. Darüber hinaus kennzeichnet tief ganze Genomsequenzierung Hunderte von Einzel-Nukleotid-Änderungen, von denen meisten nicht auf biologische Wiederholungen identisch sind, die von geringem Interesse für das Screening sind. Zum Beispiel fanden wir insgesamt 660 genomischer Veränderungen über alle drei Chromosomen zwischen zwei elf1Δ P und fünf verschiedene S-Stämme (Abbildung 3). Wir waren nicht allgemein darauf bedacht identisch Mutationen zwischen sequenzierten biologische repliziert von jedem Stamm, was darauf hindeutet, dass entweder neue Mutationen auftreten, während der Kultivierung der elf1Δ Zellen vor genomic Bibliotheksbau oder zufällige Fehler während der Bibliotheksbau und Sequenzierung eingeführt. Daher ist das Isolieren von Mutationen, die in biologischer Replikate konsistent sind ein wichtiger Aspekt bei der erfolgreichen Identifizierung von Unterdrücker mit Sequenzierung des gesamten Genoms.
Wir identifizieren und bestätigt zwei Unterdrücker in CDS Regionen in fünf aufeinander folgende S-Stämmen. Obwohl Mutationen in SPBPJ4664.02 in S-A1 und A2-S Stämme nachgewiesen wurden, ist es unwahrscheinlich, dass SPBPJ4664.02 eine gültige Suppressor da S-A1 und A2-S kein Suppressor auf dem gleichen gen enthalten sind nicht mit einander (ergänzende (Daten nicht gezeigt). Wir bestätigten auch nicht rli1 in S-A3, die nicht mit der S-Phänotyp als mit elf1Δbackcrossed Co trennen. Alternativ haben wir bestimmte Mutationen in nicht-kodierenden Regionen S-A1, S-A2 und A3-S gefunden. Es ist möglich, dass diese veränderten nichtcodierende genomischen Regionen elf1Δ Phänotypus, lindern die in unseren zukünftigen Studien angesprochen werden sollen. Verglichen mit traditionellen Methoden wie ein Gestänge-Assay, der Jahre zu eine genetische Mutation Karte erfolgen kann, haben wir zwei Entstörer innerhalb von zwei Monaten nach der Bestätigung, dass eine monogene Element S Phänotyp verursacht identifiziert. Mit der schnellen Entwicklung des gesamten Genoms Technologie sind wir optimistisch, dass diese Methode wird effizienter, konsequente Genmutationen in absehbarer Zukunft zu identifizieren.
Dieses Protokoll bietet schrittweise Anweisungen zum Suppressor Mutationen für jedes Gen des Interesses mit einem langsam wachsenden Defekt in der flüssigen Kultur erfolgreich zu identifizieren. Die Einfachheit des Assays ermöglicht die groß angelegten Screenings mehrere genetische Hintergründe von Interesse mit wenig Hands-on Training. Es gibt Zimmer weiter automatisieren mithilfe eines liquid Handling-Roboters, die täglichen Verdünnungen durchzuführen. Da Labor Manipulation von Mikroorganismen unweigerlich Wachstum in der flüssigen Kultur, ein Verfahren benötigt, die von Natur aus selektiv für Fitness, schlagen wir vor, dass dieses Protokoll auf andere große Populationsmodell Organismen wie Bakterien breit angewendet werden kann und andere Hefearten.
Offenlegungen
Die Autoren erklären keine Vermerke, die von den Herstellern der Instrumente in dieser Methode und keine finanziellen Interessenkonflikte.
Danksagungen
Diese Arbeit wurde durch das National Institute of General Medical Sciences, Zuschüsse 1R15GM119105-01 bis K.Z unterstützt. Wir bedanken uns bei allen Rezensenten für aufschlussreiche Kommentare. Wir danken auch Alicia Anderson, James Tucker und Glen Marrs, Elizabeth schwarz für die Diskussion und Kommentare zu dieser Handschrift.
Materialien
| Name | Company | Catalog Number | Comments |
| Adenine, Powder | Acros Organics | 147441000 | Use at 75 mg/L to make liquid and solid rich media (YEA) |
| Bacteriology Petri Dish | Corning, Falcon | C351029 | 100 ×15 mm, use to grow strains to single colonies on solid rich media |
| D-Glucose Anhydrous, Powder | Fisher Chemical | D16-1 | Use at 30 g/L to make liquid and solid rich media (YEA) |
| Difco Agar, Granuated | Becton, Dickinson and Co. | 214530 | Use at 20 g/L to make solid rich media (YEA) |
| DNA extraction buffer | 2% Triton X-100, 1% SDS, 100 mM NaCl, 10 mM Tris-Cl (pH 8.0), 1mM Na2-EDTA | ||
| Focused-ultrasonicator | Covaris Inc. | S220 | Alternatively, use QSonica Q800R sonicator/DNA and chromatin shearing system |
| Gen5 Data Collection and Analysis Software | Biotek, Inc. | GEN5SECURE | Or equivalent, must be compatible with the micro-plate reader, use to export data readings from the micro-plate reader |
| Hydrochloric Acid 1N, Liquid | Fisher Chemical | SA48-4 | Use to adjust pH to 5.5 in liquid and solid rich media |
| Liquid Rich Media (liquid YEA) | 30 g/L D-Glucose, 5 g/L Yeast Extract, 75 mg/L Adenine, pH adjusted to 5.5 with 1 M HCl | ||
| Microplate Reader, Synergy H1 Hybrid Multi-Mode Reader | Biotek, Inc. | BTH1MG | Or equivalent, must read visible light at 600 nm wavelength range |
| Rich Media agar plates (YEA plates) | 30 g/L D-Glucose, 5 g/L Yeast Extract, 75 mg/L Adenine, 20 g/L Agar, pH adjusted to 5.5 with 1 M HCl. | ||
| RNase A/T1 mix | Thermo Fisher Scientific | EN0551 | Use according to manufacturer recommendation |
| Sterile Polystyrene Inoculating Loop | Corning, Inc. | OS101 | Or equivalent, use to transfer colonies from agar plates to 96-well plate |
| Sterile workspace and burners | |||
| Tissue Culture Plate, 96-well Optical Flat Bottom with Low Evaporation Lid | Corning, Falcon | C353072 | Or equivalent, must have optical flat bottom for micro-plate ready |
| TruSeq DNA PCR-Free LT/HT Library Prep Kit | Illumina, Inc. | 20015962 | Use to prepare the whole-genome sequencing library |
| Yeast Extract, Powder | Fisher Chemical | BP1422-500 | Use at 5 g/L to make liquid and solid rich media (YEA) |
Referenzen
- McKay, J. K., Latta, R. G. Adaptive population divergence: Markers, QTL and traits. Trends in Ecology and Evolution. 17 (6), 285-291 (2002).
- Wood, V., Harris, M. A., et al. PomBase: A comprehensive online resource for fission yeast. Nucleic Acids Research. 40 (D1), (2012).
- de Visser, J. A. G. M., Cooper, T. F., Elena, S. F. The causes of epistasis. Proceedings of the Royal Society B: Biological Sciences. 278 (1725), 3617-3624 (2011).
- Sailer, Z. R., Harms, M. J. Detecting high-order epistasis in nonlinear genotype-phenotype maps. Genetics. 205 (3), 107911088(2017).
- Kuzmin, E., Costanzo, M., Andrews, B., Boone, C. Synthetic genetic arrays: Automation of yeast genetics. Cold Spring Harbor Protocols. 2016 (4), 326-332 (2016).
- Tong, A. H. Y., Boone, C. Synthetic genetic array analysis in Saccharomyces cerevisiae. Methods in Molecular Biology. 313 (1), 171-192 (2006).
- Dixon, S. J., Costanzo, M., Baryshnikova, A., Andrews, B., Boone, C. Systematic Mapping of Genetic Interaction Networks. Annual Review of Genetics. 43 (1), 601-625 (2009).
- Boone, C., Bussey, H., Andrews, B. J. Exploring genetic interactions and networks with yeast. Nature Reviews Genetics. 8 (6), 437-449 (2007).
- Bai, X., Yang, Z., Jiang, H., Lin, S., Zon, L. I. Genetic suppressor screens in haploids. Methods in Cell Biology. , 129-136 (2011).
- Manson, M. D. Allele-specific suppression as a tool to study protein-protein interactions in bacteria. Methods. 20 (1), 18-34 (2000).
- Motter, A. E., Gulbahce, N., Almaas, E., Barabási, A. L. Predicting synthetic rescues in metabolic networks. Molecular Systems Biology. 4, 168(2008).
- Peterson, R. T., Shaw, S. Y., et al. Chemical suppression of a genetic mutation in a zebrafish model of aortic coarctation. Nature Biotechnology. 22 (5), 595-599 (2004).
- Giorgini, F., Guidetti, P., Nguyen, Q., Bennett, S. C., Muchowski, P. J. A genomic screen in yeast implicates kynurenine 3-monooxygenase as a therapeutic target for Huntington disease. Nature Genetics. 37 (5), 526-531 (2005).
- Forsburg, S. L., Patton, E., et al. The art and design of genetic screens. Nature reviews. Genetics. 2 (9), 659-668 (2001).
- Johnston, D. S. The art and design of genetic screens. Genetics. 3 (March), 176-188 (2002).
- Jorgensen, E. M., Mango, S. E. The art and design of genetic screens: Caenorhabditis elegans. Nature Reviews Genetics. 3 (5), 356-369 (2002).
- Gocke, E., Müller, L. In vivo studies in the mouse to define a threshold for the genotoxicity of EMS and ENU. Mutation Research - Genetic Toxicology and Environmental Mutagenesis. 678 (2), 101-107 (2009).
- Suzuki, T., Hayashi, M., et al. A comparison of the genotoxicity of ethylnitrosourea and ethyl methanesulfonate in lacZ transgenic mice (Muta(TM)Mouse). Mutation Research - Genetic Toxicology and Environmental Mutagenesis. 395 (1), 75-82 (1997).
- Uttam, J., Alberico, C., De Stasio, E. ENU Mutagenesis. International C. elegans Meeting. , (1995).
- Putrament, A., Baranowska, H., Ejchart, A., Prazmo, W. Manganese Mutagenesis in Yeast. Methods in Cell Biology. 20, 25-34 (1978).
- Bose, J. L. Chemical and UV mutagenesis. Methods in Molecular Biology. 1373, 111-115 (2016).
- Ikehata, H., Ono, T. The Mechanisms of UV Mutagenesis. Journal of Radiation Research. 52 (2), 115-125 (2011).
- Shrivastav, N., Li, D., Essigmann, J. M. Chemical biology of mutagenesis and DNA repair: cellular responses to DNA alkylation. Carcinogenesis. 31 (1), 59-70 (2010).
- De Stasio, E. A., Dorman, S. Optimization of ENU mutagenesis of Caenorhabditis elegans. Mutation Research - Genetic Toxicology and Environmental Mutagenesis. 495 (1-2), 81-88 (2001).
- Probst, F. J., Justice, M. J. Mouse mutagenesis with the chemical supermutagen ENU. Methods in Enzymology. 477 (C), 297-312 (2010).
- Bähler, J., Wu, J. Q., et al. Heterologous modules for efficient and versatile PCR-based gene targeting in Schizosaccharomyces pombe. Yeast. 14 (10), 943-951 (1998).
- Li, H., Durbin, R. Fast and accurate short read alignment with Burrows – Wheeler transform. Bioinformatics. 25 (14), 1754-1760 (2009).
- Van der Auwera, G. A., Carneiro, M. O., et al. From fastQ data to high-confidence variant calls: The genome analysis toolkit best practices pipeline. Current Protocols in Bioinformatics. 43, 11.10.1-11.10.33 (2013).
- Mckenna, A., Hanna, M., et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Research. 20, 1297-1303 (2010).
- Li, H., Handsaker, B., et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 25 (16), 2078-2079 (2009).
- Marayati, B. F., Drayton, A. L., et al. Loss of Elongation-Like Factor 1 Spontaneously Induces Diverse, RNase H-Related Suppressor Mutations in Schizosaccharomyces pombe. Genetics. 209 (4), 967-981 (2018).
- Harris, M. A., Lock, A., Bähler, J., Oliver, S. G., Wood, V. FYPO: The fission yeast phenotype ontology. Bioinformatics. 29 (13), 1671-1678 (2013).
- Xu, X., Wang, L., Yanagida, M. Whole-Genome Sequencing of Suppressor DNA Mixtures Identifies Pathways That Compensate for Chromosome Segregation Defects in Schizosaccharomyces pombe. G3: Genes|Genomes|Genetics. 8 (3), 1031-1038 (2018).
- Marayati, B. F., Hoskins, V., et al. The fission yeast MTREC and EJC orthologs ensure the maturation of meiotic transcripts during meiosis. RNA. 22 (9), 1349-1359 (2016).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten