
Method Article
A Deep-sequencing-assisted, Spontaneous Suppressor Screen in the Fission Yeast Schizosaccharomyces pombe
In This Article
Summary
We present a simple suppressor screen protocol in fission yeast. This method is efficient, mutagen-free, and selective for mutations that often occur at a single genomic locus. The protocol is suitable for isolating suppressors that alleviate growth defects in liquid culture that are caused by a mutation or a drug.
Abstract
A genetic screen for mutant alleles that suppress phenotypic defects caused by a mutation is a powerful approach to identify genes that belong to closely related biochemical pathways. Previous methods such as the Synthetic Genetic Array (SGA) analysis, and random mutagenesis techniques using ultraviolet (UV) or chemicals like ethyl methanesulfonate (EMS) or N-ethyl-N- nitrosourea (ENU), have been widely used but are often costly and laborious. Also, these mutagen-based screening methods are frequently associated with severe side effects on the organism, inducing multiple mutations that add to the complexity of isolating the suppressors. Here, we present a simple and effective protocol to identify suppressor mutations in mutants which confer a growth defect in Schizosaccharomyces pombe. The fitness of cells with a growth deficiency in standard rich liquid media or synthetic liquid media can be monitored for recovery using an automated 96-well plate reader over an extended period. Once a cell acquires a suppressor mutation in the culture, its descendants outcompete those of the parental cells. The recovered cells that have a competitive growth advantage over the parental cells can then be isolated and backcrossed with the parental cells. The suppressor mutations are then identified using whole-genome sequencing. Using this approach, we have successfully isolated multiple suppressors that alleviate the severe growth defects caused by loss of Elf1, an AAA+ family ATPase that is important in nuclear mRNA transport and maintenance of genomic stability. There are currently over 400 genes in S. pombe with mutants conferring a growth defect. As many of these genes are uncharacterized, we propose that our method will hasten the identification of novel functional interactions with this user-friendly, high-throughput approach.
Introduction
The basis of understanding functional links between genes relies on the ability to identify the molecular mechanism(s) by which complex genetic traits diverge to produce diverse phenotypes1. In the fission yeast, Schizosaccharomyces pombe (S. pombe), the majority of protein-coding genes are dispensable for viability2. This result does not speak to the unimportance of these genes, but rather to the intricate compensatory mechanisms underlying the biochemical pathways to which such genes belong. Dissecting these compensatory mechanisms has generated epistasis maps, which have uncovered comprehensive genetic interactions and broadened our understanding of functional biochemical pathways3,4.
High-throughput methods (e.g., Synthetic Genetic Array analysis, or SGA) have been developed to identify genome-wide genetic interactions in budding yeast, and have been expanded for use in fission yeast5,6. Such approaches often rely on a library of strains containing all viable single protein-coding gene deletions (around 3,300 haploid deletion mutants covering over 92% of the fission yeast genome), and require a robotic arm to perform the genetic crosses between the strain of interest and all possible strains in the library6. Further, SGA techniques depend on the ability of library strains to have proper and efficient mating, a phenotype that is abnormal to 444 currently characterized genes in S. pombe2.
Despite the complexity of genetic interactions, comparing the phenotype of a strain carrying mutations in two genes to the phenotype of two strains carrying individual mutations of each gene can have one of two notable outcomes: 1) The double mutant phenotype is worse than the expected multiplicative parental phenotypes in the form of sickness or, in the most extreme case, lethality. This is referred to as a negative genetic interaction, and is generally a sign that the two genes act in parallel biological pathways. 2) The double mutant phenotype is better than the expected combination of parental phenotypes, also known as a positive genetic interaction. A positive genetic interaction is particularly interesting because it indicates that these genes function in the same process. Two positively interacting genes have three potential relationships: a mutant gene may up-regulate the expression of the other gene in a parallel pathway, the two genes may work in concert within the same pathway downstream of one another, or the two genes encode proteins that interact directly with each other. Therefore, positive genetic interactions can be used to map gene regulatory nodes and classify uncharacterized genes in biochemical pathways7,8.
A suppressor is a mutation that can alleviate the sickness phenotype of the mutation of another gene, typically representing a positive genetic interaction between the two genes9,10. Suppressor mutations on a different locus from that of the mutation they suppress are known as extragenic suppressors. They are especially valuable in studying non-viable genetic mutations by synthetically rescuing the lethal phenotype (also known as the Lazarus effect)11. They also have potential therapeutic applications in treating hereditary diseases12,13.
For all of these reasons, the identification of suppressor mutations in various model organisms has been widely utilized to facilitate our understanding of various biochemical pathways14,15,16. Screening for suppressors is usually based on the phenotype of the mutation in question and requires conducting random mutagenesis to isolate the mutations that would alleviate the phenotype. Almost all model organisms have established random mutagenesis methods. For example, N-ethyl-N-nitrosourea (ENU) and ethylmethanesulfonate (EMS), two mutagens that are capable of inducing point mutations in DNA, are widely employed in various models from bacteria to mice17,18,19. In addition, manganese chloride has long been used in yeasts for the ability of the manganese cation to inhibit DNA repair pathways20. Another common approach is UV-induced mutagenesis, which generates genome-wide mutagenic pyrimidine dimers21,22.
Although the utilization of chemical mutagenesis to identify suppressor mutations has been popular, the method has many drawbacks, including the use of dangerous chemicals, highly variable success rates, and the introduction of extra confounding variables presented by the negative side effects of the mutagen on multiple cellular processes23,24. Additionally, chemical mutagenesis often induces multiple mutations in the genome which adds to the complexity of using genetic and sequencing techniques to identify the exact mutation that conferred the suppressor phenotype in the organism25.
To address the shortcomings of current mutagenesis approaches, we present a method to screen for spontaneous suppressor mutations in fission yeast that does not rely on any mutagens or a deletion library. The method isolates suppressors through a positive selection assay. The principle of this method is based on the growth advantage of the mutated suppressor subpopulation in the liquid culture, which can be monitored by an automated plate reader. Mating and meiosis are used only if one would like to clean up the genetic background or confirm the presence of monogenic alleles of suppressors before whole-genome sequencing. If the suppression phenotype is caused by a single mutation, the suppressor phenotype will segregate 2:2 after backcrossing with the parental strains. The suppressor mutations can then be identified using whole-genome sequencing. We propose that this method is applicable for screening suppressors in all microorganisms that can grow to a large population in liquid culture.
Protocol
1. Strain construction and preparation
- Generate a mutation or a deletion of the gene (yfm, your favorite mutation) using standard site-directed mutagenesis (SDM) as described previously26.
- Before starting the screen, (optimally) backcross the mutant strains with a wild-type strain to clean the genetic background and generate fresh-born mutant cells as the parental strains. Streak the parental strain to individual colonies on standard rich media plates. Randomly pick eight to sixteen independent colonies (biological replicates) with the desired mutations for the plate reader assay (see 3.1).
NOTE: This protocol is effective only when the parental strains have a growth defect in liquid media (minimal or rich, with or without drug, or with temperature shifts that cause the growth defect). All parental strains should be haploid and thus able to be genetically crossed with other haploid strains with a complementary mating type.
2. Plate reader assay
- With a sterile applicator, take a small amount of each of the colonies prepared in step 1.1 (no exact amount necessary to inoculate the starting culture) and place in a 96-well polystyrene microplate. Suspend each of the colonies in 200 µL of appropriate liquid media (rich or minimal, with or without drug). Include a blank well for every row on the plate containing 200 µL of the same media (no cells).
- Run the following protocol on a plate reader detection software connected to an automated microplate-reader: Set a kinetic program for 24 h and temperature at 30 °C, with continuous fast orbital shaking (425 cpm, 3 mm amplitude). Set the optical reads to measure light scatter at a wavelength of 600nm for optical density, and set the light to read from below the plate at a reading frequency of 2 min (721 total reads over a 24-h period per well).
- After 24 h, record the final blanked optical density readings (blanked OD600) and use the following formula to determine the volume needed to dilute each of the samples down to O.D. = 0.1:

NOTE: Export the data from the plate reader software and use a spreadsheet software to insert the above formula as a function to batch process the dilution volume to be used from each experimental well. - Every 24 h, dilute each of the samples using the same media as day 0 to O.D. = 0.1 (about 1.5 x 106 cells/mL) using the formula indicated in step 2.3. Save all growth curves generated daily and note any individual colony that shows an increased growth rate, judged by a final O.D. that is significantly higher than the rest of the cohort with the same genetic background or by a growth curve that is similar to that of wild-type colonies.
NOTE: This assay usually takes about 7-14 days. Perform all steps under sterile conditions.
3. Selection of suppressor colonies and confirmation of phenotype.
- From the last day of the plate reader assay (step 2.4), save the liquid cultures that have a noticeably recovered growth rate, presumably by gaining a suppressor mutation that can alleviate the phenotype of the parental mutation. Transfer and mix 250 µL liquid culture to a cryotube containing 250 µL of 50% glycerol. Flash freeze the cells in liquid nitrogen and save the strains in -80oC indefinitely.
- To confirm that the suppressor mutation is a genetically heritable element, use standard genetic crossing methods to cross yfm P (for parental, the strain used at the beginning of the plate reader assay) with yfm S (for suppressor, the strain saved at the end of the plate reader assay). If the suppressor mutation is indeed a genetically heritable element, yfm P × yfm S should yield tetrads in which two colonies have the sickness phenotype of the parental strain and two colonies have the recovered growth rate of the suppressor strain.
- From the cross of step 3.2, pick three colonies with the suppressor phenotype (S strain) and three colonies with the parental phenotype (P strain) from the same genetic cross (3 biological replicates for each), and proceed with the genomic DNA extraction and sequencing steps below.
NOTE: Steps 3.2 and 3.3 are highly recommended, but are not required. Alternatively, one can spread recovered liquid culture collected from 3.1 on rich medium into single colonies, then randomly pick three colonies as biological triplicates for whole-genome sequencing without further genetic confirmation. In this case, three biological triplicates of parental strain should be used for genomic sequencing comparison.
4. Genomic DNA extraction, library production and sequencing.
- For DNA extraction, library preparation, and sequencing, randomly pick three biological replicates per yfm P strain, and three biological replicates of each individually arisen yfm S strains from the genetic crosses (step 3.2) or from the plates that have been spread to obtain single colonies of the S strain (step 3.3 note).
- Grow strains in 10 mL cultures in rich media to mid-log phase (O.D. = 0.5–0.8, about 0.75– 1.2 x 107 cells/mL), and use a shaking incubator to grow liquid cultures at 30 °C with continuously shaking at 250 rpm. Collect cells by centrifugation at 4 °C for 5 min at 1000 x g.
- Suspend pelleted cells in 400 µL of DNA extraction buffer (2% Triton X-100, 1% SDS, 100 mM NaCl, 10 mM Tris-Cl (pH 8.0), 1mM Na2-EDTA), then add 400 µL of glass beads and 400 µL of 25:24:1 phenol:chloroform:isoamyl alcohol. Vortex vigorously for 2 min at 4 °C.
- Add an additional 200 µL of the DNA extraction buffer and mix by inverting several times. Centrifuge for 5 min at 4 °C at 20,000 x g.
- Transfer the aqueous phase to a clean tube, add 20 µg of RNase A/T1 mix, and incubate at 37 °C for 15 min.
- Add an equal volume of 25:24:1 phenol:chloroform:isoamyl alcohol, spin for 5 min at 4 °C at 20,000 x g, then transfer the aqueous phase to a clean tube.
- Add an equal volume of chloroform, mix by inverting several times, then spin for 5 min at 4 °C at 20,000 x g, then transfer the aqueous phase to a clean tube.
- Precipitate DNA with two volumes of 100% ethanol plus 10% volume of 3 M NaOAC (pH 4.3) at -20 °C for at least 2 h, then spin for 5 min at 4 °C at 20,000 x g and collect the pellet.
- Wash pellet (precipitated DNA) twice with chilled 70% ethanol (Centrifuge at 20,000 x g, 5 min, 4 °C) and suspend the pellet in 50 µL of 10 mM Tris Buffer (pH 7.4).
- Use a library prep kit (see Table of Materials) per manufacturer's recommendations to prepare the whole-genome sequencing library.
NOTE: We recommend the kit listed in the Table of Materials because it allows the construction of the genomic library without PCR amplification, which minimizes error mutations generated during PCR amplification. In addition, during the genomic library preparation, do not allow the beads to fully dry by shortening the bead drying time to 1–2 min. - For shearing parameters during the library preparation, use a focused sonicator (see Table of Materials) and set the duty factor to 20%, peak power to 175 W, with 200 cycles per burst, and frequency sweeping mode at 5.5oC to 6 °C for 45 s. Alternatively, use a DNA and chomatin shearing system (see Table of Materials) with the following settings: 50% amplitude at 4 °C with pulse mode, 15 s on and 15 s off for 10 min, with a total processing time of 20 min.
- It is essential to handle the hazardous materials used in this step with care. Consult the appropriate Material Safety Data Sheets and institution’s Environmental Health and Safety Office for handling NaOAC, ethanol, 25:24:1 phenol:chloroform:isoamyl alcohol and chloroform.
- Sequence the resulting genomic libraries. The entire sequencing reads should cover at least three times of the entire genome with the resolution at a single nucleotide range. Paired-ended sequencing (or latest technology) is recommended.
5. Bioinformatics analysis for the identification of the suppressor mutations
- Perform the bioinformatics analysis to focus on the genomic changes that are consistently identified between parental and suppressed yfm strains in all biological replicates.
NOTE: The complete pipeline process is described below, but, additionally, two plain-text BASH- script files, fastq_to_vcf.sh and vcfprocess.sh, are included as supplemental materials to show examples of the workflow from processing of reads to VCF variant files and processing and intersection of the VCF files, respectively. - Trim short-reads using SHEAR (https://github.com/jbpease/shear) following the command lines (all other options default):
shear.py --fq1 $FASTQ1 --fq2 $FASTQ2 --out1 $OUTFQ1 --out2 $OUTFQ2 \
--barcodes1 $BARCODE --platform TruSeq --trimqual 20:20 \
--trimpolyat 0 --trimambig --filterlength 50 --filterunpaired - Map reads to the S. pombe reference genome v2.30 obtained from PomBase (ftp://ftp.ebi.ac.uk/pub/databases/pombase/pombe/Chromosome_Dumps/fasta/) using BWA v0.7.1527. Use the following command line (all other options default):
bwa mem -t 8 $GENOME $OUTFQ1 $OUTFQ2 > $SAM1 - Put alignment SAM files through GATK best practices pipeline28 for variant calling using GATK v3.629, PicardTools v2.5.0 (http://broadinstitute.github.io/picard), and SAMtools v1.3.130. Use the following command lines and parameters (all other options default):
java -Xmx30g -jar picard.jar AddOrReplaceReadGroups INPUT=$SAM1 \
OUTPUT=$BAMMARKED RGID=1 RGLB=lib01 RGPL=illumina \
RGPU=$BARCODE RGSM=$SAMPLENUMBER
samtools fixmate -O bam $BAMMARKED $BAMFIXED
samtools sort -O bam -o $BAMSORTED -T /home/peasejb/tmp $BAMFIXED
samtools index $BAMSORTED
java -Xmx30g -jar GenomeAnalysisTK.jar -T HaplotypeCaller \
-R $GENOME -I $BAMSORTED --genotyping_mode DISCOVERY \
-stand_emit_conf 10 -stand_call_conf 30 -o $VCFRAW - Compress and index VCF files using tabix:
bgzip $VCFRAW.vcf
tabix $VCFRAW.vcf.gz - Compare VCF files among parental and suppressor strains sequencing replicates using BCFtools v1.3.127. Use the following command lines and parameters (all other options left default):
bcftools isec -n+1 $VCFPARENTAL1.gz $VCFPARENTAL2.gz $VCFPARENTAL3.gz \
$VCFMUTANT1.gz $VCFMUTANT2.gz $VCFMUTANT3.gz > common_variants.list
NOTE: This command yielded a file encoded with binary patterns where sequence variants appearing in the first mutant only would be binary-encoded as "000100", the second mutant only as "000010", all three mutants as "000111," etc. These files were generated for each set of parental and mutant replicate VCF files. - Compile the variant intersection list files together with the file name appended to each line using the UNIX grep command:
grep "." *.list > all.list - Cross-reference the complete variant list with the current GFF3 annotation file (ftp://ftp.ebi.ac.uk/pub/databases/pombase/pombe/Chromosome_Dumps/gff3/schizosacchar omyces_pombe.chr.gff3) using a custom Python script (variant_characterize.py) to identify consistent SNP sites in protein-coding regions (synonymous and non-synonymous), 5′ and 3′ UTRs, and ncRNA.
python3 variant_characterize.py --list common_variants.list \
--gff schizosaccharomyces_pombe.chr.gff3 \
--fasta Schizosaccharomyces_pombe.ASM294v2.30.dna.genome.fa \
--pattern 000100 --out all.list.filter.000100
Repeat this script modifying the --pattern and the suffix of the output file (--out) using the binary
patterns: 000010, 000001, 000110, 000011, 000101, and 000111 - Combine the output from all these script runs in a tab-separated file, to be viewed as a spreadsheet. The annotated table of variants includes those that appear in either one or both mutant strain(s) relative to the background. The binary flag field denotes either appearance in a single mutant strain (000100, 000010, 000001), two mutant strains (000011, 000101, 000110), or all three mutant strains (000111).
- Analyze the output annotated lists of variants not found in the parental samples, but found in one, two, or all three mutant samples. The annotation denotes both the genomic location and class of variant (synonymous/non-synonymous in a coding region, 3'/5' UTR, non-coding, etc.). From this list of candidate mutations, an example of a strongly relevant candidates might be a non-synonymous coding variant appearing consistently in all three strains. Another type of strong candidate would be an accumulation of various non-synonymous or putative regulatory mutations in the mutant strains appearing close together or within the same gene.
Results
Slow-growing mutants show phenotypic recovery in liquid culture
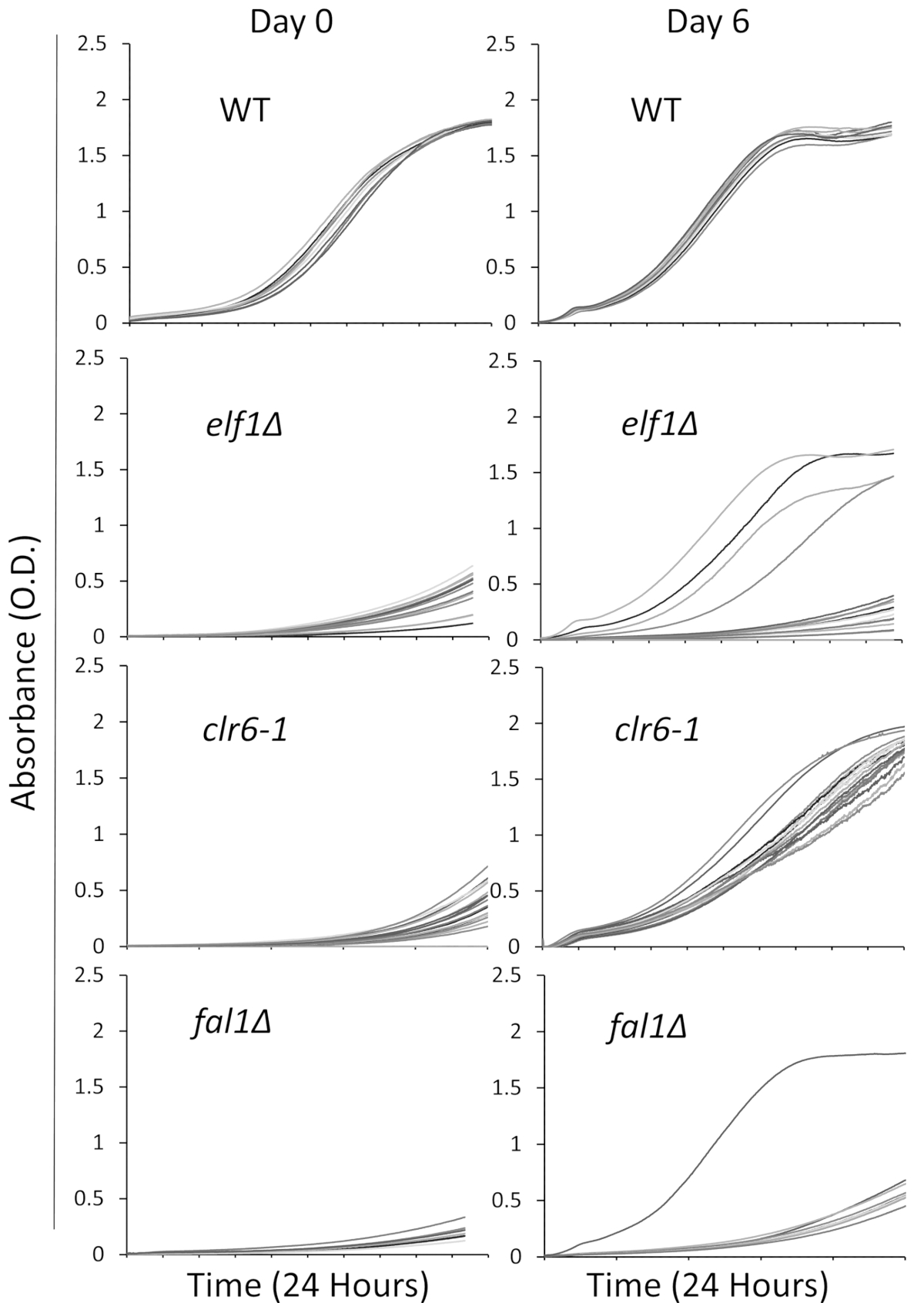
We selected three mutants involved in a variety of biological pathways with a sick, slow-growing, phenotype: AAA family ATPase Elf1, Histone Deacetylase Clr6, and Exon Junction Complex component Fal1. A wild-type strain and strains carrying mutations of these three genes that had been backcrossed with wild-type strains were streaked to individual colonies, and 16 single colonies were randomly selected to be cultured in rich liquid media using the 96-well plate as described above. Growth curves of individual colonies were recorded at the initial time point (day 0) and for 6 days with continuous monitoring using the plate reader. As expected, wild-type colonies show no noticeable changes in their growth curves throughout the experiment31 (Figure 1). Notably, four colonies with the elf1∆ background and one fal1∆ colony show a dramatic shift in growth from slow-growing to some varying levels of growth similar or close to that of wild- type colonies. Dramatically, all clr6-1 mutants show a consistent phenotypic recovery, growing at a faster rate by the end of the assay31 (Figure 1). To characterize the different phenotypes, we refer to the original strains that are slow-growing as “P strains” (or parental strains) and to the strains showing phenotypic recovery as “S strains” (or suppressed strains). Please note that Figure 1 is an example of one round of the screening experiment, and does not represent the total non-complementary suppressors identified and sequenced in the following representative results.
Phenotypic recovery is attributed to heritable traits
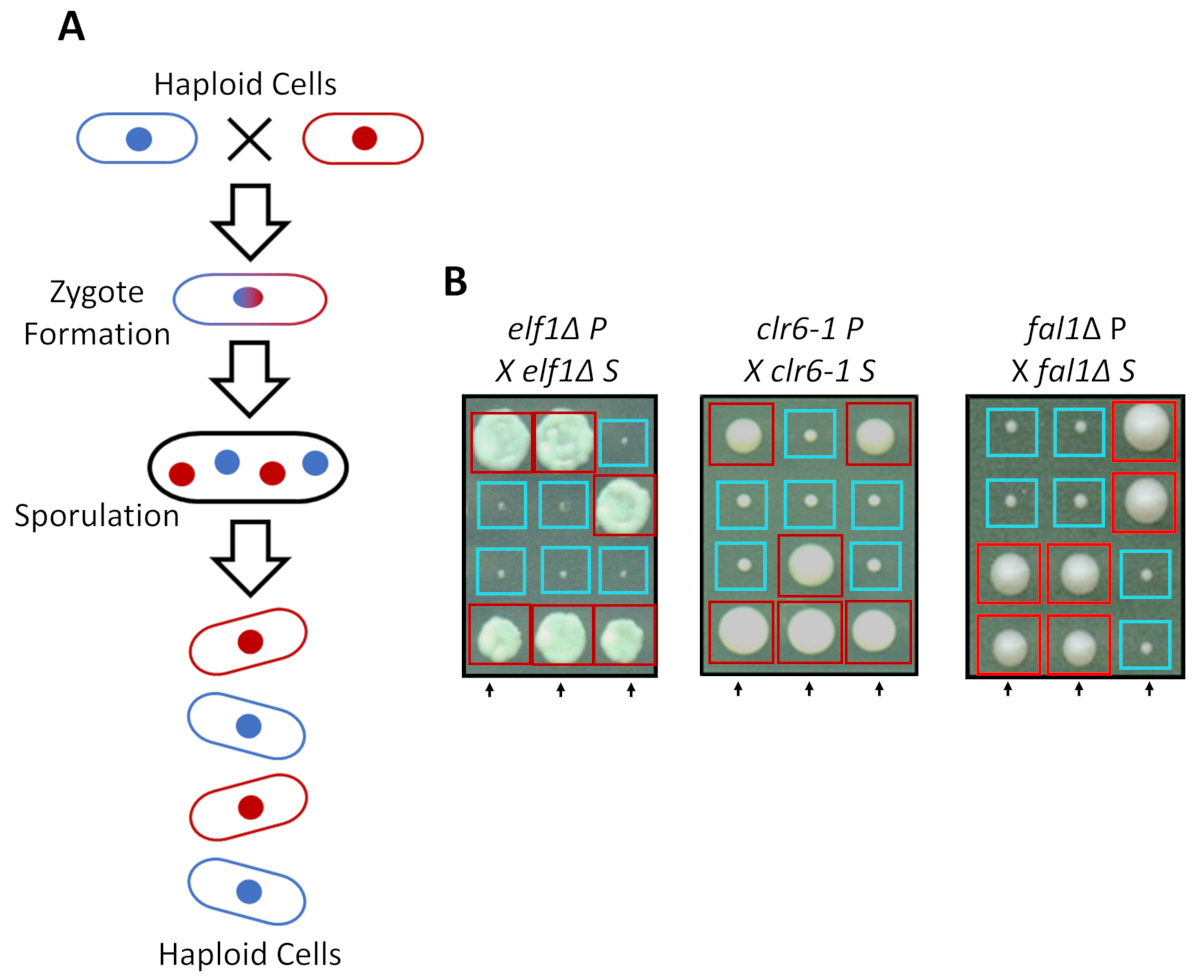
S. pombe can grow as a haploid in rich media, but two haploid strains with complementary mating types mate under nitrogen starvation. Meiosis in fission yeast entails a round of duplication followed by two rounds of cell division. The sexual cycle results in the formation of four haploid spores carrying the genetic material of the parental strain with 2:2 segregation of genetic traits following the rules of classical Mendelian genetics (Figure 2A). When grown on the same plate for the same amount of time, we confirmed the 2:2 segregation when back-crossing all suppressed strains (S strains) with their parental strains (P strains), which resulted in 2 small (growth defect) and 2 large (suppressor phenotype) colonies. Individual examples for suppressed elf1∆, clr6-1 and fal1∆ cells are shown in Figure 2B. We have confirmed that all isolated S strains carry a monogenic genetic element that suppresses the slow-growing phenotype of their P strains (data not shown).
Whole-genome sequencing successfully identifies suppressor mutations
As an example, we used paired-end whole genome sequencing to identify the genetic elements responsible for the phenotypic recovery in elf1∆ S strains. A more complete description of data analysis is available online31. Briefly, we employed biological triplicates of two independently generated elf1∆ P strains and biological duplicates of five non-complementary groups of elf1∆ S strains, each of which contains different suppressors. After we obtained a list of annotated variants from bioinformatics analysis (6.1-10), we prioritized certain classes of variants that were relevant to our analysis. We focused on identification of consistent genomic changes that were identical in all of the biological replicates of individual elf1∆ S strains compared with their parental elf1∆ P strains (Figure 3 and Supplemental Tables 1-4). We identified five nonsynonymous changes at CDS regions in all five different elf1∆ S strains, including rli1+, SPBPJ4664.02, cue2+ and rpl2702+. Both S-A1 and S-A2 contain mutated SPBPJ4664.02, although the mutations occur at different amino acids. Because SPBPJ4664.02 is a long gene (11,916 nucleotides) with hundreds of repeats, the mutations were unable to be confirmed by performing PCR followed by sequencing. S-A3 contains a deletion mutant in rli1 that is consistent in both biological duplicates. However, the mutant did not co-segregate with the S phenotype in elf1∆ background. We identified a cue2 mutant (cue2-1) in S-B1, with the amino acids 396-400 missing. S-B2 contains an rpl2702 mutant (rpl2702-1), which changes amino acid at position 45 from Glycine to Aspartate31. Both cue2-1 and rpl2702-1 were confirmed as elf1∆ suppressors as shown below.
Genetic confirmation of identified suppressor mutations verifies the heritability of the recovery phenotype
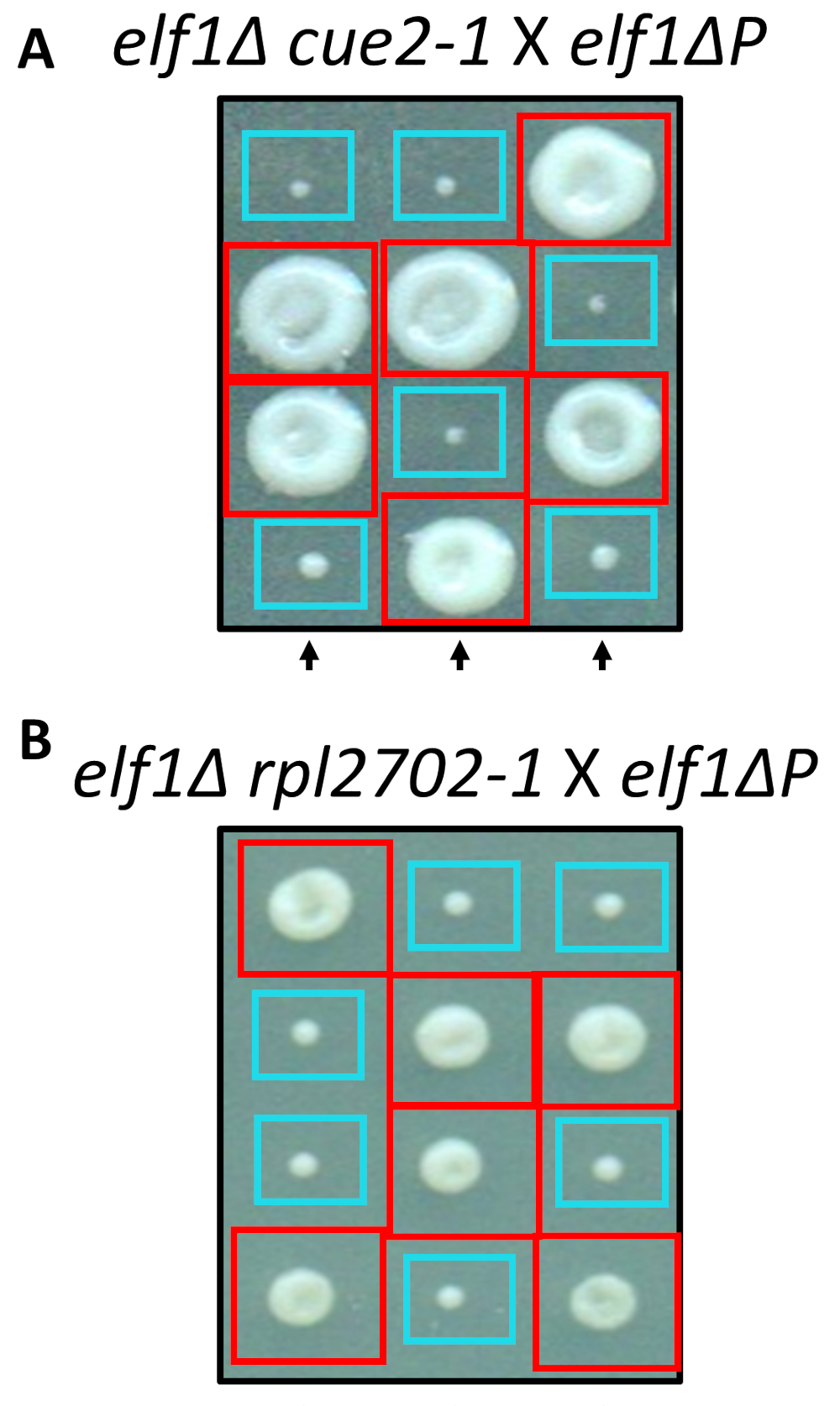
Two of the identified nonsynonymous changes, cue2-1 and rpl2702-1, were reconstructed in the lab using standard protocols for site-directed mutagenesis. Double mutant strains cue2-1 elf1∆ P and rpl2702-1 elf1∆ P were crossed with the complimentary elf1∆ P strain31 (Figure 4). If the nonsynonymous mutations, identified through this screen, were sufficient to suppress elf1∆ P, then the resulting tetrads would show a 2:2 small to large ratio in the colonies resulting from the 4 spores in each tetrad. Indeed, genetic crossing showed that the identified suppressor mutations are successful in suppressing the slow-growing phenotype of elf1∆ P and are heritable.

Figure 1: Phenotypic recovery can be monitored by recording growth curves in a plate reader. Sixteen single colonies of wild-type (WT), elf1∆, clr6-1, and fal1∆ were placed in a 96-well plate. Growth curves were recorded over a 24-h period and colonies were re-diluted daily in rich media. The growth defect is evident by the low absorbance (O.D.) at the end of the 24 h period on day 0. Phenotypically recovered strains are those that display a growth curve similar, or close to, that of wild-type over the 24-h period on day 6. Four colonies of elf1∆, one colony of fal1∆, and all colonies of clr6-1 showed various levels of phenotypic recovery after 6 days. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Genetic crossing can confirm that the phenotypic recovery is attributed to a single heritable allele. (A) When fission yeast cells are subjected to nitrogen starvation, two haploid cells with a complementary mating type can generate a zygote which sporulates to generate a tetrad of 4 spores. The parental genetic materials will segregate during meiosis following the rules of Mendelian genetics. (B) Phenotypically recovered colonies (labeled S, for suppressed) with indicated parental genotypes were back-crossed with their complimentary parental colony (which shows no phenotypic recovery, labeled P, for parental). Genetic crosses showing 2:2 small (poor fitness) to large (recovered fitness) colonies demonstrate that the phenotypic recovery is heritable and can be attributed to a single genetic element. Red boxes are colonies carrying the suppressor allele, and blue boxes are colonies carrying the parental allele. This figure has been modified from Marayati et al., 201831. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Analysis of genome-wide sequencing data to identify genetic elements responsible of phenotypic recovery. Three biological replicates of two parental “P” strains (P-A and P-B), and two biological replicates of five phenotypically recovered switched “S” strains (S-A1, S-A2, and S- A3 recovered from P-A; S-B1 and S-B2 from P-B ), were sequenced and the mutations were organized as a list of each mutation in the recovered strain as compared to the parental strain genome it was derived from (e.g., P-A vs. S-A1, etc.). The total number of detected mutations across the entire genome of all such pairwise comparisons was 660. A total of 44 mutations were identified when only mutations that occur in both biological replicates of the same “S” strain were selected. Out of 44 mutations, 12 mutations were insertion/deletion (INDEL) or non- synonymous mutations. Out of the 12 INDEL or non-synonymous mutations, five occurred in the protein coding sequence. The five mutations potentially correlate with the single genetic element responsible for the phenotypically recovered strains: a non-synonymous mutation in SPBPJ4664.02 found in S-A1 and S-A2, INDEL in rli1 found in S-A3, INDEL in cue2 found in S-B1, and non-synonymous mutations in rpl2702 found in S-B2. Detailed sequence information on the mutations and the filtered background is included in Supplemental Tables 1-4. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Confirmation of the suppressors identified through whole-genome sequencing. Results from whole-genome sequencing were confirmed by independently generating the mutations and performing genetic crosses to confirm the phenotypic recovery by crossing an elf1∆ cue2-1 strain with an elf1∆ P strain, and elf1∆ rpl2702-1 with elf1∆ P strain. Three representative vertical tetrads are shown. Red boxes are double-mutant colonies (elf1 cue2-1, or elf1 rpl2702-1); blue boxes are elf1∆ colonies. This figure has been modified from Marayati et al., 201831. Please click here to view a larger version of this figure.
{kind=link}
Supplemental Table 1. Please click here to download this table.
Supplemental Table 2. Please click here to download this table.
Supplemental Table 3. Please click here to download this table.
Supplemental Table 4. Please click here to download this table.
Supplemental Coding Files. Please click here to download the files.
Discussion
The protocol described here represents a novel and simple screen for spontaneous suppressor mutations detectable through phenotypic recovery of mutations conferring slow growth in fission yeast, a phenotype characteristic of over 400 genes in S. pombe, the function of many of which remains unknown2,32. Previous methods have taken other approaches to screen for suppressor mutations in microorganisms, including the use of mutagens21, or the application of a temperature shift in temperature-sensitive mutant backgrounds33. In contrast, this protocol shows that phenotypic recovery occurs without additional environmental/chemical interference and highlights the fitness advantage of the rise of suppression mutations eventually taking over the resources available in liquid culture. This screen allows the isolation of both bypass suppressors or interaction suppressors because it is effective for both loss-of-function mutations such as elf1∆ or fal1∆ and point mutations such as clr6-1, as long as the mutants demonstrate fitness defects in liquid culture.
So far, all recovered S strains that we have investigated have demonstrated varying degrees of phenotypic recovery. As detected through genetic crossing, the recovered phenotype is attributable to a single genetic element and is heritable (examples shown in Figure 2). This is one of the most significant advantages of this method compared to chemical-based or UV-based suppressor screens, which often target multiple genomic loci. It is common to observe one or two colonies recovered out of 16 colonies/strain (around 10%) within a week. However, we did notice that certain mutants, such as the loss-of-function of Rrp6, the nuclear-specific exosome subunit, never recovered to the almost wild-type growth rate observed in elf1Δ cells31. It is likely that the function of Rrp6 can only be partially compensated by suppressors, unlike the function of the other mutants tested, including fal1∆, which was shown to cause a severe meiotic defect through its important function in regulatory splicing34. We believe that alternative suppressor screening methods would be subject to the same problem when yfg has unique, non-replaceable roles in cell growth.
Before performing genomic sequencing, it is optimal to back-cross the phenotypically recovered colonies, identified from the plate-reader, with the parental strains to clear the genetic background and obtain biological replicates. In addition, deep whole-genome sequencing identifies hundreds of single nucleotide changes, most of which are not identical between biological replicates that are of little interest for the screening. For example, we found a total of 660 genomic alterations across all three chromosomes between the two elf1Δ P and the five different S strains (Figure 3). We did not commonly observe identical mutations between sequenced biological replicates of each strain, suggesting that either new mutations may arise during the culturing of elf1Δ cells before genomic library construction or random errors may be introduced during the library construction and sequencing. Hence, isolating mutations that are consistent across biological replicates is an important aspect in the successful identification of suppressors using whole-genome sequencing.
We identified and confirmed two suppressors in CDS regions in five sequenced S strains. Although mutations in SPBPJ4664.02 were detected in both S-A1 and S-A2 strains, it is unlikely that SPBPJ4664.02 is a valid suppressor because S-A1 and S-A2 do not contain a suppressor on the same gene as they are not complementary with each other (data not shown). We also did not confirm rli1 in S-A3, which did not co-segregate with the S phenotype when backcrossed with elf1Δ. Alternatively, we found specific mutations in non-coding regions in S-A1, S-A2 and S-A3. It is possible that these altered non-coding genomic regions alleviate the elf1Δ phenotype, which will be addressed in our future studies. Compared to traditional methods such as a linkage assay, which may take years to map a genetic mutation, we identified two suppressors within two months after confirming that a monogenic element caused the S phenotype. With the fast development of whole-genome sequencing technology, we are optimistic that this method will be more efficient to identify consistent genetic mutations in the foreseeable future.
In summary, this protocol provides step-by-step directions to successfully identify suppressor mutations for any gene of interest with a slow-growing defect in liquid culture. The simplicity of this assay allows for the large-scale screening of multiple genetic backgrounds of interest with little hands-on training. There is room to further automate the process by using a liquid handling robot to perform the daily dilutions. Since laboratory manipulation of microorganisms inevitably requires growth in liquid culture, a process that is inherently selective for fitness, we propose that this protocol can be broadly applied to other large-population model organisms such as bacteria and other yeast species.
Disclosures
The authors declare no endorsements by the manufacturers of the instruments used in this method, and no competing financial interests.
Acknowledgements
This work was supported by the National Institute of General Medical Sciences, grants 1R15GM119105-01 to K.Z. We thank all reviewers for insightful comments. We also thank James Tucker, Alicia Anderson, Elizabeth Black and Glen Marrs for the discussion and comments on this manuscript.
Materials
| Name | Company | Catalog Number | Comments |
| Adenine, Powder | Acros Organics | 147441000 | Use at 75 mg/L to make liquid and solid rich media (YEA) |
| Bacteriology Petri Dish | Corning, Falcon | C351029 | 100 ×15 mm, use to grow strains to single colonies on solid rich media |
| D-Glucose Anhydrous, Powder | Fisher Chemical | D16-1 | Use at 30 g/L to make liquid and solid rich media (YEA) |
| Difco Agar, Granuated | Becton, Dickinson and Co. | 214530 | Use at 20 g/L to make solid rich media (YEA) |
| DNA extraction buffer | 2% Triton X-100, 1% SDS, 100 mM NaCl, 10 mM Tris-Cl (pH 8.0), 1mM Na2-EDTA | ||
| Focused-ultrasonicator | Covaris Inc. | S220 | Alternatively, use QSonica Q800R sonicator/DNA and chromatin shearing system |
| Gen5 Data Collection and Analysis Software | Biotek, Inc. | GEN5SECURE | Or equivalent, must be compatible with the micro-plate reader, use to export data readings from the micro-plate reader |
| Hydrochloric Acid 1N, Liquid | Fisher Chemical | SA48-4 | Use to adjust pH to 5.5 in liquid and solid rich media |
| Liquid Rich Media (liquid YEA) | 30 g/L D-Glucose, 5 g/L Yeast Extract, 75 mg/L Adenine, pH adjusted to 5.5 with 1 M HCl | ||
| Microplate Reader, Synergy H1 Hybrid Multi-Mode Reader | Biotek, Inc. | BTH1MG | Or equivalent, must read visible light at 600 nm wavelength range |
| Rich Media agar plates (YEA plates) | 30 g/L D-Glucose, 5 g/L Yeast Extract, 75 mg/L Adenine, 20 g/L Agar, pH adjusted to 5.5 with 1 M HCl. | ||
| RNase A/T1 mix | Thermo Fisher Scientific | EN0551 | Use according to manufacturer recommendation |
| Sterile Polystyrene Inoculating Loop | Corning, Inc. | OS101 | Or equivalent, use to transfer colonies from agar plates to 96-well plate |
| Sterile workspace and burners | |||
| Tissue Culture Plate, 96-well Optical Flat Bottom with Low Evaporation Lid | Corning, Falcon | C353072 | Or equivalent, must have optical flat bottom for micro-plate ready |
| TruSeq DNA PCR-Free LT/HT Library Prep Kit | Illumina, Inc. | 20015962 | Use to prepare the whole-genome sequencing library |
| Yeast Extract, Powder | Fisher Chemical | BP1422-500 | Use at 5 g/L to make liquid and solid rich media (YEA) |
References
- McKay, J. K., Latta, R. G. Adaptive population divergence: Markers, QTL and traits. Trends in Ecology and Evolution. 17 (6), 285-291 (2002).
- Wood, V., Harris, M. A., et al. PomBase: A comprehensive online resource for fission yeast. Nucleic Acids Research. 40 (D1), (2012).
- de Visser, J. A. G. M., Cooper, T. F., Elena, S. F. The causes of epistasis. Proceedings of the Royal Society B: Biological Sciences. 278 (1725), 3617-3624 (2011).
- Sailer, Z. R., Harms, M. J. Detecting high-order epistasis in nonlinear genotype-phenotype maps. Genetics. 205 (3), 107911088(2017).
- Kuzmin, E., Costanzo, M., Andrews, B., Boone, C. Synthetic genetic arrays: Automation of yeast genetics. Cold Spring Harbor Protocols. 2016 (4), 326-332 (2016).
- Tong, A. H. Y., Boone, C. Synthetic genetic array analysis in Saccharomyces cerevisiae. Methods in Molecular Biology. 313 (1), 171-192 (2006).
- Dixon, S. J., Costanzo, M., Baryshnikova, A., Andrews, B., Boone, C. Systematic Mapping of Genetic Interaction Networks. Annual Review of Genetics. 43 (1), 601-625 (2009).
- Boone, C., Bussey, H., Andrews, B. J. Exploring genetic interactions and networks with yeast. Nature Reviews Genetics. 8 (6), 437-449 (2007).
- Bai, X., Yang, Z., Jiang, H., Lin, S., Zon, L. I. Genetic suppressor screens in haploids. Methods in Cell Biology. , 129-136 (2011).
- Manson, M. D. Allele-specific suppression as a tool to study protein-protein interactions in bacteria. Methods. 20 (1), 18-34 (2000).
- Motter, A. E., Gulbahce, N., Almaas, E., Barabási, A. L. Predicting synthetic rescues in metabolic networks. Molecular Systems Biology. 4, 168(2008).
- Peterson, R. T., Shaw, S. Y., et al. Chemical suppression of a genetic mutation in a zebrafish model of aortic coarctation. Nature Biotechnology. 22 (5), 595-599 (2004).
- Giorgini, F., Guidetti, P., Nguyen, Q., Bennett, S. C., Muchowski, P. J. A genomic screen in yeast implicates kynurenine 3-monooxygenase as a therapeutic target for Huntington disease. Nature Genetics. 37 (5), 526-531 (2005).
- Forsburg, S. L., Patton, E., et al. The art and design of genetic screens. Nature reviews. Genetics. 2 (9), 659-668 (2001).
- Johnston, D. S. The art and design of genetic screens. Genetics. 3 (March), 176-188 (2002).
- Jorgensen, E. M., Mango, S. E. The art and design of genetic screens: Caenorhabditis elegans. Nature Reviews Genetics. 3 (5), 356-369 (2002).
- Gocke, E., Müller, L. In vivo studies in the mouse to define a threshold for the genotoxicity of EMS and ENU. Mutation Research - Genetic Toxicology and Environmental Mutagenesis. 678 (2), 101-107 (2009).
- Suzuki, T., Hayashi, M., et al. A comparison of the genotoxicity of ethylnitrosourea and ethyl methanesulfonate in lacZ transgenic mice (Muta(TM)Mouse). Mutation Research - Genetic Toxicology and Environmental Mutagenesis. 395 (1), 75-82 (1997).
- Uttam, J., Alberico, C., De Stasio, E. ENU Mutagenesis. International C. elegans Meeting. , (1995).
- Putrament, A., Baranowska, H., Ejchart, A., Prazmo, W. Manganese Mutagenesis in Yeast. Methods in Cell Biology. 20, 25-34 (1978).
- Bose, J. L. Chemical and UV mutagenesis. Methods in Molecular Biology. 1373, 111-115 (2016).
- Ikehata, H., Ono, T. The Mechanisms of UV Mutagenesis. Journal of Radiation Research. 52 (2), 115-125 (2011).
- Shrivastav, N., Li, D., Essigmann, J. M. Chemical biology of mutagenesis and DNA repair: cellular responses to DNA alkylation. Carcinogenesis. 31 (1), 59-70 (2010).
- De Stasio, E. A., Dorman, S. Optimization of ENU mutagenesis of Caenorhabditis elegans. Mutation Research - Genetic Toxicology and Environmental Mutagenesis. 495 (1-2), 81-88 (2001).
- Probst, F. J., Justice, M. J. Mouse mutagenesis with the chemical supermutagen ENU. Methods in Enzymology. 477 (C), 297-312 (2010).
- Bähler, J., Wu, J. Q., et al. Heterologous modules for efficient and versatile PCR-based gene targeting in Schizosaccharomyces pombe. Yeast. 14 (10), 943-951 (1998).
- Li, H., Durbin, R. Fast and accurate short read alignment with Burrows – Wheeler transform. Bioinformatics. 25 (14), 1754-1760 (2009).
- Van der Auwera, G. A., Carneiro, M. O., et al. From fastQ data to high-confidence variant calls: The genome analysis toolkit best practices pipeline. Current Protocols in Bioinformatics. 43, 11.10.1-11.10.33 (2013).
- Mckenna, A., Hanna, M., et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Research. 20, 1297-1303 (2010).
- Li, H., Handsaker, B., et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 25 (16), 2078-2079 (2009).
- Marayati, B. F., Drayton, A. L., et al. Loss of Elongation-Like Factor 1 Spontaneously Induces Diverse, RNase H-Related Suppressor Mutations in Schizosaccharomyces pombe. Genetics. 209 (4), 967-981 (2018).
- Harris, M. A., Lock, A., Bähler, J., Oliver, S. G., Wood, V. FYPO: The fission yeast phenotype ontology. Bioinformatics. 29 (13), 1671-1678 (2013).
- Xu, X., Wang, L., Yanagida, M. Whole-Genome Sequencing of Suppressor DNA Mixtures Identifies Pathways That Compensate for Chromosome Segregation Defects in Schizosaccharomyces pombe. G3: Genes|Genomes|Genetics. 8 (3), 1031-1038 (2018).
- Marayati, B. F., Hoskins, V., et al. The fission yeast MTREC and EJC orthologs ensure the maturation of meiotic transcripts during meiosis. RNA. 22 (9), 1349-1359 (2016).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved