
Method Article
Recopilación y procesamiento de datos de cristalografía de neutrones para modelar átomos de hidrógeno en estructuras de proteínas
En este artículo
Resumen
La cristalografía de proteínas de neutrones es una técnica estructural que permite la localización de átomos de hidrógeno, proporcionando así importantes detalles mecanicistas de la función de la proteína. Presentamos aquí el flujo de trabajo para el montaje de un cristal de proteína, la recopilación de datos de difracción de neutrones, el refinamiento de la estructura y el análisis de los mapas de densidad de longitud de dispersión de neutrones.
Resumen
La cristalografía de neutrones es una técnica estructural que permite determinar las posiciones de los átomos de hidrógeno dentro de macromoléculas biológicas, proporcionando información mecánicamente importante sobre los estados de protonación e hidratación sin inducir daño por radiación. La difracción de rayos X, por el contrario, proporciona solo información limitada sobre la posición de los átomos de luz y el haz de rayos X induce rápidamente el daño por radiación de cofactores fotosensibles y centros metálicos. Aquí se presenta el flujo de trabajo empleado para las líneas de haz IMAGINE y MaNDi en el Laboratorio Nacional de Oak Ridge (ORNL) para obtener una estructura de difracción de neutrones una vez que se ha cultivado un cristal de proteína de tamaño adecuado (> 0,1 mm3). Demostramos el montaje de cristales de proteínas hidrogenadas en capilares de cuarzo para la recopilación de datos de difracción de neutrones. También se presenta el proceso de intercambio de vapor de los cristales montados con tampón que contiene D2O para garantizar el reemplazo de átomos de hidrógeno en sitios intercambiables con deuterio. La incorporación de deuterio reduce el fondo derivado de la dispersión incoherente de los átomos de hidrógeno y evita la cancelación de densidad causada por su longitud de dispersión coherente negativa. Las estrategias de alineación de muestras y recolección de datos a temperatura ambiente se ilustran utilizando la recolección de datos cuasi-Laue en IMAGINE en el Reactor de Isótopos de Alto Flujo (HFIR). Además, el montaje de cristales y la congelación rápida en nitrógeno líquido para la recopilación de criodatos para atrapar intermedios de reacción lábil se demuestran en el instrumento de tiempo de vuelo MaNDi en la fuente de neutrones de espalación (SNS). También se abordará la preparación de los archivos de datos de coordenadas y difracción del modelo y la visualización de los mapas de densidad de longitud de dispersión de neutrones (SLD). Finalmente se discutirá el refinamiento de la estructura contra datos de neutrones solo o contra datos conjuntos de rayos X / neutrones para obtener una estructura de todos los átomos de la proteína de interés. El proceso de determinación de una estructura neutrónica se demostrará utilizando cristales del polisacárido lítico monooxigenasa Neurospora crassa LPMO9D, una metaloproteína que contiene cobre implicada en la degradación de polisacáridos recalcitrantes a través de la escisión oxidativa del enlace glicosídico.
Introducción
La cristalografía macromolecular de neutrones es una técnica que proporciona una ventana única a la estructura y la química subyacente de las proteínas. Conceptualmente similar a la difracción de rayos X, la difracción de neutrones proporciona detalles atomísticos de la estructura macromolecular, sin embargo, la interacción de neutrones con núcleos permite la localización de átomos de luz, a menudo difíciles de detectar con difracción de rayos X1. Durante la difracción de rayos X, los rayos X se dispersan desde la nube de electrones, haciendo que los átomos de luz como el hidrógeno (H) sean poco visibles en los mapas de densidad de electrones que no tienen una resolución cercana a la sub-Ångström2. En contraste, la intensidad de dispersión de los neutrones depende de interacciones complejas con el núcleo, con isótopos del mismo elemento que muestran diferentes longitudes de dispersión. Por lo tanto, los átomos de luz y sus isótopos, como el hidrógeno (1H) y el deuterio (2H o D), tienen una visibilidad comparable a los átomos de carbono, nitrógeno y oxígeno de la columna vertebral en los mapas de densidad de longitud de dispersión de neutrones (SLD). Además, dado que la magnitud de la dispersión de neutrones es independiente del número de electrones, la dispersión de elementos ligeros no se ve oscurecida por elementos pesados cuando están muy cerca unos de otros, como se observa en la dispersión de rayos X. La visibilidad mejorada de H y su isótopo D al emplear la difracción de neutrones proporciona información valiosa sobre el estado de protonación de residuos, cofactores y ligandos catalíticamente importantes y ayuda a la orientación de las moléculas de agua, revelando información importante sobre los mecanismos catalíticos y la química de las proteínas3. La difracción neutrónica también ofrece la ventaja de ser una técnica no destructiva, especialmente adecuada para muestras biológicas sensibles a la ionización como proteínas con centros metálicos o cofactores redox fotosensibles2. El objetivo principal de este artículo es proporcionar una visión general del flujo de trabajo para obtener una estructura cristalina de proteína de neutrones de alta calidad. Remitimos al lector interesado a Podjarny et al.4, Blakeley5, Blakeley et al.6 y O'Dell et al.3 para una excelente visión general de la difracción de proteínas neutrónicas y Ashkar et al.7 para otras aplicaciones de dispersión de neutrones.
Los neutrones se generan principalmente durante las reacciones nucleares empleando cualquiera de dos procesos: fisión nuclear en fuentes de reactores o espalación en fuentes basadas en aceleradores8. Las fuentes de reactores proporcionan un haz de neutrones continuo mediante el empleo de la fisión nuclear del isótopo 235U, mientras que las fuentes de neutrones de espalación producen un haz de neutrones pulsados bombardeando un objetivo, por ejemplo, un metal líquido como el mercurio, con protones9. El Laboratorio Nacional de Oak Ridge (ORNL) en Oak Ridge, Tennessee, alberga una fuente de neutrones de estado estacionario en el reactor de isótopos de alto flujo (HFIR) y una fuente pulsada de 60 Hz en la fuente de neutrones de espalación (SNS). La línea de haz IMAGINE, ubicada en el HFIR, es un difractómetro de neutrones optimizado para macromoléculas biológicas (Figura suplementaria 1)10. IMAGINE emplea un detector de placas de imagen de neutrones para medir datos cuasi-Laue utilizando un paso de banda estrecho en el rango de 2.8 – 4.5 Å de cristales individuales con bordes de celda unitaria <150 Å. El difractómetro de neutrones macromoleculares (MaNDi), ubicado en el SNS, es un difractómetro de neutrones Laue de tiempo de vuelo (TOF) equipado con un marco de matriz de detectores esféricos (DAF) (Figura suplementaria 2)11. MaNDi mide datos de cristales individuales con bordes de celdas unitarias en el rango de 10 – 300 Å empleando un ancho de banda sintonizable de 2 longitudes de onda Å entre 2.0 – 6.0 Å12.
El proceso de generación de neutrones es altamente intensivo en energía, lo que resulta en flujos de haz de neutrones relativamente débiles cuando se contrasta con los flujos de haz de rayos X en fuentes de sincrotrón13. Para garantizar unas relaciones señal-ruido suficientes durante la recogida de datos, es necesario cultivar cristales de tamaño y calidad adecuados14. Normalmente, se necesitan cristales con volúmenes > 0,1 mm3 para recopilar datos con estadísticas adecuadas15. Además de los flujos más bajos, deben tenerse en cuenta las propiedades inherentes a la interacción entre los neutrones y los núcleos de la muestra16. La longitud de dispersión de los neutrones difiere para los isótopos del mismo elemento, una propiedad que puede explotarse ventajosamente en la dispersión de neutrones de ángulo pequeño (SANS) para enmascarar o resaltar regiones de una muestra, un proceso conocido como coincidencia de contraste17. En experimentos de difracción, la longitud de dispersión de neutrones coherente negativa de H (-3.741 fm para 1H) puede conducir a la cancelación de las características del mapa de densidad de dispersión de neutrones ya que las longitudes de dispersión de neutrones coherentes de otros átomos biológicamente relevantes, incluido el carbono (6.6511 fm para 12C), nitrógeno (9.37 fm para 14N), oxígeno (5.803 fm para 16O), fósforo (5.13 fm para 31P) y azufre (2.804 fm para 32S), son positivos (Tabla 1)12,14. Además, la gran longitud de dispersión incoherente de H (25.274 fm), aumenta el fondo durante la recopilación de datos, obstaculizando la calidad del conjunto de datos y comprometiendo la resolución de los datos7. Para eludir estas limitaciones introducidas por H es necesario, para la difracción de neutrones, intercambiar H por su isótopo deuterio, 2H(D), que tiene una longitud de dispersión de neutrones coherente positiva (6.671 fm) y una longitud de dispersión incoherente significativamente menor (4.04 fm)19. Esto se puede lograr mediante perdeuteración, un proceso en el que la proteína es expresada por organismos cultivados en medios totalmente deuterados asegurando la incorporación completa de D en los sitios H20. También es posible deuterar parcialmente la proteína reemplazando H con D únicamente en los sitios intercambiables (grupos titulables) mientras que los sitios no intercambiables unidos al carbono permanecen hidrogenados21. Esto se puede lograr mediante el crecimiento de cristales de proteína hidrogenada en licor madre deuterado22. Sin embargo, lo más común es que el intercambio H/D de proteínas hidrogenadas se realice mediante el intercambio de vapor después del crecimiento de cristales adecuadamente grandes en tampón basado en H2O23. En tales casos, los cristales se montan en un capilar de cuarzo y se equilibran con vapor con un licor madre a base de D20.
Los flujos de neutrones limitados en las fuentes de neutrones dan como resultado tiempos de recopilación de datos más largos, que van desde días hasta varias semanas24. En ORNL, tanto IMAGINE como MaNDi emplean un paso de banda de longitud de onda estrecha en el rango de 2 a 6 Å para optimizar la recopilación de datos25. Los datos se pueden recopilar a temperatura ambiente o a criotemperatura. La recopilación de criodatos puede mejorar potencialmente la calidad de los datos y abre la posibilidad de congelar los intermedios catalíticos. Tras la recopilación de datos de difracción de neutrones, normalmente se recopila un conjunto de datos de rayos X en el mismo cristal a la misma temperatura o en un cristal cultivado en condiciones idénticas26. La recolección de datos a la misma temperatura permite realizar un refinamiento de la estructura contra datos de rayos X y neutrones, evitando cualquier artefacto potencial inducido por la temperatura, como cambios en la visibilidad y posición de las aguas o la ocupación de residuos con conformaciones alternativas27. El refinamiento conjunto de los datos de neutrones de rayos X aumenta la relación datos-parámetro y proporciona la ventaja de permitir que las coordenadas de la columna vertebral de la proteína se refinen contra los datos de rayos X, mientras que los datos de difracción de neutrones se utilizan para refinar la posición de los átomos H/D28. Esto es particularmente útil cuando se utilizan muestras parcialmente deuteradas, donde la cancelación de densidad debido a los átomos de H en sitios no intercambiables en la proteína está presente. Aunque el número de estructuras de rayos X supera con creces el número de estructuras de neutrones depositadas en el Banco de Datos de Proteínas (PDB), los paquetes de software diseñados inicialmente para el refinamiento de los datos de rayos X se han ampliado para abarcar también los datos de neutrones3,29,30. Después de la recopilación de datos, los modelos se pueden refinar utilizando paquetes de refinamiento como phenix.refine, CNSsolve (nCNS) o SHELXL28,31,32,33. Durante el proceso de refinamiento, se pueden visualizar mapas de densidad de dispersión de neutrones para su ajuste manual utilizando COOT34. Siguiendo la solución de estructura, las coordenadas y los archivos de datos de difracción de neutrones y/o rayos X pueden enviarse al PDB, quien validará y depositará el modelo, poniéndolo a disposición del público18,29,30.
El análisis estructural de proteínas es un enfoque multifacético en el que se utilizan numerosas técnicas para sondear su función y mecanismo35. La cristalografía de proteínas de neutrones proporciona información química valiosa para ampliar y complementar los hallazgos de estudios adicionales como la difracción de rayos X, la espectroscopia, la resonancia magnética nuclear (RMN) o la difracción de electrones microcristalinos (microED)36. La difracción de proteínas de neutrones está en una posición única para proporcionar información sobre los mecanismos enzimáticos, ya que los átomos de H son fundamentales para su química. La ausencia de daño por radiación inducido por neutrones los convierte en una sonda excepcionalmente adecuada para el estudio de las metaloproteínas37. Presentamos aquí un ejemplo representativo del proceso de difracción de proteínas neutrónicas desde la preparación de la muestra hasta la recolección, refinamiento y análisis de datos (Figura 1). Se han cultivado cristales de tamaño suficiente para experimentos de difracción de neutrones de la metaloproteína Neurospora crassa LPMO9D (NcLPMO9D). Nc LPMO9D es una metaloproteína que contiene cobre implicada en la degradación de la celulosa recalcitrante por inserción de átomos de oxígeno en el enlace glicosídico38,39. El sitio activo NcLPMO9D contiene un centro de cobre mononuclear dentro de un característico "histidina-brace" compuesto por la histidina N-terminal y una segunda histidina conservada (Figura suplementaria 3)40. El N-terminal de los LPMOs fúngicos está metilado, pero la modificación post-transicional no ocurre durante la expresión recombinante en la levadura. En el estado de reposo De NcLPMO9D, el centro de cobre está presente en un estado de oxidación Cu2+ y se activa mediante la reducción de un solo electrón a Cu1+, permitiendo que el oxígeno molecular se una y se active al reducirse rápidamente a una especie de superóxido41,42. La reacción global de NcLPMO9D requiere la adición adicional de un electrón y dos protones para formar el producto polisacárido hidroxilado43. No se ha identificado la identidad de las especies de oxígeno activado responsables de la extracción del átomo de hidrógeno (HAA) del sustrato polisacárido y actualmente se están realizando estudios estructurales y computacionales intensivos44,45. Dada la química redox en el sitio activo de NcLPMO9D, la mitigación del daño por radiación es particularmente pertinente. Ilustramos aquí la recolección de datos de temperatura ambiente y criotemperatura en cristales NcLPMO9D para determinar la estructura NcLPMO9D en el estado de reposo y en la forma reducida activada, respectivamente46. Se hará hincapié en el montaje de cristales de proteínas, la configuración del instrumento de línea de haz para la recopilación de datos, la preparación de los archivos de datos y coordenadas y los pasos de refinamiento necesarios para modelar una estructura de neutrones de todos los átomos.
Protocolo
1. Evaluación del tamaño del cristal
- Mida el tamaño de los cristales utilizando un microscopio equipado con luz normal y polarizada. Seleccione cristales con un volumen mínimo de ~0,1 mm3 (Figura suplementaria 4).
- Etiquete los pozos con cristales suficientemente grandes y observe las condiciones de cristalización utilizadas para generar estos cristales.
2. Preparación del tampón de cristalización deuterado
- Disuelva los componentes del tampón de cristalización en D2O para generar un búfer de cristalización deuterado.
- Ajuste el pH del tampón calculando el pD de la solución utilizando la siguiente ecuación:
 (1)
(1)
donde pHmeas es el pH medido con un electrodo de vidrio estándar. El pH original del tampón de cristalización NcLPMO9D era 6.0, por lo tanto, usaremos un pHmeas de 5.6 para el tampón de cristalización deuterado en pD 6.0. - Sumergir el electrodo del medidor de pH en D2O durante diez minutos antes de su uso (Figura suplementaria 5).
- Ajuste el pHmeas a 5.6 mediante el uso de la base NaOD o el ácido DCl.
3. Cosecha de cristales
- Coloque portaobjetos de vidrio redondo siliconado de 22 mm junto a la bandeja de cristalización de la que se cosecharán los cristales. Utilice un portaobjetos de vidrio limpio por cristal (Figura 2A).
- Abra la caja de sándwich sellada que contiene los cristales de proteína en la placa de vidrio siliconado de gran volumen de 9 pocillos.
- Retire 10-20 μL de la solución del depósito de cristalización con una micropipeta y coloque la solución en el portaobjetos de vidrio (Figura 2B).
- Cose el cristal utilizando un micropótano de tamaño apropiado y coloque el cristal en la gota de solución del depósito en el portaobjetos de vidrio para eliminar los desechos que a menudo se cosechan junto con el cristal (Figura 2C y Figura 2D).
NOTA: Será necesario trabajar rápidamente ya que las gotas de pequeño volumen pueden evaporarse. Los cristales cosechados también corren el riesgo de secarse cuando se exponen a la atmósfera. Puede ser necesario agregar alguna solución reservorio a la gota de proteína para evitar que los cristales se sequen en pequeños volúmenes de gota de cristalización.
4. Montaje en cristal
NOTA: Los protocolos de montaje capilar varían según las preferencias de los experimentalistas. Para evitar daños en los cristales, los capilares que deben acortarse deben puntuarse con una piedra de corte o papel de lija para garantizar una rotura suave.
- Llene un extremo de un capilar de cuarzo de 2 mm de diámetro y 50 mm de longitud con tampón de reservorio por acción capilar o canalizando directamente ~ 10 μL de tampón de reservorio en el capilar (Figura 3A).
NOTA: Se anima a los usuarios a hacer uso de tubos capilares de cuarzo porque, además de su resistencia mecánica, es esencial limitar la absorción del haz de neutrones y reducir las contribuciones de fondo del capilar. Los capilares de vidrio introducen un alto fondo y absorben neutrones, comprometiendo la calidad de los datos. - Coloque suavemente el cristal en el tampón del depósito en el capilar de cuarzo utilizando el bucle de montaje (Figura 3B y Figura 3C).
- Golpee suavemente el tubo para mover el tampón del depósito y el cristal sumergido en el mismo por el capilar (Figura 3D).
- Coloque el cristal no más cerca de 13,5 mm y no más de 27,5 mm de un extremo del capilar; este será el extremo de montaje (Figura suplementaria 6).
- Aspire la solución tampón alrededor del cristal con una punta de pipeta larga y delgada, dejando el cristal ligeramente húmedo. No toque el cristal (Figura suplementaria 7A).
- Seque las paredes capilares con una mecha de papel delgada (Figura suplementaria 7B).
- Pipetear 20-50 μL de solución tampón deuterada en el extremo del capilar opuesto al extremo de montaje (Figura 4A).
- Derrita la cera de abeja con una "varita" de calor e inserte suavemente el capilar en esta cera de abeja derretida. Repita hasta que se forme un sello hermético (Figura 4B y Figura 4C).
- Pipetear una cantidad muy pequeña de tampón deuterado, aproximadamente 5 μL en el extremo de montaje del capilar para actuar como un "disipador de calor" para la cera de abeja caliente. Sumerja este extremo en cera de abeja derretida para generar un sello hermético como se describió anteriormente para formar un capilar sellado en ambos extremos (Figura 4D).
- Inspeccione el cristal montado con un microscopio después del montaje para garantizar la estanqueidad (Figura 4E).
- Asegure cuidadosamente los cristales montados con toallitas Kim en un recipiente como un tubo Falcon de 15 ml o una placa de Petri y guárdelo horizontalmente a la temperatura a la que se cultivaron los cristales (Figura 4F).
5. Intercambio de vapor
- Reemplace el búfer deuterado con un tampón nuevo dos días después del montaje del cristal.
- Derrita el sello de cera más alejado del cristal con un bucle de calentamiento y use una pipeta y mechas de papel para eliminar el tampón.
- Vuelva a llenar el capilar con 20-50 μL de solución tampón deuterada y selle con cera.
- Repita el reemplazo del tampón deuterado dos veces más a intervalos de cuatro días para asegurarse de que el intercambio de vapor tampón deuterado esté completo y permita el intercambio de vapor durante al menos dos semanas.
6. Difracción de proteínas de neutrones
NOTA: Se anima a los lectores interesados en los detalles de la línea de haz IMAGINE a consultar Meilleur et al. 2013, Meilleur et al. 201810,47.
- Recopilación de datos a temperatura ambiente en la línea de haz IMAGINE en HFIR
- Montaje de muestras
- Asegure el cristal montado en capilar en el goniómetro con masilla.
- Monte el goniómetro en la varilla de muestra y centre el cristal en el haz utilizando la estación de alineación fuera de línea.
- Asegure la varilla de muestra en la etapa de muestra del instrumento (Figura suplementaria 1B).
- Asegúrese de que la cabina experimental esté desocupada y abra el obturador de la línea de haz para la recopilación de datos de neutrones.
- Recogida de datos
- Abra el programa de adquisición de datos en la computadora de control de la línea de haz y haga clic en la pestaña Configuración para configurar la estrategia de recopilación de datos. En Parámetros del experimento , escriba el nombre de ejemplo junto a Nombre de muestra e introduzca el número de propuesta junto a Propuesta. En Nombre de imagen , seleccione Plantilla de carpeta y establezca el destino de los datos adquiridos que se guardarán y seleccione Prefijo de imagen y escriba el nombre de marco correspondiente (Figura suplementaria 8).
- Abra la GUI de Optics y haga clic en 2.78 para λmin y 4.78 para λmax para establecer el rango cuasi-Laue para la recopilación de datos (Figura suplementaria 9).
- Cambie a la pestaña Recopilar y, en Siguiente parámetros de exploración , inserte el tiempo de exposición en segundos en Exposición, el número de fotogramas en N fotogramas y los ángulos para la recopilación de datos en Δ φ/Fotograma. Asigne un nombre al fotograma que se va a recopilar en Prefijo de imagen e inicie la recopilación de datos haciendo clic en el botón Iniciar escaneo (Figura suplementaria 10).
- Los neutrones difractados serán detectados por el detector de placas de imagen. Al final de cada exposición, se leerá la placa de imagen y se mostrará el patrón en la GUI de adquisición de datos (Figura suplementaria 11).
- Los fotogramas son indexados, integrados, normalizados en longitud de onda y escalados utilizando Lauegen, Lscale50 y Scala por el científico responsable de la línea de haz, que proporcionará al usuario el archivo de reflexión fusionado después de la recopilación de datos (Figura suplementaria 12).
NOTA: La recolección de datos tanto en IMAGINE como en MaNDi se realizará en modo cuasi-Laue, utilizando metodología y software desarrollado para la recolección de datos de difracción de Laue desarrollado por Helliwell et al.48 y Nieh et al.49. - Recopile un conjunto de datos de rayos X correspondiente en el mismo cristal a la misma temperatura después de la recopilación de datos de difracción de neutrones (Figura suplementaria 13).
NOTA: Un cristal cultivado a partir de la misma gota o en las mismas condiciones de cristalización también se puede utilizar para recopilar datos de difracción de rayos X para el refinamiento de neutrones / rayos X conjuntos.
- Montaje de muestras
- Recopilación de datos criogénicos en la línea de haz MaNDi en el SNS
NOTA: Se alienta a los lectores interesados en los detalles de la línea de haz a consultar Coates et al. (2015), Meilleur et al. 201810,11.- Montaje de muestras
- Prepare la solución de remojo de ascorbato deuterado para la reducción de los cristales y el crioprotector deuterado. Coloque gotas de 20 μL de cada una de estas soluciones en pozos de gota sentados en una placa de cristalización.
NOTA: La solución crioprotectora suele ser el crioprotector que ha demostrado ser eficaz para la recopilación de datos de difracción de rayos X a criotemperatura preparada en D2O. Este crioprotector se puede optimizar aún más (por ejemplo, la concentración) para la recopilación de datos de neutrones si es necesario. - Seleccione bucles para el montaje de muestras y conéctelos a una criobase magnética siguiendo las directrices de MaNDi (Figura suplementaria 14).
- Llene un cryo-Dewar de espuma con nitrógeno líquido. Coloque una funda de crioprotección de metal dentro del nitrógeno líquido para enfriar (Figura suplementaria 15A).
- Retire los tapones de cera de ambos extremos del capilar y toque el capilar para mover el tapón intermedio de modo que el cristal montado se sumerja en el tampón. Lave el cristal en la gota de solución del depósito de 20 μL en el pozo de gota sentada (Figura suplementaria 15B y Figura suplementaria 15C).
NOTA: Este paso no es necesario si el intercambio de H/D se realizó mediante el equilibrio de las gotas de cristalización contra el tampón deuterado o mediante el remojo directo del cristal en el tampón deuterado. - Sumerja el cristal en la solución de remojo de ascorbato durante dos horas. Monte el cristal en un microloop unido a una criobase magnética. Sumerja el cristal montado en el crioprotector durante 10 segundos y sumerja el cristal y el soporte criogénico en el nitrógeno líquido para congelarlo (Figura suplementaria 15D).
- Una vez que el cristal esté congelado, use una pinza criogénica preenfriada y monte el cristal en la etapa de muestra MaNDi equipada con una crio-corriente. Abra suavemente la pinza criogénica y asegúrese de que el cristal permanezca en la crio-corriente (Figura suplementaria 2C).
- Prepare la solución de remojo de ascorbato deuterado para la reducción de los cristales y el crioprotector deuterado. Coloque gotas de 20 μL de cada una de estas soluciones en pozos de gota sentados en una placa de cristalización.
- Recogida de datos
- Abra el software de recopilación de datos en el que la información del experimento se habrá rellenado automáticamente.
- Haga clic en el centro del cristal para centrarlo con el goniómetro controlado por computadora (Figura suplementaria 16).
- En la Tabla, configure la estrategia de recopilación de datos ingresando los ángulos para la recopilación de datos en "phi", así como la exposición total al haz de neutrones por fotograma en Valor (Figura suplementaria 17).
- Haga clic en Enviar para iniciar la recopilación de datos.
- A medida que se recopilen los datos, el neutrón difractado será visible (Figura suplementaria 17). Los puntos de difracción se aclararán a medida que aumente el tiempo de exposición, mejorando la relación señal-ruido (Figura 5).
- Los fotogramas serán indexados, integrados, normalizados en longitud de onda y escalados utilizando Mantid y Lauenorm por el científico responsable de la línea de haz, que proporcionará al usuario el archivo de intensidad fusionado después de la recopilación de datos51.
- Montaje de muestras
7. Refinamiento de la estructura
- Refinamiento de datos de rayos X y neutrones conjuntos
- Preparación de la estructura
- Refinar los datos de rayos X para obtener una estructura de proteína utilizando el paquete de software phenix.refine y Coot para la construcción manual para obtener una estructura completa.
- Abra CCP4 y seleccione el programa Convertir a/modificar/extender MTZ para que coincida con los indicadores de datos libres de R de los datos de neutrones con los de los datos de rayos X. Seleccione esta opción para importar el archivo de reflexión en formato MTZ . En Cargar el archivo MTZ de neutrones obtenido y Cargar el archivo MTZ de rayos X en Importar MTZ libreR (Figura suplementaria 18). Asigne un nombre para el nuevo archivo MTZ en Salida y haga clic en Ejecutar.
- Abra el paquete de software Phenix y haga clic en ReadySet en Herramientas de refinamiento. Junto al archivo PDB , cargue el archivo de coordenadas PDB refinado contra los datos de rayos X. Seleccione Agregar hidrógenos al modelo si está ausente y seleccione H / D en sitios intercambiables, H en otro lugar del menú desplegable. Seleccione Agregar deuterios a moléculas de disolvente y deje las opciones restantes en sus valores predeterminados (Figura suplementaria 19A).
NOTA: Si se utiliza una proteína perdeuterada, seleccione la opción Agregar hidrógenos al modelo si está ausente y seleccione H / D en sitios intercambiables, D en otro lugar.
- Refinamiento de la estructura
- Abra el programa phenix.refine en la pestaña Refinamiento para configurar el refinamiento utilizando datos de rayos X y neutrones. En la pestaña Configurar de Archivos de entrada , ingrese el archivo PDB de la estructura de rayos X que se ha procesado con ReadySet y el archivo de restricciones CIF necesario para cualquier ligando relevante. Cargue el archivo MTZ desde los datos de neutrones con las banderas Rfree asignadas mediante CCP4 y asígnelas como "Datos de neutrones" y "Neutron R-free" en el encabezado Tipo de datos. Cargue el archivo MTZ desde los datos de rayos X y asígnelo como "datos de rayos X" y "sin rayos X R" en el encabezado Tipo de datos. El grupo Espacio y las etiquetas Datos se rellenarán automáticamente una vez que se hayan cargado los datos (Figura suplementaria 19B).
NOTA: Al realizar el refinamiento e ingresar la información del cristal, use la celda unitaria determinada a partir de los datos de rayos X. - En la pestaña Configurar , en la configuración de Refinamiento , mantenga la estrategia de refinamiento estándar. Aumentar el número de ciclos a cinco (Figura suplementaria 20)
- Seleccione Todos los parámetros, haga clic en Avanzado y seleccione Hidrógenos.... Cambie el modelo de refinamiento de hidrógeno a individual y apague el adp de conducción de fuerza (Figura suplementaria 20).
- Seleccione Todos los parámetros y abra la opción Buscar parámetros . Busque la palabra nuclear y seleccione Usar las distancias nucleares para X-H/D (Figura suplementaria 20).
- Seleccione Ejecutar para iniciar el refinamiento.
- Abra el programa phenix.refine en la pestaña Refinamiento para configurar el refinamiento utilizando datos de rayos X y neutrones. En la pestaña Configurar de Archivos de entrada , ingrese el archivo PDB de la estructura de rayos X que se ha procesado con ReadySet y el archivo de restricciones CIF necesario para cualquier ligando relevante. Cargue el archivo MTZ desde los datos de neutrones con las banderas Rfree asignadas mediante CCP4 y asígnelas como "Datos de neutrones" y "Neutron R-free" en el encabezado Tipo de datos. Cargue el archivo MTZ desde los datos de rayos X y asígnelo como "datos de rayos X" y "sin rayos X R" en el encabezado Tipo de datos. El grupo Espacio y las etiquetas Datos se rellenarán automáticamente una vez que se hayan cargado los datos (Figura suplementaria 19B).
- Construcción de modelos
- Después del refinamiento en Phenix, haga clic en Abrir en Focha en la pestaña Resultados para visualizar la densidad de electrones de rayos X y los mapas SLD de neutrones. Haga clic en la pestaña Administrador de pantallas y, en Mapas, haga clic en Eliminar mapa junto a los mapas _neutron para eliminar los mapas de neutrones (Figura suplementaria 21). Haga clic en archivo > Abrir MTZ, mmCIF fcf o phs.... Seleccione los archivos de refinamiento actuales y abra el archivo .mtz. Para la opción Amplitudes y Fases, seleccione los datos 2FOFC WT_no_fill_neutron en el menú desplegable. Repita esto y abra los datos FOFC WT_neutron. Abra el Administrador de pantallas y cambie a Desplazarse para los mapas de neutrones y rayos X 2FOFCWT y luego desplácese para disminuir el rmsd de los mapas 2FOFCWT a 1.00 (Figura suplementaria 21). Cambie a Scroll para los mapas FOFCWT de neutrones y rayos X y desplácese para disminuir el rmsd de los mapas FOFCWT a 3.00.
- Realice una inspección visual de los residuos para determinar si el modelo se ajusta a los datos. Determine la orientación correcta y la ocupación H /D de todos los sitios intercambiables mediante el análisis de los picos del mapa de densidad de diferencia. Esto incluye los grupos hidroxilo de serinas, treoninas y tirosinas; el nitrógeno de histidina, glutamina, asparagina y lisina; el sulfhidrilo de la cisteína; los grupos carboxilo de aspartato y glutamato; grupos amida de la columna vertebral; ligandos; cofactores y cualquier residuo funcionalizado potencial (Figura 6).
- Reorientar las moléculas de agua de acuerdo con la densidad de neutrones y las interacciones de enlaces de hidrógeno rotándolas utilizando la función Rotar zona/cadena/molécula (Figura suplementaria 22A y Figura suplementaria 22B). Ajuste las posiciones de los sitios intercambiables H/D de residuos de proteínas utilizando la herramienta Editar ángulos de Chi y la función Rotar zona/cadena/molécula de traducción (Figura suplementaria 22C y Figura suplementaria 22D).
- Preparación de la estructura
- Refinamiento de la estructura: refinamiento de solo datos de neutrones
- Preparación de la estructura
- Abra Phenix y seleccione "Reemplazo molecular" y seleccione "Phaser-MR (con todas las funciones)" para derivar la fase de las intensidades escaladas proporcionadas por el científico del instrumento mediante reemplazo molecular para generar un archivo de coordenadas iniciales en formato pdb. Ingrese la estructura pdb inicial en la pestaña "Entrada y opciones generales " y complete las opciones Conjuntos, contenido de ASU y Procedimiento de búsqueda .
- Abra Herramientas de modelo y seleccione Herramientas de PDB. Inserte el archivo pdb como archivo de entrada. Vaya a la pestaña Opciones y, en Eliminar selección de átomos , seleccione los nombres del disolvente, ligandos, cofactores y metales. Esto elimina todas las moléculas de agua, cofactores, ligandos e iones metálicos para generar un modelo mínimo. Además, seleccione Quitar conformadores alternativos del modelo (Figura suplementaria 23).
- Seleccione Refinamiento en Phenix y abra ReadySet. Introduzca el archivo de coordenadas pdb editado junto al archivo PDB. Seleccione Agregar hidrógenos al modelo si está ausente y seleccione H/D en sitios intercambiables, H en otro lugar del menú desplegable de opciones de refinamiento de neutrones (Figura suplementaria 23).
- Refinamiento de la estructura
- Abra el programa phenix.refine en la pestaña Refinamiento en Phenix. En la ficha Configurar , introduzca el archivo PDB que se ha procesado con ReadySet en el cuadro Archivos de entrada . En el cuadro Archivos de entrada , cargue el archivo MTZ de los datos de neutrones y asígnelo como datos de rayos X y sin rayos X en la columna Tipo de datos aunque se trate de datos de neutrones. La configuración de refinamiento establecida en el siguiente paso se utilizará para tratar el archivo de reflexión como datos de neutrones. El grupo Espacio y las etiquetas Datos se rellenarán automáticamente una vez que se hayan cargado los datos (Figura suplementaria 24A).
- En la pestaña Configurar , en la configuración de Refinamiento , mantenga la estrategia de refinamiento estándar. Aumente el número de ciclos a cinco. En Otras opciones , seleccione neutrón en el menú desplegable de la tabla Dispersión . Anule la selección de la opción Actualizar aguas (Figura suplementaria 24B).
- Seleccione Todos los parámetros>Aprobación>Hidrógenos. En la nueva ventana, seleccione Individual en el menú desplegable Modelo de refinamiento de hidrógeno y desactive el adp de conducción de fuerza (Figura suplementaria 20).
- Seleccione Todos los parámetros > Parámetros de búsqueda... opción. Busque la palabra nuclear y seleccione Usar las distancias nucleares para X-H/D (Figura suplementaria 20).
- Seleccione Ejecutar para iniciar el refinamiento.
NOTA: Después del refinamiento inicial, será necesario inspeccionar visualmente los mapas SLD de neutrones y realizar la construcción manual de modelos en Coot. Puede ser necesario insertar ligandos/cofactores presentes en el modelo. Los refinamientos posteriores requerirán el archivo de restricciones CIF necesario para cualquier ligando relevante y estos deben cargarse en la pestaña Configurar de phenix.refine.
- Construcción de modelos
- Después del refinamiento en Phenix, haga clic en Abrir en Focha en la pestaña Resultados para visualizar los mapas y la estructura SLD de neutrones. Haga clic en la pestaña Administrador de pantallas y, en Mapas, haga clic en Eliminar mapa para eliminar los mapas 2FOFCCWT y FOFCWT (Figura suplementaria 25). Haga clic en Archivo > Abrir MTZ, mmCIF fcf o phs.... Seleccione la carpeta de refinamiento actual y seleccione el archivo .mtz. Para la opción Amplitudes y Fases, seleccione los datos 2FOFC WT_no_fill en el menú desplegable. Repita haciendo clic en Archivo > Abrir MTZ, mmCIF fcf o phs... y seleccione los datos FOFCWT en el menú desplegable para la opción Amplitudes y Fases. Abra el Administrador de pantallas y cambie a Desplazarse por el mapa 2FOFC WT_no_fill luego desplácese para disminuir el rmsd de los datos 2FOFC WT_no_fill a 1.00 (Figura suplementaria 25). Cambie a Desplazarse por el mapa FOFCWT y desplácese para disminuir el rmsd de los datos FOFCWT a 3.00.
- Realice una inspección visual de la estructura de la proteína para determinar si el modelo se ajusta al mapa SLD de neutrones.
- Como se describe en 7.1.3.2, determinar la orientación correcta y la ocupación H/D de los residuos y grupos con sitios intercambiables H/D. Ajuste las posiciones de los residuos con la herramienta Girar traducir y Editar ángulos de Chi (Figura 6). Si es necesario, se puede utilizar Real Space Refine Zone . Fijar átomos D que explotan del residuo manualmente utilizando el editor de texto para insertar las coordenadas atómicas correctas
NOTA: Real Space Refine Zone no está optimizada para mapas SLD de neutrones en Focha y puede dar lugar a longitudes de enlace irregulares para átomos unidos a D, denominados residuos explosivos (Figura suplementaria 26). Es preferible editar manualmente las coordenadas atómicas necesarias y evitar el uso de real Space Refine Zone. - Inserte y reoriente las moléculas de agua de acuerdo con la densidad de neutrones. Para agregar aguas en Coot, seleccione el icono Colocar átomo en el puntero y seleccione insertar una molécula de agua (Figura suplementaria 27A). La focha insertará un átomo de O en esta posición por defecto.
- Para agregar átomos D a los átomos O de las aguas insertadas en Coot use Phenix. Abra el menú Refinamiento y haga clic en ReadySet. Junto a Opciones de refinamiento de neutrones , seleccione solo la opción Agregar deuterios a las moléculas de disolvente. Anule la selección de Agregar hidrógenos al modelo si está ausente (Figura suplementaria 27B y Figura suplementaria 27C).
NOTA: La construcción de modelos utilizando datos de solo neutrones difiere de la construcción de modelos de una estructura conjunta de rayos X / neutrones porque no hay datos de rayos X que contribuyan al refinamiento de las coordenadas de la columna vertebral y los átomos más pesados. En un refinamiento conjunto, el mapa de densidad de electrones se utiliza inicialmente para determinar la columna vertebral de la proteína y las coordenadas de la cadena lateral. Este modelo se utiliza posteriormente en un refinamiento conjunto de datos de rayos X / neutrones en el que la orientación y ocupación de los átomos de H / D se deriva del mapa SLD de neutrones. En un refinamiento solo de neutrones, toda la estructura se deriva del análisis de los mapas SLD de neutrones, lo que requiere la construcción de las moléculas de agua, la columna vertebral, las cadenas laterales y los ligandos, además de los átomos de H / D (Figura 6). La relación datos-parámetro es baja en refinamientos contra los datos de neutrones solos y se debe tener precaución para no sobreajustar los datos.
- Preparación de la estructura
Resultados
Los datos de difracción de neutrones en cristales de una polisacárido lítico monooxigenasa de Neurospora crassa (NcLPMO9D) se recogieron en IMAGINE en el HFIR a temperatura ambiente y en MaNDi en el SNS bajo crio-condiciones siguiendo el protocolo descrito anteriormente. Se utilizaron cristales de la proteína hidrogenada cultivada en tampón basado en H2O con un volumen superior a 0,1 mm3 (ejemplo ilustrativo de cristales grandes se muestra en la Figura suplementaria 4 y las figuras posteriores). Los cristales se montaron en capilares de cuarzo y el intercambio de vapor con el tampón basado en D2O se realizó durante tres semanas antes de la recopilación de datos (Figura 4).
La recolección de datos a temperatura ambiente se realizó en la línea de haz IMAGINE (Figura 1). Una prueba de haz blanco de cuatro horas condujo a una difracción de alta resolución que sugiere que el cristal era de tamaño y calidad adecuados para que se recopilara un conjunto de datos completo. Además de proporcionar información preliminar sobre la calidad de difracción del cristal, la exposición inicial al paso de banda ancha se puede utilizar para indexar el patrón de difracción y determinar la matriz de orientación del cristal. Dado el grupo espacial P21 del cristal, se implementó una estrategia de recolección de datos de 18 cuadros con un tiempo de recolección de 20 horas por fotograma. Al igual que con la recopilación de datos de difracción de rayos X, los grupos de espacio de mayor simetría requieren menos marcos (es decir, menos cobertura angular) para recopilar un conjunto de datos completo. Los datos se recogieron en modo cuasi-Laue utilizando un rango de longitud de onda de 2,8 – 4,0 Å. Después de la recopilación de datos, los datos se indexaron, se integraron y se fusionaron para dar un archivo SLD de neutrones en formato MTZ a una resolución de 2,14 Å. Los datos se evaluaron como de calidad suficiente siguiendo pautas similares para el análisis de datos de rayos X, aunque se consideró aceptable una completitud del 80 % y un CC1/2 de al menos 0,3, ya que la difracción de proteínas de neutrones es una técnica de flujo limitado.
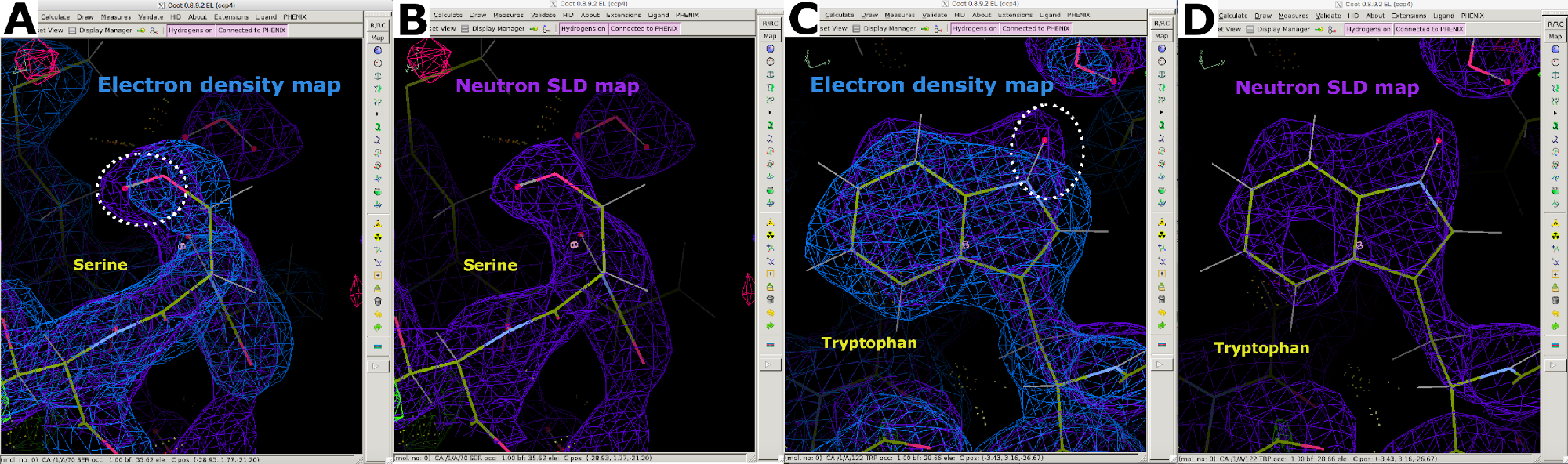
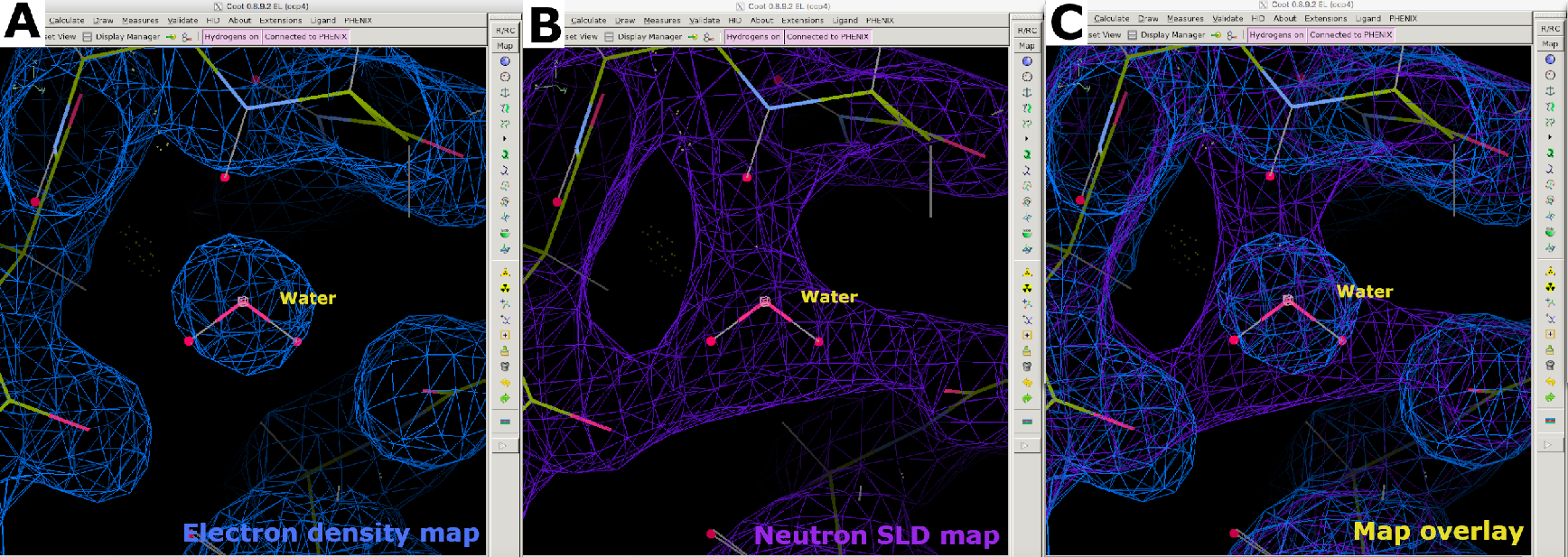
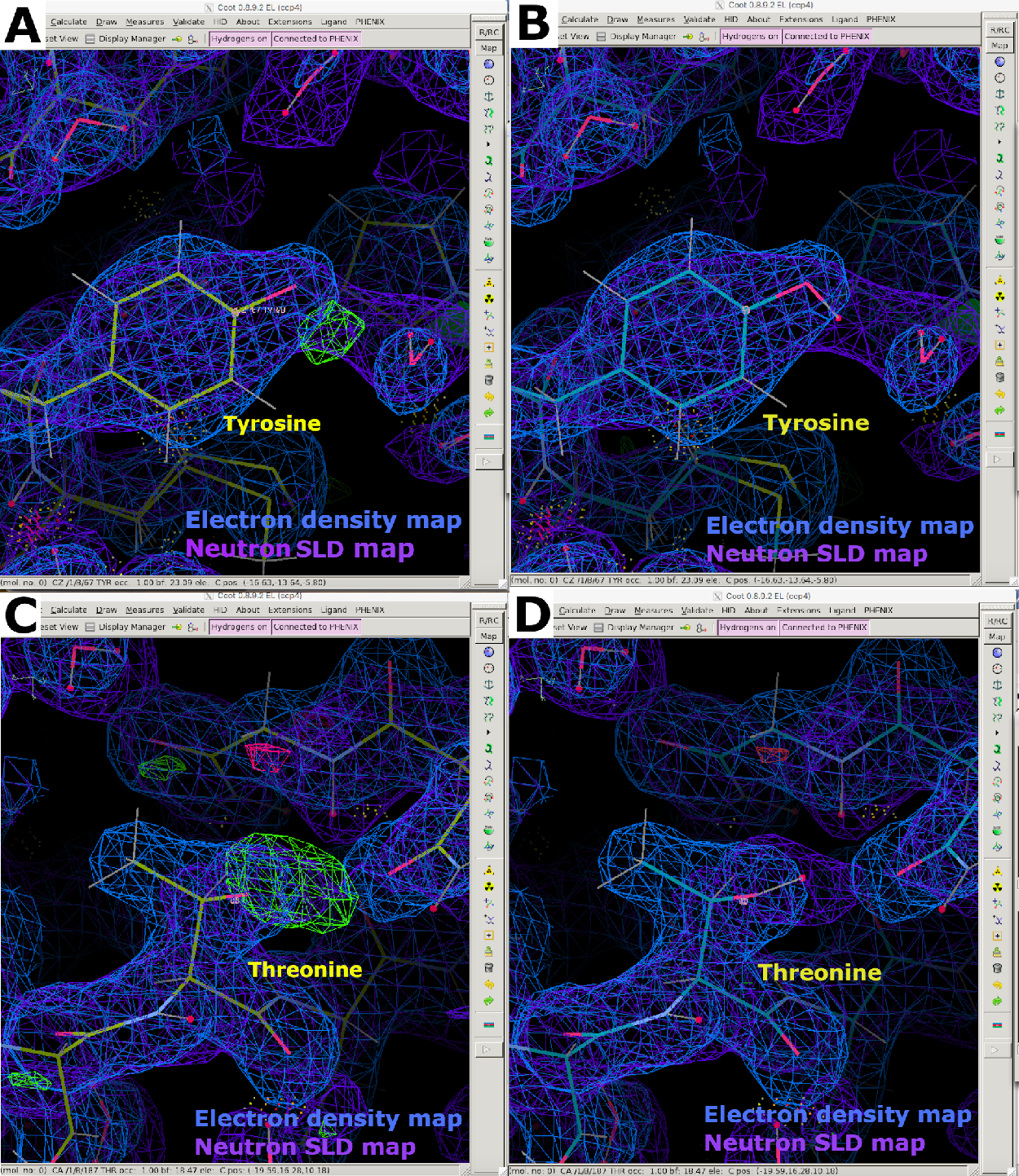
Después de la recopilación de datos de difracción de neutrones a temperatura ambiente, se utilizó el mismo cristal para recopilar un conjunto de datos de difracción de rayos X a temperatura ambiente a una resolución de 1,90 Å (Figura suplementaria 13). Los datos de rayos X se utilizaron para determinar las posiciones de los átomos "más pesados", incluidos C, N, O y S. La estructura refinada contra los datos de rayos X solos se utilizó como modelo de partida para realizar un refinamiento conjunto contra los datos de rayos X y neutrones. Phenix ReadySet se utilizó para agregar átomos de H en sitios no intercambiables, átomos de H y D en sitios intercambiables y átomos de D a moléculas de agua del modelo de rayos X inicial. Después de esta preparación del modelo, se realizaron refinamientos iterativos contra ambos conjuntos de datos (Figura suplementaria 19 y Figura suplementaria 20). La construcción interactiva de modelos se realizó en Coot inspeccionando visualmente los mapas de densidad para orientar las cadenas laterales y las moléculas de agua en consecuencia (Figura suplementaria 22). Los datos de neutrones se utilizaron principalmente para determinar los estados de protonación y las orientaciones de las moléculas de agua. La comparación del mapa de densidad de electrones de residuos como la serina y el triptófano y el correspondiente mapa SLD de neutrones ilustran la información que se puede obtener sobre los estados de protonación en los sitios intercambiables H/D de la difracción de proteínas de neutrones (Figura 7). Una superposición de mapas SLD de electrones y neutrones para moléculas de agua también indica que, si bien las interacciones de enlaces de hidrógeno se pueden inferir a partir de los datos de rayos X, los neutrones proporcionan información clara sobre la orientación de estos enlaces de hidrógeno (Figura 8). Se generaron mapas de omisión de neutrones SLD FO-FC para determinar los estados de protonación y la orientación H/D de las cadenas laterales. Se ilustran los mapas SLD de neutrones obtenidos para residuos de tirosina y treonina, en los que los mapas de neutrones Fo-FC indican claramente picos positivos que significan la presencia de H/D (Figura 9). Los datos de difracción de neutrones recopilados también proporcionaron información valiosa sobre múltiples estados de protonación, como el grupo -ND3+ de Lys (Figura 10). Las estadísticas de refinamiento (Rwork y Rfree) se supervisaron de cerca durante la optimización del modelo para evitar el ajuste excesivo. Las estadísticas finales dieron un Rwork de rayos X de 12.77% y un Rfree de 18.21%, y un Rwork de neutrones de 14.48% y un Rfree de 21.41% con 389 moléculas de agua presentes (Figura Suplementaria 28).
Los datos de criotemperatura se recopilaron en NcLPMO9D después de un remojo de ascorbato para reducir el sitio activo de cobre de CuII a CuI en la línea de haz MaNDi (Figura suplementaria 2 y Figura suplementaria 15)45. Los datos se recopilaron utilizando el modo TOF Laue después de una prueba de difracción de neutrones utilizando una exposición de 4 horas para verificar la calidad de la difracción. Dado el grupo espacial del cristal, se ideó una estrategia de recolección de datos de 18 marcos con una dosis de recolección de 80 Coulombs por marco. Los datos se recogieron en modo TOF-Laue en un rango de longitud de onda de 2,0 – 4,0 Å. Después de la recolección de datos, los datos fueron indexados, integrados, escalados y fusionados para dar un archivo de reflexión en formato MTZ a una resolución de 2.40 Å51,52.
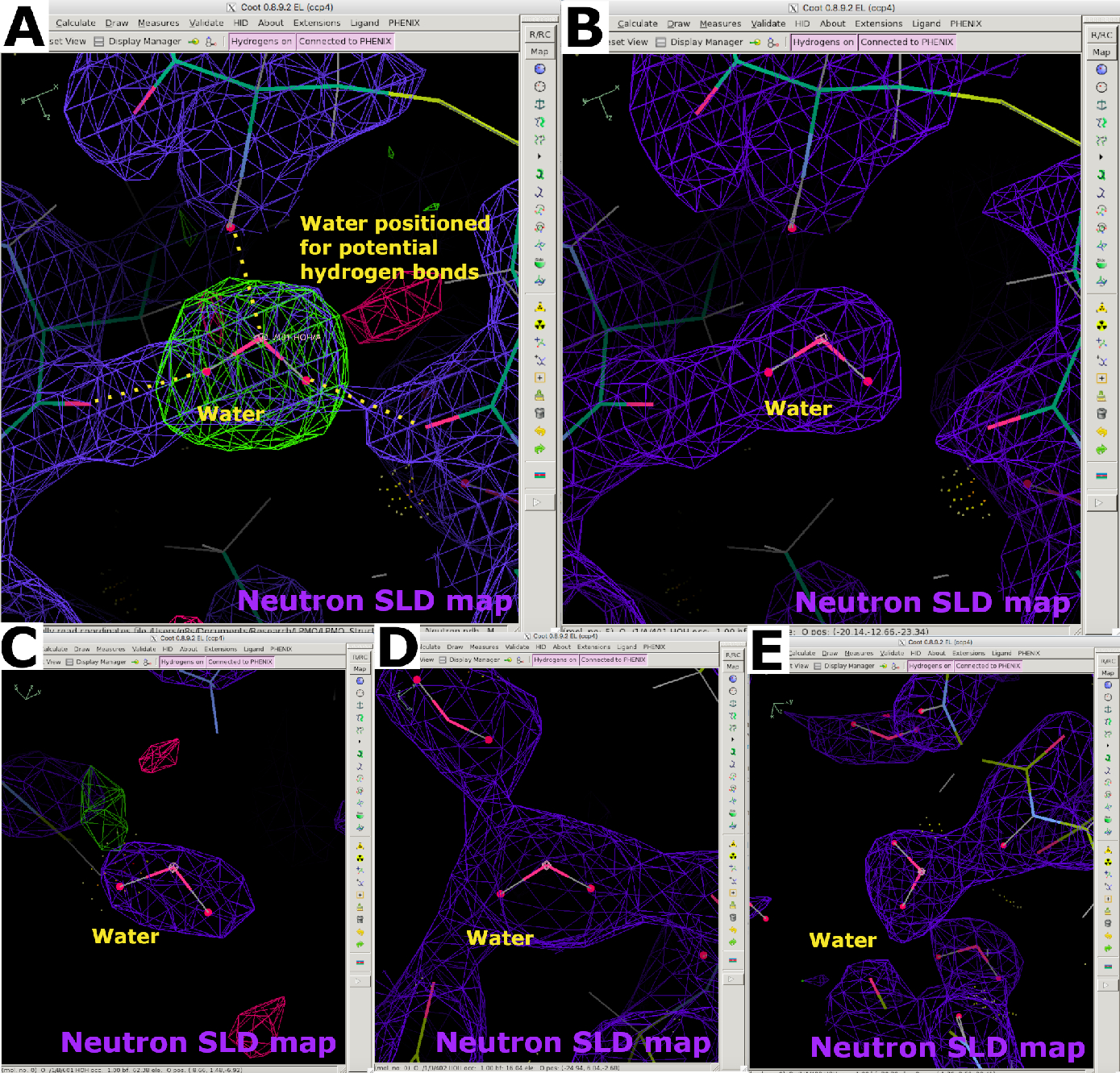
Después de la recopilación de datos, se utilizó el conjunto de datos de difracción de neutrones 2.40 Å criotemperatura NcLPMO9D para el refinamiento de datos solo con neutrones. Los datos de neutrones se escalonaron mediante reemplazo molecular utilizando PDB 5TKH como modelo de partida. Phenix ReadySet se utilizó para agregar átomos de H en sitios no intercambiables y átomos de H / D con ocupaciones parciales en sitios intercambiables. Las moléculas de agua se eliminaron del modelo inicial con herramientas PDB (Figura suplementaria 23). La preparación del modelo fue seguida por el refinamiento con phenix.refine utilizando la tabla de dispersión de neutrones (Figura suplementaria 24). La construcción de modelos interactivos se realizó en Coot, con moléculas de agua que se agregaron utilizando los picos positivos del mapa FO-Fc y se posicionaron de acuerdo con las posibles interacciones de enlaces de hidrógeno (Figura 11A y Figura 11B). Al analizar los mapas SLD de neutrones, las moléculas de agua son claramente visibles si están altamente ordenadas, sin embargo, su densidad puede ser esférica o elipsoidal si no están bien ordenadas (Figura 11C-E). Se utilizaron mapas SLD de neutrones para proporcionar información valiosa sobre la orientación de residuos como la asparagina, en la que diferenciar entre los grupos carbonilo y amino puede ser un desafío cuando se utilizan solo datos de difracción de rayos X (Figura 12A y Figura 12B). Los picos en los mapas de omisión SLD de neutrones FO-FC también fueron muy informativos para determinar los estados de protonación de los residuos de histidina en la posición N δ o N ε (Figura 12C y Figura 12D). El estado de protonación de los residuos con múltiples sitios intercambiables H/D también se puede determinar utilizando mapas SLD de neutrones. Esto se ilustró claramente con un mapa de salto SLD de neutrones FO-FC de arginina, que se sabe que tiene una carga positiva (Figura 12E y Figura 12F). Como anteriormente, el sobreajuste se evitó mediante el monitoreo de Rwork y Rfree. Las estadísticas finales dieron un Rwork de 22,58% y un Rfree de 30,84% (Figura Suplementaria 29). Dado que la difracción de proteínas de neutrones es una técnica de flujo limitado en la que se debe tener en cuenta la longitud de dispersión negativa y el gran factor de dispersión incoherente de H, se puede esperar que un refinamiento de solo datos de neutrones tenga estadísticas más pobres que un refinamiento conjunto de datos de rayos X / neutrones con menos moléculas de agua visibles (Figura suplementaria 28 y Figura suplementaria 29).
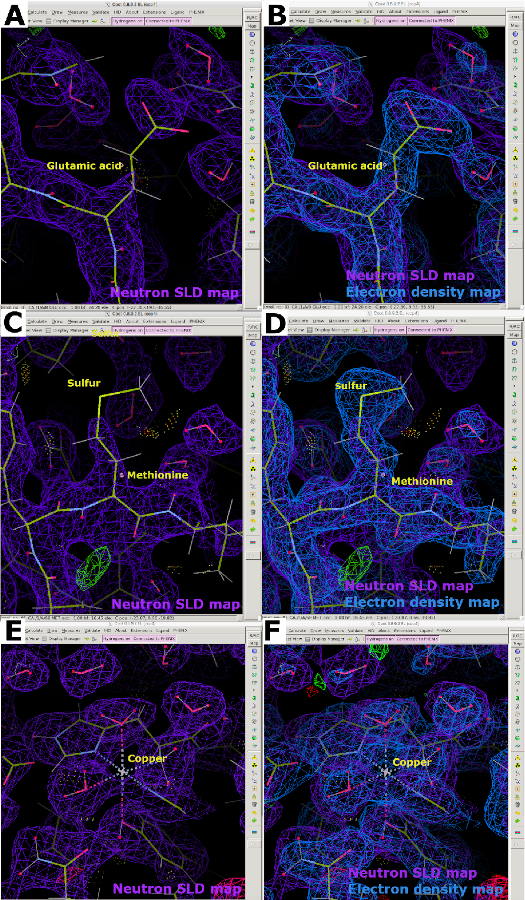
Al analizar los mapas SLD de neutrones, se hará evidente que la cancelación de densidad debido a la longitud de dispersión de neutrones negativos de H ocurrirá para las proteínas hidrogenadas que fueron sometidas a intercambio de vapor con el tampón de cristalización que contiene D2O. Debido a esta razón, los mapas SLD de neutrones en los que los átomos de H no intercambiables están unidos al carbono parecen incompletos en comparación con su contraparte del mapa de densidad de electrones (Figura 13A). El efecto de la cancelación es a menudo más evidente en resoluciones más pobres, por lo que es imperativo obtener cristales de proteína de alta calidad. Por lo tanto, es preferible realizar un refinamiento conjunto de una muestra con datos de rayos X y neutrones en el que los datos de rayos X se puedan utilizar para determinar la posición de la columna vertebral de la proteína (Figura 13B). Además, los átomos de azufre en la cisteína y la metionina pueden ser poco visibles, lo que requiere datos de rayos X para la ubicación exacta del átomo (Figura 13C y Figura 13D). Los metales con longitudes de dispersión de neutrones débiles también pueden ser difíciles de modelar en mapas SLD de neutrones, como es evidente en nuestros mapas LPMO9D. Por lo tanto, la recopilación de un conjunto de datos de rayos X de dosis baja (libre de daño por radiación) en el mismo cristal es útil, ya que permite el posicionamiento del átomo metálico utilizando mapas de densidad de electrones (Figura 13E y Figura 13F).

Figura 1: Diagrama de flujo del flujo de trabajo de cristalografía de proteínas de neutrones. Producción de proteínas. Para obtener una estructura neutrónica, primero se expresa la proteína. La expresión bacteriana en medios basados en H2O o D2O se utiliza típicamente para producir un alto rendimiento de proteína recombinante hidrogenada o perdeuterada, respectivamente. La proteína se purifica en tampón basado en H2O y luego se cristaliza en tampón de cristalización basado en H2O o D2O para hacer crecer cristales a un tamaño mínimo de 0,1 mm3. Preparación de la muestra: Antes de la recopilación de datos de difracción de neutrones, los cristales cultivados con H2O se someten a un intercambio de H / D para intercambiar los átomos de H titulables de proteínas con D. El intercambio de H / D se puede realizar mediante el remojo directo de los cristales en el tampón de cristalización deuterado, el equilibrio de la gota de cristalización con un depósito basado en D2O, o montando los cristales en capilares de cuarzo para el intercambio de vapor con el tampón de cristalización deuterada. Recopilación de datos de neutrones: Después del intercambio H/D, los cristales potenciales se examinan para determinar la calidad de la difracción. Los cristales con una resolución mínima de 2,5 Å se consideran adecuados para la recopilación de un conjunto de datos completo. Los cristales se montan en capilares de cuarzo para la recolección de datos a temperatura ambiente o se congelan en un crio-bucle para la recolección de datos a temperatura criogénica. Un conjunto de datos de rayos X se recopila en el mismo cristal (o idéntico) a la misma temperatura. Construcción de modelos: El refinamiento se realiza utilizando phenix.refine contra datos de neutrones y rayos X o solo contra los datos de neutrones. La construcción manual del modelo de la estructura de la proteína se realiza en Focha utilizando los mapas SLD de neutrones. Estructura completa: Una vez completada la estructura de la proteína, el modelo de coordenadas se valida y se deposita en el Banco de Datos de Proteínas. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 2: Recolección de cristales de proteínas. (A) Los cristales se manejan bajo un microscopio. (B) Se abre la caja de sándwich sellada que contiene la placa de vidrio siliconado. El amortiguador del depósito se canaliza sobre portaobjetos de vidrio siliconado. (C) Un cristal se cosecha con un microloop. (D) El cristal se coloca en una gota de licor madre para lavar cualquier residuo que a menudo se cosecha junto con el cristal. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 3: Transferencia de cristal a capilar de cuarzo. (A) El extremo de un capilar de cuarzo se llena con un tampón de reservorio. (B) El cristal se transfiere al capilar de cuarzo y (C) se sumerge en el tampón del reservorio. (D) El cristal se transporta por el capilar utilizando el tampón del reservorio. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 4: Sellado del capilar de cuarzo. (A) Se agrega un tampón deuterado al final del capilar para formar un "tapón". (B) La cera se funde con una "varita". (C) El capilar se coloca en la cera derretida para sellar. (D) Se forman tapones de cera en ambos extremos para sellar el capilar. (E) El cristal después del montaje. (F) El capilar sellado se coloca en una placa de Petri y se mantiene en su lugar con masilla. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 5: Aumento de la señal al ruido del patrón de difracción de neutrones. A medida que avanza la recopilación de datos, los puntos difractados se vuelven más intensos. (NOTA: las imágenes de difracción en vivo presentadas aquí son para ilustración y fueron tomadas de diferentes cristales). Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 6: Construcción de modelos interactivos utilizando datos de neutrones en Coot. (A) Un pico positivo de densidad SLD de neutrones FO-FC (verde) que indique serina debe reorientarse editando ángulos chi. El mapa SLD de neutrones 2FO-FC se muestra en púrpura y el mapa de densidad de electrones 2FO-FC se muestra en azul. (B) Serina correctamente posicionada. (C) Picos de densidad SLD de neutrones FO-FC positivos y negativos (verde y rojo, respectivamente) que indican que el triptófano debe rotarse / traducirse para que coincida con el pico de densidad de diferencia. (D) Triptófano correctamente orientado. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 7: Información adicional de mapas SLD de neutrones. (A) El mapa de densidad de electrones 2FO-FC (azul) muestra las posiciones de los átomos "más pesados" en la serina. (B) El mapa SLD de neutrones 2FO-FC (púrpura) muestra claramente la posición del átomo D "más ligero" en la serina. (C) El mapa de densidad de electrones 2FO-FC (azul) muestra las posiciones de los átomos "más pesados" en triptófano. (D) El mapa SLD de neutrones 2FO-FC (púrpura) muestra claramente la posición del átomo D "más ligero" en triptófano. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 8: Posicionamiento de la molécula de agua. (A) La forma esférica de una característica de mapa de densidad de electrones 2FO-FC (azul) para el agua. (B) El mapa SLD de neutrones 2FO-FC (púrpura) proporciona información sobre la orientación del agua y la interacción del enlace de hidrógeno. (C) Superposición cartográfica de mapas SLD de electrones y neutrones del agua. El mapa SLD de neutrones 2FO-FC se muestra en púrpura y el mapa de densidad de electrones 2FO-FC se muestra en azul. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 9: Mapas SLD FO-FComit de neutrones. (A) El mapa SLD de neutrones FO-FC (verde) proporciona información clara sobre la orientación H/D de los residuos de tirosina. El mapa SLD de neutrones 2FO-FC se muestra en púrpura y el mapa de densidad de electrones 2FO-FC se muestra en azul. (B) Residuo de tirosina con orientación H/D correcta. (C) El mapa SLD de neutrones FO-FC (verde) proporciona información clara sobre la orientación H/D de los residuos de treonina. (D) Residuo de treonina con orientación H/D correcta. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 10: Múltiples estados de protonación mostrados con mapas SLD de neutrones. (A) El mapa de densidad de electrones 2FO-FC (azul) solo proporciona la posición del átomo N del grupo lisina ε-amonio. (B-E) El mapa de omisión SLD de neutrones FO-FC (verde) demuestra claramente el grupo -NH3 cargado positivamente. El mapa SLD de neutrones 2FO-FC se muestra en púrpura y el mapa de densidad de electrones 2FO-FC se muestra en azul. (F) Superposición de mapas SLD de densidad de electrones y neutrones. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 11: Aparición de moléculas de agua en mapas SLD de neutrones. (A) Las moléculas de agua se posicionan de acuerdo con los mapas SLD de neutrones FO-FC (verdes) y los enlaces potenciales de hidrógeno. El mapa SLD de neutrones 2FO-FC se muestra en púrpura. (B) Molécula de agua correctamente posicionada. (C-E) Las diversas formas de los mapas SLD de neutrones para moléculas de agua dependen de factores B e interacciones de enlaces de hidrógeno. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 12: Información sobre la orientación y protonación de aminoácidos proporcionada por los mapas SLD de neutrones. (A) Los picos del mapa de neutrones SLD FO-FC (verde) indican una orientación incorrecta de un residuo de asparagina. El mapa SLD de neutrones 2FO-FC se muestra en púrpura y el mapa de densidad de electrones 2FO-FC se muestra en azul. (B) Mapa SLD de neutrones 2FO-FC (púrpura) de la orientación correcta de la asparagina. (C) El pico del mapa de neutrones SLD FO-FC (verde) indica una sola protonación de la histidina en N ε. (D) Mapa SLD de neutrones 2FO-FC (púrpura) de histidina N ε -protonación. (E) Neutron SLD FO-FC omitir los picos del mapa (verde) confirman la carga positiva de la arginina. (F) Mapa SLD de neutrones 2FO-FC (púrpura) de arginina cargada positivamente. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 13: Mapas SLD de neutrones discontinuos. (A) Mapa SLD de neutrones 2FO-FC (púrpura) de una proteína hidrogenada intercambiada por vapor H/D. El ácido glutámico muestra la cancelación del mapa SLD de neutrones debido a la longitud de dispersión negativa de los átomos de H no intercambiables. (B) Un mapa de densidad de electrones 2FO-FC superpuesto (azul) muestra claramente la densidad del ácido glutámico. (C) El átomo de azufre en la metionina es poco visible en los mapas SLD de neutrones 2FO-FC (púrpura). (D) Un mapa de densidad de electrones superpuesto muestra claramente la densidad de la metionina. (E) Los átomos metálicos, aquí cobre, son poco visibles en los mapas SLD de neutrones 2FO-FC (púrpura). (F) Un mapa de densidad de electrones 2FO-FC superpuesto (azul) muestra claramente la densidad del átomo de cobre coordinado. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
| Isótopo | Longitud de dispersión coherente (fm) | Longitud de dispersión incoherente (fm) |
| 1H | -3.741 | 25.274 |
| 2H | 6.671 | 4.04 |
| 12C | 6.6511 | 0 |
| 14N | 9.37 | 2 |
| 16O | 5.803 | 0 |
| 23Na | 3.63 | 3.59 |
| 24Mg | 5.66 | 0 |
| 31P | 5.13 | 0.2 |
| 32S | 2.804 | 0 |
| 35Cl | 11.65 | 6.1 |
| 39 mil | 3.74 | 1.4 |
| 40Ca | 4.8 | 0 |
| 55Mn | -3.73 | 1.79 |
| 56fe | 9.94 | 0 |
| 63Cu | 6.43 | 0.22 |
| 64Zn | 5.22 | 0 |
Tabla 1: Longitudes de dispersión de neutrones y valores de dispersión incoherentes. Adaptado de Sears, 199216.
Figura complementaria 1: El instrumento IMAGINE en el reactor de isótopos de alto flujo. (A) El instrumento IMAGINE en la sala de guía de neutrones fríos. (B) Muestra montada en un capilar de cuarzo unido con masilla al goniómetro. La mesa de muestras y detectores se cierra para colocar el cristal y la placa de imagen cilíndrica en el haz de neutrones. Modificado con permiso de la Unión Internacional de Cristalografía53. Imágenes proporcionadas con permiso de Genevieve Martin, Laboratorio Nacional de Oak Ridge. Haga clic aquí para descargar esta figura.
Figura suplementaria 2: El instrumento MaNDi en la fuente de neutrones de espalación. (A) La matriz de detectores de cámaras MaNDi Anger. Reproducido con el permiso de la Unión Internacional de Cristalografía11. (B) Etapa de muestra móvil MaNDi. (C) Muestra montada en capilar de cuarzo montada en el goniómetro en MaNDi para la recopilación de datos a temperatura ambiente. Imágenes proporcionadas con permiso de Genevieve Martin, Laboratorio Nacional de Oak Ridge. Haga clic aquí para descargar esta figura.
Figura complementaria 3: Estructura del polisacárido lítico monooxigenasa NcLPMO9D. El sitio activo de cobre NcLPMO9D se encuentra en una superficie plana de unión a polisacáridos. El cobre está coordinado por dos residuos de histidina en un "corsé de histidina" clásico, así como un residuo de tirosina axial. Haga clic aquí para descargar esta figura.
Figura suplementaria 4: Cristal con volumen suficiente en bandeja de cristalización de gota sentada. (A) Los cristales grandes se cultivan en gotas sentadas instaladas en placas de vidrio siliconado de 9 pocillos. (B y C) Los cristales se miden para identificar aquellos con > volumen 0,1 mm3. Haga clic aquí para descargar esta figura.
Figura complementaria 5: Medidor de pH configurado para lecturas tampón deuteradas. El electrodo de pH se empapa en D2O antes de su uso. NaOD y DCl se utilizan para ajustar el pH de los tampones deuterados. Haga clic aquí para descargar esta figura.
Figura complementaria 6: Directrices de montaje de muestras maNDi. Dimensiones máximas del capilar de cuarzo y posición de la muestra para la recolección de datos a temperatura ambiente.
Reproducido de: https://neutrons.ornl.gov/mandi/sample-environment Haga clic aquí para descargar esta figura.
Figura suplementaria 7: Eliminación del exceso de colchón. (A) El exceso de tampón se aspira desde el capilar de cuarzo con puntas microcapilares. (B) El tampón restante se retira con una mecha de papel delgada para secar completamente el capilar. Haga clic aquí para descargar esta figura.
Figura complementaria 8: La GUI de adquisición de datos. Ventana de entrada de los "Parámetros del experimento" para la recopilación de datos. Haga clic aquí para descargar esta figura.
Figura suplementaria 9: La GUI de Óptica. Selección del rango cuasi-Laue para la recopilación de datos y el monitoreo de la tasa de recuento de neutrones. Haga clic aquí para descargar esta figura.
Figura complementaria 10: Recopilación de datos en la GUI de adquisición de datos. El tiempo de exposición, el número de fotogramas y los ángulos para la recopilación de datos se especifican en la pestaña "Recopilar". La recopilación de datos se inicia mediante "Iniciar escaneo". Haga clic aquí para descargar esta figura.
Figura complementaria 11: Neutrones difractados detectados y mostrados. Al final del tiempo de exposición, se lee el detector de placas de imagen sensible a neutrones y se muestra el patrón de difracción en la GUI de adquisición de datos. Haga clic aquí para descargar esta figura.
Figura complementaria 12: Procesamiento de datos tras difracción de neutrones. Los fotogramas se indexan, integran, normalizan la longitud de onda y se escalan utilizando Lauegen, Lscale y Scala para generar un archivo de reflexión combinado después de la recopilación de datos. Haga clic aquí para descargar esta figura.
Figura complementaria 13: Recopilación de datos de rayos X. Generador de rayos X de origen doméstico configurado con cristal de cuarzo montado en capilar para la recopilación de datos a temperatura ambiente. Haga clic aquí para descargar esta figura.
Figura complementaria 14: Directrices de montaje para la recopilación de criodatos de MaNDi. Dimensiones de CrystalCaps y altura de pasador para la recolección de crio-datos en MaNDi.
Reproducido de: https://neutrons.ornl.gov/mandi/sample-environment Haga clic aquí para descargar esta figura.
Figura complementaria 15: Congelación por flash para la recopilación de datos de difracción de crione neutrones. (A) Configuración para remojo de cristales, cosecha con un microloop y congelación en nitrógeno líquido utilizando un recipiente compatible con crioterapia como una espuma Dewar. El cristal montado se transfiere directamente al criogoniómetro de línea de haz utilizando pinzas de pasador criogénico preenfriadas. (B) El sello de cera se funde para la eliminación del cristal. (C) El cristal se enjuaga hasta el extremo del capilar de cuarzo para la cosecha. (D) El cristal se empapa secuencialmente en tampón de remojo de ascorbato y luego crioprotector seguido de congelación instantánea en nitrógeno líquido. Haga clic aquí para descargar esta figura.
Figura complementaria 16: Interfaz de alineación de muestra. La alineación del cristal en el haz de neutrones, representada por la cruz azul, se realiza mediante el centrado de apuntar y hacer clic. Haga clic aquí para descargar esta figura.
Figura complementaria 17: La GUI CSS para la recopilación de datos. La estrategia de recopilación de datos, incluidas las dosis de exposición y los ángulos, se cargan en la GUI de CSS. A medida que avanza la recopilación de datos, los neutrones difractados detectados en el detector en tiempo real se mostrarán en el panel superior. Haga clic aquí para descargar esta figura.
Figura suplementaria 18: Coincidencia de banderas libres de R en CCP4. Las banderas libres de R de los datos de neutrones se combinan con las banderas libres de R de los datos de rayos X recopilados en el mismo cristal o en un cristal idéntico para el refinamiento de las articulaciones. Haga clic aquí para descargar esta figura.
Figura complementaria 19: Preparación y refinamiento de la estructura. (A) La herramienta Phenix ReadySet se utiliza para agregar ocupación H/D dual en sitios intercambiables. (B) Tanto los datos de neutrones como los datos de rayos X se utilizan para un refinamiento conjunto, mientras que el modelo de entrada inicial se refinó contra el conjunto de datos de rayos X recopilado en el mismo cristal o un cristal idéntico. Haga clic aquí para descargar esta figura.
Figura complementaria 20: Configuración de los ajustes de refinamiento. El modelo de refinamiento, así como las distancias nucleares, están configurados para el refinamiento conjunto de datos de rayos X / neutrones. Haga clic aquí para descargar esta figura.
Figura complementaria 21: Selección de datos para la construcción de modelos de fochas. La salida del archivo MTZ phenix que contiene datos de rayos X y neutrones sin rellenar se abre en Coot para generar mapas SLD de electrones y neutrones para la construcción de modelos interactivos. Haga clic aquí para descargar esta figura.
Figura complementaria 22: Construcción de modelos interactivos en Focha durante un refinamiento conjunto. (A) Un pico de densidad SLD de neutrones FO-FC positivo y negativo (verde y rojo, respectivamente) que indica que el agua debe reorientarse por rotación/traslación. El mapa SLD de neutrones 2FO-FC se muestra en púrpura y el mapa de densidad de electrones 2FO-FC se muestra en azul. (B) Agua correctamente posicionada. (C) Un pico positivo del mapa SLD de neutrones FO-FC (verde) indica que la treonina debe girarse para que coincida con el pico de densidad de diferencia mediante la edición de ángulos chi. (D) Treonina correctamente orientada. Haga clic aquí para descargar esta figura.
Figura complementaria 23: Preparación de la estructura para el refinamiento de datos solo con neutrones. El archivo de coordenadas iniciales se prepara para el refinamiento mediante la eliminación del átomo de agua en PDBTools y mediante la adición de ocupación dual H / D en sitios intercambiables. Haga clic aquí para descargar esta figura.
Figura complementaria 24: Refinamiento de solo datos de neutrones. (A) Se cargan los datos de neutrones, así como el modelo de partida preparado. (B) La configuración para el refinamiento de datos de neutrones utiliza la tabla de dispersión de neutrones. Haga clic aquí para descargar esta figura.
Figura complementaria 25: Selección de datos para la construcción de modelos de fochas. Los datos de neutrones sin rellenar se abren en Coot para la construcción de modelos interactivos. Haga clic aquí para descargar esta figura.
Figura suplementaria 26: Refinamiento del espacio real en focha para residuos deuterados. (A) Picos de densidad SLD de neutrones FO-FC positivos y negativos (verde y rojo, respectivamente) que indican que un residuo de arginina debe moverse para ajustarse al pico de densidad FO-FC. El mapa SLD de neutrones 2FO-FC se muestra en púrpura y el mapa de densidad de electrones 2FO-FC se muestra en azul. (B) La utilización de Real Space Refine da como resultado la "explosión" de átomos D debido a la falta de bibliotecas de restricción de geometría de focha. (C) Los átomos D no se mueven con el resto de los átomos residuales. (D) Las posiciones del átomo D se pueden fijar manualmente utilizando un editor de texto. Haga clic aquí para descargar esta figura.
Figura complementaria 27: Adición de moléculas de agua. (A) Las moléculas de agua se pueden agregar manualmente a los picos positivos de densidad del mapa SLD de neutrones FO-FC (verde). Las moléculas de agua insertadas estarán representadas inicialmente por un átomo de O en La focha. (B) Phenix ReadySet se utiliza para agregar átomos D a los átomos de O para moléculas de agua. (C) La molécula de agua deuterada se agrega con éxito. Haga clic aquí para descargar esta figura.
Figura complementaria 28: Estadísticas de refinamiento. Estadísticas finales de refinamiento de datos después del refinamiento conjunto de rayos X/neutrones. Haga clic aquí para descargar esta figura.
Figura complementaria 29: Estadísticas de refinamiento. Estadísticas finales de refinamiento de datos después del refinamiento de solo datos de neutrones. Haga clic aquí para descargar esta figura.
Discusión
La cristalografía de proteínas de neutrones es una técnica altamente sensible para sondear los estados de protonación y la orientación de la molécula de agua en las proteínas. Esta información arroja luz sobre los mecanismos catalíticos de las proteínas, ya que los cambios en la protonación y las interacciones de enlace de hidrógeno a menudo son fundamentales para la química enzimática10. La cristalografía de proteínas de neutrones, aunque es una técnica informativa, tiene una serie de factores que deben tenerse en cuenta antes de planificar la realización de un experimento de difracción de neutrones, a saber:
- El requisito de grandes cristales de proteínas para la recopilación de datos.
- Las propiedades de dispersión del hidrógeno y otros elementos, como los iones metálicos.
- Limitaciones en el refinamiento de la estructura y el software de construcción de modelos cuando se trabaja con muestras deuteradas.
La cristalografía de proteínas de neutrones es una técnica de flujo limitado. A diferencia de los conjuntos de datos de difracción de rayos X, se esperan factores R más altos y menor integridad, redundancia y relaciones señal-ruido para los conjuntos de datos de neutrones debido a las limitaciones inherentes a la técnica (flujo limitado, cuasi-Laue, longitudes de onda más largas). La recopilación de datos de un solo fotograma suele ser de 12 a 18 horas. El éxito de un experimento depende en gran medida del tamaño y la calidad de la muestra, siendo cristales de 0,1 mm3 a menudo el requisito mínimo3. La difracción de neutrones requiere la producción de grandes cantidades de proteínas para establecer gotas de cristalización que van desde 10 a 800 μL. El volumen mínimo para cultivar cristales suficientemente grandes se puede estimar utilizando una calculadora de volumen dados los parámetros de cristal y muestra (https://neutrons.ornl.gov/imagine). El crecimiento de cristales grandes se ha logrado con mayor frecuencia por difusión de vapor3. La cristalización de gotas colgantes permite el crecimiento de cristales en grandes gotas que van desde 10-25 μL, mientras que las gotas más grandes que van hasta ~ 50 μL se pueden configurar utilizando equipos de caída sentada disponibles comercialmente14,54. Las placas de vidrio siliconadas de nueve pocillos se pueden usar para configurar gotas muy grandes, con volúmenes de hasta 800 μL. Estas placas de vidrio se colocan en "cajas de sándwich" disponibles comercialmente en Hampton Research. Otras técnicas de cristalización incluyen la cristalización por lotes, en la que el límite del tamaño de la gota es dictado por el recipiente. La configuración del experimento de cristalización por lotes puede variar desde microlitros hasta mililitros55. La cristalización también se puede realizar mediante la técnica de diálisis en la que la proteína se equilibra con el precipitante a través de una membrana de diálisis o por contradifusión a lo largo de un gradiente de concentración de precipitantes o a través de un tapón poroso como la agarosa56,57. La siembra ofrece otra alternativa para obtener cristales del volumen deseado. La micro y macrosiembra se han empleado con éxito para el crecimiento de grandes cristales, incluido el cristal grande de NcLPMO9D45. Algunos conocimientos del diagrama de fases de proteínas, incluida la influencia de la temperatura en la solubilidad, ayudan en el crecimiento de grandes cristales.
Al planificar un experimento de difracción de neutrones, la optimización de la preparación de proteínas para maximizar la relación señal-ruido durante la recopilación de datos de difracción es esencial7. Para eludir la cancelación de densidad y la alta dispersión incoherente causada por los átomos de H, los mapas SLD de neutrones se pueden mejorar intercambiando átomos de H por su isótopo D, que posee una longitud de dispersión coherente positiva y una longitud de dispersión incoherente baja. Para lograr esto, se realiza el intercambio de vapor del cristal de proteína hidrogenada contra el tampón de cristalización deuterado. Esto asegura el intercambio H/D de moléculas de disolvente y los átomos de proteína H lábiles y titulables23. El intercambio de vapor se realiza montando el cristal hidrogenado en un capilar de cuarzo con "tapones" tampón de cristalización deuterados basados en D2O y representa una técnica efectiva y suave que se aplica con mayor frecuencia14,23,35. El intercambio puede tardar varias semanas y preferiblemente requiere que el búfer deuterado se cambie con frecuencia para garantizar el máximo intercambio H / D. El intercambio H/D también se puede realizar remojando directamente el cristal en tampón deuterado. Para evitar colocar el cristal bajo estrés debido a la exposición a D2O, el proceso de remojo debe realizarse gradualmente aumentando gradualmente la relación D2O: H2O58. Además de esto, la cristalización de proteínas hidrogenadas también se puede realizar en tampón deuterado para el intercambio de H/D en sitios H lábiles22,59. Cabe señalar, sin embargo, que el tampón basado en D2O tiene un efecto sobre la solubilidad de la proteína que requiere un ajuste adicional de las condiciones conocidas basadas en H2O3,59. También se ha observado que los tampones basados en D2O conducen a cristales más pequeños en algunos casos59. El intercambio completo de átomos de H titulables y unidos al carbono a D se puede lograr expresando proteínas en medios deuterados para generar una muestra perdeuterada20. Los mapas SLD de neutrones resultantes de la muestra perdeuterada se mejorarán significativamente, ya no mostrando la cancelación de densidad de la contraparte de la muestra hidrogenada. Esto es beneficioso cuando se caracteriza H/D unido en sitios no intercambiables en una proteína o cofactor. Sin embargo, la expresión de proteína perdeuterada es a la vez alta en costo y baja en rendimiento60. El Centro de Biología Molecular Estructural (CSMB) del Laboratorio Nacional de Oak Ridge (ORNL) ofrece una instalación de deuteración para los usuarios que buscan generar una muestra perdeuterada (https://www.ornl.gov/facility/csmb). La expresión perdeuterada se realiza típicamente en un biorreactor en la escala de 1 L que produce ~ 50 mg de proteína purificada61.
Después de la recopilación de datos de difracción de neutrones, se realiza el refinamiento y la construcción de modelos interactivos. El refinamiento se puede ejecutar utilizando múltiples suites de software, incluyendo phenix.refine, nCNS o SHELXL28,31,32,33. La suite Phenix es el software más utilizado para el refinamiento de los datos de difracción de neutrones junto con Coot, que se utiliza para construir manualmente el modelo a partir de los mapas SLD de neutrones34. Aunque tanto Phenix como Coot permiten el procesamiento de datos de difracción de neutrones, pueden carecer de ciertas características necesarias para procesar la idiosincrasia asociada con los datos de neutrones y las muestras deuteradas. Por ejemplo, Coot no contiene optimización geométrica para residuos deuterados, lo que puede provocar complicaciones durante la construcción del modelo, ya que la función "Real Space Refine" da como resultado residuos "explosivos" (Figura suplementaria 26)62. Esto se puede resolver generando archivos de retención para todos los residuos deuterados. Sin embargo, este es un proceso intensivo y tales bibliotecas no están actualmente disponibles públicamente. Al realizar refinamientos en Phenix, los sitios H / D intercambiables se establecerán inicialmente en 0.50 de ocupación para H y D. A medida que se realicen refinamientos, la ocupación de H y D se refinará de acuerdo con los mapas SLD de neutrones. Durante la construcción interactiva de modelos, los mapas Fo-Fc de densidad de diferencia son muy informativos para evaluar las ocupaciones H / D. Los mapas se pueden utilizar para determinar qué sitios poseen una alta ocupación D, lo que es particularmente informativo en el sitio activo donde los estados de protonación son catalíticamente relevantes63. Las situaciones ambiguas surgen, sin embargo, cuando el H:D la ocupación es cercana a 0.70:0.30, lo que resulta en la cancelación completa de la señal en los mapas SLD de neutrones64. También debe tenerse en cuenta que los conjuntos de datos de neutrones cuasi-Laue a menudo tienen una completitud de alrededor del 80%, que es inferior a la observada rutinariamente ≥ 98% para los datos de difracción de rayos X. Al refinar los datos de difracción de neutrones en Phenix, las amplitudes observadas faltantes (Fo) se calculan a partir del modelo para completar la lista de reflexión, introduciendo así el sesgo del modelo. Para tener en cuenta este posible sesgo, los mapas "no_fill" deben examinarse durante la construcción de modelos interactivos en lugar de los mapas "llenos".
Los usuarios pueden optar por realizar un refinamiento conjunto de datos de rayos X / neutrones de su estructura, o un refinamiento de datos de neutrones solamente. Visualizar mapas SLD de neutrones, particularmente a menor resolución, puede ser inicialmente desconcertante, especialmente para una proteína hidrogenada en la que H todavía está presente en sitios no intercambiables a pesar del intercambio de vapor H / D. Esto da lugar a cancelaciones de mapas de densidad de neutrones, dando la impresión de mapas discontinuos65,66. La recopilación de un conjunto de datos de rayos X correspondiente complementa ventajosamente estas cancelaciones en un refinamiento conjunto (Figura 13A y Figura 13B). Una estrategia de refinamiento conjunto generalmente implica refinar las coordenadas de la columna vertebral de la proteína contra los datos de rayos X, mientras que los datos de difracción de neutrones se utilizan para refinar la posición y ocupación de los átomos de H / D en sitios intercambiables28. Dado que la introducción de la ocupación conjunta de H/D en sitios intercambiables aumenta el número de parámetros que se refinan, un refinamiento conjunto con datos de rayos X también aumenta la relación datos-parámetro. Un refinamiento conjunto requiere que se recolecte un conjunto de datos de rayos X correspondiente a la misma temperatura en el mismo cristal o en un cristal cultivado en las mismas condiciones. Para los datos de difracción de neutrones recopilados a temperatura ambiente (300K), el conjunto de datos de rayos X correspondiente debe recopilarse a temperatura ambiente utilizando una estrategia de recopilación de datos de dosis baja para limitar el daño por radiación. Las muestras perdeuteradas, por el contrario, proporcionan mapas SLD de neutrones mejorados y continuos, ya que no poseen la misma magnitud de cancelación de señal H / D. Sin embargo, la longitud de dispersión de neutrones de ciertos elementos, incluidos los metales y el azufre, los hace poco visibles en los mapas SLD de neutrones, incluso si la proteína ha sido perdeuterada (Figura 13C-F)18. Si un metal necesita ser caracterizado, es mejor utilizar la difracción de rayos X en un refinamiento conjunto o aplicar técnicas espectroscópicas para complementar los experimentos de difracción. Los refinamientos de datos solo con neutrones a menudo se realizan cuando el conjunto de datos de neutrones tiene alta resolución o si se utilizó una proteína perdeuterada. Además, el refinamiento de los datos de solo neutrones es particularmente útil si se está estudiando una proteína altamente sensible al daño por radiación, ya que una estructura derivada de rayos X puede poseer artefactos inducidos por radiación. Si se va a realizar un refinamiento de solo datos de neutrones, se debe determinar si el conjunto de datos de neutrones correspondiente tiene suficiente integridad y resolución.
ORNL ofrece dos instalaciones para la recopilación de datos de difracción de neutrones: la línea de haz IMAGINE en el HFIR, así como la línea de haz MaNDi en el SNS36,67. Si bien ambos instrumentos proporcionan medios efectivos para recopilar un conjunto de datos de difracción de neutrones que emplea principios similares, cada instrumento tiene especificaciones únicas que deben tenerse en cuenta al solicitar el tiempo de haz. IMAGINE recopila datos cuasi-Laue y está optimizado para la recopilación de datos a temperatura ambiente en cristales con celdas unitarias de hasta ~ 100 Å. MaNDi se puede utilizar para la recopilación de datos de temperatura ambiente y criotemperatura empleando la recolección TOF-Laue en cristales con celdas unitarias de hasta ~ 300 Å. Antes de recopilar un conjunto de datos completo, se realiza una prueba en el cristal para evaluar la calidad del patrón de difracción obtenido en el que el cristal se expone al haz de neutrones durante un solo marco. Si el cristal es de calidad suficiente, se recopilará, indexará, integrará, escalará y fusionará un conjunto de datos completos de difracción de neutrones en un proceso análogo al procesamiento de datos de rayos X. IMAGINE hace uso de Lauegen y Lscale y MaNDi utiliza el paquete Mantid y emplea el ajuste de perfiles tridimensionales48,50,51,68,69,70. A los científicos que se conviertan en usuarios de cualquiera de estas instalaciones se les proporcionará un conjunto de datos en formato MTZ o HKL para su posterior análisis.
La difracción de neutrones es una técnica no destructiva y altamente sensible para sondear el estado de protonación y las interacciones de enlaces de hidrógeno de macromoléculas biológicas. Es particularmente útil para proteínas fotosensibles y metaloproteínas. Se deben tener en cuenta varias consideraciones con respecto a la técnica, así como el procesamiento de los datos antes de realizar un experimento, sin embargo, el resultado arroja resultados que pueden dar una visión valiosa del mecanismo catalítico de la proteína de interés. La cristalografía de proteínas de neutrones complementa los estudios computacionales, estructurales, bioquímicos y espectroscópicos, lo que la convierte en una herramienta valiosa en la caja de herramientas del biólogo de técnicas utilizadas para caracterizar macromoléculas biológicas.
Divulgaciones
Los autores no tienen nada que revelar.
Agradecimientos
Los experimentos de expresión, purificación y cristalización de proteínas se llevaron a cabo en el Centro de Biología Molecular Estructural (CSMB), una instalación de usuario de investigación biológica y ambiental del Departamento de Energía de los Estados Unidos en el Laboratorio Nacional de Oak Ridge. Los datos de difracción de neutrones se recopilaron en BL-11B MaNDi en la Fuente de Neutrones de Espalación (SNS) en ORNL, que está patrocinada por la División de Instalaciones de Usuarios Científicos, Oficina de Ciencias Básicas de la Energía, Departamento de Energía de los Estados Unidos. Los autores agradecen a Brendan Sullivan por su ayuda con la reducción de datos. Los datos de difracción de rayos X se recopilaron en las instalaciones del Centro de Innovación de Educación, Tecnología e Investigación Molecular (METRIC) de la Universidad Estatal de Carolina del Norte, que cuenta con el apoyo del Estado de Carolina del Norte. GCS reconoce el apoyo en parte de la Fundación Nacional de Investigación (NRF), Sudáfrica y el programa de Oportunidades para Graduados (GO!) en ORNL. FM reconoce el apoyo de USDA NIFA Hatch 211001.
Materiales
| Name | Company | Catalog Number | Comments |
| Absorbent Paper Points Size #30-#40, 60 mm length | DiaDent/DiaVet | 218-292 | |
| Capillary wax | Hampton | HR4-328 | |
| CCP4 | Version 7.0.077 | ||
| Conical Centrifuge Tubes (15 mL) | Corning | CLS430790 | |
| Conical Centrifuge Tubes (50mL) | Corning | CLS430828 | |
| Coot | Version 0.8.9.2 | ||
| CrystalCap ALS | Hampton | HR4-779 | |
| Curved-Tip Forceps | Mitegen | TW-CTF-1 | |
| Deuterium chloride solution, 35 wt. % in D2O, ≥99 atom % D | Sigma-Aldrich | 543047 | |
| Deuterium oxide 99.9 atom % D | Sigma-Aldrich | 151882 | |
| Dual Thickness MicroLoops 1000 µm | Mitegen | M5-L18SP-1000 | |
| FiveEasy pH meter F20-Std-Kit | Mettler Toledo | 30266626 | |
| Foam Dewars Standard Vessel 800 ml | Spearlab | M-FD-800 | |
| Four Color Mounting Clay | Hampton | HR4-326 | |
| HEPES, BioUltra, for molecular biology, ≥99.5% (T), | Sigma-Aldrich | 54457 | |
| High flux rotating anode X-ray diffractomemeter with EIGER 4M detector | Rigaku, Oxford Cryostream and Dectris | XtaLAB Synergy-R | Home source X-ray diffractometer |
| Magnetic Wand Straight | Mitegen | M-R-1013198 | |
| Microloader, tip for filling Femtotips and other glass microcapillaries (for research use only), 0.5 – 20 µL, 100 mm, light gray, 192 pcs. (2 racks × 96 pcs.) | Eppendorf | 930001007 | |
| Microtubes volume 1.5 mL | Eppendorf | Z606340 | |
| Petri Dishes with Clear Lid 100 mm diameter | Fischerbrand | FB0875713 | |
| Phenix | Version 1.14-3260 | ||
| Pin Tong 18 mm | Mitegen | M-R-1013196 | |
| Pipette Volume 0.1-2.5 μL | Eppendorf Research | Z683779 | |
| Pipette Volume 100-1000 μL | Eppendorf Research | Z683825 | |
| Pipette Volume 10-100 μL | Eppendorf Research | Z683809 | |
| Pipette Volume 20-200 μL | Eppendorf Research | Z683817 | |
| Poly(ethylene glycol) BioXtra, average mol wt 3,350, powder | Sigma-Aldrich | P4338 | |
| Quartz Capillary , 1.00 mm inner diameter, 80 mm length | Hampton | HR6-146 | Thin-walled capillary |
| Research Stereomicroscope System | Olympus | SZX16 | |
| Reusable B3 (SSRL/SAM Style) Goniometer Bases | Mitegen | GB-B3-R | |
| Round - Miniature Hollow Glass Tubing (VitroTubes) Clear Fused Quartz / 1.00 mm inner diameter, 100 mm length | VitroCom | CV1012 | Thick-walled capillary |
| Sandwich Box with cover | Hampton | HR3-132 | |
| Siliconized 9 Well Glass Plate | Hampton | HR3-134 | |
| Sitting Drop Crystallization Plate (24 Big Well) | Mitegen | XQ-P-24S-A | |
| Sodium deuteroxide solution, 40 wt. % in D2O, 99 atom % D | Sigma-Aldrich | 176788 | |
| Thick Siliconized circle cover slides (22 mm x 0.96 mm) | Hampton | HR3-247 | |
| Universal Pipet Tips, 0.1 - 10 µL | VWR | 76322-528 | |
| Universal Pipet Tips, 1 - 100 µL | VWR | 76322-136 | |
| Universal Pipet Tips, 100 - 1000 µL | VWR | 76322-154 | |
| Universal Pipet Tips, 20 - 200 µL | VWR | 76322-150 | |
| Universal Pipet Tips, 1 - 20 µL | VWR | 76322-134 | |
| Wax pen | Hampton | HR4-342 |
Referencias
- Neumann, P., Tittmann, K. Marvels of enzyme catalysis at true atomic resolution: distortions, bond elongations, hidden flips, protonation states and atom identities. Current Opinion in Structural Biology. 29, 122-133 (2014).
- Pynn, R. Neutron Scattering-A Non-destructive Microscope for Seeing Inside Matter. Neutron Applications in Earth, Energy and Environmental Sciences. , 15-36 (2009).
- O'Dell, W. B., Bodenheimer, A. M., Meilleur, F. Neutron protein crystallography: A complementary tool for locating hydrogens in proteins. Archives of Biochemistry and Biophysics. 602, 48-60 (2016).
- Niimura, N., Podjarny, A. . Neutron Protein Crystallography: Hydrogen, Protons, and Hydration in Bio-macromolecules. , (2011).
- Blakeley, M. P. P. Neutron macromolecular crystallography. Crystallography Reviews. 15 (3), 157-218 (2009).
- Blakeley, M. P., Cianci, M., Helliwell, J. R., Rizkallah, P. J. Synchrotron and neutron techniques in biological crystallography. Chemical Society Reviews. 33 (8), 548-557 (2004).
- Ashkar, R., et al. Neutron scattering in the biological sciences: progress and prospects. Acta Crystallographica Section D: Structural Biology. 74 (12), (2018).
- Teixeira, S. C. M., et al. New sources and instrumentation for neutrons in biology. Chemical Physics. 345 (2-3), 133-151 (2008).
- Furrer, A., Mesot, J., Strässle, T. . Neutron Scattering in Condensed Matter Physics. , (2009).
- Meilleur, F., Coates, L., Cuneo, M. J., Kovalevsky, A., Myles, D. A. A. The neutron macromolecular crystallography instruments at Oak Ridge national laboratory: Advances, challenges, and opportunities. Crystals. 8 (10), 1-10 (2018).
- Coates, L., et al. The Macromolecular Neutron Diffractometer MaNDi at the Spallation Neutron Source. Journal of Applied Crystallography. 48, 1302-1306 (2015).
- Coates, L., Stoica, A. D., Hoffmann, C., Richards, J., Cooper, R. The macromolecular neutron diffractometer (MaNDi) at the Spallation Neutron Source, Oak Ridge: enhanced optics design, high-resolution neutron detectors and simulated diffraction. Journal of Applied Crystallography. 43 (3), 570-577 (2010).
- Koetzle, T. F., Piccoli, P. M. B., Schultz, A. J. Single-crystal neutron diffraction studies of hydrogen-bonded systems: Two recent examples from IPNS. Nuclear Instruments and Methods in Physics Research, Section A: Accelerators, Spectrometers, Detectors and Associated Equipment. 600 (1), 260-262 (2009).
- Ng, J. D., et al. Large-volume protein crystal growth for neutron macromolecular crystallography. Acta Crystallographica Section F:Structural Biology Communications. 71, 358-370 (2015).
- Blakeley, M. P., Langan, P., Niimura, N., Podjarny, A. Neutron crystallography: opportunities, challenges, and limitations. Current Opinion in Structural Biology. 18 (5), 593-600 (2008).
- Sears, V. F. Neutron scattering lengths and cross sections. Neutron News. 3 (3), 26-37 (1992).
- Weik, M., Patzelt, H., Zaccai, G., Oesterhelt, D. Localization of glycolipids in membranes by in vivo labeling and neutron diffraction. Molecular Cell. 1 (3), 411-419 (1998).
- Helliwell, J. R. Fundamentals of neutron crystallography in structural biology. Methods in Enzymology. 634, (2020).
- Niimura, N., Bau, R. Neutron protein crystallography: Beyond the folding structure of biological macromolecules. Acta Crystallographica Section A: Foundations of Crystallography. 64 (1), 12-22 (2008).
- Hazemann, I., et al. High-resolution neutron protein crystallography with radically small crystal volumes: Application of perdeuteration to human aldose reductase. Acta Crystallographica Section D: Biological Crystallography. 61 (10), 1413-1417 (2005).
- Niimura, N., Chatake, T., Ostermann, A., Kurihara, K., Tanaka, I. High resolution neutron protein crystallography. Hydrogen and hydration in proteins. Zeitschrift für Kristallographie - Crystalline Materials. 218 (2), (2003).
- Meilleur, F., Contzen, J., Myles, D. A. A., Jung, C. Structural stability and dynamics of hydrogenated and perdeuterated cytochrome P450cam (CYP101). Biochemistry. 43 (27), 8744-8753 (2004).
- Bennett, B. C., Gardberg, A. S., Blair, M. D., Dealwis, C. G. On the determinants of amide backbone exchange in proteins: A neutron crystallographic comparative study. Acta Crystallographica Section D: Biological Crystallography. 64 (7), 764-783 (2008).
- Meilleur, F., Kovalevsky, A., Myles, D. A. A. IMAGINE: The neutron protein crystallography beamline at the high flux isotope reactor. Methods in Enzymology. 634, (2020).
- Wang, X. P., et al. A suite-level review of the neutron single-crystal diffraction instruments at Oak Ridge National Laboratory. Review of Scientific Instruments. 89 (9), 092802 (2018).
- Wlodawer, A., Hendrickson, W. A. A procedure for joint refinement of macromolecular structures with X-ray and neutron diffraction data from single crystals. Acta Crystallographica Section A. 38 (2), 239-247 (1982).
- Halle, B. Biomolecular cryocrystallography: Structural changes during flash-cooling. Proceedings of the National Academy of Sciences of the United States of America. 101 (14), 4793-4798 (2004).
- Adams, P. D., Mustyakimov, M., Afonine, P. V., Langan, P. Generalized X-ray and neutron crystallographic analysis: More accurate and complete structures for biological macromolecules. Acta Crystallographica Section D: Biological Crystallography. 65 (6), 567-573 (2009).
- Berman, H. M., et al. The Protein Data Bank. Acta Crystallographica Section D Biological Crystallography. 58 (6), 899-907 (2002).
- Liebschner, D., Afonine, P. V., Moriarty, N. W., Langan, P., Adams, P. D. Evaluation of models determined by neutron diffraction and proposed improvements to their validation and deposition. Acta Crystallographica Section D: Structural Biology. 74, 800-813 (2018).
- Afonine, P. V., Mustyakimov, M., Grosse-Kunstleve, R. W., Moriarty, N. W., Langan, P., Adams, P. D. Joint X-ray and neutron refinement with phenix.refine. Acta Crystallographica Section D: Biological Crystallography. 66 (11), 1153-1163 (2010).
- Brünger, A. T., et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallographica Section D: Biological Crystallography. 54 (5), 905-921 (1998).
- Gruene, T., Hahn, H. W., Luebben, A. V., Meilleur, F., Sheldrick, G. M. Refinement of macromolecular structures against neutron data with SHELXL2013. Journal of Applied Crystallography. 47 (1), 462-466 (2014).
- Emsley, P., Lohkamp, B., Scott, W. G., Cowtan, K. Features and development of Coot. Acta Crystallographica Section D Biological Crystallography. 66 (4), 486-501 (2010).
- Lakey, J. H. Neutrons for biologists: A beginner's guide, or why you should consider using neutrons. Journal of the Royal Society Interface. 6, (2009).
- Schröder, G. C., O'Dell, W. B., Myles, D. A. A., Kovalevsky, A., Meilleur, F. IMAGINE: Neutrons reveal enzyme chemistry. Acta Crystallographica Section D: Structural Biology. 74, 778-786 (2018).
- Halsted, T. P., et al. Catalytically important damage-free structures of a copper nitrite reductase obtained by femtosecond X-ray laser and room-temperature neutron crystallography. IUCrJ. 6, 761-772 (2019).
- Meier, K. K., et al. Oxygen Activation by Cu LPMOs in Recalcitrant Carbohydrate Polysaccharide Conversion to Monomer Sugars. Chemical Reviews. 118 (5), 2593-2635 (2018).
- Beeson, W. T., Vu, V. V., Span, E. A., Phillips, C. M., Marletta, M. A. Cellulose Degradation by Polysaccharide Monooxygenases. Annual Review of Biochemistry. 84 (1), 923-946 (2015).
- Walton, P. H., Davies, G. J. On the catalytic mechanisms of lytic polysaccharide monooxygenases. Current Opinion in Chemical Biology. 31, 195-207 (2016).
- Bertini, L., et al. Catalytic Mechanism of Fungal Lytic Polysaccharide Monooxygenases Investigated by First-Principles Calculations. Inorganic Chemistry. 57 (1), 86-97 (2018).
- Hedegård, E. D., Ryde, U. Molecular mechanism of lytic polysaccharide monooxygenases. Chemical Science. 9 (15), 3866-3880 (2018).
- Hangasky, J. A., Detomasi, T. C., Marletta, M. A. Glycosidic Bond Hydroxylation by Polysaccharide Monooxygenases. Trends in Chemistry. 1 (2), 198-209 (2019).
- Bacik, J. P., et al. Neutron and Atomic Resolution X-ray Structures of a Lytic Polysaccharide Monooxygenase Reveal Copper-Mediated Dioxygen Binding and Evidence for N-Terminal Deprotonation. Biochemistry. 56 (20), 2529-2532 (2017).
- O'Dell, W. B., Agarwal, P. K., Meilleur, F. Oxygen Activation at the Active Site of a Fungal Lytic Polysaccharide Monooxygenase. Angewandte Chemie - International Edition. 56 (3), 767-770 (2017).
- O'Dell, W. B., Swartz, P. D., Weiss, K. L., Meilleur, F. Crystallization of a fungal lytic polysaccharide monooxygenase expressed from glycoengineered Pichia pastoris for X-ray and neutron diffraction. Acta Crystallographica Section:F Structural Biology Communications. 73 (2), 70-78 (2017).
- Meilleur, F., et al. The IMAGINE instrument: First neutron protein structure and new capabilities for neutron macromolecular crystallography. Acta Crystallographica Section D: Biological Crystallography. 69 (10), 2157-2160 (2013).
- Helliwell, J. R., et al. The recording and analysis of synchrotron X-radiation Laue diffraction photographs. Journal of Applied Crystallography. 22 (5), 483-497 (1989).
- Nieh, Y. P., et al. Accurate and highly complete synchrotron protein crystal Laue diffraction data using the ESRF CCD and the Daresbury Laue software. Journal of Synchrotron Radiation. 6 (5), 995-1006 (1999).
- Arzt, S., Campbell, J. W., Harding, M. M., Hao, Q., Helliwell, J. R. LSCALE - The new normalization, scaling and absorption correction program in the Daresbury Laue software suite. Journal of Applied Crystallography. 32 (3), 554-562 (1999).
- Sullivan, B., et al. Improving the accuracy and resolution of neutron crystallographic data by three-dimensional profile fitting of Bragg peaks in reciprocal space. Acta Crystallographica Section D: Structural Biology. 74 (11), 1085-1095 (2018).
- Sullivan, B., et al. BraggNet: integrating Bragg peaks using neural networks. Journal of Applied Crystallography. 52 (4), 854-863 (2019).
- Schröder, G. C., O'Dell, W. B., Myles, D. A. A., Kovalevsky, A., Meilleur, F. IMAGINE: Neutrons reveal enzyme chemistry. Acta Crystallographica Section D: Structural Biology. 74, (2018).
- Blum, M. M., et al. Preliminary time-of-flight neutron diffraction study on diisopropyl fluorophosphatase (DFPase) from Loligo vulgaris. Acta Crystallographica Section F: Structural Biology and Crystallization Communications. 63 (1), 42-45 (2007).
- Tomanicek, S. J., et al. Neutron and X-ray crystal structures of a perdeuterated enzyme inhibitor complex reveal the catalytic proton network of the Toho-1 β-lactamase for the acylation reaction. Journal of Biological Chemistry. 288 (7), 4715-4722 (2013).
- Metcalfe, C., et al. The tuberculosis prodrug isoniazid bound to activating peroxidases. Journal of Biological Chemistry. 283 (10), 6193-6200 (2008).
- Hughes, R. C., et al. Inorganic pyrophosphatase crystals from Thermococcus thioreducens for X-ray and neutron diffraction. Acta Crystallographica Section F: Structural Biology and Crystallization Communications. 68 (12), 1482-1487 (2012).
- Bennett, B. C., Meilleur, F., Myles, D. A. A., Howell, E. E., Dealwis, C. G. Preliminary neutron diffraction studies of Escherichia coli dihydrofolate reductase bound to the anticancer drug methotrexate. Acta Crystallographica Section D: Biological Crystallography. 61 (5), 574-579 (2005).
- Snell, E. H., et al. Optimizing crystal volume for neutron diffraction: D-xylose isomerase. European Biophysics Journal. 35 (7), 621-632 (2006).
- Golden, E., Attwood, P. V., Duff, A. P., Meilleur, F., Vrielink, A. Production and characterization of recombinant perdeuterated cholesterol oxidase. Analytical Biochemistry. 485, 102-108 (2015).
- Munshi, P., et al. Rapid visualization of hydrogen positions in protein neutron crystallographic structures. Acta Crystallographica Section D: Biological Crystallography. 68 (1), 35-41 (2012).
- Logan, D. T. Interactive model building in neutron macromolecular crystallography. Methods in Enzymology. 634, (2020).
- Koruza, K., Lafumat, B., Végvári, W., Knecht, S. Z., Fisher, Deuteration of human carbonic anhydrase for neutron crystallography: Cell culture media, protein thermostability, and crystallization behavior. Archives of Biochemistry and Biophysics. 645, 26-33 (2018).
- Fisher, S. J., et al. Perdeuteration: Improved visualization of solvent structure in neutron macromolecular crystallography. Acta Crystallographica Section D: Biological Crystallography. 70 (12), 3266-3272 (2014).
- Chen, J. C. H., Hanson, B. L., Fisher, S. Z., Langan, P., Kovalevsky, a. Y. Direct observation of hydrogen atom dynamics and interactions by ultrahigh resolution neutron protein crystallography. Proceedings of the National Academy of Sciences. 109 (38), 15301-15306 (2012).
- Cuypers, M. G., et al. Near-atomic resolution neutron crystallography on perdeuterated Pyrococcus furiosus rubredoxin: Implication of hydronium ions and protonation state equilibria in redox changes. Angewandte Chemie - International Edition. 52 (3), 1022-1025 (2013).
- Coates, L., Sullivan, B. The macromolecular neutron diffractometer at the spallation neutron source. Methods in Enzymology. 634, (2020).
- Ren, Z., Kingman, N. G., Borgstahl, G. E. O., Getzoff, E. D., Moffat, K. Quantitative Analysis of Time-Resolved Laue Diffraction Patterns. Journal of Applied Crystallography. 29 (3), 246-260 (1996).
- Campbell, J. W., Hao, Q., Harding, M. M., Nguti, N. D., Wilkinson, C. LAUEGEN version 6.0 and INTLDM. Journal of Applied Crystallography. 31 (3), 496-502 (1998).
- Arnold, O., et al. Mantid - Data analysis and visualization package for neutron scattering and μ SR experiments. Nuclear Instruments and Methods in Physics Research, Section A: Accelerators, Spectrometers, Detectors and Associated Equipment. 764, 156-166 (2014).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados