
Method Article
Neutronenkristallographie Datenerhebung und -verarbeitung zur Modellierung von Wasserstoffatomen in Proteinstrukturen
In diesem Artikel
Zusammenfassung
Die Neutronenproteinkristallographie ist eine Strukturtechnik, die die Lokalisierung von Wasserstoffatomen ermöglicht und damit wichtige mechanistische Details der Proteinfunktion liefert. Wir stellen hier den Workflow für die Montage eines Proteinkristalls, die Sammlung von Neutronenbeugungsdaten, die Strukturverfeinerung und die Analyse der Neutronenstreulängendichtekarten vor.
Zusammenfassung
Die Neutronenkristallographie ist eine Strukturtechnik, die es ermöglicht, die Positionen von Wasserstoffatomen in biologischen Makromolekülen zu bestimmen und mechanistisch wichtige Informationen über Protonierungs- und Hydratationszustände zu liefern, ohne Strahlenschäden zu verursachen. Die Röntgenbeugung hingegen liefert nur begrenzte Informationen über die Position von Lichtatomen und der Röntgenstrahl induziert schnell Strahlungsschäden an lichtempfindlichen Cofaktoren und Metallzentren. Hier wird der Workflow vorgestellt, der für die IMAGINE- und MaNDi-Beamlines am Oak Ridge National Laboratory (ORNL) verwendet wird, um eine Neutronenbeugungsstruktur zu erhalten, sobald ein Proteinkristall geeigneter Größe (> 0,1 mm3) gezüchtet wurde. Wir demonstrieren die Montage von hydrierten Proteinkristallen in Quarzkapillaren zur Erfassung von Neutronenbeugungsdaten. Ebenfalls vorgestellt wird der Dampfaustauschprozess der montierten Kristalle mit D2O-haltigem Puffer, um den Ersatz von Wasserstoffatomen an austauschbaren Stellen durch Deuterium sicherzustellen. Der Einbau von Deuterium reduziert den Hintergrund, der durch die inkohärente Streuung von Wasserstoffatomen entsteht, und verhindert eine Dichteunterdrückung, die durch ihre negative kohärente Streulänge verursacht wird. Strategien zur Probenausrichtung und Zur Erfassung von Raumtemperaturdaten werden anhand der Quasi-Laue-Datenerfassung bei IMAGINE am High Flux Isotope Reactor (HFIR) veranschaulicht. Darüber hinaus wird die Kristallmontage und das schnelle Einfrieren in flüssigem Stickstoff zur Kryodatenerfassung zur Erfassung labiler Reaktionszwischenprodukte am MaNDi-Flugzeitinstrument an der Spallationsneutronenquelle (SNS) demonstriert. Die Aufbereitung der Modellkoordinaten- und Beugungsdatendateien und die Visualisierung der SLD-Karten (Neutron Scattering Length Density) werden ebenfalls behandelt. Die Strukturverfeinerung gegen reine Neutronendaten oder gegen gemeinsame Röntgen-/Neutronendaten, um eine rein atomare Struktur des interessierenden Proteins zu erhalten, wird abschließend diskutiert. Der Prozess der Bestimmung einer Neutronenstruktur wird anhand von Kristallen der lytischen Polysaccharid-Monooxygenase Neurospora crassa LPMO9D demonstriert, einem kupferhaltigen Metalloprotein, das am Abbau widerspenstiger Polysaccharide durch oxidative Spaltung der glykosidischen Bindung beteiligt ist.
Einleitung
Die makromolekulare Neutronenkristallographie ist eine Technik, die ein einzigartiges Fenster in die Struktur und zugrunde liegende Chemie von Proteinen bietet. Konzeptionell ähnlich wie die Röntgenbeugung liefert die Neutronenbeugung atomistische Details der makromolekularen Struktur, jedoch ermöglicht die Wechselwirkung von Neutronen mit Kernen die Lokalisierung leichter Atome, die mit Röntgenbeugung oft schwer nachzuweisen sind1. Während der Röntgenbeugung streuen Röntgenstrahlen aus der Elektronenwolke, wodurch leichte Atome wie Wasserstoff (H) in Elektronendichtekarten, die keine auflösung nahe unter Ångström haben, schlecht sichtbar sind2. Im Gegensatz dazu hängt die Streuintensität von Neutronen von komplexen Wechselwirkungen mit dem Kern ab, wobei Isotope desselben Elements unterschiedliche Streulängen aufweisen. Daher haben leichte Atome und ihre Isotope wie Wasserstoff (1H) und Deuterium (2H oder D) eine vergleichbare Sichtbarkeit wie die Kohlenstoff-, Stickstoff- und Sauerstoffatome des Rückgrats in SLD-Karten (Neutron Scattering Length Density). Da die Größe der Neutronenstreuung unabhängig von der Anzahl der Elektronen ist, wird die Streuung von leichten Elementen nicht durch schwere Elemente verdeckt, wenn sie sich in unmittelbarer Nähe zueinander befinden, wie dies bei der Röntgenstreuung beobachtet wird. Die verbesserte Sichtbarkeit von H und seinem Isotop D bei der Neutronenbeugung liefert wertvolle Informationen über den Protonierungszustand von katalytisch wichtigen Resten, Cofaktoren und Liganden und unterstützt die Orientierung von Wassermolekülen und enthüllt wichtige Informationen über katalytische Mechanismen und Proteinchemie3. Die Neutronenbeugung bietet auch den Vorteil, dass es sich um eine zerstörungsfreie Technik handelt, die sich besonders für ionisationsempfindliche biologische Proben eignet, wie Proteine mit Metallzentren oder photosensitive Redoxcofaktoren2. Das Hauptaugenmerk dieses Artikels liegt darauf, einen Überblick über den Workflow zu geben, um eine hochwertige Neutronenproteinkristallstruktur zu erhalten. Wir verweisen den interessierten Leser auf Podjarny et al.4, Blakeley5, Blakeley et al.6 und O'Dell et al.3 für einen hervorragenden Überblick über die Neutronenproteinbeugung und Ashkar et al.7 für weitere Anwendungen der Neutronenstreuung.
Neutronen entstehen hauptsächlich bei Kernreaktionen, bei denen einer von zwei Prozessen zum Einsatz kommt: Kernspaltung an Reaktorquellen oder Spallation an beschleunigerbasierten Quellen8. Reaktorquellen liefern einen kontinuierlichen Neutronenstrahl durch Kernspaltung des 235U-Isotops, während Spallationsneutronenquellen einen gepulsten Neutronenstrahl erzeugen, indem sie ein Ziel, beispielsweise ein flüssiges Metall wie Quecksilber, mit Protonen bombardieren9. Das Oak Ridge National Laboratory (ORNL) in Oak Ridge, Tennessee, beherbergt sowohl eine stationäre Neutronenquelle am High Flux Isotope Reactor (HFIR) als auch eine gepulste 60 Hz-Quelle an der Spallation Neutron Source (SNS). Die IMAGINE-Beamline am HFIR ist ein für biologische Makromoleküle optimiertes Neutronendiffraktometer (Ergänzende Abbildung 1)10. IMAGINE verwendet einen Neutronenbildplattendetektor, um Quasi-Laue-Daten mit einem schmalen Bandpass im Bereich von 2,8 – 4,5 Å von Einkristallen mit Einheitszellenrändern <150 Å zu messen. Das Makromolekulare Neutronendiffraktometer (MaNDi) am SNS ist ein Laue-Neutronendiffraktometer (Time-of-Flight, TOF), das mit einem sphärischen Detektorarray-Rahmen (DAF) ausgestattet ist (ergänzende Abbildung 2)11. MaNDi misst Daten von Einkristallen mit Einheitszellenkanten im Bereich von 10 – 300 Å unter Verwendung einer abstimmbaren Bandbreite von 2 Å-Wellenlängen zwischen 2,0 – 6,0 Å12.
Der Prozess der Erzeugung von Neutronen ist sehr energieintensiv, was im Vergleich zu Röntgenstrahlflüssen an Synchrotronquellen zu relativ schwachen Neutronenstrahlflüssen führt13. Um ausreichende Signal-Rausch-Verhältnisse während der Datenerfassung zu gewährleisten, ist es notwendig, Kristalle geeigneter Größe und Qualität zu züchten14. Typischerweise werden Kristalle mit Volumina > 0,1 mm3 benötigt, um Daten mit angemessenen Statistiken zu sammeln15. Neben geringeren Flussströmen müssen inhärente Eigenschaften der Wechselwirkung zwischen Neutronen und den Probenkernen berücksichtigt werden16. Die Streulänge von Neutronen unterscheidet sich bei Isotopen desselben Elements, eine Eigenschaft, die bei der Kleinwinkel-Neutronenstreuung (SANS) vorteilhaft ausgenutzt werden kann, um Bereiche einer Probe zu maskieren oder hervorzuheben – ein Prozess, der als Kontrastabgleich17 bekannt ist. In Beugungsexperimenten kann die negativ kohärente Neutronenstreulänge von H (-3,741 fm für 1H) zur Aufhebung der Merkmale der Neutronenstreudichte führen, da die kohärenten Neutronenstreulängen anderer biologisch relevanter Atome, einschließlich Kohlenstoff (6,6511 fm für 12C), Stickstoff (9,37 fm für 14N), Sauerstoff (5,803 fm für 16O), Phosphor (5,13 fm für 31P) und Schwefel (2,804 fm für 32S) sind positiv (Tabelle 1)12,14. Darüber hinaus erhöht die große inkohärente Streulänge von H (25.274 fm) den Hintergrund während der Datenerfassung, was die Qualität des Datensatzes beeinträchtigt und die Datenauflösung beeinträchtigt7. Um diese durch H eingeführten Einschränkungen zu umgehen, ist es für die Neutronenbeugung notwendig, H gegen sein Isotop Deuterium 2H(D) auszutauschen, das eine positive kohärente Neutronenstreulänge (6.671 fm) und eine signifikant geringere inkohärente Streulänge (4.04 fm)19 aufweist. Dies kann durch Perdeuteration erreicht werden, ein Prozess, bei dem das Protein von Organismen exprimiert wird, die in vollständig deuterierten Medien gezüchtet werden, um die vollständige Aufnahme von D an H-Stellen zu gewährleisten20. Es ist auch möglich, Protein teilweise zu deuterieren, indem H durch D nur an den austauschbaren Stellen (titrierbare Gruppen) ersetzt wird, während die nicht austauschbaren kohlenstoffgebundenen Stellen hydriert bleiben21. Dies kann durch das Wachstum von hydrierten Proteinkristallen in deuterierter Mutterlauge erreicht werden22. Am häufigsten wird der H/D-Austausch von hydrierten Proteinen jedoch durch Dampfaustausch nach dem Wachstum von entsprechend großen Kristallen in H2O-basiertem Buffer23 durchgeführt. In solchen Fällen werden Kristalle in einer Quarzkapillare montiert und mit einer D20-basierten Mutterlauge dampfgleich.
Die begrenzten Neutronenflüsse an Neutronenquellen führen zu längeren Datenerfassungszeiten von Tagen bis zu mehreren Wochen24. Bei ORNL verwenden sowohl IMAGINE als auch MaNDi einen schmalen Wellenlängenbandpass im Bereich von 2–6 Å, um die Datenerfassung zu optimieren25. Die Daten können bei Raumtemperatur oder bei Kryotemperatur gesammelt werden. Die Kryo-Datenerfassung kann potenziell die Datenqualität verbessern und eröffnet die Möglichkeit für katalytische Zwischenprodukte, die einfrieren. Nach der Erfassung von Neutronenbeugungsdaten wird ein Röntgendatensatz typischerweise auf demselben Kristall bei gleicher Temperatur oder auf einem Unter identischen Bedingungen gezüchteten Kristall gesammelt26. Die Datenerfassung bei gleicher Temperatur ermöglicht eine Strukturverfeinerung sowohl gegen Röntgen- als auch gegen Neutronendaten, wodurch mögliche temperaturinduzierte Artefakte wie Änderungen der Sichtbarkeit und Position von Gewässern oder die Besetzung von Rückständen mit abwechselnden Konformationen27 verhindert werden. Die gemeinsame Verfeinerung der Röntgenneutronendaten erhöht das Daten-zu-Parameter-Verhältnis und bietet den Vorteil, dass die Protein-Backbone-Koordinaten gegen die Röntgendaten verfeinert werden können, während die Neutronenbeugungsdaten zur Verfeinerung der Position der H/D-Atome verwendet werden28. Dies ist besonders nützlich bei der Verwendung von teilweise deuterierten Proben, bei denen eine Dichtestornierung aufgrund von H-Atomen an nicht austauschbaren Stellen auf dem Protein vorliegt. Obwohl die Anzahl der Röntgenstrukturen die Anzahl der in der Protein Data Bank (PDB) hinterlegten Neutronenstrukturen bei weitem übersteigt, wurden Softwarepakete, die ursprünglich für die Verfeinerung von Röntgendaten entwickelt wurden, um auch Neutronendaten zu umfassen3,29,30. Nach der Datenerfassung können Modelle mit Refinement-Paketen wie phenix.refine, CNSsolve (nCNS) oder SHELXL28,31,32,33 verfeinert werden. Während des Verfeinerungsprozesses können Neutronenstreudichtekarten für die manuelle Anpassung mit COOT34 visualisiert werden. Nach der Strukturlösung können die Koordinaten und die Neutronen- und/oder Röntgenbeugungsdatendateien an die PDB übermittelt werden, die das Modell validiert und hinterlegt und öffentlich zugänglich macht18,29,30.
Die Strukturanalyse von Proteinen ist ein facettenreicher Ansatz, bei dem zahlreiche Techniken eingesetzt werden, um ihre Funktion und ihren Mechanismus zu untersuchen35. Die Neutronenproteinkristallographie liefert wertvolle chemische Erkenntnisse, um Erkenntnisse aus zusätzlichen Studien wie Röntgenbeugung, Spektroskopie, Kernspinresonanz (NMR) oder Mikrokristallelektronenbeugung (microED)36 zu erweitern und zu ergänzen. Die Neutronenproteinbeugung ist einzigartig positioniert, um Einblicke in enzymatische Mechanismen zu geben, da H-Atome für ihre Chemie von zentraler Bedeutung sind. Das Fehlen von Durch Neutronen verursachten Strahlenschäden macht sie zu einer Sonde, die sich hervorragend für die Untersuchung von Metalloproteinen eignet37. Wir präsentieren hier ein repräsentatives Beispiel für den Prozess der Neutronenproteinbeugung von der Probenvorbereitung bis zur Datenerhebung, Verfeinerung und Analyse (Abbildung 1). Aus dem Metalloprotein Neurospora crassa LPMO9D (NcLPMO9D) wurden Kristalle von ausreichender Größe für Neutronenbeugungsexperimente gezüchtet. NC LPMO9D ist ein kupferhaltiges Metalloprotein, das am Abbau widerspenstiger Cellulose durch Sauerstoffatomeinfügung an der glykosidischen Bindung beteiligt ist38,39. Die aktive Stelle NcLPMO9D enthält ein mononukleäres Kupferzentrum innerhalb einer charakteristischen "Histidin-Klammer", die aus dem N-terminalen Histidin und einem zweiten konservierten Histidin besteht (ergänzende Abbildung 3)40. Das N-terminale Pilz-LPMOs ist methyliert, aber die Post-Transitional-Modifikation tritt während der rekombinanten Expression in Hefe nicht auf. Im NcLPMO9D-Ruhezustand liegt das Kupferzentrum in einer Cu2+-Oxidationsstufe vor und wird durch Einzelelektronenreduktion zu Cu1+ aktiviert, so dass molekularer Sauerstoff binden und aktiviert werden kann, indem er schnell zu einer Superoxidspezies reduziert wird41,42. Die gesamte NcLPMO9D-Reaktion erfordert eine weitere Zugabe von einem Elektron und zwei Protonen, um das hydroxylierte Polysaccharidprodukt zu bilden43. Die Identität der aktivierten Sauerstoffspezies, die für die Wasserstoffatomabstraktion (HAA) aus dem Polysaccharidsubstrat verantwortlich ist, wurde nicht identifiziert, und es laufen derzeit intensive Struktur- und Computerstudien44,45. Angesichts der Redoxchemie am aktiven Standort NcLPMO9D ist die Minderung von Strahlenschäden besonders wichtig. Wir veranschaulichen hier die Erfassung von Raumtemperatur- und Kryotemperaturdaten an NcLPMO9D-Kristallen zur Bestimmung der NcLPMO9D-Struktur im Ruhezustand bzw. in aktivierter reduzierter Form46. Der Schwerpunkt liegt auf der Proteinkristallmontage, dem Aufbau des Beamline-Instruments für die Datenerfassung, der Aufbereitung der Daten- und Koordinatendateien und den Verfeinerungsschritten, die zur Modellierung einer rein atomaren Neutronenstruktur erforderlich sind.
Protokoll
1. Bewertung der Kristallgröße
- Messen Sie die Größe der Kristalle mit einem Mikroskop, das mit normalem und polarisiertem Licht ausgestattet ist. Wählen Sie Kristalle mit einem Mindestvolumen von ~0,1 mm3 (Ergänzende Abbildung 4).
- Markieren Sie Vertiefungen mit ausreichend großen Kristallen und notieren Sie die Kristallisationsbedingungen, die zur Erzeugung dieser Kristalle verwendet werden.
2. Herstellung von deuteriertem Kristallisationspuffer
- Kristallisationspufferkomponenten in D2O auflösen, um einen deuterierten Kristallisationspuffer zu erzeugen.
- Passen Sie den pH-Wert des Puffers an, indem Sie den pD-Wert der Lösung mit der folgenden Gleichung berechnen:
 (1)
(1)
wobei pHmeas der pH-Wert ist, der mit einer Standardglaselektrode gemessen wird. Der ursprüngliche pH-Wert des NcLPMO9D-Kristallisationspuffers betrug 6,0, daher werden wir einen pH-Wert von 5,6 für den deuterierten Kristallisationspuffer bei pD 6,0 verwenden. - Tauchen Sie die pH-Meter-Elektrode vor gebrauchsweise zehn Minuten lang in D2O ein (ergänzende Abbildung 5).
- Stellen Sie den pHmeas auf 5,6 ein, indem Sie die Base NaOD oder die Säure DCl verwenden.
3. Kristallgewinnung
- Platzieren Sie silikonisierte 22 mm runde Glasobjektträger neben der Kristallisationsschale, aus der Kristalle geerntet werden. Verwenden Sie pro Kristall einen Objektträger aus sauberem Glas (Abbildung 2A).
- Öffnen Sie die versiegelte Sandwichbox mit den Proteinkristallen in der 9-Well-Großvolumen-Silikonglasplatte.
- Entfernen Sie 10-20 μL aus der Kristallisationsbehälterlösung mit einer Mikropipette und legen Sie die Lösung auf den Glasobjektträger (Abbildung 2B).
- Ernten Sie den Kristall mit einem Mikroloop geeigneter Größe und legen Sie den Kristall in den Reservoirlösungstropfen auf den Glasobjektträger, um Ablagerungen zu entfernen, die häufig zusammen mit dem Kristall geerntet werden (Abbildung 2C und Abbildung 2D).
HINWEIS: Es ist notwendig, schnell zu arbeiten, da die Tröpfchen mit kleinem Volumen verdunsten können. Geerntete Kristalle laufen auch Gefahr, auszutrocknen, wenn sie der Atmosphäre ausgesetzt werden. Es kann notwendig sein, dem Proteintropfen etwas Reservoirlösung hinzuzufügen, um zu verhindern, dass Kristalle in kleinen Kristallisationstropfenvolumina austrocknen.
4. Kristallmontage
HINWEIS: Die Kapillarmontageprotokolle variieren je nach den Präferenzen der Experimentatoren. Um Schäden an Kristallen zu vermeiden, sollten Kapillaren, die gekürzt werden müssen, mit einem Schneidstein oder Schleifpapier versehen werden, um einen reibungslosen Bruch zu gewährleisten.
- Füllen Sie ein Ende einer Quarzkapillare mit einem Durchmesser von 2 mm und einer Länge von 50 mm mit Reservoirpuffer durch Kapillarwirkung oder durch direktes Pipettieren von ~ 10 μL Reservoirpuffer in die Kapillare (Abbildung 3A).
HINWEIS: Den Benutzern wird empfohlen, Quarzkapillarrohre zu verwenden, da es neben ihrer mechanischen Festigkeit unerlässlich ist, die Neutronenstrahlabsorption zu begrenzen und die Hintergrundbeiträge der Kapillare zu verringern. Glaskapillaren führen zu einem hohen Hintergrund und absorbieren Neutronen, wodurch die Datenqualität beeinträchtigt wird. - Legen Sie den Kristall vorsichtig in den Reservoirpuffer in der Quarzkapillare mit der Montageschleife (Abbildung 3B und Abbildung 3C).
- Klopfen Sie vorsichtig auf das Rohr, um den Reservoirpuffer und den darin eingetauchten Kristall durch die Kapillare zu bewegen (Abbildung 3D).
- Platzieren Sie den Kristall nicht näher als 13,5 mm und nicht weiter 27,5 mm von einem Ende der Kapillare; Dies ist das Montageende (ergänzende Abbildung 6).
- Saugen Sie die Pufferlösung mit einer langen, dünnen Pipettenspitze um den Kristall herum ab und lassen Sie den Kristall leicht nass. Berühren Sie den Kristall nicht (ergänzende Abbildung 7A).
- Trocknen Sie die Kapillarwände mit einem dünnen Papierdocht (Ergänzende Abbildung 7B).
- 20-50 μL deuterierte Pufferlösung werden in das Ende der Kapillare gegenüber dem Montageende pipettiert (Abbildung 4A).
- Bienenwachs mit einem Hitzestab schmelzen und die Kapillare vorsichtig in dieses geschmolzene Bienenwachs einführen. Wiederholen Sie diesen Vorgang, bis sich eine luftdichte Dichtung bildet (Abbildung 4B und Abbildung 4C).
- Pipettieren Sie eine sehr kleine Menge deuterierten Puffers, etwa 5 μL im Montageende der Kapillare, um als "Kühlkörper" für das heiße Bienenwachs zu fungieren. Tauchen Sie dieses Ende in geschmolzenes Bienenwachs, um eine luftdichte Versiegelung wie zuvor beschrieben zu erzeugen, um eine Kapillare zu bilden, die an beiden Enden versiegelt ist (Abbildung 4D).
- Untersuchen Sie den montierten Kristall nach der Montage mit einem Mikroskop, um die Luftdichtheit sicherzustellen (Abbildung 4E).
- Sichern Sie die montierten Kristalle vorsichtig mit Kim-Tüchern in einem Behälter wie einem 15-ml-Falcon-Röhrchen oder einer Petrischale und lagern Sie sie horizontal bei der Temperatur, bei der die Kristalle gezüchtet wurden (Abbildung 4F).
5. Dampfaustausch
- Ersetzen Sie den deuterierten Puffer zwei Tage nach der Kristallmontage durch einen frischen Puffer.
- Schmelzen Sie die Wachsdichtung am weitesten vom Kristall entfernt mit einer Heizschleife und verwenden Sie eine Pipette und Papierdochte, um den Puffer zu entfernen.
- Füllen Sie die Kapillare mit 20-50 μL deuterierter Pufferlösung auf und verschließen Sie sie mit Wachs.
- Wiederholen Sie den deuterierten Pufferaustausch noch zweimal in Abständen von vier Tagen, um sicherzustellen, dass der deuterierte Pufferdampfaustausch abgeschlossen ist, und den Dampfaustausch für mindestens zwei Wochen zu ermöglichen.
6. Neutronenproteinbeugung
HINWEIS: Leser, die an den Besonderheiten der IMAGINE-Strahllinie interessiert sind, werden gebeten, Meilleur et al. 2013, Meilleur et al. 201810,47 zu konsultieren.
- Raumtemperatur-Datenerfassung an der IMAGINE-Beamline am HFIR
- Probenmontage
- Befestigen Sie den an der Kapillare befestigten Kristall mit Kitt auf dem Goniometer.
- Montieren Sie das Goniometer auf dem Probenstab und zentrieren Sie den Kristall im Strahl mit der Offline-Ausrichtungsstation.
- Befestigen Sie den Probenstab auf dem Instrumenten-Abtasttisch (ergänzende Abbildung 1B).
- Stellen Sie sicher, dass der Versuchsstall geräumt ist, und öffnen Sie den Beamline-Verschluss für die Neutronendatenerfassung.
- Datensammlung
- Öffnen Sie das Datenerfassungsprogramm auf dem Beamline-Steuerungscomputer und klicken Sie auf die Registerkarte Setup , um die Datenerfassungsstrategie einzurichten. Geben Sie unter Experimentparameter den Beispielnamen neben Beispielname ein , und geben Sie die Vorschlagsnummer neben Vorschlag ein. Wählen Sie unter Bildbenennung die Option Ordnervorlage aus, legen Sie das Ziel für die zu speichernden erfassten Daten fest, und wählen Sie Bildpräfix und geben Sie den relevanten Rahmennamen ein (Ergänzende Abbildung 8).
- Öffnen Sie die Optics GUI und klicken Sie auf 2.78 für λmin und 4.78 für λmax , um den Quasi-Laue-Bereich für die Datenerfassung einzustellen (Ergänzende Abbildung 9).
- Wechseln Sie zur Registerkarte Sammeln und geben Sie unter Nächste Scanparameter die Belichtungszeit in Sekunden unter Belichtung, die Anzahl der Frames unter N Frames und die Winkel für die Datenerfassung unter Δ φ/Frame ein. Benennen Sie den zu erfassenden Frame unter Bildpräfix und starten Sie die Datenerfassung, indem Sie auf die Schaltfläche Scan starten klicken (ergänzende Abbildung 10).
- Die gebeugten Neutronen werden vom Image Plate Detector detektiert. Am Ende jeder Belichtung wird die Bildplatte gelesen und das Muster auf der Datenerfassungs-GUI angezeigt (Ergänzende Abbildung 11).
- Frames werden mit Lauegen, Lscale50 und Scala vom verantwortlichen Beamline Scientist indiziert, integriert, wellenlängennormalisiert und skaliert (Ergänzende Abbildung 12).
HINWEIS: Die Datenerhebung sowohl bei IMAGINE als auch bei MaNDi erfolgt im Quasi-Laue-Modus unter Verwendung von Methoden und Software, die für die Laue-Beugungsdatenerhebung entwickelt wurden, wie sie von Helliwell et al.48 und Nieh et al.49 entwickelt wurden. - Sammeln Sie nach der Erhebung von Neutronenbeugungsdaten einen entsprechenden Röntgendatensatz auf demselben Kristall bei gleicher Temperatur (ergänzende Abbildung 13).
HINWEIS: Ein Kristall, der aus demselben Tropfen oder unter den gleichen Kristallisationsbedingungen gezüchtet wurde, kann auch zum Sammeln von Röntgenbeugungsdaten für die gemeinsame Neutronen- / Röntgenverfeinerung verwendet werden.
- Probenmontage
- Kryo-Datenerfassung an der MaNDi-Beamline am SNS
HINWEIS: Leser, die an den Besonderheiten der Strahllinie interessiert sind, werden gebeten, Coates et al. (2015), Meilleur et al. 201810,11 zu konsultieren.- Probenmontage
- Bereiten Sie die deuterierte Ascorbat-Einweichlösung für die Reduktion der Kristalle und des deuterierten Kryoprotektors vor. Geben Sie 20 μL Tropfen jeder dieser Lösungen in sitzende Tropfentöpfe in einer Kristallisationsplatte.
HINWEIS: Die kryoprotektive Lösung ist in der Regel das Kryoprotektivum, das sich bei der Erfassung von Röntgenbeugungsdaten bei Kryotemperatur, die in D2O hergestellt wurden, als wirksam erwiesen hat. Dieses Kryoprotektivum kann bei Bedarf für die Neutronendatenerfassung weiter optimiert werden (z.B. Konzentration). - Wählen Sie Schlaufen für die Probenmontage aus und befestigen Sie sie gemäß den MaNDi-Richtlinien an einer magnetischen Kryobasis (ergänzende Abbildung 14).
- Füllen Sie einen Schaum-Kryo-Dewar mit flüssigem Stickstoff. Legen Sie eine Metall-Kryoschutzhülse in den flüssigen Stickstoff, um abzukühlen (ergänzende Abbildung 15A).
- Entfernen Sie die Wachsstopfen von beiden Enden der Kapillare und klopfen Sie auf die Kapillare, um den Pufferstopfen so zu bewegen, dass der montierte Kristall in Puffer eingetaucht wird. Waschen Sie den Kristall in den 20-μL-Reservoirlösungstropfen in der sitzenden Tropfenmulde (Ergänzende Abbildung 15B und Ergänzende Abbildung 15C).
HINWEIS: Dieser Schritt ist nicht erforderlich, wenn der H/D-Austausch durch Äquilibrierung der Kristallisationstropfen gegen deuterierten Puffer oder durch direktes Einweichen des Kristalls in deuteriertem Puffer durchgeführt wurde. - Tauchen Sie den Kristall für zwei Stunden in die Ascorbat-Einweichlösung. Montieren Sie den Kristall in einem Mikroloop, der an einer magnetischen Kryobasis befestigt ist. Tauchen Sie den montierten Kristall für 10 Sekunden in das Kryoprotektivum und tauchen Sie den Kristall und die Kryohalterung in den flüssigen Stickstoff, um einzufrieren (ergänzende Abbildung 15D).
- Sobald der Kristall eingefroren ist, verwenden Sie eine vorgekühlte Kryo-Pin-Zange und montieren Sie den Kristall auf der MaNDi-Probenstufe, die mit einem Kryostrom ausgestattet ist. Öffnen Sie vorsichtig die Kryostiftzange und stellen Sie sicher, dass der Kristall im Kryostrom verbleibt (ergänzende Abbildung 2C).
- Bereiten Sie die deuterierte Ascorbat-Einweichlösung für die Reduktion der Kristalle und des deuterierten Kryoprotektors vor. Geben Sie 20 μL Tropfen jeder dieser Lösungen in sitzende Tropfentöpfe in einer Kristallisationsplatte.
- Datensammlung
- Öffnen Sie die Datenerfassungssoftware, in der die Experimentinformationen automatisch ausgefüllt werden.
- Klicken Sie auf die Mitte des Kristalls, um ihn mit dem computergesteuerten Goniometer zu zentrieren (Ergänzende Abbildung 16).
- Legen Sie unter Tabelle die Datenerfassungsstrategie fest, indem Sie die Winkel für die Datenerfassung unter "Phi" sowie die gesamte Neutronenstrahlbelichtung pro Bild unter Wert eingeben (ergänzende Abbildung 17).
- Klicken Sie auf Senden , um die Datenerfassung zu starten.
- Wenn die Daten gesammelt werden, wird das gebeugte Neutron sichtbar sein (ergänzende Abbildung 17). Beugungspunkte werden mit zunehmender Belichtungszeit klarer, wodurch das Signal-Rausch-Verhältnis verbessert wird (Abbildung 5).
- Die Frames werden vom verantwortlichen Beamline-Wissenschaftler indiziert, integriert, wellenlängennormalisiert und mit Mantid und Lauenorm skaliert, der dem Benutzer nach der Datenerfassung die zusammengeführte Intensitätsdatei zur Verfügung stellt51.
- Probenmontage
7. Verfeinerung der Struktur
- Gemeinsame Verfeinerung von Röntgen- und Neutronendaten
- Strukturvorbereitung
- Verfeinern Sie die Röntgendaten, um eine Proteinstruktur zu erhalten, indem Sie das Phenix.refine-Softwarepaket und Coot für den manuellen Aufbau verwenden, um eine fertige Struktur zu erhalten.
- Öffnen Sie CCP4 und wählen Sie das Programm Konvertieren in/modifizieren/erweitern , um die R-freien Datenflags der Neutronendaten mit denen der Röntgendaten abzugleichen. Wählen Sie diese Option, um die Reflexionsdatei im MTZ-Format zu importieren. Unter In die erhaltene Neutronen-MTZ-Datei hochladen und die Röntgen-MTZ-Datei unter Import FreeR MTZ hochladen (Ergänzende Abbildung 18). Geben Sie unter Aus einen Namen für die neue MTZ-Datei ein , und klicken Sie auf Ausführen.
- Öffnen Sie das Phenix-Softwarepaket und klicken Sie unter Refinement Tools auf ReadySet . Neben der PDB-Datei laden Sie die PDB-Koordinatendatei hoch, die gegen die Röntgendaten verfeinert wurde. Wählen Sie die Option Wasserstoff zum Modell hinzufügen, falls nicht vorhanden , und wählen Sie H/D an austauschbaren Standorten, H an anderer Stelle aus dem Dropdown-Menü aus. Wählen Sie Deuterium zu Lösungsmittelmolekülen hinzufügen aus, und lassen Sie die verbleibenden Optionen auf ihren Standardwerten (Ergänzende Abbildung 19A).
HINWEIS: Wenn ein perdeuteriertes Protein verwendet wird, wählen Sie die Option Wasserstoffe zum Modell hinzufügen, falls nicht vorhanden , und wählen Sie H/D an austauschbaren Stellen, D an anderer Stelle.
- Verfeinerung der Struktur
- Öffnen Sie das Programm phenix.refine unter der Registerkarte Verfeinerung, um die Verfeinerung mit Röntgen- und Neutronendaten einzurichten. Geben Sie auf der Registerkarte Konfigurieren in Eingabedateien die PDB-Datei aus der mit ReadySet verarbeiteten Röntgenstruktur und die erforderliche CIF-Restrains-Datei für alle relevanten Liganden ein. Laden Sie die MTZ-Datei aus den Neutronendaten mit den Rfree-Flags hoch, die mit CCP4 zugewiesen wurden, und weisen Sie diese unter der Überschrift Datentyp als "Neutronendaten" und "Neutron R-frei" zu. Laden Sie die MTZ-Datei aus den Röntgendaten hoch und ordnen Sie diese unter der Überschrift Datentyp als "Röntgendaten" und "Röntgen-R-frei" zu. Die Beschriftungen Space-Gruppe und Daten werden automatisch ausgefüllt, sobald die Daten hochgeladen wurden (ergänzende Abbildung 19B).
HINWEIS: Wenn Sie die Verfeinerung durchführen und die Kristallinformationen eingeben, verwenden Sie die aus den Röntgendaten ermittelte Elementarzelle. - Behalten Sie unter der Registerkarte Konfigurieren in den Einschränkungseinstellungen die Standardeinschränkungsstrategie bei. Erhöhen Sie die Anzahl der Zyklen auf fünf (Ergänzende Abbildung 20)
- Wählen Sie Alle Parameter, klicken Sie auf Erweitert und wählen Sie Wasserstoff.... Ändern Sie das Wasserstoff-Verfeinerungsmodell in individuell und schalten Sie den Force Riding ADP aus (Ergänzende Abbildung 20).
- Wählen Sie Alle Parameter aus, und öffnen Sie die Option Parameter suchen . Suchen Sie nach dem Wort nuklear , und wählen Sie die Option Kernabstände für X-H/D verwenden aus (Ergänzende Abbildung 20).
- Wählen Sie Ausführen aus, um die Einschränkung zu initiieren.
- Öffnen Sie das Programm phenix.refine unter der Registerkarte Verfeinerung, um die Verfeinerung mit Röntgen- und Neutronendaten einzurichten. Geben Sie auf der Registerkarte Konfigurieren in Eingabedateien die PDB-Datei aus der mit ReadySet verarbeiteten Röntgenstruktur und die erforderliche CIF-Restrains-Datei für alle relevanten Liganden ein. Laden Sie die MTZ-Datei aus den Neutronendaten mit den Rfree-Flags hoch, die mit CCP4 zugewiesen wurden, und weisen Sie diese unter der Überschrift Datentyp als "Neutronendaten" und "Neutron R-frei" zu. Laden Sie die MTZ-Datei aus den Röntgendaten hoch und ordnen Sie diese unter der Überschrift Datentyp als "Röntgendaten" und "Röntgen-R-frei" zu. Die Beschriftungen Space-Gruppe und Daten werden automatisch ausgefüllt, sobald die Daten hochgeladen wurden (ergänzende Abbildung 19B).
- Modellbau
- Klicken Sie nach der Verfeinerung in Phenix auf Der Registerkarte Ergebnisse auf In Blässhuhn öffnen, um die Röntgenelektronendichte- und Neutronen-SLD-Karten zu visualisieren. Klicken Sie auf die Registerkarte Display-Manager und unter Karten auf Karte löschen neben den _neutron Karten, um die Neutronenkarten zu entfernen (Ergänzende Abbildung 21). Klicken Sie auf Datei > MTZ, mmCIF fcf oder phs öffnen.... Wählen Sie die aktuellen Einschränkungsdateien aus, und öffnen Sie die MTZ-Datei. Wählen Sie für die Option Amplituden und Phasen die 2FOFC WT_no_fill_neutron Daten aus dem Dropdown-Menü aus. Wiederholen Sie dies und öffnen Sie die FOFC WT_neutron Daten. Öffnen Sie den Display-Manager und wechseln Sie zu Scrollen für die Neutronen- und Röntgen-2FOFCWT-Karten und scrollen Sie dann, um den effektiven Wert der 2FOFCWT-Karten auf 1,00 zu verringern (ergänzende Abbildung 21). Wechseln Sie zu Scrollen für die Neutronen- und Röntgen-FOFCWT-Karten und scrollen Sie, um den effektiven Wert der FOFCWT-Karten auf 3,00 zu verringern.
- Führen Sie eine visuelle Inspektion der Rückstände durch, um festzustellen, ob das Modell zu den Daten passt. Bestimmen Sie die korrekte Ausrichtung und H/D-Belegung aller austauschbaren Standorte, indem Sie die Spitzen der Differenzdichtekarte analysieren. Dazu gehören die Hydroxylgruppen von Serinen, Threoninen und Tyrosinen; der Stickstoff von Histidin, Glutamin, Asparagin und Lysin; das Sulfhydryl von Cystein; die Carboxylgruppen von Aspartat und Glutamat; Rückgratamidgruppen; Liganden; Cofaktoren und mögliche funktionalisierte Reste (Abbildung 6).
- Richten Sie Wassermoleküle entsprechend der Neutronendichte und den Wasserstoffbrückenbindungswechselwirkungen neu aus, indem Sie sie mit der Funktion Rotate Translate Zone/Chain/Molecule drehen (ergänzende Abbildung 22A und ergänzende Abbildung 22B). Passen Sie die Positionen der austauschbaren H/D-Stellen von Proteinresten mit dem Werkzeug Chi-Winkel bearbeiten und der Funktion "Zone/Kette/Molekül drehen" umwandeln (ergänzende Abbildung 22C und ergänzende Abbildung 22D).
- Strukturvorbereitung
- Strukturverfeinerung – Neutronendatenverfeinerung
- Strukturvorbereitung
- Öffnen Sie Phenix und wählen Sie "Molekularer Ersatz" und wählen Sie "Phaser-MR (voll funktionsfähig)", um die Phase der skalierten Intensitäten abzuleiten, die der Instrumentenwissenschaftler durch molekularen Ersatz zur Verfügung stellt, um eine Startkoordinatendatei im pdb-Format zu generieren. Geben Sie die Start-pdb-Struktur in die Registerkarte "Eingabe- und allgemeine Optionen " ein und vervollständigen Sie die Optionen Ensembles, ASU-Inhalte und Suchprozedur .
- Öffnen Sie die Modellwerkzeuge , und wählen Sie PDB-Werkzeuge aus. Fügen Sie die pdb-Datei als Eingabedatei ein. Navigieren Sie zur Registerkarte Optionen , und wählen Sie unter Atomauswahl entfernen die Namen des Lösungsmittels, der Liganden, der Cofaktoren und der Metalle aus. Dadurch werden alle Wassermoleküle, Cofaktoren, Liganden und Metallionen entfernt, um ein minimales Modell zu erstellen. Wählen Sie zusätzlich die Option Alternative Konformitätsvarianten aus dem Modell entfernen (Ergänzende Abbildung 23).
- Wählen Sie Verfeinerung in Phenix und öffnen Sie ReadySet. Geben Sie die bearbeitete pdb-Koordinatendatei neben der PDB-Datei ein. Wählen Sie die Option Wasserstoffe zum Modell hinzufügen, falls nicht vorhanden , und wählen Sie H/D an austauschbaren Stellen, H an anderer Stelle aus dem Dropdown-Menü der Neutronenverfeinerungsoptionen (Ergänzende Abbildung 23).
- Verfeinerung der Struktur
- Öffnen Sie das Programm phenix.refine unter der Registerkarte Verfeinerung in Phenix. Geben Sie auf der Registerkarte Konfigurieren im Feld Eingabedateien die PDB-Datei ein, die mit ReadySet verarbeitet wurde. Laden Sie im Feld Eingabedateien die MTZ-Datei aus den Neutronendaten hoch und weisen Sie diese unter der Spalte Datentyp als Röntgendaten und Röntgen-R-frei zu, obwohl es sich um Neutronendaten handelt. Die im nächsten Schritt eingerichtete Verfeinerungskonfiguration wird verwendet, um die Reflexionsdatei als Neutronendaten zu behandeln. Die Beschriftungen Space und Daten werden automatisch ausgefüllt, sobald die Daten hochgeladen wurden (Ergänzende Abbildung 24A).
- Behalten Sie unter der Registerkarte Konfigurieren in den Einschränkungseinstellungen die Standardeinschränkungsstrategie bei. Erhöhen Sie die Anzahl der Zyklen auf fünf. Wählen Sie unter Andere Optionen die Option Neutron aus dem Dropdown-Menü Streutabelle aus. Deaktivieren Sie die Option Wasser aktualisieren (ergänzende Abbildung 24B).
- Wählen Sie Alle Parameter>Advanced>Hydrogens. Im neuen Fenster wählen Sie Einzelperson aus dem Dropdown-Menü Wasserstoffverfeinerungsmodell und deaktivieren Sie den Force Riding ADP (Ergänzende Abbildung 20).
- Wählen Sie Alle Parameter > Suchparameter... Option. Suchen Sie nach dem Wort nuklear , und wählen Sie die Option Kernabstände für X-H/D verwenden aus (Ergänzende Abbildung 20).
- Wählen Sie Ausführen aus, um die Einschränkung zu initiieren.
HINWEIS: Nach der anfänglichen Verfeinerung ist es notwendig, die Neutronen-SLD-Karten visuell zu inspizieren und eine manuelle Modellerstellung in Coot durchzuführen. Es kann notwendig sein, im Modell vorhandene Liganden/Cofaktoren einzufügen. Nachfolgende Verfeinerungen erfordern die erforderliche CIF-Beschränkungsdatei für alle relevanten Liganden, und diese sollten auf der Registerkarte Konfigurieren von phenix.refine hochgeladen werden.
- Modellbau
- Klicken Sie nach der Verfeinerung in Phenix auf Der Registerkarte Ergebnisse auf In Blässhuhn öffnen, um die Neutronen-SLD-Karten und -Strukturen zu visualisieren. Klicken Sie auf die Registerkarte Display-Manager und unter Karten auf Karte löschen, um sowohl die 2FOFCWT- als auch die FOFCWT-Karten zu löschen (ergänzende Abbildung 25). Klicken Sie auf Datei > MTZ, mmCIF fcf oder phs öffnen.... Wählen Sie den aktuellen Einschränkungsordner und dann die MTZ-Datei aus. Wählen Sie für die Option Amplituden und Phasen die 2FOFC WT_no_fill Daten aus dem Dropdown-Menü aus. Wiederholen Sie den Vorgang, indem Sie auf Datei > MTZ, mmCIF fcf oder phs... öffnen und die FOFCWT-Daten aus dem Dropdown-Menü für die Option Amplituden und Phasen auswählen. Öffnen Sie den Display-Manager, wechseln Sie zu Scrollen für die 2FOFC WT_no_fill-Karte und scrollen Sie dann, um den rmsd der 2FOFC WT_no_fill-Daten auf 1,00 zu verringern (Ergänzende Abbildung 25). Wechseln Sie zu Scrollen für die FOFCWT-Karte und scrollen Sie, um den rmsd der FOFCWT-Daten auf 3,00 zu verringern.
- Führen Sie eine visuelle Inspektion der Proteinstruktur durch, um festzustellen, ob das Modell zur Neutronen-SLD-Karte passt.
- Wie in 7.1.3.2 beschrieben, ist die korrekte Orientierung und H/D-Belegung von Rückständen und Gruppen mit H/D-Austauschstellen zu bestimmen. Passen Sie die Rückstandspositionen mit dem Umsetzwerkzeug drehen (Rotate Translate ) und Bearbeiten von Chi-Winkeln (Abbildung 6) an. Bei Bedarf kann Real Space Refine Zone verwendet werden. Korrigieren Sie D-Atome, die aus dem Rückstand explodieren, manuell mit dem Texteditor, um die richtigen Atomkoordinaten einzufügen
HINWEIS: Real Space Refine Zone ist nicht für Neutronen-SLD-Karten in Coot optimiert und kann zu unregelmäßigen Bindungslängen für an D gebundene Atome führen, die als explodierende Reste bezeichnet werden (ergänzende Abbildung 26). Es ist vorzuziehen, die erforderlichen atomaren Koordinaten manuell zu bearbeiten und die Verwendung der Real Space Refine Zone zu vermeiden. - Einsetzen und Neuausrichten von Wassermolekülen entsprechend der Neutronendichte. Um Wasser in Blässhuhn hinzuzufügen, wählen Sie das Symbol Atom am Zeiger platzieren und wählen Sie, ob ein Wassermolekül eingefügt werden soll (ergänzende Abbildung 27A). Coot fügt standardmäßig ein O-Atom an dieser Position ein.
- Um D-Atome zu den O-Atomen von Wasser hinzuzufügen, die in Coot eingefügt werden, verwenden Sie Phenix. Öffnen Sie das Menü Verfeinerung und klicken Sie auf ReadySet. Wählen Sie neben Neutronenverfeinerungsoptionen nur die Option Deuterium zu Lösungsmittelmolekülen hinzufügen aus. Deaktivieren Sie Wasserstoff zum Modell hinzufügen, falls nicht vorhanden (Ergänzende Abbildung 27B und Ergänzende Abbildung 27C).
HINWEIS: Die Modellbildung mit reinen Neutronendaten unterscheidet sich von der Modellbildung einer gemeinsamen Röntgen- / Neutronenstruktur, da es keine Röntgendaten gibt, die zur Verfeinerung der Koordinaten des Rückgrats und der schwereren Atome beitragen. In einer gemeinsamen Verfeinerung wird die Elektronendichtekarte zunächst verwendet, um die Proteinrückgrat- und Seitenkettenkoordinaten zu bestimmen. Dieses Modell wird anschließend in einer gemeinsamen Röntgen-/Neutronendatenverfeinerung verwendet, bei der die Orientierung und Belegung von H/D-Atomen aus der Neutronen-SLD-Karte abgeleitet wird. Bei einer reinen Neutronenverfeinerung wird die gesamte Struktur aus der Analyse der Neutronen-SLD-Karten abgeleitet, die neben den H/D-Atomen auch den Aufbau der Wassermoleküle, des Rückgrats, der Seitenketten und der Liganden erfordert (Abbildung 6). Das Daten-zu-Parameter-Verhältnis ist niedrig in Verfeinerungen gegenüber Neutronendaten allein und es sollte darauf geachtet werden, die Daten nicht zu überpassen.
- Strukturvorbereitung
Ergebnisse
Neutronenbeugungsdaten an Kristallen einer lytischen Polysaccharidmonooxygenase aus Neurospora crassa (NcLPMO9D) wurden auf IMAGINE am HFIR bei Raumtemperatur und an MaNDi am SNS unter Kryo-Bedingungen nach dem oben beschriebenen Protokoll gesammelt. Es wurden Kristalle des hydrierten Proteins verwendet, das in H2O-basiertem Puffer mit einem Volumen von mehr als 0,1 mm3 gezüchtet wurde (illustratives Beispiel für große Kristalle sind in ergänzender Abbildung 4 und Abbildungen danach dargestellt). Kristalle wurden in Quarzkapillaren montiert und der Dampfaustausch mit dem D2O-basierten Puffer wurde drei Wochen vor der Datenerfassung durchgeführt (Abbildung 4).
Die Raumtemperaturdatenerfassung wurde auf der IMAGINE-Beamline durchgeführt (Abbildung 1). Ein vierstündiger Weißstrahltest führte zu einer hochauflösenden Beugung, was darauf hindeutet, dass der Kristall von geeigneter Größe und Qualität für die Erfassung eines vollständigen Datensatzes war. Zusätzlich zur Bereitstellung vorläufiger Informationen über die Beugungsqualität des Kristalls kann die anfängliche Breitbandbelichtung verwendet werden, um das Beugungsmuster zu indizieren und die Kristallorientierungsmatrix zu bestimmen. Angesichts der P21-Raumgruppe des Kristalls wurde eine Datenerfassungsstrategie von 18 Frames mit einer Erfassungszeit von 20 Stunden pro Frame implementiert. Wie bei der Datenerfassung von Röntgenbeugungsdaten benötigen Raumgruppen mit höherer Symmetrie weniger Frames (d. h. weniger Winkelabdeckung), um einen vollständigen Datensatz zu sammeln. Die Daten wurden im Quasi-Laue-Modus mit einem Wellenlängenbereich von 2,8 – 4,0 Å erhoben. Nach der Datenerfassung wurden die Daten indiziert, integriert und zu einer Neutronen-SLD-Datei im MTZ-Format mit einer Auflösung von 2,14 Å zusammengeführt. Die Daten wurden nach ähnlichen Richtlinien für die Röntgendatenanalyse als von ausreichender Qualität bewertet, obwohl eine Vollständigkeit von 80 % und ein CC1/2 von mindestens 0,3 als akzeptabel angesehen wurden, da die Neutronenproteinbeugung eine flussbegrenzte Technik ist.
Nach der Erfassung von Neutronenbeugungsdaten bei Raumtemperatur wurde derselbe Kristall verwendet, um einen Röntgenbeugungsdatensatz bei Raumtemperatur mit einer Auflösung von 1,90 Å zu sammeln (ergänzende Abbildung 13). Die Röntgendaten wurden verwendet, um die Positionen der "schwereren" Atome einschließlich C, N, O und S zu bestimmen. Allein die gegen die Röntgendaten verfeinerte Struktur wurde dann als Ausgangsmodell verwendet, um eine gemeinsame Verfeinerung gegen die Röntgen- und Neutronendaten durchzuführen. Phenix ReadySet wurde verwendet, um H-Atome an nicht austauschbaren Stellen, H- und D-Atome an austauschbaren Stellen und D-Atome zu Wassermolekülen des Startröntgenmodells hinzuzufügen. Im Anschluss an diese Modellerstellung wurden iterative Verfeinerungen an beiden Datensätzen durchgeführt (Ergänzende Abbildung 19 und Ergänzende Abbildung 20). Die interaktive Modellbildung wurde in Coot durchgeführt, indem die Dichtekarten visuell inspiziert wurden, um Seitenketten und Wassermoleküle entsprechend auszurichten (ergänzende Abbildung 22). Die Neutronendaten wurden hauptsächlich zur Bestimmung von Protonierungszuständen und Wassermolekülorientierungen verwendet. Der Vergleich der Elektronendichtekarte von Resten wie Serin und Tryptophan und die entsprechende Neutronen-SLD-Karte veranschaulichen die Informationen, die über Protonierungszustände an H/D-Austauschstellen durch Neutronenproteinbeugung gewonnen werden können (Abbildung 7). Eine Kartenüberlagerung von Elektronen- und Neutronen-SLD-Karten für Wassermoleküle zeigt auch, dass Wasserstoffbrückenbindungswechselwirkungen zwar aus Röntgendaten abgeleitet werden können, Neutronen jedoch klare Informationen über die Ausrichtung dieser Wasserstoffbrückenbindungen liefern (Abbildung 8). Neutronen-SLD-FO-FC-Auslasskarten wurden erzeugt, um Protonierungszustände und H/ D-Orientierung von Seitenketten zu bestimmen. Abgebildet sind die Neutronen-SLD-Karten für Tyrosin- und Threoninreste, in denen die Neutronen-Fo-FC-Karten eindeutig positive Peaks anzeigen, die das Vorhandensein von H/D bedeuten (Abbildung 9). Die gesammelten Neutronenbeugungsdaten lieferten auch wertvolle Informationen über mehrere Protonierungszustände, wie die -ND3+-Gruppe von Lys (Abbildung 10). Verfeinerungsstatistiken (Rwork und Rfree) wurden während der Modelloptimierung genau überwacht, um eine Überanpassung zu verhindern. Endgültige Statistiken ergaben einen Röntgen-Rwork von 12,77 % und einen Rfree von 18,21 % sowie einen Neutronen-Rwork von 14,48 % und einen Rfree von 21,41 % bei 389 vorhandenen Wassermolekülen (ergänzende Abbildung 28).
Kryotemperaturdaten wurden auf NcLPMO9D nach einem Ascorbat-Einweichen gesammelt, um das aktive Kupferzentrum von CuII auf CuI auf der MaNDi-Beamline zu reduzieren (ergänzende Abbildung 2 und ergänzende Abbildung 15)45. Die Daten wurden im TOF-Laue-Modus nach einem Neutronenbeugungstest mit einer 4-stündigen Exposition gesammelt, um die Qualität der Beugung zu überprüfen. Angesichts der Raumgruppe des Kristalls wurde eine Datenerfassungsstrategie von 18 Frames mit einer Sammeldosis von 80 Coulomb pro Frame entwickelt. Die Daten wurden im TOF-Laue-Modus in einem Wellenlängenbereich von 2,0 – 4,0 Å gesammelt. Nach der Datenerfassung wurden die Daten indiziert, integriert, skaliert und zusammengeführt, um eine Reflexionsdatei im MTZ-Format mit einer Auflösung von 2,40 Å51,52 zu erhalten.
Nach der Datenerhebung wurde der 2,40 Å Kryotemperatur-Neutronenbeugungsdatensatz NcLPMO9D für die Verfeinerung reiner Neutronendaten verwendet. Die Neutronendaten wurden durch molekularen Ersatz unter Verwendung von PDB 5TKH als Ausgangsmodell phasenweise ausgewertet. Phenix ReadySet wurde verwendet, um H-Atome an nicht austauschbaren Stellen und H/D-Atome mit Teilbelegungen an austauschbaren Stellen hinzuzufügen. Wassermoleküle wurden mit PDB-Werkzeugen aus dem Ausgangsmodell entfernt (Ergänzende Abbildung 23). Auf die Modellerstellung folgte die Verfeinerung mit phenix.refine unter Verwendung der Neutronenstreutabelle (Ergänzende Abbildung 24). Die interaktive Modellbildung wurde in Coot durchgeführt, wobei Wassermoleküle unter Verwendung der positiven Peaks der FO-Fc-Karte hinzugefügt und entsprechend potenzieller Wasserstoffbrückenbindungswechselwirkungen positioniert wurden (Abbildung 11A und Abbildung 11B). Bei der Analyse von Neutronen-SLD-Karten sind Wassermoleküle deutlich sichtbar, wenn sie hochgeordnet sind, ihre Dichte kann jedoch sphärisch oder ellipsoidisch sein, wenn sie nicht gut geordnet sind (Abbildung 11C-E). Neutronen-SLD-Karten wurden verwendet, um wertvolle Informationen über die Orientierung von Resten wie Asparagin zu liefern, in denen die Unterscheidung zwischen den Carbonyl- und Aminogruppen bei alleiniger Verwendung von Röntgenbeugungsdaten eine Herausforderung darstellen kann (Abbildung 12A und Abbildung 12B). Peaks in FO-FC neutron SLD omit maps waren auch sehr informativ bei der Bestimmung der Protonierungszustände von Histidinresten an der N δ- oder N ε-Position (Abbildung 12C und Abbildung 12D). Der Protonierungszustand von Resten mit mehreren H/D-Austauschstellen kann auch mit Hilfe von Neutronen-SLD-Karten bestimmt werden. Dies wurde deutlich mit einer FO-FC-Neutronen-SLD-Wegfallkarte von Arginin veranschaulicht, von dem bekannt ist, dass es eine positive Ladung aufweist (Abbildung 12E und Abbildung 12F). Wie bisher wurde eine Überanpassung durch die Überwachung von Rwork und Rfree verhindert. Endgültige Statistiken ergaben einen Rwork von 22,58% und einen Rfree von 30,84% (ergänzende Abbildung 29). Da es sich bei der Neutronenproteinbeugung um eine flussbegrenzte Technik handelt, bei der die negative Streulänge und der große inkohärente Streufaktor von H berücksichtigt werden müssen, kann davon ausgegangen werden, dass eine Verfeinerung der Neutronendaten schlechtere Statistiken aufweist als eine gemeinsame Röntgen- / Neutronendatenverfeinerung mit weniger sichtbaren Wassermolekülen (ergänzende Abbildung 28 und ergänzende Abbildung 29).
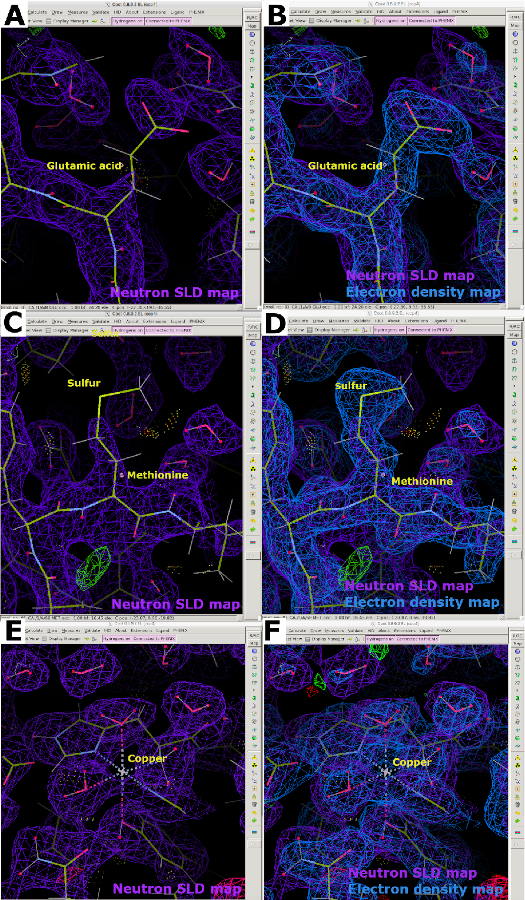
Bei der Analyse von Neutronen-SLD-Karten wird deutlich, dass bei hydrierten Proteinen, die einem Dampfaustausch mit D2O-haltigem Kristallisationspuffer unterzogen wurden, eine Dichteaufhebung aufgrund der negativen Neutronenstreulänge von H auftritt. Aus diesem Grund erscheinen Neutronen-SLD-Karten, in denen nicht austauschbare H-Atome an Kohlenstoff gebunden sind, im Vergleich zu ihrem Elektronendichte-Map-Gegenstück unvollständig (Abbildung 13A). Der Effekt der Aufhebung ist bei schlechteren Auflösungen oft deutlicher, so dass es unerlässlich ist, Proteinkristalle von hoher Qualität zu erhalten. Es ist daher vorzuziehen, eine gemeinsame Verfeinerung einer Probe sowohl mit Röntgen- als auch mit Neutronendaten durchzuführen, bei der die Röntgendaten verwendet werden können, um die Position des Proteinrückgrats zu bestimmen (Abbildung 13B). Darüber hinaus sind Schwefelatome in Cystein und Methionin möglicherweise schlecht sichtbar, so dass Röntgendaten für die genaue Platzierung der Atome erforderlich sind (Abbildung 13C und Abbildung 13D). Metalle mit schwachen Neutronenstreulängen können auch in Neutronen-SLD-Karten schwer zu modellieren sein, wie in unseren LPMO9D-Karten ersichtlich ist. Die Erfassung eines Röntgendatensatzes mit niedriger Dosis (frei von Strahlenschäden) auf demselben Kristall ist daher nützlich, da er die Positionierung von Metallatomen unter Verwendung von Elektronendichtekarten ermöglicht (Abbildung 13E und Abbildung 13F).

Abbildung 1: Flussdiagramm des Arbeitsablaufs der Neutronenproteinkristallographie. Proteinproduktion. Um eine Neutronenstruktur zu erhalten, wird zunächst Protein exprimiert. Die bakterielle Expression in H2O- oder D2O-basierten Medien wird typischerweise verwendet, um eine hohe Ausbeute an hydriertem bzw. perdeuteriertem rekombinantem Protein herzustellen. Das Protein wird in H2O-basiertem Puffer gereinigt und dann entweder in H2O- oder D2O-basiertem Kristallisationspuffer kristallisiert, um Kristalle auf eine Mindestgröße von 0,1 mm3 zu züchten. Probenvorbereitung: Vor der Erfassung von Neutronenbeugungsdaten durchlaufen H2O-gezüchtete Kristalle einen H/D-Austausch, um die proteintitierbaren H-Atome mit D auszutauschen. H/D-Austausch kann durch direktes Einweichen der Kristalle in deuterierten Kristallisationspuffer, Ausgleich des Kristallisationstropfens mit einem D2O-basierten Reservoir oder durch Montage der Kristalle in Quarzkapillaren für den Dampfaustausch mit deuteriertem Kristallisationspuffer erfolgen. Neutronendatenerfassung: Nach dem H/D-Austausch werden potentielle Kristalle gescreent, um die Beugungsqualität zu bestimmen. Kristalle mit einer Mindestauflösung von 2,5 Å gelten als geeignet für einen vollständigen Datensatz, der gesammelt werden soll. Kristalle werden in Quarzkapillaren zur Datenerfassung bei Raumtemperatur montiert oder in einer Kryoschleife für die Datenerfassung bei kryogener Temperatur schockgefroren. Ein Röntgendatensatz wird auf demselben (oder einem identischen) Kristall bei gleicher Temperatur gesammelt. Modellbau: Die Verfeinerung erfolgt mit phenix.refine gegen Neutronen- und Röntgendaten oder nur gegen die Neutronendaten. Der manuelle Modellaufbau der Proteinstruktur wird in Coot unter Verwendung der Neutronen-SLD-Karten durchgeführt. Komplette Struktur: Nach Fertigstellung der Proteinstruktur wird das Koordinatenmodell validiert und in der Proteindatenbank hinterlegt. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 2: Proteinkristalle ernten. (A) Kristalle werden unter einem Mikroskop behandelt. (B) Der versiegelte Sandwichkasten mit der silikonisierten Glasplatte wird geöffnet. Reservoirpuffer wird auf silikonisierte Glasobjektträger pipettiert. (C) Ein Kristall wird mit einem Mikroloop geerntet. (D) Der Kristall wird in einen Tropfen Mutterlauge gegeben, um alle Ablagerungen abzuwaschen, die oft zusammen mit dem Kristall geerntet werden. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 3: Transfer von Kristall zu Quarzkapillar. (A) Das Ende einer Quarzkapillare ist mit Reservoirpuffer gefüllt. (B) Der Kristall wird in die Quarzkapillare überführt und (C) in den Reservoirpuffer eingetaucht. (D) Der Kristall wird mit Hilfe von Reservoirpuffer nach unten kapillar getragen. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 4: Abdichtung der Quarzkapillare. (A) Deuterierter Puffer wird am Ende der Kapillare zugegeben, um einen "Pfropfen" zu bilden. (B) Wachs wird mit einem "Zauberstab" geschmolzen. (C) Die Kapillare wird zum Verschließen in das geschmolzene Wachs gegeben. (D) An beiden Enden werden Wachsstopfen gebildet, um die Kapillare abzudichten. (E) Der Kristall nach der Montage. (F) Die versiegelte Kapillare wird in eine Petrischale gegeben und mit Kitt an Ort und Stelle gehalten. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 5: Erhöhtes Signal-Rauschen des Neutronenbeugungsmusters. Mit fortschreitender Datenerfassung werden gebeugte Flecken intensiver. (HINWEIS: Die hier gezeigten Live-Beugungsbilder dienen zur Veranschaulichung und wurden aus verschiedenen Kristallen entnommen.) Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 6: Interaktive Modellbildung unter Verwendung von Neutronendaten in Coot. (A) Ein positiver FO-FC Neutronen-SLD-Dichtepeak (grün), der Serin anzeigt, muss durch Editieren von Chi-Winkeln neu ausgerichtet werden. Die 2FO-FC-Neutronen-SLD-Karte wird in lila und die 2FO-FC-Elektronendichtekarte in Blau angezeigt. (B) Korrekt positioniertes Serin. (C) Positive und negative FO-FC-Neutronen-SLD-Dichtepeaks (grün bzw. rot), die darauf hinweisen, dass Tryptophan gedreht/übersetzt werden muss, um dem Differenzdichtepeak zu entsprechen. (D) Korrekt ausgerichtetes Tryptophan. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 7: Zusätzliche Informationen aus Neutronen-SLD-Karten. (A) 2FO-FC Elektronendichtekarte (blau) zeigt die Positionen der "schwereren" Atome in Serin an. (B) Die 2FO-FC-Neutronen-SLD-Karte (violett) zeigt deutlich die Position des "helleren" D-Atoms in Serin. (C) 2FO-FC-Elektronendichtekarte (blau) zeigt die Positionen der "schwereren" Atome in Tryptophan an. (D) Die 2FO-FC-Neutronen-SLD-Karte (violett) zeigt deutlich die Position des "helleren" D-Atoms in Tryptophan. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
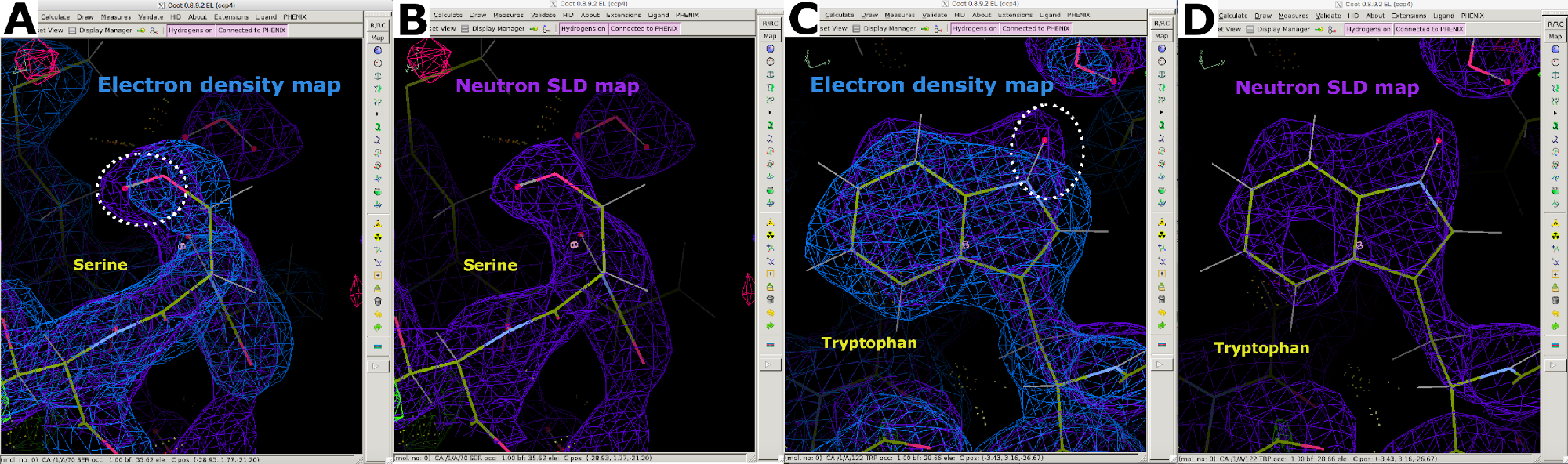{kind=link}

Abbildung 8: Positionierung von Wassermolekülen. (A) Die sphärische Form einer 2FO-FC-Elektronendichtekarte (blau) für Wasser. (B) Die 2FO-FC Neutronen-SLD-Karte (violett) liefert Informationen über die Wasserorientierung und die Wasserstoffbrückenbindungswechselwirkung. (C) Kartenüberlagerung von Elektronen- und Neutronen-SLD-Karten von Wasser. Die 2FO-FC-Neutronen-SLD-Karte wird in lila und die 2FO-FC-Elektronendichtekarte in Blau angezeigt. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
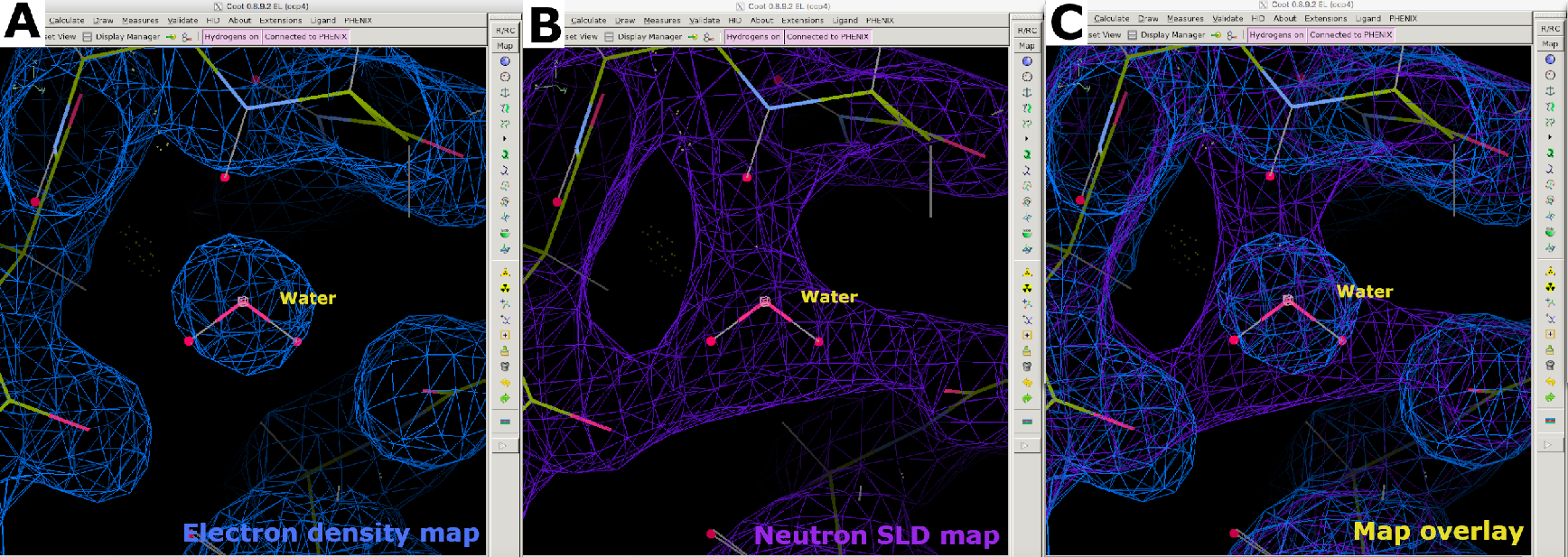{kind=link}

Abbildung 9: Neutronen-SLD-FO-FComit-Karten. (A) Die FO-FC-Neutronen-SLD-Karte (grün) liefert klare Informationen über die H/D-Orientierung von Tyrosinresten. Die 2FO-FC-Neutronen-SLD-Karte wird in lila und die 2FO-FC-Elektronendichtekarte in Blau angezeigt. (B) Tyrosinrest mit korrekter H/D-Orientierung. (C) FO-FC Neutronen-SLD-Karte (grün) liefert klare Informationen über die H/D-Orientierung von Threoninresten. (D) Threoninrest mit korrekter H/D-Orientierung. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
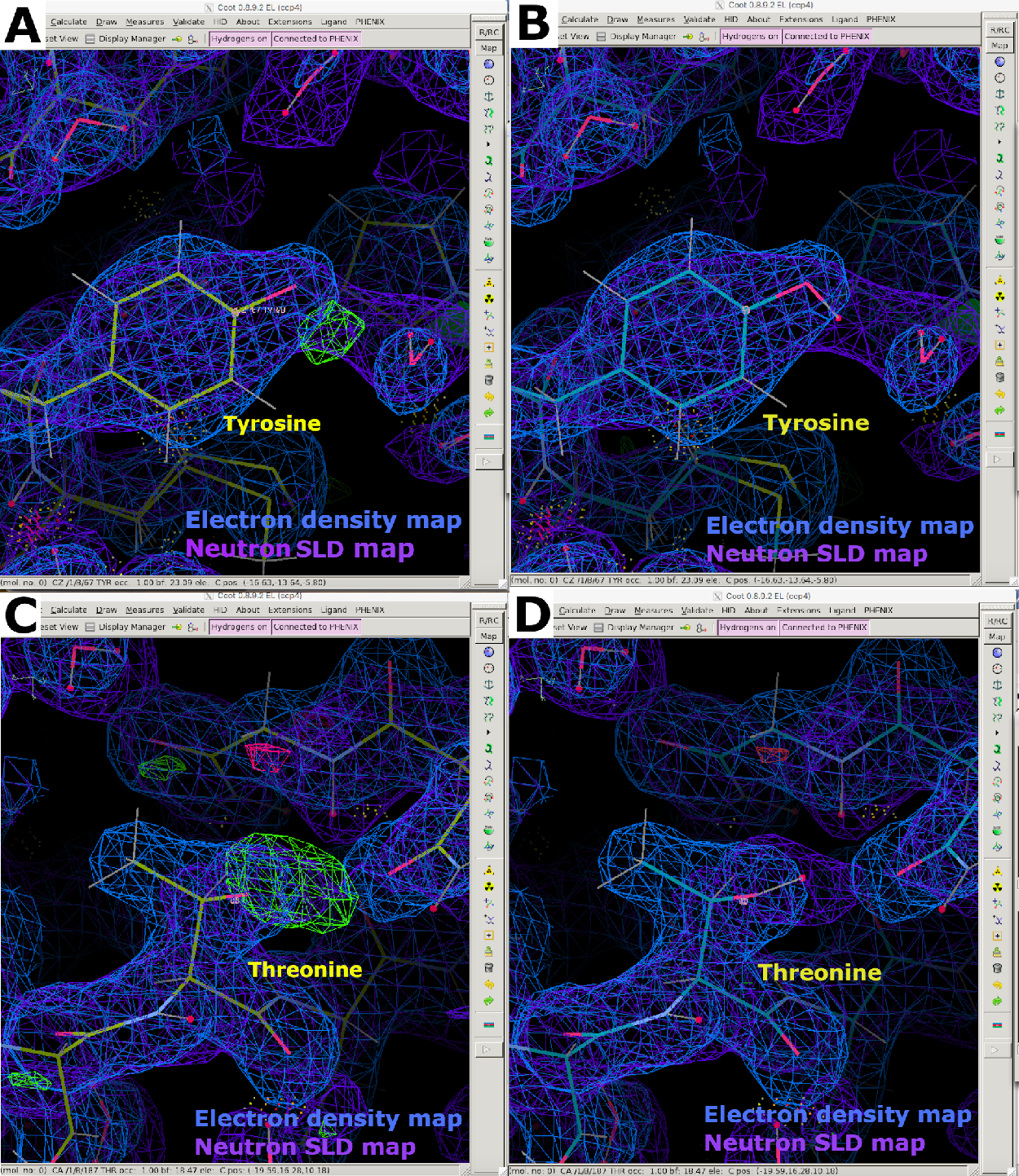{kind=link}

Abbildung 10: Mehrere Protonierungszustände, dargestellt mit Neutronen-SLD-Karten. (A) Die 2FO-FC-Elektronendichtekarte (blau) liefert nur die Position des N-Atoms der Lysin-ε-Ammonium-Gruppe. (B-E) Die FO-FC-Neutronen-SLD-Wegweiskarte (grün) zeigt deutlich die positiv geladene -NH3-Gruppe. Die 2FO-FC-Neutronen-SLD-Karte wird in lila und die 2FO-FC-Elektronendichtekarte in Blau angezeigt. (F) Überlagerung von Elektronendichte- und Neutronen-SLD-Karten. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 11: Aussehen von Wassermolekülen in Neutronen-SLD-Karten. (A) Wassermoleküle werden gemäß FO-FC-Neutronen-SLD-Karten (grün) und potenziellen Wasserstoffbrückenbindungen positioniert. Die 2FO-FC Neutronen-SLD-Karte ist violett dargestellt. (B) Korrekt positioniertes Wassermolekül. (C-E) Die verschiedenen Formen von Neutronen-SLD-Karten für Wassermoleküle in Abhängigkeit von B-Faktoren und Wasserstoffbrückenbindungswechselwirkungen. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
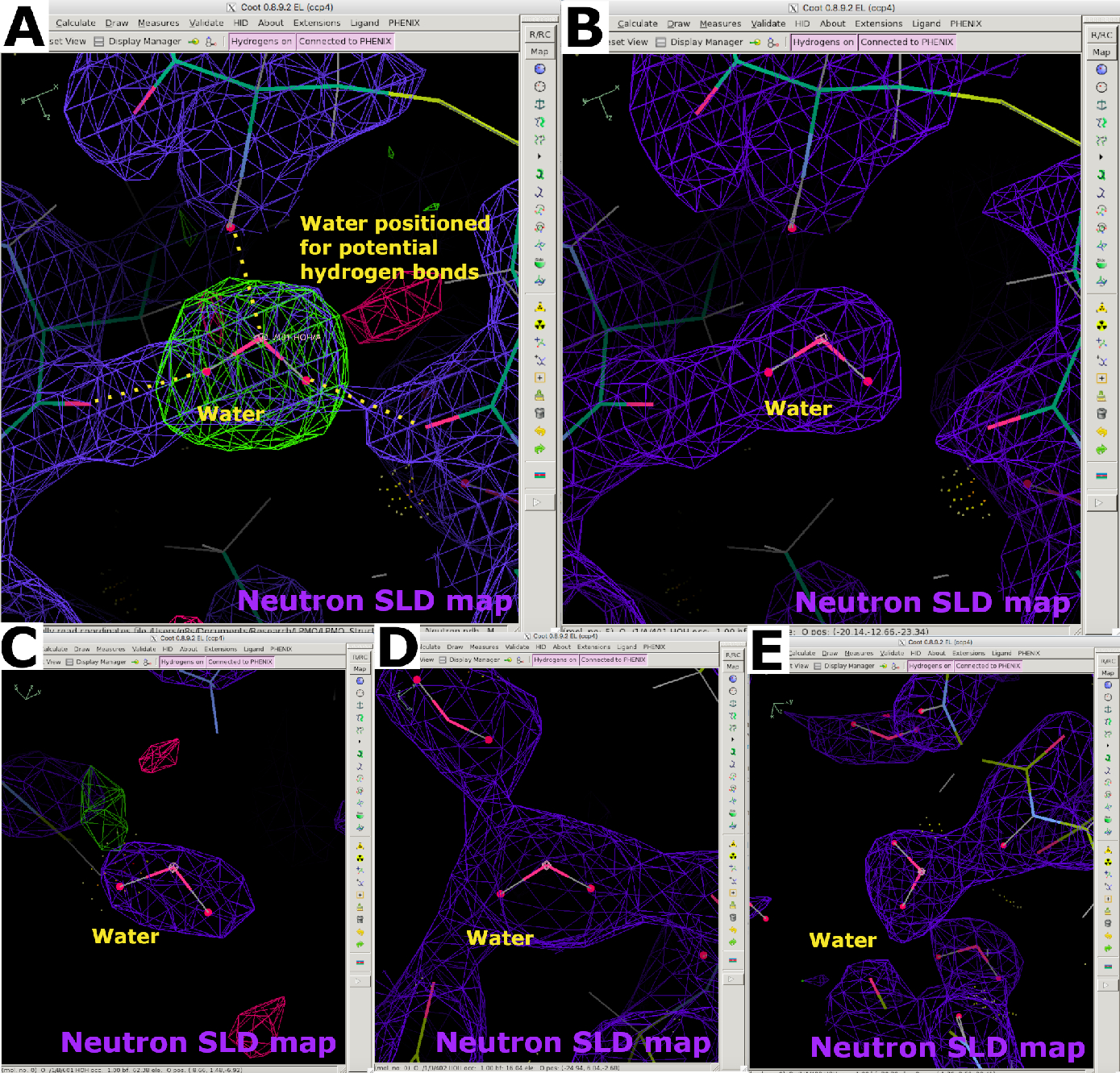{kind=link}

Abbildung 12: Informationen über Aminosäureorientierung und Protonierung durch Neutronen-SLD-Karten. (A) Die Neutronen-SLD-FO-FC-Kartenpeaks (grün) zeigen eine falsche Orientierung eines Asparaginrests an. Die 2FO-FC-Neutronen-SLD-Karte wird in lila und die 2FO-FC-Elektronendichtekarte in Blau angezeigt. (B) 2FO-FC Neutronen-SLD-Karte (violett) der korrekten Asparaginorientierung. (C) Der Neutronen-SLD-FO-FC-Kartenpeak (grün) zeigt eine einzelne Protonierung des Histidins bei N ε an. (D) 2FO-FC Neutronen-SLD-Karte (violett) von Histidin N ε-Protonierung. (E) Neutron SLD FO-FC lassen Kartenpeaks weg (grün) bestätigen die positive Ladung von Arginin. (F) 2FO-FC Neutronen-SLD-Karte (violett) von positiv geladenem Arginin. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 13: Diskontinuierliche Neutronen-SLD-Karten. (A) 2FO-FC Neutronen-SLD-Karte (violett) eines hydrierten, Dampf-H/D-ausgetauschten Proteins. Glutaminsäure zeigt neutronen-SLD-Kartenunterdrückung aufgrund der negativen Streulänge von nicht austauschbaren H-Atomen. (B) Eine überlagerte 2FO-FC-Elektronendichtekarte (blau) zeigt deutlich die Dichte der Glutaminsäure an. (C) Das Schwefelatom in Methionin ist in 2FO-FC Neutronen-SLD-Karten (violett) schlecht sichtbar. (D) Eine überlagerte Elektronendichtekarte zeigt deutlich die Dichte des Methionins. (E) Metallatome, hier Kupfer, sind in Neutronen-2FO-FC-SLD-Karten (violett) schlecht sichtbar. (F) Eine überlagerte 2FO-FC-Elektronendichtekarte (blau) zeigt deutlich die Dichte des koordinierten Kupferatoms. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
| Isotop | Kohärente Streulänge (fm) | Inkohärente Streulänge (fm) |
| 1H | -3.741 | 25.274 |
| 2H | 6.671 | 4.04 |
| 12C | 6.6511 | 0 |
| 14N | 9.37 | 2 |
| 16O | 5.803 | 0 |
| 23Na | 3.63 | 3.59 |
| 24 Mg | 5.66 | 0 |
| 31P | 5.13 | 0.2 |
| 32ER | 2.804 | 0 |
| 35Cl | 11.65 | 6.1 |
| 39K | 3.74 | 1.4 |
| 40Ca | 4.8 | 0 |
| 55 Mio. | -3.73 | 1.79 |
| 56Fe | 9.94 | 0 |
| 63Cu | 6.43 | 0.22 |
| 64Zn | 5.22 | 0 |
Tabelle 1: Neutronenstreulängen und inkohärente Streuwerte. Adaptiert von Sears, 199216.
Ergänzende Abbildung 1: Das IMAGINE-Instrument am High Flux Isotope Reactor. (A) Das IMAGINE-Instrument in der Kalten Neutronenleiterhalle. (B) Probe in einer Quarzkapillare montiert, die mit Kitt am Goniometer befestigt ist. Der Proben- und Detektortisch schließt sich, um den Kristall und die zylindrische Bildplatte im Neutronenstrahl zu positionieren. Geändert mit Genehmigung der International Union of Crystallography53. Bilder mit Genehmigung von Genevieve Martin, Oak Ridge National Laboratory. Bitte klicken Sie hier, um diese Abbildung herunterzuladen.
Ergänzende Abbildung 2: Das MaNDi-Instrument an der Spallationsneutronenquelle. (A) Das MaNDi Anger Kamera-Detektor-Array. Reproduziert mit Genehmigung der International Union of Crystallography11. (B) MaNDi bewegliche Probenstufe. (C) In Quarzkapillare montierte Probe, die auf dem Goniometer bei MaNDi zur Erfassung von Raumtemperaturdaten montiert ist. Bilder mit Genehmigung von Genevieve Martin, Oak Ridge National Laboratory. Bitte klicken Sie hier, um diese Abbildung herunterzuladen.
Ergänzende Abbildung 3: Struktur der lytischen Polysaccharid-Monooxygenase NcLPMO9D. Die kupferaktive Stelle NcLPMO9D befindet sich auf einer flachen Polysaccharid-Bindungsfläche. Das Kupfer wird durch zwei Histidinreste in einem klassischen "Histidin-Korsett" sowie einem axialen Tyrosinrest koordiniert. Bitte klicken Sie hier, um diese Abbildung herunterzuladen.
Ergänzende Abbildung 4: Kristall mit ausreichendem Volumen in sitzender Tropfenkristallisationsschale. (A) Große Kristalle werden in sitzenden Tropfen gezüchtet, die in 9-well silikonisierten Glasplatten angeordnet sind. (B und C) Kristalle werden gemessen, um diejenigen mit einem Volumen > 0,1 mm3 zu identifizieren. Bitte klicken Sie hier, um diese Abbildung herunterzuladen.
Ergänzende Abbildung 5: pH-Messgerät für deuterierte Pufferablesungen. Die pH-Elektrode wird vor Gebrauch in D2O eingeweicht. NaOD und DCl werden verwendet, um den pH-Wert von deuterierten Puffern anzupassen. Bitte klicken Sie hier, um diese Abbildung herunterzuladen.
Ergänzende Abbildung 6: MaNDi-Richtlinien für die Probenmontage. Maximale Abmessungen der Quarzkapillare und Probenposition für die Erfassung von Raumtemperaturdaten.
Reproduziert aus: https://neutrons.ornl.gov/mandi/sample-environment Bitte klicken Sie hier, um diese Abbildung herunterzuladen.
Ergänzende Abbildung 7: Entfernung des überschüssigen Puffers. (A) Überschüssiger Puffer wird mit Mikrokapillarspitzen aus der Quarzkapillare abgesaugt. (B) Der verbleibende Puffer wird mit einem dünnen Papierdocht entfernt, um die Kapillare vollständig zu trocknen. Bitte klicken Sie hier, um diese Abbildung herunterzuladen.
Ergänzende Abbildung 8: Die Datenerfassungs-GUI. Eingabefenster der "Experimentparameter" zur Datenerfassung. Bitte klicken Sie hier, um diese Abbildung herunterzuladen.
Ergänzende Abbildung 9: Die grafische Benutzeroberfläche der Optik. Auswahl des Quasi-Laue-Bereichs zur Datenerfassung und Überwachung der Neutronenzählrate. Bitte klicken Sie hier, um diese Abbildung herunterzuladen.
Ergänzende Abbildung 10: Datenerfassung in der Datenerfassungs-GUI. Die Belichtungszeit, die Anzahl der Frames und die Winkel für die Datenerfassung sind auf der Registerkarte "Sammeln" angegeben. Die Datenerfassung wird dann über "Scan starten" initiiert. Bitte klicken Sie hier, um diese Abbildung herunterzuladen.
Ergänzende Abbildung 11: Gebeugte Neutronen detektiert und dargestellt. Am Ende der Belichtungszeit wird der neutronenempfindliche Bildplattendetektor ausgelesen und das Beugungsmuster in der Datenerfassungs-GUI angezeigt. Bitte klicken Sie hier, um diese Abbildung herunterzuladen.
Ergänzende Abbildung 12: Datenverarbeitung nach Neutronenbeugung. Frames werden indiziert, integriert, Wellenlänge normalisiert und skaliert mit Lauegen, Lscale und Scala, um nach der Datenerfassung eine zusammengeführte Reflexionsdatei zu generieren. Bitte klicken Sie hier, um diese Abbildung herunterzuladen.
Ergänzende Abbildung 13: Erhebung von Röntgendaten. Heimquelle Röntgengenerator mit Quarzkapillar-montiertem Kristall zur Erfassung von Raumtemperaturdaten. Bitte klicken Sie hier, um diese Abbildung herunterzuladen.
Ergänzende Abbildung 14: Montagerichtlinien für die MaNDi-Kryodatenerhebung. Abmessungen der CrystalCaps und Stifthöhe für die Kryodatenerfassung bei MaNDi.
Reproduziert aus: https://neutrons.ornl.gov/mandi/sample-environment Bitte klicken Sie hier, um diese Abbildung herunterzuladen.
Ergänzende Abbildung 15: Schockgefrieren zur Erfassung von Kryo-Neutronenbeugungsdaten. (A) Einrichtung für das Einweichen von Kristallen, das Ernten mit einem Mikroloop und das Einfrieren in flüssigem Stickstoff mit einem kryokompatiblen Behälter wie einem Schaum-Dewar. Der montierte Kristall wird mit einer vorgekühlten Kryostiftzange direkt auf das Beamline-Kryo-Goniometer übertragen. (B) Die Wachsdichtung wird zur Kristallentfernung geschmolzen. (C) Der Kristall wird zur Ernte bis zum Ende der Quarzkapillar gespült. (D) Der Kristall wird nacheinander in Ascorbat-Einweichpuffer und dann kryoprotektiv getränkt, gefolgt von einem Schockgefrieren in flüssigem Stickstoff. Bitte klicken Sie hier, um diese Abbildung herunterzuladen.
Ergänzende Abbildung 16: Beispielausrichtungsschnittstelle. Die Kristallausrichtung im Neutronenstrahl, dargestellt durch das blaue Kreuz, erfolgt durch Punkt- und Klickzentrierung. Bitte klicken Sie hier, um diese Abbildung herunterzuladen.
Ergänzende Abbildung 17: Die CSS-GUI für die Datenerfassung. Die Datenerfassungsstrategie, einschließlich Expositionsdosen und -winkel, wird in die CSS-GUI hochgeladen. Während der Datenerfassung werden die gebeugten Neutronen, die auf dem Echtzeitdetektor detektiert wurden, im oberen Bereich angezeigt. Bitte klicken Sie hier, um diese Abbildung herunterzuladen.
Ergänzende Abbildung 18: Übereinstimmende R-freie Flags in CCP4. Die R-freien Flaggen der Neutronendaten werden mit den R-freien Flaggen von Röntgendaten abgeglichen, die auf demselben oder einem identischen Kristall zur gemeinsamen Verfeinerung gesammelt wurden. Bitte klicken Sie hier, um diese Abbildung herunterzuladen.
Ergänzende Abbildung 19: Strukturvorbereitung und -verfeinerung. (A) Das Phenix ReadySet-Tool wird verwendet, um eine doppelte H/D-Belegung an austauschbaren Standorten hinzuzufügen. (B) Sowohl die Neutronendaten als auch die Röntgendaten werden für eine gemeinsame Verfeinerung verwendet, während das ursprüngliche Eingangsmodell gegen den Röntgendatensatz verfeinert wurde, der auf demselben Kristall oder einem identischen Kristall gesammelt wurde. Bitte klicken Sie hier, um diese Abbildung herunterzuladen.
Ergänzende Abbildung 20: Konfiguration der Einschränkungseinstellungen. Das Verfeinerungsmodell sowie die Kernabstände sind für die gemeinsame Verfeinerung von Röntgen-/Neutronendaten konfiguriert. Bitte klicken Sie hier, um diese Abbildung herunterzuladen.
Ergänzende Abbildung 21: Datenauswahl für den Bau des Blässhuhnmodells. Die Phenix-MTZ-Dateiausgabe mit Röntgen- und ungefüllten Neutronendaten wird in Coot geöffnet, um Elektronen- und Neutronen-SLD-Karten für die interaktive Modellbildung zu erstellen. Bitte klicken Sie hier, um diese Abbildung herunterzuladen.
Ergänzende Abbildung 22: Interaktiver Modellbau in Coot während einer gemeinsamen Verfeinerung. (A) Ein positiver und negativer FO-FC Neutronen-SLD-Dichtepeak (grün bzw. rot), der anzeigt, dass das Wasser durch Rotation/Translation neu ausgerichtet werden muss. Die 2FO-FC-Neutronen-SLD-Karte wird in lila und die 2FO-FC-Elektronendichtekarte in Blau angezeigt. (B) Richtig positioniertes Wasser. (C) Ein positiver FO-FC Neutronen-SLD-Kartenpeak (grün) zeigt an, dass Threonin gedreht werden muss, um den Differenzdichtepeak durch Bearbeiten von Chi-Winkeln zu erreichen. (D) Korrekt orientiertes Threonin. Bitte klicken Sie hier, um diese Abbildung herunterzuladen.
Ergänzende Abbildung 23: Strukturvorbereitung für die reine Neutronendatenverfeinerung. Die Startkoordinatendatei wird für die Verfeinerung durch Wasseratomentfernung in PDBTools und durch Hinzufügen einer doppelten H/D-Belegung an austauschbaren Stellen vorbereitet. Bitte klicken Sie hier, um diese Abbildung herunterzuladen.
Ergänzende Abbildung 24: Verfeinerung nur von Neutronendaten. (A) Neutronendaten werden ebenso hochgeladen wie das vorbereitete Startmodell. (B) Die Einstellungen für die Neutronendatenverfeinerung verwenden die Neutronenstreutabelle. Bitte klicken Sie hier, um diese Abbildung herunterzuladen.
Ergänzende Abbildung 25: Datenauswahl für den Bau des Blässhuhnmodells. Die ungefüllten Neutronendaten werden in Coot zur interaktiven Modellbildung geöffnet. Bitte klicken Sie hier, um diese Abbildung herunterzuladen.
Ergänzende Abbildung 26: Realraumverfeinerung in Blässhuhn für deuterierte Reste. (A) Positive und negative FO-FC-Neutronen-SLD-Dichtepeaks (grün bzw. rot), die darauf hinweisen, dass ein Argininrest bewegt werden muss, um ihn an den FO-FC-Dichtepeak anzupassen. Die 2FO-FC-Neutronen-SLD-Karte wird in lila und die 2FO-FC-Elektronendichtekarte in Blau angezeigt. (B) Die Verwendung von Real Space Refine führt zu "explodierenden" D-Atomen aufgrund fehlender Blässhuhngeometrie-Beschränkungsbibliotheken. (C) Die D-Atome bewegen sich nicht mit dem Rest der Restatome. (D) Die D-Atompositionen können manuell mit einem Texteditor fixiert werden. Bitte klicken Sie hier, um diese Abbildung herunterzuladen.
Ergänzende Abbildung 27: Zugabe von Wassermolekülen. (A) Wassermoleküle können manuell zu den positiven FO-FC Neutronen-SLD-Kartendichtespitzen (grün) hinzugefügt werden. Die eingefügten Wassermoleküle werden zunächst durch ein O-Atom in Blässhuhn dargestellt. (B) Phenix ReadySet wird verwendet, um D-Atome zu den O-Atomen für Wassermoleküle hinzuzufügen. (C) Das deuterierte Wassermolekül wird erfolgreich zugegeben. Bitte klicken Sie hier, um diese Abbildung herunterzuladen.
Ergänzende Abbildung 28: Verfeinerungsstatistik. Abschließende Datenverfeinerungsstatistik nach gemeinsamer Röntgen-/Neutronenveredelung. Bitte klicken Sie hier, um diese Abbildung herunterzuladen.
Ergänzende Abbildung 29: Verfeinerungsstatistik. Abschließende Datenverfeinerungsstatistik nach Neutronendatenverfeinerung. Bitte klicken Sie hier, um diese Abbildung herunterzuladen.
Diskussion
Die Neutronenproteinkristallographie ist eine hochempfindliche Technik zur Untersuchung von Protonierungszuständen und der Orientierung von Wassermolekülen in Proteinen. Diese Informationen geben Aufschluss über die katalytischen Mechanismen von Proteinen, da Änderungen der Protonierungs- und Wasserstoffbrückenbindungswechselwirkungen oft von zentraler Bedeutung für die Enzymchemie sind10. Die Neutronenproteinkristallographie, obwohl eine informative Technik, hat eine Reihe von Faktoren, die vor der Planung eines Neutronenbeugungsexperiments berücksichtigt werden sollten, nämlich:
- Der Bedarf an großen Proteinkristallen für die Datenerfassung.
- Die Streueigenschaften von Wasserstoff und anderen Elementen, wie Metallionen.
- Einschränkungen in der Strukturverfeinerungs- und Modellerstellungssoftware bei der Arbeit mit deuterierten Samples.
Die Neutronenproteinkristallographie ist eine flussbegrenzte Technik. Im Gegensatz zu Röntgenbeugungsdatensätzen werden für Neutronendatensätze aufgrund der technikimmanenten Einschränkungen (Flussbegrenzung, Quasi-Laue, längere Wellenlängen) höhere R-Faktoren und geringere Vollständigkeit, Redundanz und Signal-Rausch-Verhältnisse erwartet. Die Datenerfassung eines einzelnen Frames dauert in der Regel 12 – 18 Stunden. Der Erfolg eines Experiments hängt stark von der Probengröße und -qualität ab, wobei Kristalle von 0,1 mm3 oft die Mindestanforderung sind3. Die Neutronenbeugung erfordert die Produktion großer Mengen an Protein, um Kristallisationstropfen im Bereich von 10 bis 800 μL aufzubauen. Das Mindestvolumen für die Züchtung ausreichend großer Kristalle kann mit einem Volumenrechner unter Berücksichtigung der Kristall- und Probenparameter (https://neutrons.ornl.gov/imagine) geschätzt werden. Das Wachstum großer Kristalle wurde am häufigsten durch Dampfdiffusion erreicht3. Die Hängetropfenkristallisation ermöglicht das Wachstum von Kristallen in großen Tropfen im Bereich von 10-25 μL, während größere Tropfen im Bereich von bis zu ~ 50 μL mit handelsüblichen Sitztropfengeräten eingerichtet werden können14,54. Silikonisierte Neun-Well-Glasplatten können verwendet werden, um sehr große Tropfen mit Volumina von bis zu 800 μL aufzustellen. Diese Glasplatten werden in "Sandwich-Boxen" platziert, die im Handel bei Hampton Research erhältlich sind. Weitere Kristallisationstechniken umfassen die Batchkristallisation, bei der die Grenze der Tropfengröße vom Gefäß vorgegeben wird. Das Batch-Kristallisationsexperiment kann von Mikrolitern bis millilitern reichen55. Die Kristallisation kann auch unter Verwendung der Dialysetechnik durchgeführt werden, bei der das Protein über eine Dialysemembran oder durch Gegendiffusion entlang eines Fällungsmittelkonzentrationsgradienten oder durch einen porösen Pfropfen wie Agarose56,57 mit dem Fällungsmittel ausgeglichen wird. Seeding bietet eine weitere Alternative, um Kristalle des gewünschten Volumens zu erhalten. Mikro- und Makroseierung wurden erfolgreich für große Kristallwuchse eingesetzt, einschließlich großer Kristalle von NcLPMO9D45. Einige Kenntnisse des Proteinphasendiagramms, einschließlich des Einflusses der Temperatur auf die Löslichkeit, helfen bei großem Kristallwachstum.
Bei der Planung eines Neutronenbeugungsexperiments ist die Optimierung der Proteinvorbereitung zur Maximierung des Signal-Rausch-Verhältnisses während der Beugungsdatenerhebung unerlässlich7. Um die Dichteunterdrückung und die hohe inkohärente Streuung durch H-Atome zu umgehen, können Neutronen-SLD-Karten verbessert werden, indem H-Atome gegen sein Isotop D ausgetauscht werden, das eine positive kohärente Streulänge und eine geringe inkohärente Streulänge besitzt. Um dies zu erreichen, wird ein Dampfaustausch des hydrierten Proteinkristalls gegen deuterierten Kristallisationspuffer durchgeführt. Dies gewährleistet den H/D-Austausch von Lösungsmittelmolekülen und den labilen, titrierbaren Protein-H-Atomen23. Der Dampfaustausch erfolgt durch Montage des hydrierten Kristalls in einer Quarzkapillare mit D2O-basierten, deuterierten Kristallisationspuffer-"Plugs" und stellt eine effektive, schonende Technik dar, die am häufigsten angewendet wird14,23,35. Der Austausch kann mehrere Wochen dauern und erfordert vorzugsweise, dass der deuterierte Puffer häufig gewechselt wird, um einen maximalen H/D-Austausch zu gewährleisten. Der H / D-Austausch kann auch durchgeführt werden, indem der Kristall direkt in deuteriertem Puffer eingeweicht wird. Um zu vermeiden, dass der Kristall aufgrund von D2O-Exposition unter Stress gesetzt wird, sollte der Einweichvorgang schrittweise durchgeführt werden, indem das D2O: H2O-Verhältnis schrittweise erhöht wird58. Darüber hinaus kann die Kristallisation von hydriertem Protein auch in deuteriertem Puffer für den H/D-Austausch an labilen H-Stellen durchgeführt werden22,59. Es sollte jedoch beachtet werden, dass D2O-basierter Puffer eine Wirkung auf die Proteinlöslichkeit hat, die eine weitere Anpassung der bekannten H2O-basierten Bedingungen erfordert3,59. Es wurde auch beobachtet, dass D2O-basierte Puffer in einigen Fällen zu kleineren Kristallen führen59. Der vollständige Austausch von titrierbaren und kohlenstoffgebundenen H-Atomen zu D kann erreicht werden, indem Proteine in deuterierten Medien exprimiert werden, um eine perdeuterierte Probe zu erzeugen20. Die resultierenden Neutronen-SLD-Karten der perdeuterierten Probe werden signifikant verbessert und zeigen nicht mehr die Dichteaufhebung des hydrierten Probengegenstücks an. Dies ist vorteilhaft bei der Charakterisierung von H / D, das an nicht austauschbaren Stellen in einem Protein oder Cofaktor gebunden ist. Die Expression von perdeuteriertem Protein ist jedoch sowohl kostenintensiv als auch ertragsarm60. Das Oak Ridge National Laboratory (ORNL) Center for Structural Molecular Biology (CSMB) bietet eine Deuterationseinrichtung für Benutzer, die eine perdeuterierte Probe (https://www.ornl.gov/facility/csmb) erzeugen möchten. Die perdeuterierte Expression wird typischerweise in einem Bioreaktor auf der 1-l-Skala durchgeführt, der ~ 50 mg gereinigtes Protein ergibt61.
Nach der Erfassung der Neutronenbeugungsdaten wird die Verfeinerung und interaktive Modellbildung durchgeführt. Die Verfeinerung kann mit mehreren Software-Suiten ausgeführt werden, einschließlich phenix.refine, nCNS oder SHELXL28,31,32,33. Die Phenix-Suite ist die am häufigsten verwendete Software zur Verfeinerung von Neutronenbeugungsdaten in Verbindung mit Coot, mit der das Modell manuell aus den Neutronen-SLD-Karten erstellt wird34. Obwohl sowohl Phenix als auch Coot die Verarbeitung von Neutronenbeugungsdaten ermöglichen, fehlen ihnen möglicherweise bestimmte Merkmale, die notwendig sind, um die mit Neutronendaten und deuterierten Proben verbundenen Eigenheiten zu verarbeiten. Zum Beispiel enthält Coot keine Geometrieoptimierung für deuterierte Reste, was zu Komplikationen beim Modellaufbau führen kann, da das Feature "Real Space Refine" zu "explodierenden" Residuen führt (ergänzende Abbildung 26) 62. Dies kann gelöst werden, indem Rückhaltedateien für alle deuterierten Rückstände generiert werden. Dies ist jedoch ein intensiver Prozess und solche Bibliotheken sind derzeit nicht öffentlich zugänglich. Bei der Durchführung von Verfeinerungen in Phenix werden austauschbare H/D-Standorte zunächst auf 0,50 Belegung für H und D eingestellt. Wenn Verfeinerungen durchgeführt werden, wird die Belegung von H und D gemäß den Neutronen-SLD-Karten verfeinert. Während des interaktiven Modellbaus sind Fo-Fc-Karten mit Differenzdichte sehr informativ bei der Beurteilung der H / D-Belegung. Anhand von Karten kann festgestellt werden, welche Standorte eine hohe D-Belegung aufweisen, was besonders an dem aktiven Standort, an dem Protonierungszustände katalytisch relevant sind, aufschlussreich ist63. Mehrdeutige Situationen treten jedoch auf, wenn die H: D Die Belegung liegt in der Nähe von 0,70:0,30, was zu einer vollständigen Signalunterdrückung in Neutronen-SLD-Karten führt64. Es sollte auch berücksichtigt werden, dass Quasi-Laue-Neutronen-Datensätze oft eine Vollständigkeit von etwa 80% aufweisen, was niedriger ist als die routinemäßig beobachteten ≥ 98% für Röntgenbeugungsdaten. Bei der Verfeinerung von Neutronenbeugungsdaten in Phenix werden daher die fehlenden beobachteten Amplituden (Fo) aus dem Modell berechnet, um die Reflexionsliste zu vervollständigen und so eine Modellverzerrung einzuführen. Um dieser potenziellen Verzerrung Rechnung zu tragen, sollten "no_fill" -Karten während der interaktiven Modellerstellung im Gegensatz zu "gefüllten" Karten untersucht werden.
Benutzer können wählen, ob sie eine gemeinsame Röntgen- / Neutronendatenverfeinerung ihrer Struktur oder eine Verfeinerung nur für Neutronendaten durchführen möchten. Die Visualisierung von Neutronen-SLD-Karten, insbesondere bei niedrigerer Auflösung, kann insbesondere für ein hydriertes Protein, in dem H trotz H/D-Dampfaustausch noch an nicht austauschbaren Stellen vorhanden ist, zunächst beunruhigend sein. Dies führt zu Neutronendichtekartenaufhebungen, die den Eindruck von diskontinuierlichen Karten erwecken65,66. Das Sammeln eines entsprechenden Röntgendatensatzes ergänzt diese Aufhebungen vorteilhaft in einer gemeinsamen Verfeinerung (Abbildung 13A und Abbildung 13B). Eine gemeinsame Verfeinerungsstrategie beinhaltet typischerweise die Verfeinerung der Protein-Backbone-Koordinaten anhand der Röntgendaten, während die Neutronenbeugungsdaten verwendet werden, um die Position und Belegung der H / D-Atome an austauschbaren Stellen zu verfeinern28. Da die Einführung einer gemeinsamen H/D-Belegung an austauschbaren Standorten die Anzahl der zu verfeinernden Parameter erhöht, erhöht eine gemeinsame Verfeinerung mit Röntgendaten auch das Daten-zu-Parameter-Verhältnis. Eine gemeinsame Verfeinerung erfordert, dass ein entsprechender Röntgendatensatz bei gleicher Temperatur auf demselben Kristall oder einem unter den gleichen Bedingungen gezüchteten Kristall gesammelt wird. Für Neutronenbeugungsdaten, die bei Raumtemperatur (300K) gesammelt werden, sollte der entsprechende Röntgendatensatz bei Raumtemperatur unter Verwendung einer Niedrigdosis-Datenerfassungsstrategie gesammelt werden, um Strahlenschäden zu begrenzen. Perdeuterierte Proben hingegen liefern verbesserte und kontinuierliche Neutronen-SLD-Karten, da sie nicht die gleiche Größe der H/D-Signalunterdrückung besitzen. Die Neutronenstreulänge bestimmter Elemente, einschließlich Metalle und Schwefel, macht sie jedoch in Neutronen-SLD-Karten schlecht sichtbar, selbst wenn das Protein perdeuteriert wurde (Abbildung 13C-F)18. Wenn ein Metall charakterisiert werden muss, ist es am besten, die Röntgenbeugung in einer Gelenkverfeinerung zu verwenden oder spektroskopische Techniken anzuwenden, um Beugungsexperimente zu ergänzen. Reine Neutronendatenverfeinerungen werden häufig durchgeführt, wenn der Neutronendatensatz eine hohe Auflösung aufweist oder wenn ein perdeuteriertes Protein verwendet wurde. Darüber hinaus ist die Verfeinerung reiner Neutronendaten besonders nützlich, wenn ein Protein untersucht wird, das sehr empfindlich auf Strahlungsschäden reagiert, da eine von Röntgenstrahlen abgeleitete Struktur strahlungsinduzierte Artefakte besitzen kann. Soll eine reine Neutronendatenverfeinerung durchgeführt werden, muss festgestellt werden, ob der entsprechende Neutronendatensatz eine ausreichende Vollständigkeit und Auflösung aufweist.
ORNL bietet zwei Möglichkeiten zur Erfassung von Neutronenbeugungsdaten: die IMAGINE-Beamline am HFIR sowie die MaNDi-Beamline am SNS36,67. Während beide Instrumente effektive Mittel zum Sammeln eines Neutronenbeugungsdatensatzes bieten, der ähnliche Prinzipien anwendet, hat jedes Instrument einzigartige Spezifikationen, die bei der Beantragung der Strahlzeit berücksichtigt werden sollten. IMAGINE sammelt Quasi-Laue-Daten und ist für die Raumtemperatur-Datenerfassung an Kristallen mit Elementarzellen bis ~100 Å optimiert. MaNDi kann für die Erfassung von Raumtemperatur- und Kryotemperaturdaten unter Verwendung der TOF-Laue-Sammlung auf Kristallen mit Einheitszellen bis zu ~ 300 Å verwendet werden. Vor dem Sammeln eines vollständigen Datensatzes wird ein Test am Kristall durchgeführt, um die Qualität des erhaltenen Beugungsmusters zu bewerten, in dem der Kristall dem Neutronenstrahl für ein einzelnes Bild ausgesetzt wird. Ist der Kristall von ausreichender Qualität, wird analog zur Röntgendatenverarbeitung ein vollständiger Neutronenbeugungsdatensatz gesammelt, indiziert, integriert, skaliert und in einem Prozess zusammengeführt. IMAGINE verwendet Lauegen und Lscale und MaNDi verwendet das Mantid-Paket und verwendet dreidimensionale Profilanpassungen48,50,51,68,69,70. Wissenschaftler, die an einer dieser Einrichtungen nutzer werden, erhalten einen Datensatz im MTZ- oder HKL-Format zur weiteren Analyse.
Die Neutronenbeugung ist eine zerstörungsfreie, hochempfindliche Technik zur Untersuchung des Protonierungszustands und der Wasserstoffbrückenbindungswechselwirkungen biologischer Makromoleküle. Es ist besonders nützlich für photosensible Proteine und Metalloproteine. Mehrere Überlegungen bezüglich der Technik sowie der Verarbeitung der Daten müssen vor der Durchführung eines Experiments berücksichtigt werden, aber das Ergebnis liefert Ergebnisse, die wertvolle Einblicke in den katalytischen Mechanismus des interessierenden Proteins geben können. Die Neutronenproteinkristallographie ergänzt computergestützte, strukturelle, biochemische und spektroskopische Studien und ist damit ein wertvolles Werkzeug in der Toolbox des Biologen von Techniken zur Charakterisierung biologischer Makromoleküle.
Offenlegungen
Die Autoren haben nichts preiszugeben.
Danksagungen
Experimente zur Proteinexpression, -reinigung und -kristallisation wurden am Center for Structural Molecular Biology (CSMB), einer Nutzereinrichtung des US-Ministeriums für biologische und Umweltforschung am Oak Ridge National Laboratory, durchgeführt. Neutronenbeugungsdaten wurden an BL-11B MaNDi an der Spallationsneutronenquelle (SNS) am ORNL gesammelt, die von der Scientific User Facilities Division, Office of Basic Energy Sciences, U.S. Department of Energy gesponsert wird. Die Autoren danken Brendan Sullivan für die Unterstützung bei der Datenreduktion. Röntgenbeugungsdaten wurden an den Einrichtungen des Molecular Education, Technology and Research Innovation Center (METRIC) an der North Carolina State University gesammelt, die vom Bundesstaat North Carolina unterstützt wird. GCS würdigt die Unterstützung zum Teil durch die National Research Foundation (NRF), Südafrika und das Graduate Opportunities (GO!) FM bestätigt die Unterstützung von USDA NIFA Hatch 211001.
Materialien
| Name | Company | Catalog Number | Comments |
| Absorbent Paper Points Size #30-#40, 60 mm length | DiaDent/DiaVet | 218-292 | |
| Capillary wax | Hampton | HR4-328 | |
| CCP4 | Version 7.0.077 | ||
| Conical Centrifuge Tubes (15 mL) | Corning | CLS430790 | |
| Conical Centrifuge Tubes (50mL) | Corning | CLS430828 | |
| Coot | Version 0.8.9.2 | ||
| CrystalCap ALS | Hampton | HR4-779 | |
| Curved-Tip Forceps | Mitegen | TW-CTF-1 | |
| Deuterium chloride solution, 35 wt. % in D2O, ≥99 atom % D | Sigma-Aldrich | 543047 | |
| Deuterium oxide 99.9 atom % D | Sigma-Aldrich | 151882 | |
| Dual Thickness MicroLoops 1000 µm | Mitegen | M5-L18SP-1000 | |
| FiveEasy pH meter F20-Std-Kit | Mettler Toledo | 30266626 | |
| Foam Dewars Standard Vessel 800 ml | Spearlab | M-FD-800 | |
| Four Color Mounting Clay | Hampton | HR4-326 | |
| HEPES, BioUltra, for molecular biology, ≥99.5% (T), | Sigma-Aldrich | 54457 | |
| High flux rotating anode X-ray diffractomemeter with EIGER 4M detector | Rigaku, Oxford Cryostream and Dectris | XtaLAB Synergy-R | Home source X-ray diffractometer |
| Magnetic Wand Straight | Mitegen | M-R-1013198 | |
| Microloader, tip for filling Femtotips and other glass microcapillaries (for research use only), 0.5 – 20 µL, 100 mm, light gray, 192 pcs. (2 racks × 96 pcs.) | Eppendorf | 930001007 | |
| Microtubes volume 1.5 mL | Eppendorf | Z606340 | |
| Petri Dishes with Clear Lid 100 mm diameter | Fischerbrand | FB0875713 | |
| Phenix | Version 1.14-3260 | ||
| Pin Tong 18 mm | Mitegen | M-R-1013196 | |
| Pipette Volume 0.1-2.5 μL | Eppendorf Research | Z683779 | |
| Pipette Volume 100-1000 μL | Eppendorf Research | Z683825 | |
| Pipette Volume 10-100 μL | Eppendorf Research | Z683809 | |
| Pipette Volume 20-200 μL | Eppendorf Research | Z683817 | |
| Poly(ethylene glycol) BioXtra, average mol wt 3,350, powder | Sigma-Aldrich | P4338 | |
| Quartz Capillary , 1.00 mm inner diameter, 80 mm length | Hampton | HR6-146 | Thin-walled capillary |
| Research Stereomicroscope System | Olympus | SZX16 | |
| Reusable B3 (SSRL/SAM Style) Goniometer Bases | Mitegen | GB-B3-R | |
| Round - Miniature Hollow Glass Tubing (VitroTubes) Clear Fused Quartz / 1.00 mm inner diameter, 100 mm length | VitroCom | CV1012 | Thick-walled capillary |
| Sandwich Box with cover | Hampton | HR3-132 | |
| Siliconized 9 Well Glass Plate | Hampton | HR3-134 | |
| Sitting Drop Crystallization Plate (24 Big Well) | Mitegen | XQ-P-24S-A | |
| Sodium deuteroxide solution, 40 wt. % in D2O, 99 atom % D | Sigma-Aldrich | 176788 | |
| Thick Siliconized circle cover slides (22 mm x 0.96 mm) | Hampton | HR3-247 | |
| Universal Pipet Tips, 0.1 - 10 µL | VWR | 76322-528 | |
| Universal Pipet Tips, 1 - 100 µL | VWR | 76322-136 | |
| Universal Pipet Tips, 100 - 1000 µL | VWR | 76322-154 | |
| Universal Pipet Tips, 20 - 200 µL | VWR | 76322-150 | |
| Universal Pipet Tips, 1 - 20 µL | VWR | 76322-134 | |
| Wax pen | Hampton | HR4-342 |
Referenzen
- Neumann, P., Tittmann, K. Marvels of enzyme catalysis at true atomic resolution: distortions, bond elongations, hidden flips, protonation states and atom identities. Current Opinion in Structural Biology. 29, 122-133 (2014).
- Pynn, R. Neutron Scattering-A Non-destructive Microscope for Seeing Inside Matter. Neutron Applications in Earth, Energy and Environmental Sciences. , 15-36 (2009).
- O'Dell, W. B., Bodenheimer, A. M., Meilleur, F. Neutron protein crystallography: A complementary tool for locating hydrogens in proteins. Archives of Biochemistry and Biophysics. 602, 48-60 (2016).
- Niimura, N., Podjarny, A. . Neutron Protein Crystallography: Hydrogen, Protons, and Hydration in Bio-macromolecules. , (2011).
- Blakeley, M. P. P. Neutron macromolecular crystallography. Crystallography Reviews. 15 (3), 157-218 (2009).
- Blakeley, M. P., Cianci, M., Helliwell, J. R., Rizkallah, P. J. Synchrotron and neutron techniques in biological crystallography. Chemical Society Reviews. 33 (8), 548-557 (2004).
- Ashkar, R., et al. Neutron scattering in the biological sciences: progress and prospects. Acta Crystallographica Section D: Structural Biology. 74 (12), (2018).
- Teixeira, S. C. M., et al. New sources and instrumentation for neutrons in biology. Chemical Physics. 345 (2-3), 133-151 (2008).
- Furrer, A., Mesot, J., Strässle, T. . Neutron Scattering in Condensed Matter Physics. , (2009).
- Meilleur, F., Coates, L., Cuneo, M. J., Kovalevsky, A., Myles, D. A. A. The neutron macromolecular crystallography instruments at Oak Ridge national laboratory: Advances, challenges, and opportunities. Crystals. 8 (10), 1-10 (2018).
- Coates, L., et al. The Macromolecular Neutron Diffractometer MaNDi at the Spallation Neutron Source. Journal of Applied Crystallography. 48, 1302-1306 (2015).
- Coates, L., Stoica, A. D., Hoffmann, C., Richards, J., Cooper, R. The macromolecular neutron diffractometer (MaNDi) at the Spallation Neutron Source, Oak Ridge: enhanced optics design, high-resolution neutron detectors and simulated diffraction. Journal of Applied Crystallography. 43 (3), 570-577 (2010).
- Koetzle, T. F., Piccoli, P. M. B., Schultz, A. J. Single-crystal neutron diffraction studies of hydrogen-bonded systems: Two recent examples from IPNS. Nuclear Instruments and Methods in Physics Research, Section A: Accelerators, Spectrometers, Detectors and Associated Equipment. 600 (1), 260-262 (2009).
- Ng, J. D., et al. Large-volume protein crystal growth for neutron macromolecular crystallography. Acta Crystallographica Section F:Structural Biology Communications. 71, 358-370 (2015).
- Blakeley, M. P., Langan, P., Niimura, N., Podjarny, A. Neutron crystallography: opportunities, challenges, and limitations. Current Opinion in Structural Biology. 18 (5), 593-600 (2008).
- Sears, V. F. Neutron scattering lengths and cross sections. Neutron News. 3 (3), 26-37 (1992).
- Weik, M., Patzelt, H., Zaccai, G., Oesterhelt, D. Localization of glycolipids in membranes by in vivo labeling and neutron diffraction. Molecular Cell. 1 (3), 411-419 (1998).
- Helliwell, J. R. Fundamentals of neutron crystallography in structural biology. Methods in Enzymology. 634, (2020).
- Niimura, N., Bau, R. Neutron protein crystallography: Beyond the folding structure of biological macromolecules. Acta Crystallographica Section A: Foundations of Crystallography. 64 (1), 12-22 (2008).
- Hazemann, I., et al. High-resolution neutron protein crystallography with radically small crystal volumes: Application of perdeuteration to human aldose reductase. Acta Crystallographica Section D: Biological Crystallography. 61 (10), 1413-1417 (2005).
- Niimura, N., Chatake, T., Ostermann, A., Kurihara, K., Tanaka, I. High resolution neutron protein crystallography. Hydrogen and hydration in proteins. Zeitschrift für Kristallographie - Crystalline Materials. 218 (2), (2003).
- Meilleur, F., Contzen, J., Myles, D. A. A., Jung, C. Structural stability and dynamics of hydrogenated and perdeuterated cytochrome P450cam (CYP101). Biochemistry. 43 (27), 8744-8753 (2004).
- Bennett, B. C., Gardberg, A. S., Blair, M. D., Dealwis, C. G. On the determinants of amide backbone exchange in proteins: A neutron crystallographic comparative study. Acta Crystallographica Section D: Biological Crystallography. 64 (7), 764-783 (2008).
- Meilleur, F., Kovalevsky, A., Myles, D. A. A. IMAGINE: The neutron protein crystallography beamline at the high flux isotope reactor. Methods in Enzymology. 634, (2020).
- Wang, X. P., et al. A suite-level review of the neutron single-crystal diffraction instruments at Oak Ridge National Laboratory. Review of Scientific Instruments. 89 (9), 092802 (2018).
- Wlodawer, A., Hendrickson, W. A. A procedure for joint refinement of macromolecular structures with X-ray and neutron diffraction data from single crystals. Acta Crystallographica Section A. 38 (2), 239-247 (1982).
- Halle, B. Biomolecular cryocrystallography: Structural changes during flash-cooling. Proceedings of the National Academy of Sciences of the United States of America. 101 (14), 4793-4798 (2004).
- Adams, P. D., Mustyakimov, M., Afonine, P. V., Langan, P. Generalized X-ray and neutron crystallographic analysis: More accurate and complete structures for biological macromolecules. Acta Crystallographica Section D: Biological Crystallography. 65 (6), 567-573 (2009).
- Berman, H. M., et al. The Protein Data Bank. Acta Crystallographica Section D Biological Crystallography. 58 (6), 899-907 (2002).
- Liebschner, D., Afonine, P. V., Moriarty, N. W., Langan, P., Adams, P. D. Evaluation of models determined by neutron diffraction and proposed improvements to their validation and deposition. Acta Crystallographica Section D: Structural Biology. 74, 800-813 (2018).
- Afonine, P. V., Mustyakimov, M., Grosse-Kunstleve, R. W., Moriarty, N. W., Langan, P., Adams, P. D. Joint X-ray and neutron refinement with phenix.refine. Acta Crystallographica Section D: Biological Crystallography. 66 (11), 1153-1163 (2010).
- Brünger, A. T., et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallographica Section D: Biological Crystallography. 54 (5), 905-921 (1998).
- Gruene, T., Hahn, H. W., Luebben, A. V., Meilleur, F., Sheldrick, G. M. Refinement of macromolecular structures against neutron data with SHELXL2013. Journal of Applied Crystallography. 47 (1), 462-466 (2014).
- Emsley, P., Lohkamp, B., Scott, W. G., Cowtan, K. Features and development of Coot. Acta Crystallographica Section D Biological Crystallography. 66 (4), 486-501 (2010).
- Lakey, J. H. Neutrons for biologists: A beginner's guide, or why you should consider using neutrons. Journal of the Royal Society Interface. 6, (2009).
- Schröder, G. C., O'Dell, W. B., Myles, D. A. A., Kovalevsky, A., Meilleur, F. IMAGINE: Neutrons reveal enzyme chemistry. Acta Crystallographica Section D: Structural Biology. 74, 778-786 (2018).
- Halsted, T. P., et al. Catalytically important damage-free structures of a copper nitrite reductase obtained by femtosecond X-ray laser and room-temperature neutron crystallography. IUCrJ. 6, 761-772 (2019).
- Meier, K. K., et al. Oxygen Activation by Cu LPMOs in Recalcitrant Carbohydrate Polysaccharide Conversion to Monomer Sugars. Chemical Reviews. 118 (5), 2593-2635 (2018).
- Beeson, W. T., Vu, V. V., Span, E. A., Phillips, C. M., Marletta, M. A. Cellulose Degradation by Polysaccharide Monooxygenases. Annual Review of Biochemistry. 84 (1), 923-946 (2015).
- Walton, P. H., Davies, G. J. On the catalytic mechanisms of lytic polysaccharide monooxygenases. Current Opinion in Chemical Biology. 31, 195-207 (2016).
- Bertini, L., et al. Catalytic Mechanism of Fungal Lytic Polysaccharide Monooxygenases Investigated by First-Principles Calculations. Inorganic Chemistry. 57 (1), 86-97 (2018).
- Hedegård, E. D., Ryde, U. Molecular mechanism of lytic polysaccharide monooxygenases. Chemical Science. 9 (15), 3866-3880 (2018).
- Hangasky, J. A., Detomasi, T. C., Marletta, M. A. Glycosidic Bond Hydroxylation by Polysaccharide Monooxygenases. Trends in Chemistry. 1 (2), 198-209 (2019).
- Bacik, J. P., et al. Neutron and Atomic Resolution X-ray Structures of a Lytic Polysaccharide Monooxygenase Reveal Copper-Mediated Dioxygen Binding and Evidence for N-Terminal Deprotonation. Biochemistry. 56 (20), 2529-2532 (2017).
- O'Dell, W. B., Agarwal, P. K., Meilleur, F. Oxygen Activation at the Active Site of a Fungal Lytic Polysaccharide Monooxygenase. Angewandte Chemie - International Edition. 56 (3), 767-770 (2017).
- O'Dell, W. B., Swartz, P. D., Weiss, K. L., Meilleur, F. Crystallization of a fungal lytic polysaccharide monooxygenase expressed from glycoengineered Pichia pastoris for X-ray and neutron diffraction. Acta Crystallographica Section:F Structural Biology Communications. 73 (2), 70-78 (2017).
- Meilleur, F., et al. The IMAGINE instrument: First neutron protein structure and new capabilities for neutron macromolecular crystallography. Acta Crystallographica Section D: Biological Crystallography. 69 (10), 2157-2160 (2013).
- Helliwell, J. R., et al. The recording and analysis of synchrotron X-radiation Laue diffraction photographs. Journal of Applied Crystallography. 22 (5), 483-497 (1989).
- Nieh, Y. P., et al. Accurate and highly complete synchrotron protein crystal Laue diffraction data using the ESRF CCD and the Daresbury Laue software. Journal of Synchrotron Radiation. 6 (5), 995-1006 (1999).
- Arzt, S., Campbell, J. W., Harding, M. M., Hao, Q., Helliwell, J. R. LSCALE - The new normalization, scaling and absorption correction program in the Daresbury Laue software suite. Journal of Applied Crystallography. 32 (3), 554-562 (1999).
- Sullivan, B., et al. Improving the accuracy and resolution of neutron crystallographic data by three-dimensional profile fitting of Bragg peaks in reciprocal space. Acta Crystallographica Section D: Structural Biology. 74 (11), 1085-1095 (2018).
- Sullivan, B., et al. BraggNet: integrating Bragg peaks using neural networks. Journal of Applied Crystallography. 52 (4), 854-863 (2019).
- Schröder, G. C., O'Dell, W. B., Myles, D. A. A., Kovalevsky, A., Meilleur, F. IMAGINE: Neutrons reveal enzyme chemistry. Acta Crystallographica Section D: Structural Biology. 74, (2018).
- Blum, M. M., et al. Preliminary time-of-flight neutron diffraction study on diisopropyl fluorophosphatase (DFPase) from Loligo vulgaris. Acta Crystallographica Section F: Structural Biology and Crystallization Communications. 63 (1), 42-45 (2007).
- Tomanicek, S. J., et al. Neutron and X-ray crystal structures of a perdeuterated enzyme inhibitor complex reveal the catalytic proton network of the Toho-1 β-lactamase for the acylation reaction. Journal of Biological Chemistry. 288 (7), 4715-4722 (2013).
- Metcalfe, C., et al. The tuberculosis prodrug isoniazid bound to activating peroxidases. Journal of Biological Chemistry. 283 (10), 6193-6200 (2008).
- Hughes, R. C., et al. Inorganic pyrophosphatase crystals from Thermococcus thioreducens for X-ray and neutron diffraction. Acta Crystallographica Section F: Structural Biology and Crystallization Communications. 68 (12), 1482-1487 (2012).
- Bennett, B. C., Meilleur, F., Myles, D. A. A., Howell, E. E., Dealwis, C. G. Preliminary neutron diffraction studies of Escherichia coli dihydrofolate reductase bound to the anticancer drug methotrexate. Acta Crystallographica Section D: Biological Crystallography. 61 (5), 574-579 (2005).
- Snell, E. H., et al. Optimizing crystal volume for neutron diffraction: D-xylose isomerase. European Biophysics Journal. 35 (7), 621-632 (2006).
- Golden, E., Attwood, P. V., Duff, A. P., Meilleur, F., Vrielink, A. Production and characterization of recombinant perdeuterated cholesterol oxidase. Analytical Biochemistry. 485, 102-108 (2015).
- Munshi, P., et al. Rapid visualization of hydrogen positions in protein neutron crystallographic structures. Acta Crystallographica Section D: Biological Crystallography. 68 (1), 35-41 (2012).
- Logan, D. T. Interactive model building in neutron macromolecular crystallography. Methods in Enzymology. 634, (2020).
- Koruza, K., Lafumat, B., Végvári, W., Knecht, S. Z., Fisher, Deuteration of human carbonic anhydrase for neutron crystallography: Cell culture media, protein thermostability, and crystallization behavior. Archives of Biochemistry and Biophysics. 645, 26-33 (2018).
- Fisher, S. J., et al. Perdeuteration: Improved visualization of solvent structure in neutron macromolecular crystallography. Acta Crystallographica Section D: Biological Crystallography. 70 (12), 3266-3272 (2014).
- Chen, J. C. H., Hanson, B. L., Fisher, S. Z., Langan, P., Kovalevsky, a. Y. Direct observation of hydrogen atom dynamics and interactions by ultrahigh resolution neutron protein crystallography. Proceedings of the National Academy of Sciences. 109 (38), 15301-15306 (2012).
- Cuypers, M. G., et al. Near-atomic resolution neutron crystallography on perdeuterated Pyrococcus furiosus rubredoxin: Implication of hydronium ions and protonation state equilibria in redox changes. Angewandte Chemie - International Edition. 52 (3), 1022-1025 (2013).
- Coates, L., Sullivan, B. The macromolecular neutron diffractometer at the spallation neutron source. Methods in Enzymology. 634, (2020).
- Ren, Z., Kingman, N. G., Borgstahl, G. E. O., Getzoff, E. D., Moffat, K. Quantitative Analysis of Time-Resolved Laue Diffraction Patterns. Journal of Applied Crystallography. 29 (3), 246-260 (1996).
- Campbell, J. W., Hao, Q., Harding, M. M., Nguti, N. D., Wilkinson, C. LAUEGEN version 6.0 and INTLDM. Journal of Applied Crystallography. 31 (3), 496-502 (1998).
- Arnold, O., et al. Mantid - Data analysis and visualization package for neutron scattering and μ SR experiments. Nuclear Instruments and Methods in Physics Research, Section A: Accelerators, Spectrometers, Detectors and Associated Equipment. 764, 156-166 (2014).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten