Se requiere una suscripción a JoVE para ver este contenido. Inicie sesión o comience su prueba gratuita.
Method Article
Siguiendo la dinámica de variantes estructurales en poblaciones evolucionadas experimentalmente
En este artículo
Resumen
Desarrollamos un método rentable para seguir la dinámica del alelo de polimorfismo de un solo nucleótido que puede adaptarse fácilmente a los archivos congelados de evolución experimental. Una técnica de PCR triplete se combinó con electroforesis capilar paralela automatizada para cuantificar la frecuencia relativa de un alelo de inserción en el transcurso de la evolución experimental.
Resumen
Ahora se sabe que las variantes estructurales (SV) (es decir, deleciones, inserciones, duplicaciones e inversiones) desempeñan un papel importante en la variación fenotípica y, en consecuencia, en procesos como la determinación de enfermedades o la adaptación a un nuevo entorno. Sin embargo, las variantes de un solo nucleótido reciben mucha más atención que las SV, probablemente porque son más fáciles de detectar y sus efectos fenotípicos son más fáciles de predecir. El desarrollo de tecnologías de secuenciación profunda de lectura corta y larga ha mejorado considerablemente la detección de SV, pero la cuantificación de su frecuencia a partir de datos de secuenciación agrupada (poolseq) sigue siendo técnicamente compleja y costosa.
Aquí, presentamos un método bastante simple y económico, que permite a los investigadores seguir la dinámica de la frecuencia del alelo SV. Como ejemplo de aplicación, seguimos la frecuencia de inserción de una secuencia de inserción (IS) en poblaciones de bacterias en evolución experimental. Este método se basa en el diseño de tripletes de cebadores alrededor de los bordes de la variante estructural, de modo que los amplicones producidos por la amplificación de los alelos de tipo salvaje (WT) y derivados difieren en tamaño en al menos un 5%, y que su eficiencia de amplificación es similar. La cantidad de cada amplicón se determina entonces mediante electroforesis capilar paralela y se normaliza a una curva de calibración. Este método puede extenderse fácilmente a la cuantificación de la frecuencia de otras variantes estructurales (deleciones, duplicaciones e inversiones) y a enfoques de agrupamiento de poblaciones naturales, incluidas las poblaciones de patógenos dentro de los pacientes.
Introducción
Las variantes estructurales (SV) son alteraciones de la secuencia genómica, que generalmente afectan a 50 pb o más. Las cuatro categorías de SV descritas son inserciones grandes, eliminaciones grandes, inversiones y duplicaciones. Hasta hace poco, se ha prestado más atención a las variantes de un solo nucleótido (SNV) que a las variantes estructurales, en términos de sus efectos fenotípicos y su papel como determinantes genéticos de la enfermedad, o su contribución a la adaptación. Esto se debe probablemente a que es más fácil detectar SNV y predecir sus efectos fenotípicos. Sin embargo, las tecnologías de secuenciación profunda de lectura corta y larga han mejorado fuertemente la detección de SV, al menos en genomas individuales o clonales1. Paralelamente, se han caracterizado mejor sus efectos fenotípicos y se han documentado muchos ejemplos de su implicación como determinantes genéticos de la enfermedad humana 2,3 o adaptación a un nuevo entorno4.
Las deleciones e inserciones, a menudo debidas a inserciones de elementos genéticos móviles (MGE), son mucho más perjudiciales que los polimorfismos de nucleótido único (SNP) y conducen a mutaciones de cambio de marco y modificaciones de la estructura de las proteínas. Las deleciones e inserciones de MGE dentro de los genes casi siempre resultan en la inactivación de genes, y las inserciones en regiones no codificantes pueden conducir a la represión o expresión constitutiva de genes adyacentes cuando las secuencias de inserción (IS) contienen secuencias promotoras o de terminación5. Si bien la eliminación de genes esenciales conduce a claros efectos perjudiciales sobre la aptitud bacteriana, la pérdida de genes no esenciales es beneficiosa en algunos casos. A pesar de sus costos inherentes, las duplicaciones también pueden ser ventajosas y participar en la adaptación, ya que conducen a un cambio en la dosis de genes; Un aumento en la actividad de una proteína específica puede ser ventajoso dependiendo de las condiciones6.
Las poblaciones de evolución experimental microbiana generalmente se inician con clones. Esta ausencia inicial de diversidad genética, combinada con el "ambiente cerrado" característico de los tubos de ensayo, conduce a un potencial muy limitado de evolución por ganancia de genes a través de la transferencia horizontal de genes y la recombinación. En estas condiciones específicas, la contribución a la adaptación de deleciones, duplicaciones e inserción intragenómica de MGE es particularmente importante; Las bacterias a menudo se adaptan a través de mutaciones de pérdida de función (principalmente debido a deleciones o inserciones de MGE), afectando genes que no son útiles en ambientes artificiales de monocultivo estables, a menudo ricos en nutrientes,7. En el experimento de evolución de E. coli de más larga duración, las inserciones de IS150 son particularmente frecuentes entre las poblaciones evolucionadas después de 50.000 generaciones, con elementos IS que representan el 35% de las mutaciones que alcanzan alta frecuencia en poblaciones que conservan su tasa de mutación puntual ancestral8.
Estudios de evolución y resecuencia que combinan la evolución experimental y las tecnologías de secuenciación de próxima generación (NGS) para investigar cómo las bacterias se adaptan, a nivel fenotípico y genómico, a diferentes condiciones ambientales y tensiones, como diferentes fuentes de carbono y energía, antibióticos y estrés osmótico 9,10,11 . Estos estudios suelen obtener información genómica sobre las poblaciones evolucionadas o clones únicamente en el punto final experimental y, en algunos casos, en una serie de puntos temporales intermedios12,13,14. Estos datos proporcionan información sobre los genes y las vías involucradas en la adaptación a un entorno determinado, pero rara vez permiten a los investigadores seguir la dinámica de los alelos emergentes y amplios de novo a lo largo del tiempo.
Un enfoque para seguir estas dinámicas es elegir un número limitado de alelos segregadores de interés (debido a la función de los genes que afectan, porque barren en paralelo en poblaciones independientes, etc.) y usar la secuenciación de amplicones para cuantificar la proporción de alelos, agrupando muchos puntos de tiempo en la misma ejecución de secuenciación15. Este método se ha utilizado con éxito para seguir la dinámica de variantes de pequeño tamaño (SNPs o indeles de 1 pb) en16 poblaciones experimentales ynaturales de 17 microbios. Sin embargo, en el caso de indeles más grandes o inserciones MGE, la diferencia de tamaño de los amplicones induce diferencias en la eficiencia de PCR, que distorsionan la relación entre las proporciones de lectura y alelos. En ciertos casos, la diferencia de tamaño entre los dos alelos es superior a la longitud clásica del amplicón. Aquí, combinamos una técnica de PCR triplete con electroforesis capilar paralela automatizada para cuantificar la frecuencia relativa de un alelo de inserción basado en la discriminación de tamaño. Este enfoque permite la explotación de puntos de tiempo experimentales infrautilizados para determinar la dinámica de un alelo mutante emergente y seguir su frecuencia hasta la fijación o pérdida, de una manera rentable. Aplicamos este método para rastrear alelos mutS emergentes, mutados a través de una inserción IS10, proporcionando al genotipo mutado un fenotipo hipermutador.
Este método requiere dos alelos objetivo con una diferencia de tamaño del ≥5%. En primer lugar, los trillizos de imprimación están diseñados para producir fragmentos de tamaño similar, que comparten una imprimación común. En segundo lugar, se optimizan las condiciones de PCR y se produce una curva de calibración utilizando mezclas de gDNA de tipo salvaje (WT) y mutante. Por último, las muestras se amplifican mediante PCR, y la frecuencia relativa de cada alelo se cuantifica mediante electroforesis capilar cuantitativa paralela.
Protocolo
La configuración de este protocolo requiere un conocimiento preciso del punto de inserción, eliminación, inversión o duplicación dentro de la secuencia ancestral. Esta información generalmente se obtiene mediante secuenciación del genoma completo (WGS) de las muestras puntuales finales o intermedias. En el siguiente protocolo, se da el principio general para el caso de una mutación de inserción para cada paso, junto con un caso representativo donde se sigue la frecuencia de una inserción IS10 en el gen mutS en una población experimental de evolución de E. coli. En esta población, WGS de la población de punto final identificó la inserción de un IS10 de 1.329 pb entre las posiciones 2.463 y 2.471, lo que resultó en la duplicación de este sitio de inserción. Este método es aplicable a los otros tres tipos de SV, y las especificidades de cada caso se dan en la discusión.
1. Diseño de imprimaciones tripletes
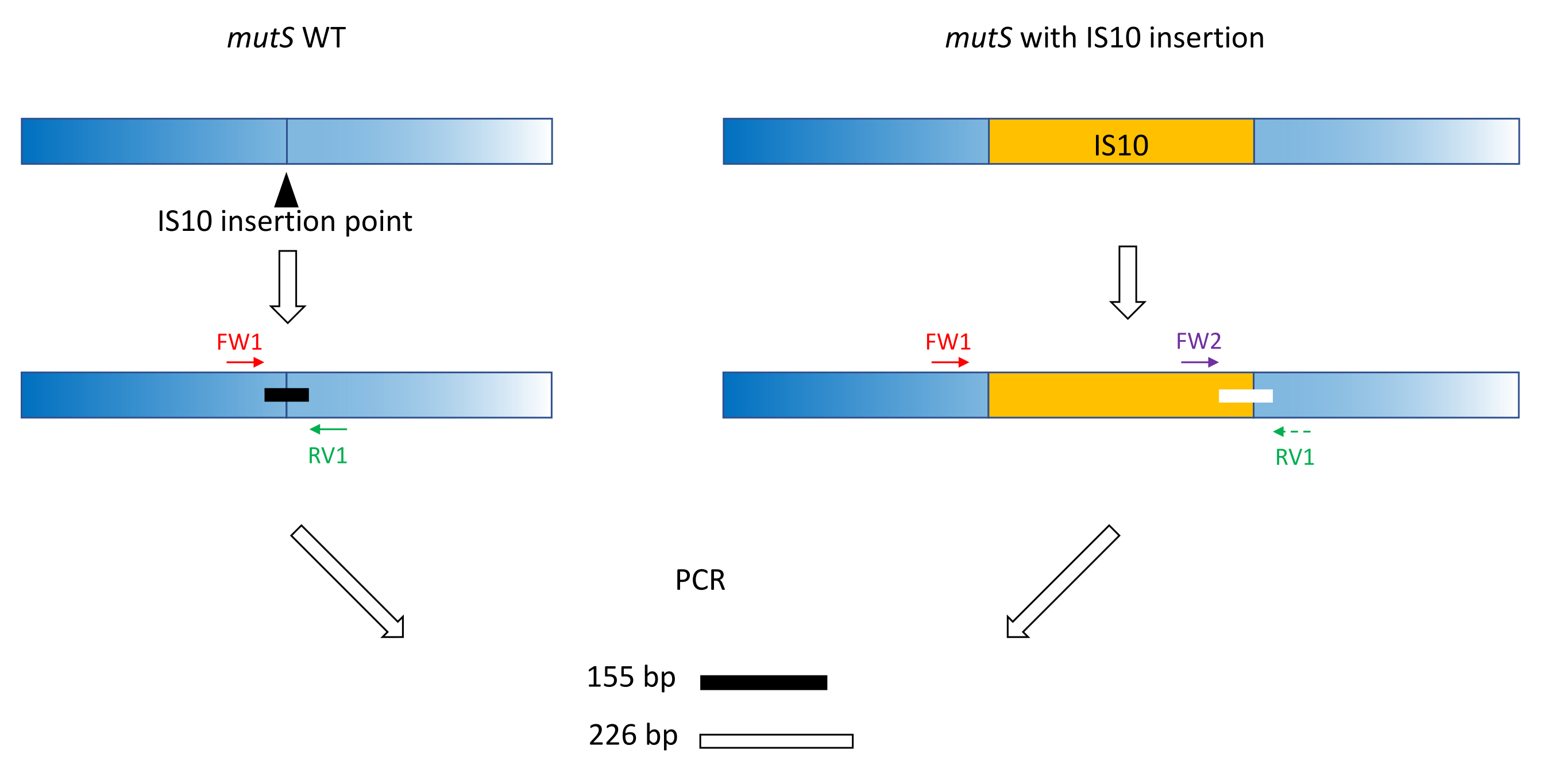
- Utilice prácticas clásicas de diseño de imprimación (18-24 pb, 40% -60% de contenido de GC, inicio / final con pares G / C cuando sea posible, diferencia de Tm < 5 ° C) para producir imprimaciones FW1 y RV1. Diseñar cebadores para amplificar un amplicón corto en el alelo WT alrededor del sitio de inserción del alelo mutante (Figura 1).
NOTA: El tamaño del amplicón puede variar de 100 pb a hasta 3.000 pb, en línea con la escala de tamaño de ADN utilizada en la electroforesis capilar. En este ejemplo, se amplificó un amplicón de 155 pb. El pequeño tamaño de fragmento elegido aquí evita la amplificación fuera del objetivo de toda la secuencia de inserción IS10 (ver sección 2). - Diseñe un segundo cebador directo FW2 dentro de la secuencia de inserción, para producir un segundo amplicón que sea aproximadamente un 5% más grande o más pequeño que el amplicón WT (Figura 1). Esta diferencia de tamaño del 5% es la diferencia de tamaño mínima que el dispositivo de electroforesis capilar paralela podría distinguir de manera confiable. Por lo tanto, diseñe los cebadores de tal manera que los dos amplicones tengan una diferencia de tamaño, que esté por encima pero lo más cerca posible del umbral relativo.
NOTA: Asegúrese de minimizar la diferencia Tm y la formación de dímeros de imprimación. En este ejemplo, se diseñó un segundo cebador directo para producir un amplicón de 226 pb, 71 pb más grande que el amplicón WT. En este ejemplo representativo, las secuencias de cebadores son las siguientes:
FW1:AAAGCATTTCGCCGAACGCC
RV1: GCGATAAATCCACTCCAGCGCC
FW2: AGTTCGCTTAGGCATGGAAG

Figura 1: Esquema del diseño del cebador triplete en el gen mutS WT y la inserción mutante mutS IS10. El triángulo negro representa el sitio de inserción IS10 en el gen mutS. El gen WT está en azul, y el IS10 está en naranja. Los cebadores FW1 y RV1 marcan el sitio de inserción IS10 y producen un amplicón WT de 155 pb. El cebador RV1 y el cebador intra-IS10 FW2 producen un segundo amplicón de 226 pb. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
2. Optimización de las condiciones de PCR
- Cultivar un cultivo nocturno de WT fijo y clones de alelos mutantes.
- Extrae el ADN usando cualquier kit.
- Cuantificar el ADN.
- Prepare la muestra de ADN diluyendo la extracción de WT y ADN mutante a 5 ng / μL. Mezcle las dos muestras de ADN en una proporción de 50/50.
- Amplíe 10 ng de las tres muestras de ADN (WT, mutante, mezcla 50/50) utilizando una mezcla maestra de PCR lista para usar 2x, cebador FW1 de 0,5 μM, cebador RV1 de 1 μM y cebador FW2 de 0,5 μM en un volumen de reacción de 20 μL. Utilice un gel de agarosa al 2% para migrar el producto de PCR mediante electroforesis clásica y determine las condiciones óptimas de PCR.
NOTA: Los tiempos de elongación deben minimizarse para evitar la formación del amplicón FW1 y RV1 en el alelo mutante. La temperatura de recocido debe ajustarse para minimizar la amplificación sesgada de los alelos y la amplificación no específica.- Para seguir el programa de este ejemplo, utilice los siguientes ajustes: 98 °C durante 10 s, seguido de 25 ciclos de 98 °C durante 1 s, 58 °C durante 15 s, 72 °C durante 8 s y un paso final de elongación a 72 °C durante 1 min.
NOTA: El tiempo de elongación se redujo a 8 s para evitar la amplificación del producto de >1.000 pb del cebador directo y el cebador RV1 en el alelo mutante (mutS con inserción IS10).
- Para seguir el programa de este ejemplo, utilice los siguientes ajustes: 98 °C durante 10 s, seguido de 25 ciclos de 98 °C durante 1 s, 58 °C durante 15 s, 72 °C durante 8 s y un paso final de elongación a 72 °C durante 1 min.
3. Curva de calibración
- Mezcle las dos muestras de ADN, WT y mutante, en proporciones 10/90, 25/75, 40/60, 50/50, 60/40, 75/25 y 90/10.
NOTA: Las réplicas biológicas se preparan a partir de cultivos bacterianos independientes durante la noche. - Amplificar utilizando condiciones de PCR optimizadas (ver sección 2).
- Cuantificar el producto amplicón.
- Diluir los productos de PCR a 0,1 ng/μL.
- Preparar el instrumento de electroforesis capilar paralela.
- Mezcle gel fresco y colorante (kit de análisis cuantitativo NGS (22, 33 o 55); Fragmento HS NGS 1-6,000 pb para este ejemplo).
NOTA: Consulte la guía de fragmentos de electroforesis capilar paralela HS NGS para obtener instrucciones detalladas (consulte la Tabla de materiales).
- Mezcle gel fresco y colorante (kit de análisis cuantitativo NGS (22, 33 o 55); Fragmento HS NGS 1-6,000 pb para este ejemplo).
- Reemplace la solución de almacenamiento capilar y el tampón de entrada, y coloque la placa tampón de enjuague en las ubicaciones correctas de los cajones del instrumento de electroforesis capilar paralela.
- Agregue 2 μL de marcador diluyente HS a 22 μL de cada muestra diluida en una placa de 96 pocillos.
- Agregue una escalera de tamaño (escalera de tamaño de ADN; rango 1-6,000 pb) de un kit de análisis cuantitativo HS NGS a un pocillo de la placa de 96 pocillos.
- Coloque la placa de 96 pocillos en el cajón correcto del instrumento de electroforesis capilar paralela y seleccione Ejecutar en el software del instrumento de electroforesis capilar paralela.
- Analice los resultados utilizando el software de análisis de datos, que detecta e identifica cada pico de la escala de tamaño, asignando cada pico de las muestras a su tamaño real conocido.
- Cuando se utiliza un kit cuantitativo, utilice el software para determinar la cantidad de ADN de cada fragmento mediante la integración del área bajo el pico, como en el análisis de datos de cromatografía. Una vez más, compare las muestras con las cantidades conocidas en los estándares para cuantificar cada pico de una muestra y calcule las proporciones entre los diferentes picos detectados en las muestras.
- Construir una curva de calibración (Figura 2), vinculando la proporción conocida del alelo mutante (mezcla de ADN) con la medida utilizando el instrumento de electroforesis capilar paralela. Esta curva de calibración permite evaluar y corregir la fiabilidad del método para detectar un sesgo de amplificación menor.
4. Preparación de la muestra
- Cultive muestras puntuales durante la noche en condiciones estándar.
- Extraer el ADN.
- Cuantificar el ADN.
- Amplificar las muestras utilizando condiciones de PCR optimizadas (ver sección 2).
- Ejecutar las muestras en el instrumento de electroforesis capilar paralela (ver pasos 3.5-3.10).
5. Cuantificación de alelos
- Extraiga cantidades de alelos mutantes de los datos del instrumento de electroforesis capilar paralela con el software y calcule las proporciones reales trazando estos valores en la curva de calibración.
Resultados
Utilizando ADN extraído de un clon ancestral y un clon hipermutador aislado de la población S2.11 en la generación 1.000, establecimos la curva de calibración que se muestra en la Figura 2. Las proporciones mutantes reales de mezclas de ADN preparadas en laboratorio y medidas por el instrumento de electroforesis capilar paralela se unieron mediante una relación lineal de pendiente 1.0706, con un R2 de 0.9705. Además, hubo un buen acuerdo entre las réplicas biológicas; La d...
Discusión
Aquí, hemos propuesto un método rentable que permite seguir la dinámica de los alelos SV adaptativos emergentes en poblaciones de evolución experimental. Este método combina las técnicas clásicas de PCR y la electroforesis capilar paralela automatizada, lo que permite determinar las cantidades relativas de dos alelos. Una vez configurado, permite la cuantificación de proporciones alélicas en muchas muestras en paralelo, y es mucho menos costoso que WGS. Este método puede verse como un equivalente a la secuencia...
Divulgaciones
Los autores no tienen conflictos de intereses que revelar.
Agradecimientos
Este trabajo fue apoyado por el ERC HGTCODONUSE (ERC-2015-CoG-682819) a S.B. Los datos utilizados en este trabajo fueron (parcialmente) producidos a través de las instalaciones técnicas GenSeq del Institut des Sciences de l'Evolution de Montpellier con el apoyo de LabEx CeMEB, un programa ANR "Investissements d'avenir" (ANR-10-LABX-04-01).
Materiales
| Name | Company | Catalog Number | Comments |
| 96 Well Skirted PCR Plate | 4titude | 4Ti - 0740 | PCR |
| Agarose molecular biology grade | Eurogentec | EP-0010-05 | Agarose gel electrophoresis |
| Agilent DNF-474 HS NGS Fragment Kit Quick Guide for the Fragment Analyzer Systems | Agilent | PDF instruction guide | |
| Buffer TBE | Panreac appliChem | A4228,5000Pc | Agarose gel electrophoresis |
| Calibrated Disposable Inoculating Loops and Needles | LABELIANS | 8175CSR40H | Bacterial culture |
| Dneasy Blood and Tissue Kit | Qiagen | 69506 | DNA extraction |
| Electrophoresis power supply | Amilabo | ST606T | Agarose gel electrophoresis |
| Fragment Analyzer Automated CE System | Agilent | Parallel capillary electrophoresis | |
| Fragment DNA Ladder | Agilent | DNF-396, range 1-6000bp | Parallel capillary electrophoresis |
| GENTAMICIN SULFATE SALT BIOREAGENT | Sigma-Aldrich | G1264-1G | Bacterial culture |
| High Sensitivity diluent marker | Agilent | DNF-373 | Parallel capillary electrophoresis |
| High Sensitivity NGS quantitative analysis kit | Agilent | DNF-474 | Parallel capillary electrophoresis |
| Ladder quick load 1 kb plus DNA ladder | NEB | N0469S | Agarose gel electrophoresis |
| LB Broth, VegitoneNutriSelect Plus | Millipore | 28713 | Bacterial culture |
| Master Mix PCR High Fidelity Phusion Flash | Thermo Fisher Scientific | F548L | PCR |
| Primers | Eurogentec | PCR | |
| Prosize data analysis software v.4 | Agilent | V.4 | Parallel capillary electrophoresis |
| Qubit assays | Invitrogen | MAN0010876 | DNA quantification |
| Qubit dsDNA HS Assay Kit | LIFE TECHNOLOGIES SAS | Q32854 | DNA quantification |
| Thermocycler | Eppendorf | Ep gradients | PCR |
| UVbox, eBOX VX5 | Vilber Lourmat | Agarose gel electrophoresis visualisation | |
| Water for injectable preparation | Aguettant | PROAMP | PCR |
Referencias
- Mahmoud, M., et al. Structural variant calling: the long and the short of it. Genome Biology. 20 (1), 246 (2019).
- Bragg, D. C., et al. Disease onset in X-linked dystonia-parkinsonism correlates with expansion of a hexameric repeat within an SVA retrotransposon in TAF1. Proceedings of the National Academy of Sciences. 114 (51), 11020-11028 (2017).
- Stransky, N., Cerami, E., Schalm, S., Kim, J. L., Lengauer, C. The landscape of kinase fusions in cancer. Nature Communications. 5, 4846 (2014).
- Tenaillon, O., et al. The molecular diversity of adaptive convergence. Science. 335 (6067), 457-461 (2012).
- Vandecraen, J., Chandler, M., Aertsen, A., Van Houdt, R. The impact of insertion sequences on bacterial genome plasticity and adaptability. Critical Reviews in Microbiology. 43 (6), 709-730 (2017).
- Andersson, D. I., Gene Hughes, D. amplification and adaptive evolution in bacteria. Annual Review of Genetics. 43, 167-195 (2009).
- Bailey, S. F., Bataillon, T. Can the experimental evolution programme help us elucidate the genetic basis of adaptation in nature. Molecular Ecology. 25 (1), 203-218 (2016).
- Consuegra, J., et al. Insertion-sequence-mediated mutations both promote and constrain evolvability during a long-term experiment with bacteria. Nature Communications. 12 (1), 980 (2021).
- Burch, C. L., Romanchuk, A., Kelly, M., Wu, Y., Jones, C. D. Genome-wide determination of barriers to horizontal gene transfer. bioRxiv. , (2022).
- Slomka, S., et al. Experimental evolution of Bacillus subtilis reveals the evolutionary dynamics of horizontal gene transfer and suggests adaptive and neutral effects. Genetics. 216 (2), 543-558 (2020).
- Choudhury, D., Saini, S. Evolution of Escherichia coli in different carbon environments for 2,000 generations. Journal of Evolutionary Biology. 32 (12), 1331-1341 (2019).
- Tenaillon, O., et al. Tempo and mode of genome evolution in a 50,000-generation experiment. Nature. 536 (7615), 165-170 (2016).
- Behringer, M. G., et al. Escherichiacoli cultures maintain stable subpopulation structure during long-term evolution. Proceedings of the National Academy of Sciences. 115 (20), 4642-4650 (2018).
- Voordeckers, K., et al. Adaptation to high ethanol reveals complex evolutionary pathways. PLoS Genetics. 11 (11), 1005635 (2015).
- Levy, S. F., et al. Quantitative evolutionary dynamics using high-resolution lineage tracking. Nature. 519 (7542), 181-186 (2015).
- Bruger, E. L., Marx, C. J. A decade of genome sequencing has revolutionized studies of experimental evolution. Current Opinion in Microbiology. 45, 149-155 (2018).
- Grubaugh, N. D., et al. An amplicon-based sequencing framework for accurately measuring intrahost virus diversity using PrimalSeq and iVar. Genome Biology. 20 (1), 8 (2019).
- Bedhomme, S., et al. Evolutionary changes after translational challenges imposed by horizontal gene transfer. Genome Biology and Evolution. 11 (3), 814-831 (2019).
- Tenaillon, O., Toupance, B., Le Nagard, H., Taddei, F., Godelle, B. Mutators, population size, adaptive landscape and the adaptation of asexual populations of bacteria. Genetics. 152 (2), 485-493 (1999).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoExplorar más artículos
This article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados