JoVE 비디오를 활용하시려면 도서관을 통한 기관 구독이 필요합니다. 전체 비디오를 보시려면 로그인하거나 무료 트라이얼을 시작하세요.
Method Article
실험적으로 진화한 개체군에서 구조적 변이의 역학을 따릅니다.
요약
우리는 실험적 진화 동결 아카이브에 쉽게 적응할 수 있는 비단일 뉴클레오티드 다형성 대립유전자 역학을 추적하는 비용 효율적인 방법을 개발했습니다. 삼중항 PCR 기술은 실험적 진화 과정에서 삽입 대립 유전자의 상대적 빈도를 정량화하기 위해 자동화된 병렬 모세관 전기영동과 결합되었습니다.
초록
구조적 변이체(SV)(즉, 결실, 삽입, 복제 및 반전)는 이제 표현형 변이에서 중요한 역할을 하는 것으로 알려져 있으며, 결과적으로 질병 결정 또는 새로운 환경에 대한 적응과 같은 과정에서 중요한 역할을 하는 것으로 알려져 있습니다. 그러나 단일 뉴클레오티드 변이체는 SV보다 훨씬 더 많은 관심을 받는데, 이는 아마도 검출하기 쉽고 표현형 효과를 예측하기 쉽기 때문일 것입니다. 단거리 및 장판독 심층 염기서열 분석 기술의 개발로 SV 검출이 크게 향상되었지만, 풀링된 염기서열 분석(poolseq) 데이터에서 SV의 빈도를 정량화하는 것은 여전히 기술적으로 복잡하고 비용이 많이 듭니다.
여기에서 우리는 연구자들이 SV 대립 유전자 빈도의 역학을 따를 수 있도록 하는 다소 간단하고 저렴한 방법을 제시합니다. 적용의 예로서, 우리는 박테리아의 실험적 진화 집단에서 삽입 서열(IS) 삽입 빈도를 따릅니다. 이 방법은 야생형(WT)과 파생 대립유전자의 증폭에 의해 생성된 앰플리콘의 크기가 5% 이상 다르고 증폭 효율이 유사하도록 구조적 변이체 경계 주변의 프라이머 삼중체 설계를 기반으로 합니다. 그런 다음 각 앰플리콘의 양은 평행 모세관 전기영동에 의해 결정되고 보정 곡선으로 정규화됩니다. 이 방법은 다른 구조적 변이(결실, 복제 및 반전)의 빈도 정량화와 환자 내 병원체 개체군을 포함한 자연 개체군의 풀-시퀀스 접근 방식으로 쉽게 확장할 수 있습니다.
서문
구조적 변이체(SV)는 게놈 서열의 변경으로, 일반적으로 50bp 이상에 영향을 미칩니다. 설명된 SV의 네 가지 범주는 큰 삽입, 큰 삭제, 반전 및 중복입니다. 최근까지 단일 뉴클레오티드 변이체(SNV)의 표현형 효과와 질병의 유전적 결정 요인으로서의 역할 또는 적응에 대한 기여 측면에서 구조적 변이체보다 단일 뉴클레오티드 변이체(SNV)에 더 많은 관심이 기울여졌습니다. 이것은 아마도 SNV를 감지하고 표현형 효과를 예측하는 것이 더 쉽기 때문일 것입니다. 그러나 단거리 및 장판독 심층 염기서열 분석 기술은 적어도 단일 개인 또는 클론 게놈에서 SV의 검출을 크게 개선했습니다1. 이와 동시에, 이들의 표현형 효과는 더 잘 특성화되었으며, 인간 질병2,3 또는 새로운 환경4에 대한 적응의 유전적 결정인자로서의 의미에 대한 많은 예가 문서화되었습니다.
종종 이동 유전 요소(MGE) 삽입으로 인한 결실 및 삽입은 단일 뉴클레오티드 다형성(SNP)보다 훨씬 더 파괴적이며 프레임시프트 돌연변이 및 단백질 구조 변형으로 이어집니다. 유전자 내의 결실 및 MGE 삽입은 거의 항상 유전자 불활성화를 초래하며, 삽입 서열(IS)이 프로모터 또는 종결 서열을 함유하는 경우 비코딩 영역으로의 삽입은 인접 유전자의 억제 또는 구성적 발현을 유발할 수 있다5. 필수 유전자의 녹아웃은 박테리아 적합성에 명백한 해로운 영향을 미치지만, 비필수 유전자의 손실은 경우에 따라 유익합니다. 고유한 비용에도 불구하고 복제는 유리할 수 있으며 유전자 투여량의 변화로 이어지기 때문에 적응에 참여할 수 있습니다. 특정 단백질의 활성의 증가는 조건에 따라 유리할 수 있다6.
미생물 실험 진화 집단은 일반적으로 클론으로 시작됩니다. 이러한 유전적 다양성의 초기 부재는 시험관의 "폐쇄된 환경" 특성과 결합되어 수평적 유전자 전달 및 재조합을 통한 유전자 획득에 의한 진화 가능성을 매우 제한적으로 만듭니다. 이러한 특정 조건에서 결실, 복제 및 게놈 내 MGE 삽입의 적응에 대한 기여가 특히 중요합니다. 박테리아는 종종 기능 상실 돌연변이(주로 결실 또는 MGE 삽입으로 인한)를 통해 적응하여 안정적이고 영양이 풍부한 단일 배양 인공 환경에서 유용하지 않은 유전자에 영향을 미칩니다7. 가장 오래 지속된 대장균 진화 실험에서, IS150 삽입은 50,000세대 후에 진화된 집단에서 특히 빈번하며, IS 요소는 조상 점 돌연변이율을 유지하는 집단에서 높은 빈도에 도달하는 돌연변이의 35%를 나타낸다8.
진화 및 재서열 연구는 실험 진화와 차세대 염기서열 분석(NGS) 기술을 결합하여 박테리아가 표현형 및 게놈 수준에서 다양한 탄소 및 에너지원, 항생제 및 삼투압 스트레스와 같은 다양한 환경 조건 및 스트레스에 어떻게 적응하는지 조사합니다 9,10,11 . 이러한 연구는 전형적으로 실험적 종점에서만, 그리고 어떤 경우에는 다수의 중간 시점(12,13,14)에서 진화된 집단 또는 클론에 대한 게놈 정보를 얻는다. 이러한 데이터는 주어진 환경에 대한 적응과 관련된 유전자와 경로에 대한 통찰력을 제공하지만 연구자가 시간이 지남에 따라 새롭게 나타나고 휩쓸리는 대립 유전자의 역학을 추적하는 것을 거의 허용하지 않습니다.
이러한 역학을 따르는 한 가지 접근법은 관심 있는 분리형 대립유전자의 제한된 수를 선택하고(영향을 미치는 유전자의 기능 때문에, 독립적인 집단에서 병렬로 스윕하기 때문에 등) 앰플리콘 시퀀싱을 사용하여 대립유전자 비율을 정량화하고 동일한 시퀀싱 실행에서 많은 시점을 통합하는 것이다15. 이 방법은 실험 16개 및 자연 미생물17 개 개체군에서 작은 크기 변이체(SNP 또는1bp 인델)의 역학을 추적하는 데 성공적으로 사용되었습니다. 그러나 더 큰 인델 또는 MGE 삽입의 경우 앰플리콘의 크기 차이가 PCR 효율 차이를 유도하여 판독과 대립 유전자 비율 사이의 관계를 왜곡합니다. 어떤 경우에는 두 대립 유전자 사이의 크기 차이가 앰플리콘의 고전적인 길이보다 우수합니다. 여기에서 우리는 삼중항 PCR 기술과 자동화된 병렬 모세관 전기영동을 결합하여 크기 차별을 기반으로 삽입 대립 유전자의 상대적 빈도를 정량화했습니다. 이 접근 방식을 사용하면 잘 사용되지 않는 실험 시점을 활용하여 새로운 돌연변이 대립 유전자의 역학을 결정하고 비용 효율적인 방식으로 고정 또는 손실에 대한 빈도를 따를 수 있습니다. 우리는 IS10 삽입을 통해 돌연변이된 새로운 mutS-대립유전자를 추적하기 위해 이 방법을 적용하여 돌연변이된 유전자형에 과돌연변이자 표현형을 제공했습니다.
이 방법은 크기가 ≥5% 차이가 나는 두 개의 표적 대립 유전자가 필요합니다. 첫째, 프라이머 삼중체는 공통 프라이머를 공유하는 유사한 크기의 단편을 생성하도록 설계되었습니다. 둘째, PCR 조건을 최적화하고 야생형(WT)과 돌연변이 gDNA의 혼합물을 사용하여 보정 곡선을 생성합니다. 마지막으로, 샘플은 PCR에 의해 증폭되고, 각 대립 유전자의 상대 주파수는 병렬 정량적 모세관 전기 영동에 의해 정량화됩니다.
프로토콜
이 프로토콜을 설정하려면 조상 서열 내의 삽입, 삭제, 반전 또는 복제 지점에 대한 정확한 지식이 필요합니다. 이 정보는 일반적으로 종점 또는 중간점 샘플의 전체 게놈 시퀀싱(WGS)에 의해 얻어집니다. 다음 프로토콜에서는 삽입 돌연변이의 경우에 대한 일반 원칙이 각 단계에 대해 제공되며, 대장균의 실험적 진화 집단에서 mutS 유전자에 IS10 삽입 빈도를 따르는 대표적인 사례가 있습니다. 이 모집단에서 종점 모집단의 WGS는 위치 2,463과 2,471 사이에 1,329bp IS10의 삽입을 식별하여 이 삽입 부위의 중복을 초래했습니다. 이 방법은 세 가지 다른 SV 유형에 적용할 수 있으며 각 경우의 특수성은 논의에서 제공됩니다.
1. 삼중항 프라이머의 설계
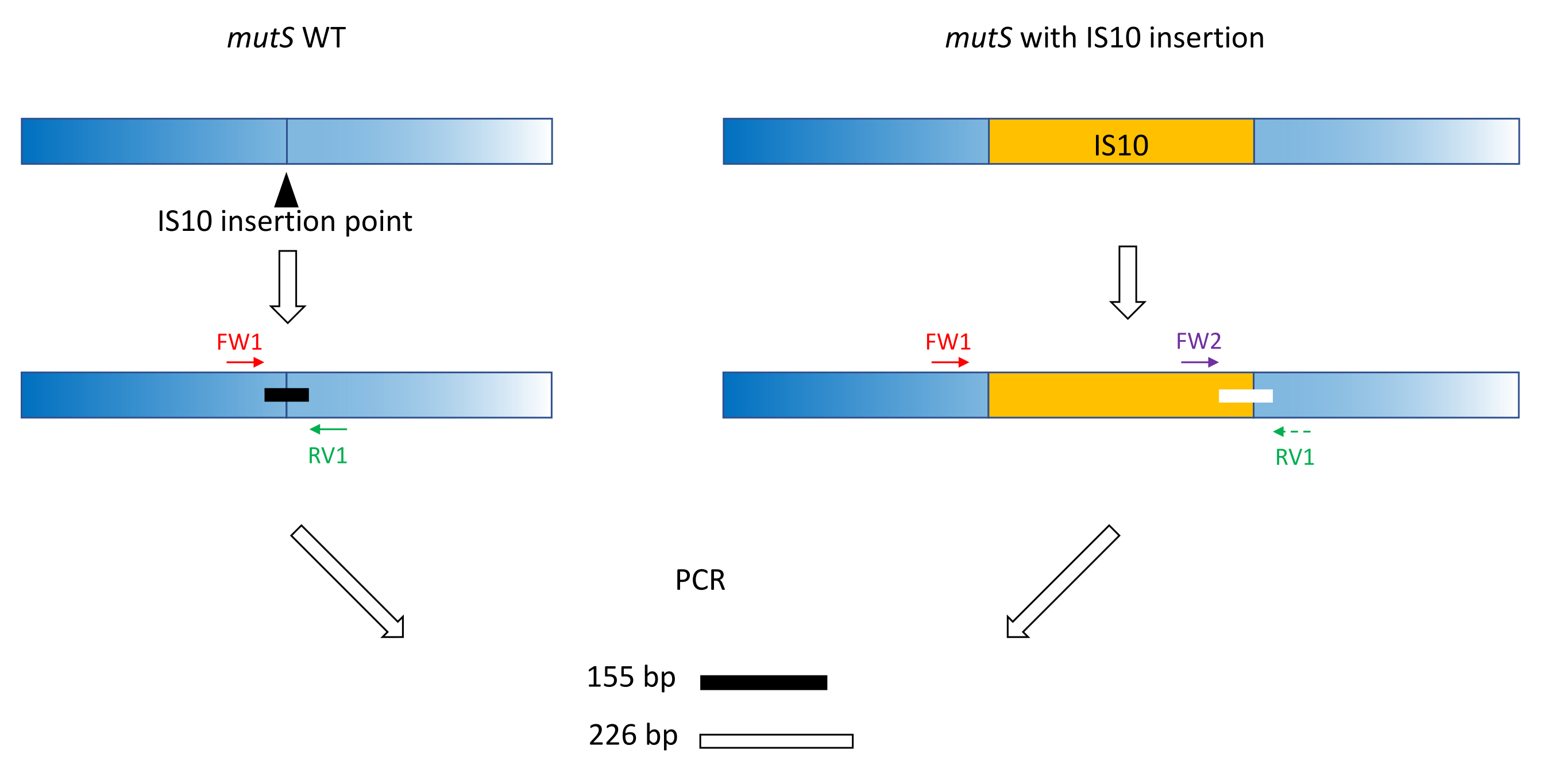
- 고전적인 프라이머 설계 방법(18-24bp, 40%-60% GC 함량, 가능한 경우 G/C 쌍으로 시작/종료,Tm 차이< 5°C)을 사용하여 프라이머 FW1 및 RV1을 생산합니다. 돌연변이 대립유전자 삽입 부위 주변의 WT 대립유전자에서 짧은 앰플리콘을 증폭하기 위한 프라이머를 설계합니다(그림 1).
참고: 앰플리콘 크기는 모세관 전기영동에 사용되는 DNA 크기 사다리에 따라 100bp에서 최대 3,000bp까지 다양할 수 있습니다. 이 예에서, 155 bp 앰플리콘이 증폭되었다. 여기에서 선택한 작은 단편 크기는 전체 IS10 삽입 시퀀스의 타겟 이탈 증폭을 방지합니다(섹션 2 참조). - 삽입 시퀀스 내에서 두 번째 정방향 프라이머 FW2를 설계하여 WT 앰플리콘보다 약 5% 크거나 작은 두 번째 앰플리콘을 생성합니다(그림 1). 이 5%의 크기 차이는 병렬 모세관 전기영동 장치가 확실하게 구별할 수 있는 최소 크기 차이입니다. 따라서 두 앰플리콘의 크기 차이가 상대 임계값보다 높지만 가능한 한 가깝도록 프라이머를 설계합니다.
참고:Tm 차이와 프라이머 이량체 형성을 최소화해야 합니다. 이 예에서, 제2 정방향 프라이머는 WT 앰플리콘보다 71 bp 더 큰 226 bp 앰플리콘을 생성하도록 설계되었다. 이러한 대표적인 예에서, 프라이머 서열은 다음과 같다.
FW1:AAAGCATTTCGCCGAACGCC
RV1: GCGATAAATCCACTCCAGCGCC
FW2: AGTTCGCTTAGGCATGGAAG

그림 1: mutS WT 유전자 및 돌연변이 mutS IS10 삽입에 대한 삼중항 프라이머 설계 스키마. 검은색 삼각형은 mutS 유전자의 IS10 삽입 부위를 나타냅니다. WT 유전자는 파란색이고 IS10은 주황색입니다. 프라이머 FW1 및 RV1은 IS10 삽입 부위에 플래그를 지정하고 155bp WT 앰플리콘을 생성합니다. RV1 프라이머와 IS10 내 프라이머 FW2는 두 번째 226bp 앰플리콘을 생성합니다. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
{kind=link}
2. PCR 조건의 최적화
- 고정된 WT 및 돌연변이 대립유전자 클론의 하룻밤 배양을 성장시킵니다.
- 모든 키트를 사용하여 DNA를 추출합니다.
- DNA를 정량화합니다.
- WT 및 돌연변이 DNA의 추출을 5ng/μL로 희석하여 DNA 샘플을 준비합니다. 두 DNA 샘플을 50/50 비율로 혼합합니다.
- 20μL 반응 부피에서 바로 사용할 수 있는 2x PCR 마스터 믹스, 0.5μM FW1 프라이머, 1μM RV1 프라이머 및 0.5μM FW2 프라이머를 사용하여 3개의 DNA 샘플(WT, 돌연변이, 50/50 믹스) 중 10ng을 증폭합니다. 2% 아가로스 겔을 사용하여 고전적인 전기영동에 의해 PCR 산물을 이동시키고 최적의 PCR 조건을 결정합니다.
참고: 돌연변이 대립유전자에 FW1 및 RV1 앰플리콘이 형성되는 것을 방지하기 위해 신장 시간을 최소화해야 합니다. 어닐링 온도는 대립 유전자의 편향된 증폭과 비특이적 증폭을 최소화하기 위해 조정되어야 합니다.- 이 예제의 프로그램을 따르려면 98°C에서 10초, 98°C에서 1초, 58°C에서 15초, 72°C에서 8초, 72°C에서 1분 동안 최종 신장 단계를 수행합니다.
참고: 돌연변이 대립유전자에 대한 정방향 프라이머 및 RV1 프라이머로부터 >1,000bp 생성물의 증폭을 방지하기 위해 신장 시간을 8초로 단축했습니다(IS10 삽입이 있는 mutS ).
- 이 예제의 프로그램을 따르려면 98°C에서 10초, 98°C에서 1초, 58°C에서 15초, 72°C에서 8초, 72°C에서 1분 동안 최종 신장 단계를 수행합니다.
3. 교정 곡선
- 두 개의 DNA 샘플인 WT와 돌연변이체를 10/90, 25/75, 40/60, 50/50, 60/40, 75/25 및 90/10 비율로 혼합합니다.
참고: 생물학적 복제물은 독립적인 하룻밤 박테리아 배양에서 준비됩니다. - 최적화된 PCR 조건을 사용하여 증폭합니다(섹션 2 참조).
- 앰플리콘 생성물을 정량화합니다.
- PCR 산물을 0.1ng/μL로 희석합니다.
- 병렬 모세관 전기영동 기기를 준비합니다.
- 신선한 겔과 염료를 혼합한다 (NGS 정량 분석 키트 (22, 33, 또는 55); HS NGS 단편은 이 예에서 1-6,000 bp).
참고: 자세한 지침은 병렬 모세관 전기영동 HS NGS 단편 가이드를 참조하십시오( 재료 표 참조).
- 신선한 겔과 염료를 혼합한다 (NGS 정량 분석 키트 (22, 33, 또는 55); HS NGS 단편은 이 예에서 1-6,000 bp).
- 모세관 저장 용액과 입구 버퍼를 교체하고 헹굼 버퍼 플레이트를 평행 모세관 전기영동 기기의 올바른 서랍 위치에 놓습니다.
- 96웰 플레이트에서 각 희석된 샘플 22μL에 HS 희석제 마커 2μL를 추가합니다.
- HS NGS 정량 분석 키트의 크기 사다리(DNA size ladder; 범위 1-6,000 bp)를 96-웰 플레이트의 하나의 웰에 추가합니다.
- 96웰 플레이트를 병렬 모세관 전기영동 기기의 올바른 서랍에 넣고 병렬 모세관 전기영동 기기 소프트웨어에서 실행을 선택합니다.
- 크기 사다리의 각 피크를 감지 및 식별하고 샘플의 각 피크를 알려진 실제 크기에 할당하는 데이터 분석 소프트웨어를 사용하여 결과를 분석합니다.
- 정량 키트를 사용하는 경우 크로마토그래피 데이터 분석에서와 같이 피크 아래 면적을 통합하여 각 단편의 DNA 양을 결정하는 소프트웨어를 사용합니다. 다시 말하지만, 표준물질에서 알려진 양과 샘플을 비교하여 샘플에서 각 피크를 정량화하고 샘플에서 검출된 서로 다른 피크 간의 비율을 계산합니다.
- 돌연변이 대립유전자(DNA 혼합)의 알려진 비율을 평행 모세관 전기영동 기기를 사용하여 측정된 비율과 연결하는 보정 곡선(그림 2)을 구성합니다. 이 검량선을 통해 경미한 증폭 편향에 대해 분석법의 신뢰성을 평가하고 보정할 수 있습니다.
4. 시료 전처리
- 표준 조건에서 하룻밤 사이에 시점 샘플을 성장시킵니다.
- DNA를 추출합니다.
- DNA를 정량화합니다.
- 최적화된 PCR 조건을 사용하여 샘플을 증폭합니다(섹션 2 참조).
- 병렬 모세관 전기영동 기기에서 샘플을 실행합니다(3.5-3.10단계 참조).
5. 대립유전자 정량화
- 소프트웨어를 사용하여 병렬 모세관 전기영동 기기 데이터에서 돌연변이 대립유전자 양을 추출하고 이러한 값을 검량선에 표시하여 실제 비율을 계산합니다.
결과
조상 클론에서 추출한 DNA와 1,000세대의 S2.11 집단에서 분리된 과돌연변이 클론을 사용하여 그림 2에 표시된 보정 곡선을 설정했습니다. 실험실에서 준비한 DNA 혼합물의 실제 돌연변이 비율과 평행 모세관 전기영동 기기로 측정한 값은 기울기 1.0706의 선형 관계와 R2 0.9705로 연결되었습니다. 또한 생물학적 복제물 사이에는 좋은 일치가 있었습니다. 표준 편차는 표준 ?...
토론
여기에서 우리는 실험적 진화 집단에서 새로운 적응 SV 대립 유전자의 역학을 따를 수 있는 비용 효율적인 방법을 제안했습니다. 이 방법은 고전적인 PCR 기술과 자동화된 병렬 모세관 전기영동을 결합하여 두 대립 유전자의 상대적인 양을 측정할 수 있습니다. 일단 설정되면 많은 샘플에서 대립 유전자 비율을 병렬로 정량화할 수 있으며 WGS보다 훨씬 저렴합니다. 이 방법은 non-SNP 돌연변이에 대한 ...
공개
저자는 공개할 이해 상충이 없습니다.
감사의 말
이 작업은 ERC HGTCODONUSE (ERC-2015-CoG-682819)에서 S.B.에 의해 지원되었습니다. 이 작업에 사용 된 데이터는 ANR "Investissements d' avenir"프로그램 (ANR-10-LABX-04-01)의 지원 인 LabEx CeMEB의 지원으로 Institut des Sciences de l' Evolution de Montpellier의 GenSeq 기술 시설을 통해 (부분적으로) 생성되었습니다.
자료
| Name | Company | Catalog Number | Comments |
| 96 Well Skirted PCR Plate | 4titude | 4Ti - 0740 | PCR |
| Agarose molecular biology grade | Eurogentec | EP-0010-05 | Agarose gel electrophoresis |
| Agilent DNF-474 HS NGS Fragment Kit Quick Guide for the Fragment Analyzer Systems | Agilent | PDF instruction guide | |
| Buffer TBE | Panreac appliChem | A4228,5000Pc | Agarose gel electrophoresis |
| Calibrated Disposable Inoculating Loops and Needles | LABELIANS | 8175CSR40H | Bacterial culture |
| Dneasy Blood and Tissue Kit | Qiagen | 69506 | DNA extraction |
| Electrophoresis power supply | Amilabo | ST606T | Agarose gel electrophoresis |
| Fragment Analyzer Automated CE System | Agilent | Parallel capillary electrophoresis | |
| Fragment DNA Ladder | Agilent | DNF-396, range 1-6000bp | Parallel capillary electrophoresis |
| GENTAMICIN SULFATE SALT BIOREAGENT | Sigma-Aldrich | G1264-1G | Bacterial culture |
| High Sensitivity diluent marker | Agilent | DNF-373 | Parallel capillary electrophoresis |
| High Sensitivity NGS quantitative analysis kit | Agilent | DNF-474 | Parallel capillary electrophoresis |
| Ladder quick load 1 kb plus DNA ladder | NEB | N0469S | Agarose gel electrophoresis |
| LB Broth, VegitoneNutriSelect Plus | Millipore | 28713 | Bacterial culture |
| Master Mix PCR High Fidelity Phusion Flash | Thermo Fisher Scientific | F548L | PCR |
| Primers | Eurogentec | PCR | |
| Prosize data analysis software v.4 | Agilent | V.4 | Parallel capillary electrophoresis |
| Qubit assays | Invitrogen | MAN0010876 | DNA quantification |
| Qubit dsDNA HS Assay Kit | LIFE TECHNOLOGIES SAS | Q32854 | DNA quantification |
| Thermocycler | Eppendorf | Ep gradients | PCR |
| UVbox, eBOX VX5 | Vilber Lourmat | Agarose gel electrophoresis visualisation | |
| Water for injectable preparation | Aguettant | PROAMP | PCR |
참고문헌
- Mahmoud, M., et al. Structural variant calling: the long and the short of it. Genome Biology. 20 (1), 246 (2019).
- Bragg, D. C., et al. Disease onset in X-linked dystonia-parkinsonism correlates with expansion of a hexameric repeat within an SVA retrotransposon in TAF1. Proceedings of the National Academy of Sciences. 114 (51), 11020-11028 (2017).
- Stransky, N., Cerami, E., Schalm, S., Kim, J. L., Lengauer, C. The landscape of kinase fusions in cancer. Nature Communications. 5, 4846 (2014).
- Tenaillon, O., et al. The molecular diversity of adaptive convergence. Science. 335 (6067), 457-461 (2012).
- Vandecraen, J., Chandler, M., Aertsen, A., Van Houdt, R. The impact of insertion sequences on bacterial genome plasticity and adaptability. Critical Reviews in Microbiology. 43 (6), 709-730 (2017).
- Andersson, D. I., Gene Hughes, D. amplification and adaptive evolution in bacteria. Annual Review of Genetics. 43, 167-195 (2009).
- Bailey, S. F., Bataillon, T. Can the experimental evolution programme help us elucidate the genetic basis of adaptation in nature. Molecular Ecology. 25 (1), 203-218 (2016).
- Consuegra, J., et al. Insertion-sequence-mediated mutations both promote and constrain evolvability during a long-term experiment with bacteria. Nature Communications. 12 (1), 980 (2021).
- Burch, C. L., Romanchuk, A., Kelly, M., Wu, Y., Jones, C. D. Genome-wide determination of barriers to horizontal gene transfer. bioRxiv. , (2022).
- Slomka, S., et al. Experimental evolution of Bacillus subtilis reveals the evolutionary dynamics of horizontal gene transfer and suggests adaptive and neutral effects. Genetics. 216 (2), 543-558 (2020).
- Choudhury, D., Saini, S. Evolution of Escherichia coli in different carbon environments for 2,000 generations. Journal of Evolutionary Biology. 32 (12), 1331-1341 (2019).
- Tenaillon, O., et al. Tempo and mode of genome evolution in a 50,000-generation experiment. Nature. 536 (7615), 165-170 (2016).
- Behringer, M. G., et al. Escherichiacoli cultures maintain stable subpopulation structure during long-term evolution. Proceedings of the National Academy of Sciences. 115 (20), 4642-4650 (2018).
- Voordeckers, K., et al. Adaptation to high ethanol reveals complex evolutionary pathways. PLoS Genetics. 11 (11), 1005635 (2015).
- Levy, S. F., et al. Quantitative evolutionary dynamics using high-resolution lineage tracking. Nature. 519 (7542), 181-186 (2015).
- Bruger, E. L., Marx, C. J. A decade of genome sequencing has revolutionized studies of experimental evolution. Current Opinion in Microbiology. 45, 149-155 (2018).
- Grubaugh, N. D., et al. An amplicon-based sequencing framework for accurately measuring intrahost virus diversity using PrimalSeq and iVar. Genome Biology. 20 (1), 8 (2019).
- Bedhomme, S., et al. Evolutionary changes after translational challenges imposed by horizontal gene transfer. Genome Biology and Evolution. 11 (3), 814-831 (2019).
- Tenaillon, O., Toupance, B., Le Nagard, H., Taddei, F., Godelle, B. Mutators, population size, adaptive landscape and the adaptation of asexual populations of bacteria. Genetics. 152 (2), 485-493 (1999).
재인쇄 및 허가
JoVE'article의 텍스트 или 그림을 다시 사용하시려면 허가 살펴보기
허가 살펴보기더 많은 기사 탐색
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. 판권 소유