Zum Anzeigen dieser Inhalte ist ein JoVE-Abonnement erforderlich. Melden Sie sich an oder starten Sie Ihre kostenlose Testversion.
Method Article
Auf der Suche nach der Dynamik struktureller Varianten in experimentell entwickelten Populationen
In diesem Artikel
Zusammenfassung
Wir haben eine kostengünstige Methode entwickelt, um die Alleldynamik von Nicht-Einzelnukleotid-Polymorphismen zu verfolgen, die leicht an experimentelle Evolutionsarchive angepasst werden kann. Eine Triplett-PCR-Technik wurde mit einer automatisierten parallelen Kapillarelektrophorese gekoppelt, um die relative Häufigkeit eines Insertionsallels im Verlauf der experimentellen Evolution zu quantifizieren.
Zusammenfassung
Es ist heute bekannt, dass strukturelle Varianten (SVs) (d. h. Deletionen, Insertionen, Duplikationen und Inversionen) eine wichtige Rolle bei der phänotypischen Variation und damit bei Prozessen wie der Bestimmung von Krankheiten oder der Anpassung an eine neue Umgebung spielen. Einzelnukleotidvarianten erhalten jedoch viel mehr Aufmerksamkeit als SVs, wahrscheinlich weil sie leichter zu erkennen sind und ihre phänotypischen Wirkungen leichter vorherzusagen sind. Die Entwicklung von Short- und Long-Read-Deep-Sequencing-Technologien hat die Detektion von SVs stark verbessert, aber die Quantifizierung ihrer Häufigkeit aus gepoolten Sequenzierungsdaten (Pooleq) ist immer noch technisch komplex und teuer.
Hier stellen wir eine recht einfache und kostengünstige Methode vor, die es Forschern ermöglicht, die Dynamik der SV-Allelfrequenz zu verfolgen. Als Anwendungsbeispiel verfolgen wir die Häufigkeit der Insertion einer Insertionssequenz (IS) in experimentellen Evolutionspopulationen von Bakterien. Diese Methode basiert auf dem Design von Tripletts von Primern um die strukturellen Variantengrenzen, so dass sich die durch Amplifikation des Wildtyps (WT) und der abgeleiteten Allele erzeugten Amplikons in der Größe um mindestens 5% unterscheiden und dass ihre Amplifikationseffizienz ähnlich ist. Die Menge jedes Amplikons wird dann durch parallele Kapillarelektrophorese bestimmt und auf eine Kalibrierkurve normiert. Diese Methode kann leicht auf die Quantifizierung der Häufigkeit anderer Strukturvarianten (Deletionen, Duplikationen und Inversionen) und auf Pool-Seq-Ansätze natürlicher Populationen, einschließlich patienteninterner Erregerpopulationen, ausgeweitet werden.
Einleitung
Strukturvarianten (SVs) sind Veränderungen der genomischen Sequenz, die im Allgemeinen 50 bp oder mehr betreffen. Die vier Kategorien der beschriebenen SVs sind große Einfügungen, große Deletionen, Inversionen und Duplikationen. Bis vor kurzem wurde Einzelnukleotidvarianten (SNVs) mehr Aufmerksamkeit geschenkt als strukturellen Varianten, was ihre phänotypischen Auswirkungen und ihre Rolle als genetische Determinanten von Krankheiten oder ihren Beitrag zur Anpassung betrifft. Dies liegt wahrscheinlich daran, dass es einfacher ist, SNVs zu erkennen und ihre phänotypischen Auswirkungen vorherzusagen. Kurz- und Langzeit-Deep-Sequencing-Technologien haben jedoch den Nachweis von SVs zumindest in einzelnen individuellen oder klonalen Genomen stark verbessert1. Parallel dazu wurden ihre phänotypischen Wirkungen besser charakterisiert, und es wurden viele Beispiele für ihre Implikation als genetische Determinanten menschlicher Krankheiten 2,3 oder Anpassung an eine neue Umgebung4 dokumentiert.
Deletionen und Insertionen, oft aufgrund von Insertionen mobiler genetischer Elemente (MGE), sind viel störender als Einzelnukleotid-Polymorphismen (SNPs) und führen zu Frameshift-Mutationen und Proteinstrukturmodifikationen. Deletionen und MGE-Insertionen innerhalb von Genen führen fast immer zu einer Geninaktivierung, und Insertionen in nicht-kodierende Regionen können zu einer Repression oder konstitutiven Expression benachbarter Gene führen, wenn Insertionssequenzen (ISs) Promotor- oder Terminationssequenzen enthalten5. Während der Knockout essentieller Gene zu deutlich nachteiligen Auswirkungen auf die bakterielle Fitness führt, ist der Verlust von nicht-essentiellen Genen in einigen Fällen von Vorteil. Trotz ihrer inhärenten Kosten können Duplikationen auch vorteilhaft sein und zur Anpassung beitragen, da sie zu einer Änderung der Gendosis führen. Eine Erhöhung der Aktivität eines bestimmten Proteins kann in Abhängigkeit von den Bedingungen vorteilhaft sein6.
Mikrobielle experimentelle Evolutionspopulationen werden in der Regel mit Klonen begonnen. Dieses anfängliche Fehlen genetischer Vielfalt, kombiniert mit der für Reagenzgläser charakteristischen "geschlossenen Umgebung", führt zu einem sehr begrenzten Potenzial der Evolution durch Gengewinn durch horizontalen Gentransfer und Rekombination. Unter diesen spezifischen Bedingungen ist der Beitrag zur Anpassung von Deletionen, Duplikationen und intragenomischer MGE-Insertion besonders wichtig; Bakterien passen sich häufig durch Funktionsverlustmutationen an (hauptsächlich aufgrund von Deletionen oder MGE-Insertionen) und beeinflussen Gene, die in stabilen, oft nährstoffreichen künstlichen Monokulturumgebungen nicht nützlich sind7. Im am längsten laufenden E. coli-Evolutionsexperiment sind IS150-Insertionen besonders häufig in Populationen, die sich nach 50.000 Generationen entwickelt haben, wobei IS-Elemente 35% der Mutationen ausmachen, die in Populationen, die ihre angestammte Punktmutationsrate beibehalten, eine hohe Häufigkeit erreichen8.
Evolve- und Resequenzierungsstudien koppeln experimentelle Evolutions- und Next-Generation-Sequencing-Technologien (NGS), um zu untersuchen, wie sich Bakterien auf phänotypischer und genomischer Ebene an unterschiedliche Umweltbedingungen und -belastungen anpassen, wie z. B. unterschiedliche Kohlenstoff- und Energiequellen, Antibiotika und osmotischen Stress 9,10,11 . Diese Studien erhalten in der Regel genomische Informationen über die entwickelten Populationen oder Klone nur am experimentellen Endpunkt und in einigen Fällen zu einer Reihe von Zwischenzeitpunkten12,13,14. Diese Daten geben Aufschluss über Gene und Signalwege, die an der Anpassung an eine bestimmte Umgebung beteiligt sind, erlauben es den Forschern jedoch selten, die Dynamik von de novo entstehenden und weitreichenden Allelen im Laufe der Zeit zu verfolgen.
Ein Ansatz, um dieser Dynamik zu folgen, besteht darin, eine begrenzte Anzahl von segregierenden Allelen von Interesse auszuwählen (aufgrund der Funktion der Gene, die sie beeinflussen, weil sie in unabhängigen Populationen parallel verlaufen usw.) und die Amplikonsequenzierung zu verwenden, um den Allelanteil zu quantifizieren, wobei viele Zeitpunkte im selben Sequenzierungslaufzusammengefasst werden 15. Diese Methode wurde erfolgreich eingesetzt, um die Dynamik kleiner Größenvarianten (SNPs oder 1 bp Indels) in experimentellen16 undnatürlichen 17 Populationen von Mikroben zu verfolgen. Bei größeren Indels oder MGE-Insertionen induziert der Größenunterschied der Amplikon jedoch PCR-Effizienzunterschiede, die die Beziehung zwischen Read- und Allel-Anteilen verzerren. In bestimmten Fällen ist der Größenunterschied zwischen den beiden Allelen der klassischen Länge des Amplikons überlegen. Hier haben wir eine Triplett-PCR-Technik mit einer automatisierten parallelen Kapillarelektrophorese gekoppelt, um die relative Häufigkeit eines Insertionsallels basierend auf der Größenunterscheidung zu quantifizieren. Dieser Ansatz ermöglicht die Nutzung von zu wenig genutzten experimentellen Zeitpunkten, um die Dynamik eines neu auftretenden mutierten Allels zu bestimmen und seine Häufigkeit bis zur Fixierung oder zum Verlust auf kostengünstige Weise zu verfolgen. Wir haben diese Methode angewendet, um neu auftretende mutS-Allele zu verfolgen, die durch eine IS10-Insertion mutiert sind und dem mutierten Genotyp einen Hypermutator-Phänotyp verleihen.
Diese Methode erfordert zwei Zielallele mit einem Größenunterschied von ≥5%. Erstens sind Primer-Tripletts so konzipiert, dass sie ähnlich große Fragmente erzeugen, die einen gemeinsamen Primer haben. Zweitens werden die PCR-Bedingungen optimiert und eine Kalibrierungskurve unter Verwendung von Mischungen aus Wildtyp- (WT) und mutierter gDNA erstellt. Schließlich werden die Proben durch PCR amplifiziert, und die relative Häufigkeit jedes Allels wird durch parallele quantitative Kapillarelektrophorese quantifiziert.
Protokoll
Das Einrichten dieses Protokolls erfordert eine genaue Kenntnis des Einfüge-, Lösch-, Inversions- oder Duplizierungspunkts innerhalb der Ahnensequenz. Diese Informationen werden in der Regel durch die Sequenzierung des gesamten Genoms (WGS) der End- oder Zwischenpunktproben gewonnen. Im folgenden Protokoll wird das allgemeine Prinzip für den Fall einer Insertionsmutation für jeden Schritt angegeben, zusammen mit einem repräsentativen Fall, in dem die Häufigkeit einer IS10-Insertion in das mutS-Gen in einer experimentellen Evolutionspopulation von E. coli verfolgt wird. In dieser Population identifizierte WGS der Endpunktpopulation die Insertion eines 1.329 bp IS10 zwischen den Positionen 2.463 und 2.471, was zur Duplikation dieser Insertionsstelle führte. Diese Methode ist auf die drei anderen SV-Typen anwendbar, und die Besonderheiten jedes Falles werden in der Diskussion angegeben.
1. Design von Triplett-Primern
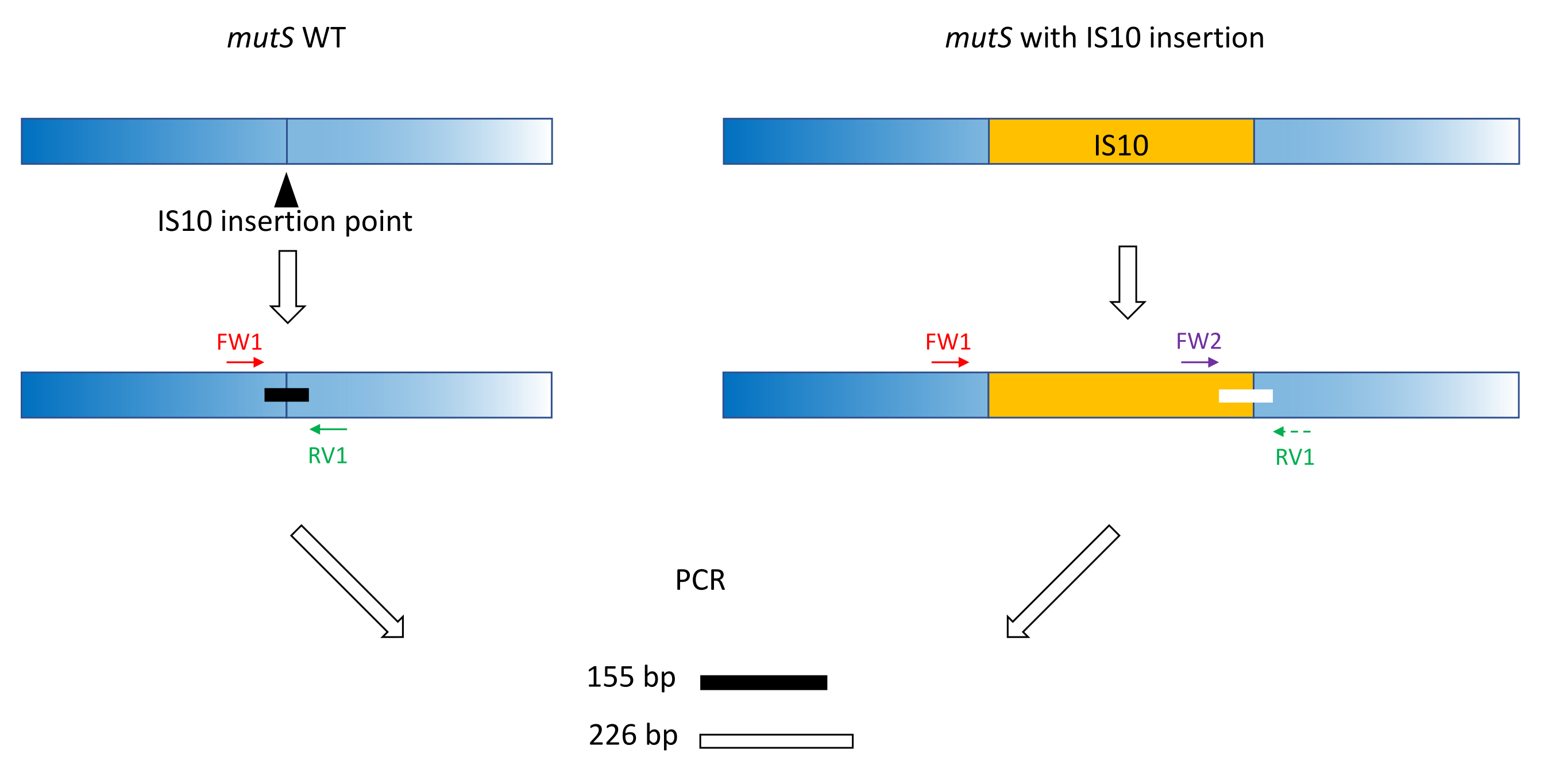
- Verwenden Sie klassische Primer-Designpraktiken (18-24 bp, 40%-60% GC-Gehalt, Beginn/Ende mit G/C-Paaren, wenn möglich, Tm Differenz < 5 °C), um die Primer FW1 und RV1 herzustellen. Entwerfen Sie Primer zur Amplifikation eines kurzen Amplikons auf dem WT-Allel um die Insertionsstelle des mutierten Allels (Abbildung 1).
HINWEIS: Die Amplikongröße kann von 100 bp bis zu 3.000 bp reichen, entsprechend der DNA-Größenleiter, die in der Kapillarelektrophorese verwendet wird. In diesem Beispiel wurde ein 155-bp-Amplikon verstärkt. Die hier gewählte kleine Fragmentgröße verhindert eine Off-Target-Amplifikation der gesamten IS10-Insertionssequenz (siehe Abschnitt 2). - Entwerfen Sie einen zweiten Vorwärtsprimer FW2 innerhalb der Insertionssequenz, um ein zweites Amplikon zu erzeugen, das etwa 5 % größer oder kleiner als das WT-Amplikon ist (Abbildung 1). Dieser Größenunterschied von 5 % ist der minimale Größenunterschied, den das parallele Kapillarelektrophoresegerät zuverlässig unterscheiden kann. Entwerfen Sie daher die Primer so, dass die beiden Amplikons einen Größenunterschied aufweisen, der über, aber so nahe wie möglich an der relativen Schwelle liegt.
HINWEIS: Achten Sie darauf,die TM-Differenz und die Primer-Dimer-Bildung zu minimieren. In diesem Beispiel wurde ein zweiter Vorwärtsprimer entwickelt, um ein 226 bp Amplikon zu erzeugen, 71 bp größer als das WT-Amplikon. In diesem repräsentativen Beispiel lauten die Primersequenzen wie folgt:
FW1:AAAGCATTTCGCCGAACGCC
RV1: GCGATAAATCCACTCCAGCGCC
FW2: AGTTCGCTTAGGCATGGAAG

Abbildung 1: Schema des Triplett-Primer-Designs auf mutS WT-Gen und mutierter mutS IS10-Insertion. Das schwarze Dreieck stellt die IS10-Insertionsstelle im mutS-Gen dar. Das WT-Gen ist blau und das IS10 orange. Die Primer FW1 und RV1 kennzeichnen die IS10-Insertionsstelle und erzeugen ein 155 bp WT-Amplikon. Der RV1-Primer und der Intra-IS10-Primer FW2 erzeugen ein zweites 226-bp-Amplikon. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
2. Optimierung der PCR-Bedingungen
- Züchten Sie über Nacht eine Kultur von festen WT- und mutierten Allelklonen.
- Extrahieren Sie die DNA mit einem beliebigen Kit.
- Quantifizieren Sie die DNA.
- Bereiten Sie die DNA-Probe vor, indem Sie die Extraktion von WT und mutierter DNA auf 5 ng/μl verdünnen. Mischen Sie die beiden DNA-Proben im Verhältnis 50/50.
- Amplifizieren Sie 10 ng der drei DNA-Proben (WT, Mutante, 50/50-Mix) mit einem 2x gebrauchsfertigen PCR-Mastermix, 0,5 μM FW1-Primer, 1 μM RV1-Primer und 0,5 μM FW2-Primer in einem 20-μl-Reaktionsvolumen. Verwenden Sie ein 2%iges Agarosegel, um das PCR-Produkt durch klassische Elektrophorese zu migrieren und die optimalen PCR-Bedingungen zu bestimmen.
HINWEIS: Die Verlängerungszeiten sollten minimiert werden, um die Bildung des FW1- und RV1-Amplikons auf dem mutierten Allel zu verhindern. Die Annealing-Temperatur sollte eingestellt werden, um eine verzerrte Amplifikation der Allele und eine unspezifische Amplifikation zu minimieren.- Um dem Programm in diesem Beispiel zu folgen, verwenden Sie die folgenden Einstellungen: 98 °C für 10 s, gefolgt von 25 Zyklen von 98 °C für 1 s, 58 °C für 15 s, 72 °C für 8 s und einem letzten Dehnungsschritt bei 72 °C für 1 min.
ANMERKUNG: Die Verlängerungszeit wurde auf 8 s reduziert, um eine Amplifikation des >1.000 bp-Produkts aus dem vorderen Primer und dem RV1-Primer auf dem mutierten Allel (mutS mit IS10-Insertion) zu verhindern.
- Um dem Programm in diesem Beispiel zu folgen, verwenden Sie die folgenden Einstellungen: 98 °C für 10 s, gefolgt von 25 Zyklen von 98 °C für 1 s, 58 °C für 15 s, 72 °C für 8 s und einem letzten Dehnungsschritt bei 72 °C für 1 min.
3. Kalibrierkurve
- Mischen Sie die beiden DNA-Proben, WT und Mutante, in den Verhältnissen 10/90, 25/75, 40/60, 50/50, 60/40, 75/25 und 90/10.
HINWEIS: Biologische Replikate werden aus unabhängigen Bakterienkulturen über Nacht hergestellt. - Amplifikation unter optimierten PCR-Bedingungen (siehe Abschnitt 2).
- Quantifizieren Sie das Amplikonprodukt.
- Verdünnen Sie die PCR-Produkte auf 0,1 ng/μl.
- Bereiten Sie das Instrument für die parallele Kapillarelektrophorese vor.
- Mischen Sie frisches Gel und Farbstoff (NGS-Kit für quantitative Analysen (22, 33 oder 55); HS NGS-Fragment 1-6.000 bp für dieses Beispiel).
HINWEIS: Detaillierte Anweisungen finden Sie in der Anleitung zur parallelen Kapillarelektrophorese HS NGS-Fragmenten (siehe Materialtabelle).
- Mischen Sie frisches Gel und Farbstoff (NGS-Kit für quantitative Analysen (22, 33 oder 55); HS NGS-Fragment 1-6.000 bp für dieses Beispiel).
- Ersetzen Sie die Kapillarspeicherlösung und den Einlasspuffer und platzieren Sie die Spülpufferplatte an den richtigen Schubladenpositionen des Parallelkapillarelektrophoresegeräts.
- Fügen Sie 2 μl HS-Verdünnungsmarker zu 22 μl jeder verdünnten Probe in einer 96-Well-Platte hinzu.
- Fügen Sie eine Größenleiter (DNA-Größenleiter; Bereich 1-6.000 bp) aus einem quantitativen HS NGS-Analysekit zu einer Vertiefung der 96-Well-Platte hinzu.
- Setzen Sie die 96-Well-Platte in die richtige Schublade des Instruments für die parallele Kapillarelektrophorese ein, und wählen Sie in der Software des Geräts für die parallele Kapillarelektrophorese die Option Ausführen aus.
- Analysieren Sie die Ergebnisse mit der Datenanalysesoftware, die jeden Peak der Größenleiter erkennt und identifiziert und jeden Peak der Proben seiner bekannten tatsächlichen Größe zuordnet.
- Wenn Sie ein quantitatives Kit verwenden, verwenden Sie die Software, um die DNA-Menge jedes Fragments zu bestimmen, indem Sie den Bereich unter dem Peak integrieren, wie bei der Chromatographie-Datenanalyse. Vergleichen Sie die Proben erneut mit bekannten Mengen in den Standards, um jeden Peak aus einer Probe zu quantifizieren, und berechnen Sie die Verhältnisse zwischen den verschiedenen Peaks, die in den Proben nachgewiesen wurden.
- Konstruieren Sie eine Kalibrierungskurve (Abbildung 2), die den bekannten Anteil des mutierten Allels (DNA-Mix) mit dem mit dem parallelen Kapillarelektrophoresegerät gemessenen Anteil verknüpft. Diese Kalibrierkurve ermöglicht es, die Zuverlässigkeit der Methode zu bewerten und auf geringfügige Amplifikationsverzerrungen zu korrigieren.
4. Probenvorbereitung
- Züchten Sie Time-Point-Proben über Nacht unter Standardbedingungen.
- Extrahieren Sie die DNA.
- Quantifizieren Sie die DNA.
- Amplifizieren Sie die Proben unter optimierten PCR-Bedingungen (siehe Abschnitt 2).
- Führen Sie die Proben im Parallel-Kapillarelektrophorese-Gerät aus (siehe Schritte 3.5-3.10).
5. Quantifizierung des Allels
- Extrahieren Sie mit der Software mutierte Allelmengen aus den Daten des Instruments für die parallele Kapillarelektrophorese und berechnen Sie die tatsächlichen Anteile, indem Sie diese Werte auf der Kalibrierungskurve darstellen.
Ergebnisse
Unter Verwendung von DNA, die aus einem angestammten Klon extrahiert wurde, und einem Hypermutator-Klon, der aus der S2.11-Population der Generation 1.000 isoliert wurde, haben wir die in Abbildung 2 gezeigte Kalibrierungskurve erstellt. Die tatsächlichen Mutantenanteile aus im Labor hergestellten DNA-Mischungen, die mit dem parallelen Kapillarelektrophorese-Instrument gemessen wurden, wurden durch eine lineare Beziehung der Steigung 1,0706 mit einem R2 von 0,9705 verknüpft. Dar...
Diskussion
Hier haben wir eine kostengünstige Methode vorgeschlagen, die es ermöglicht, die Dynamik neuer adaptiver SV-Allele in experimentellen Evolutionspopulationen zu verfolgen. Diese Methode koppelt klassische PCR-Techniken mit automatisierter paralleler Kapillarelektrophorese, so dass die relativen Mengen zweier Allele bestimmt werden können. Einmal eingerichtet, ermöglicht es die Quantifizierung von Allelanteilen in vielen Proben parallel und ist viel kostengünstiger als WGS. Diese Methode kann als Äquivalent zur Ampli...
Offenlegungen
Die Autoren haben keine Interessenkonflikte offenzulegen.
Danksagungen
Diese Arbeit wurde vom ERC HGTCODONUSE (ERC-2015-CoG-682819) an S.B. Die in dieser Arbeit verwendeten Daten wurden (teilweise) durch die technischen Einrichtungen des GenSeq des Institut des Sciences de l'Evolution de Montpellier mit Unterstützung von LabEx CeMEB, einem ANR-Programm "Investissements d'avenir" (ANR-10-LABX-04-01), erstellt.
Materialien
| Name | Company | Catalog Number | Comments |
| 96 Well Skirted PCR Plate | 4titude | 4Ti - 0740 | PCR |
| Agarose molecular biology grade | Eurogentec | EP-0010-05 | Agarose gel electrophoresis |
| Agilent DNF-474 HS NGS Fragment Kit Quick Guide for the Fragment Analyzer Systems | Agilent | PDF instruction guide | |
| Buffer TBE | Panreac appliChem | A4228,5000Pc | Agarose gel electrophoresis |
| Calibrated Disposable Inoculating Loops and Needles | LABELIANS | 8175CSR40H | Bacterial culture |
| Dneasy Blood and Tissue Kit | Qiagen | 69506 | DNA extraction |
| Electrophoresis power supply | Amilabo | ST606T | Agarose gel electrophoresis |
| Fragment Analyzer Automated CE System | Agilent | Parallel capillary electrophoresis | |
| Fragment DNA Ladder | Agilent | DNF-396, range 1-6000bp | Parallel capillary electrophoresis |
| GENTAMICIN SULFATE SALT BIOREAGENT | Sigma-Aldrich | G1264-1G | Bacterial culture |
| High Sensitivity diluent marker | Agilent | DNF-373 | Parallel capillary electrophoresis |
| High Sensitivity NGS quantitative analysis kit | Agilent | DNF-474 | Parallel capillary electrophoresis |
| Ladder quick load 1 kb plus DNA ladder | NEB | N0469S | Agarose gel electrophoresis |
| LB Broth, VegitoneNutriSelect Plus | Millipore | 28713 | Bacterial culture |
| Master Mix PCR High Fidelity Phusion Flash | Thermo Fisher Scientific | F548L | PCR |
| Primers | Eurogentec | PCR | |
| Prosize data analysis software v.4 | Agilent | V.4 | Parallel capillary electrophoresis |
| Qubit assays | Invitrogen | MAN0010876 | DNA quantification |
| Qubit dsDNA HS Assay Kit | LIFE TECHNOLOGIES SAS | Q32854 | DNA quantification |
| Thermocycler | Eppendorf | Ep gradients | PCR |
| UVbox, eBOX VX5 | Vilber Lourmat | Agarose gel electrophoresis visualisation | |
| Water for injectable preparation | Aguettant | PROAMP | PCR |
Referenzen
- Mahmoud, M., et al. Structural variant calling: the long and the short of it. Genome Biology. 20 (1), 246 (2019).
- Bragg, D. C., et al. Disease onset in X-linked dystonia-parkinsonism correlates with expansion of a hexameric repeat within an SVA retrotransposon in TAF1. Proceedings of the National Academy of Sciences. 114 (51), 11020-11028 (2017).
- Stransky, N., Cerami, E., Schalm, S., Kim, J. L., Lengauer, C. The landscape of kinase fusions in cancer. Nature Communications. 5, 4846 (2014).
- Tenaillon, O., et al. The molecular diversity of adaptive convergence. Science. 335 (6067), 457-461 (2012).
- Vandecraen, J., Chandler, M., Aertsen, A., Van Houdt, R. The impact of insertion sequences on bacterial genome plasticity and adaptability. Critical Reviews in Microbiology. 43 (6), 709-730 (2017).
- Andersson, D. I., Gene Hughes, D. amplification and adaptive evolution in bacteria. Annual Review of Genetics. 43, 167-195 (2009).
- Bailey, S. F., Bataillon, T. Can the experimental evolution programme help us elucidate the genetic basis of adaptation in nature. Molecular Ecology. 25 (1), 203-218 (2016).
- Consuegra, J., et al. Insertion-sequence-mediated mutations both promote and constrain evolvability during a long-term experiment with bacteria. Nature Communications. 12 (1), 980 (2021).
- Burch, C. L., Romanchuk, A., Kelly, M., Wu, Y., Jones, C. D. Genome-wide determination of barriers to horizontal gene transfer. bioRxiv. , (2022).
- Slomka, S., et al. Experimental evolution of Bacillus subtilis reveals the evolutionary dynamics of horizontal gene transfer and suggests adaptive and neutral effects. Genetics. 216 (2), 543-558 (2020).
- Choudhury, D., Saini, S. Evolution of Escherichia coli in different carbon environments for 2,000 generations. Journal of Evolutionary Biology. 32 (12), 1331-1341 (2019).
- Tenaillon, O., et al. Tempo and mode of genome evolution in a 50,000-generation experiment. Nature. 536 (7615), 165-170 (2016).
- Behringer, M. G., et al. Escherichiacoli cultures maintain stable subpopulation structure during long-term evolution. Proceedings of the National Academy of Sciences. 115 (20), 4642-4650 (2018).
- Voordeckers, K., et al. Adaptation to high ethanol reveals complex evolutionary pathways. PLoS Genetics. 11 (11), 1005635 (2015).
- Levy, S. F., et al. Quantitative evolutionary dynamics using high-resolution lineage tracking. Nature. 519 (7542), 181-186 (2015).
- Bruger, E. L., Marx, C. J. A decade of genome sequencing has revolutionized studies of experimental evolution. Current Opinion in Microbiology. 45, 149-155 (2018).
- Grubaugh, N. D., et al. An amplicon-based sequencing framework for accurately measuring intrahost virus diversity using PrimalSeq and iVar. Genome Biology. 20 (1), 8 (2019).
- Bedhomme, S., et al. Evolutionary changes after translational challenges imposed by horizontal gene transfer. Genome Biology and Evolution. 11 (3), 814-831 (2019).
- Tenaillon, O., Toupance, B., Le Nagard, H., Taddei, F., Godelle, B. Mutators, population size, adaptive landscape and the adaptation of asexual populations of bacteria. Genetics. 152 (2), 485-493 (1999).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten