È necessario avere un abbonamento a JoVE per visualizzare questo. Accedi o inizia la tua prova gratuita.
Method Article
Seguire la dinamica delle varianti strutturali in popolazioni evolute sperimentalmente
In questo articolo
Riepilogo
Abbiamo sviluppato un metodo economico per seguire la dinamica allelica del polimorfismo non a singolo nucleotide che può essere facilmente adattata agli archivi congelati dell'evoluzione sperimentale. Una tecnica di tripletta PCR è stata accoppiata con elettroforesi capillare parallela automatizzata per quantificare la frequenza relativa di un allele di inserzione nel corso dell'evoluzione sperimentale.
Abstract
Le varianti strutturali (SV) (cioè delezioni, inserzioni, duplicazioni e inversioni) sono ora note per svolgere un ruolo importante nella variazione fenotipica e, di conseguenza, in processi come la determinazione della malattia o l'adattamento a un nuovo ambiente. Tuttavia, le varianti a singolo nucleotide ricevono molta più attenzione delle SV, probabilmente perché sono più facili da rilevare e i loro effetti fenotipici sono più facili da prevedere. Lo sviluppo di tecnologie di sequenziamento profondo a breve e lunga lettura ha notevolmente migliorato il rilevamento delle SV, ma la quantificazione della loro frequenza dai dati di sequenziamento aggregato (poolseq) è ancora tecnicamente complessa e costosa.
Qui, presentiamo un metodo piuttosto semplice ed economico, che consente ai ricercatori di seguire la dinamica della frequenza dell'allele SV. Come esempio di applicazione, seguiamo la frequenza di inserimento di una sequenza di inserzione (IS) nelle popolazioni sperimentali evolutive di batteri. Questo metodo si basa sulla progettazione di triplette di primer attorno ai bordi delle varianti strutturali, in modo tale che gli ampliconi prodotti dall'amplificazione del wild-type (WT) e degli alleli derivati differiscano in dimensioni di almeno il 5% e che la loro efficienza di amplificazione sia simile. La quantità di ciascun amplicone viene quindi determinata mediante elettroforesi capillare parallela e normalizzata a una curva di calibrazione. Questo metodo può essere facilmente esteso alla quantificazione della frequenza di altre varianti strutturali (delezioni, duplicazioni e inversioni) e agli approcci pool-seq di popolazioni naturali, comprese le popolazioni patogene all'interno dei pazienti.
Introduzione
Le varianti strutturali (SV) sono alterazioni della sequenza genomica, che generalmente colpiscono 50 bp o più. Le quattro categorie di SV descritte sono inserimenti di grandi dimensioni, grandi cancellazioni, inversioni e duplicazioni. Fino a poco tempo fa, è stata dedicata maggiore attenzione alle varianti a singolo nucleotide (SNV) che alle varianti strutturali, in termini di effetti fenotipici e del loro ruolo come determinanti genetici della malattia, o del loro contributo all'adattamento. Ciò è probabilmente dovuto al fatto che è più facile rilevare gli SNV e prevederne gli effetti fenotipici. Tuttavia, le tecnologie di sequenziamento profondo a breve e lunga lettura hanno notevolmente migliorato la rilevazione delle SV, almeno in genomi singoli individuali o clonali1. Parallelamente, i loro effetti fenotipici sono stati meglio caratterizzati e sono stati documentati molti esempi della loro implicazione come determinanti genetici della malattia umana 2,3 o dell'adattamento ad un nuovo ambiente4.
Le delezioni e le inserzioni, spesso dovute a inserzioni di elementi genetici mobili (MGE), sono molto più dirompenti dei polimorfismi a singolo nucleotide (SNP) e portano a mutazioni frameshift e modifiche della struttura proteica. Le delezioni e le inserzioni MGE all'interno dei geni provocano quasi sempre l'inattivazione genica, e le inserzioni in regioni non codificanti possono portare alla repressione o all'espressione costitutiva di geni adiacenti quando le sequenze di inserzione (IS) contengono sequenze promotrici o di terminazione5. Mentre il knockout di geni essenziali porta a chiari effetti dannosi sulla forma fisica batterica, la perdita di geni non essenziali è vantaggiosa in alcuni casi. Nonostante i loro costi intrinseci, le duplicazioni possono anche essere vantaggiose e partecipare all'adattamento in quanto portano a un cambiamento nel dosaggio genico; Un aumento dell'attività di una proteina specifica può essere vantaggioso a seconda delle condizioni6.
Le popolazioni di evoluzione sperimentale microbica sono di solito iniziate con cloni. Questa iniziale assenza di diversità genetica, combinata con la caratteristica dell'"ambiente chiuso" delle provette, porta a un potenziale molto limitato di evoluzione per guadagno genico attraverso il trasferimento genico orizzontale e la ricombinazione. In queste condizioni specifiche, il contributo all'adattamento di delezioni, duplicazioni e inserimento intragenomico di MGE è particolarmente importante; i batteri spesso si adattano attraverso mutazioni con perdita di funzione (principalmente a causa di delezioni o inserzioni di MGE), colpendo geni che non sono utiliin ambienti artificiali stabili, spesso ricchi di sostanze nutritive, 7. Nell'esperimento di evoluzione di E. coli più longevo, le inserzioni di IS150 sono particolarmente frequenti tra le popolazioni evolute dopo 50.000 generazioni, con elementi IS che rappresentano il 35% delle mutazioni che raggiungono un'alta frequenza nelle popolazioni che mantengono il loro tasso di mutazione del punto ancestrale8.
Gli studi di evoluzione e risequenziamento accoppiano l'evoluzione sperimentale e le tecnologie di sequenziamento di nuova generazione (NGS) per studiare come i batteri si adattano, a livello fenotipico e genomico, a diverse condizioni ambientali e stress, come diverse fonti di carbonio ed energia, antibiotici e stress osmotico 9,10,11 . Questi studi tipicamente ottengono informazioni genomiche sulle popolazioni evolute o sui cloni solo nel punto finale sperimentale e, in alcuni casi, in un numero di punti temporali intermedi12,13,14. Questi dati forniscono informazioni sui geni e sui percorsi coinvolti nell'adattamento a un determinato ambiente, ma raramente consentono ai ricercatori di seguire le dinamiche degli alleli emergenti e ampi de novo nel tempo.
Un approccio per seguire queste dinamiche è quello di scegliere un numero limitato di alleli segreganti di interesse (a causa della funzione dei geni che influenzano, perché si muovono in parallelo in popolazioni indipendenti, ecc.) e utilizzare il sequenziamento amplico per quantificare la proporzione allelica, raggruppando molti punti temporali nella stessa sequenza15. Questo metodo è stato utilizzato con successo per seguire la dinamica di varianti di piccole dimensioni (SNPs o 1 bp indels) in16 e17 popolazioni naturali di microbi. Tuttavia, nel caso di indels più grandi o inserimenti MGE, la differenza di dimensioni degli ampliconi induce differenze di efficienza PCR, che distorcono la relazione tra proporzioni leggere e alleliche. In alcuni casi, la differenza di dimensioni tra i due alleli è superiore alla lunghezza classica dell'amplicone. Qui, abbiamo accoppiato una tecnica di tripletta PCR con elettroforesi capillare parallela automatizzata per quantificare la frequenza relativa di un allele di inserzione in base alla discriminazione delle dimensioni. Questo approccio consente lo sfruttamento di punti temporali sperimentali sottoutilizzati per determinare la dinamica di un allele mutante emergente e per seguire la sua frequenza di fissazione o perdita, in modo economicamente vantaggioso. Abbiamo applicato questo metodo per tracciare gli alleli mutS emergenti, mutati attraverso un'inserzione IS10, fornendo al genotipo mutato un fenotipo ipermutatore.
Questo metodo richiede due alleli bersaglio con una differenza di dimensioni del ≥5%. In primo luogo, le triplette di primer sono progettate per produrre frammenti di dimensioni simili, che condividono un primer comune. In secondo luogo, le condizioni di PCR sono ottimizzate e viene prodotta una curva di calibrazione utilizzando miscele di gDNA wild-type (WT) e mutante. Infine, i campioni vengono amplificati mediante PCR e la frequenza relativa di ciascun allele viene quantificata mediante elettroforesi capillare quantitativa parallela.
Protocollo
L'impostazione di questo protocollo richiede una conoscenza precisa del punto di inserimento, cancellazione, inversione o duplicazione all'interno della sequenza ancestrale. Queste informazioni sono solitamente ottenute mediante sequenziamento dell'intero genoma (WGS) dei campioni finali o intermedi. Nel seguente protocollo, il principio generale per il caso di una mutazione di inserzione è dato per ogni fase, insieme a un caso rappresentativo in cui viene seguita la frequenza di un'inserzione IS10 nel gene mutS in una popolazione evolutiva sperimentale di E. coli. In questa popolazione, WGS della popolazione endpoint ha identificato l'inserimento di un IS10 di 1.329 bp tra le posizioni 2.463 e 2.471, con conseguente duplicazione di questo sito di inserimento. Questo metodo è applicabile agli altri tre tipi di SV e le specificità di ciascun caso sono fornite nella discussione.
1. Progettazione di primer a tripletta
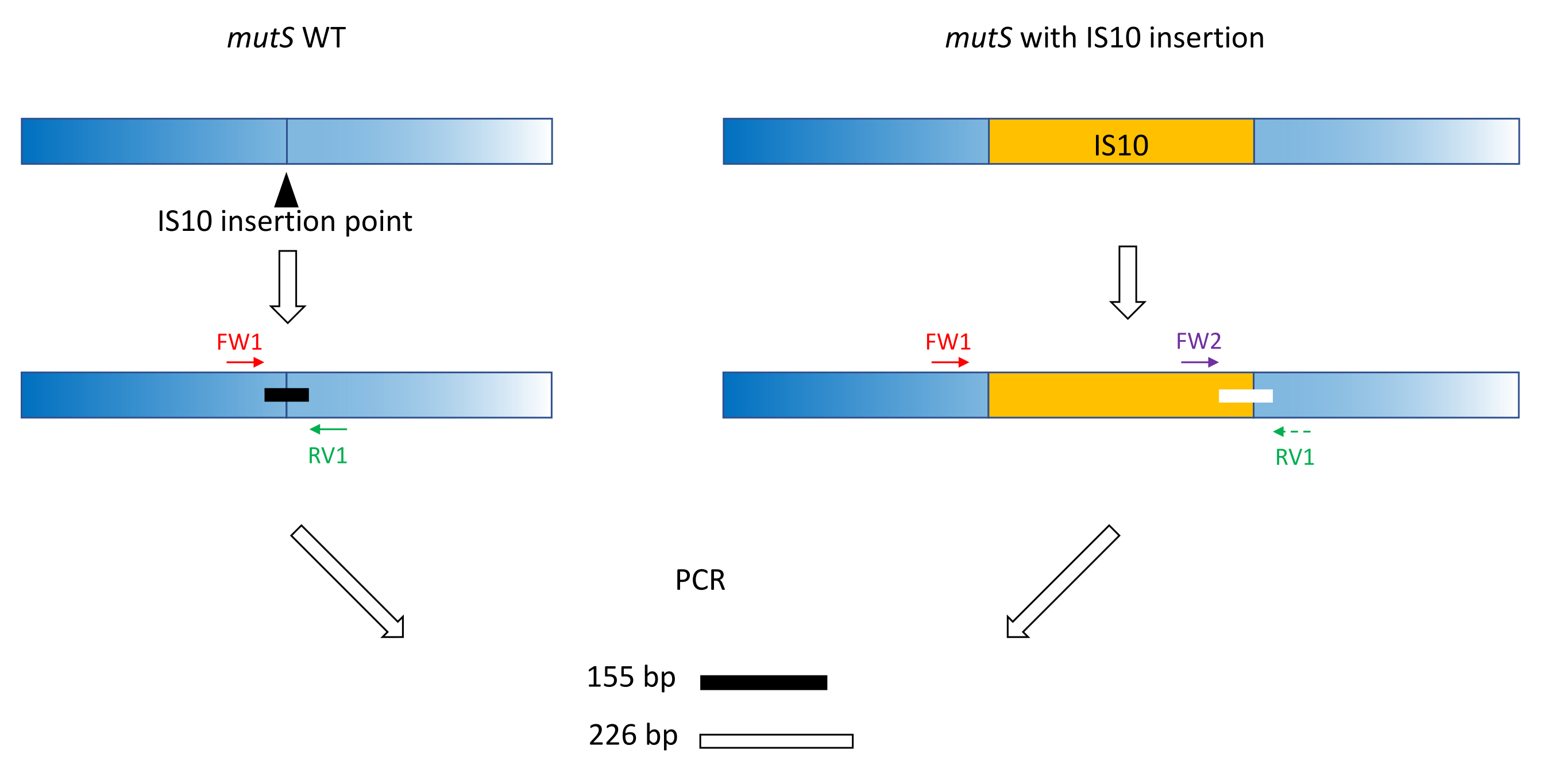
- Utilizzare le classiche pratiche di progettazione dei primer (18-24 bp, contenuto GC 40%-60%, inizio/fine con coppie G/C ove possibile, differenza Tm < 5 °C) per produrre primer FW1 e RV1. Progettare primer per amplificare un breve amplicone sull'allele WT attorno al sito di inserimento dell'allele mutante (Figura 1).
NOTA: La dimensione dell'amplicone può variare da 100 bp a 3.000 bp, in linea con la scala dimensionale del DNA utilizzata nell'elettroforesi capillare. In questo esempio, un amplicone da 155 bp è stato amplificato. La piccola dimensione del frammento qui scelta impedisce l'amplificazione fuori bersaglio dell'intera sequenza di inserimento IS10 (vedere sezione 2). - Progettare un secondo primer FW2 in avanti all'interno della sequenza di inserimento, per produrre un secondo amplicone che sia circa il 5% più grande o più piccolo dell'amplicone WT (Figura 1). Questa differenza di dimensioni del 5% è la differenza di dimensioni minima che il dispositivo di elettroforesi capillare parallela potrebbe distinguere in modo affidabile. Pertanto, progettare i primer in modo tale che i due ampliconi abbiano una differenza di dimensioni, che è superiore ma il più vicino possibile alla soglia relativa.
NOTA: Assicurarsi di ridurre al minimo la differenza di Tm e la formazione di dimero primer. In questo esempio, un secondo primer in avanti è stato progettato per produrre un amplicone da 226 bp, 71 bp più grande dell'amplicone WT. In questo esempio rappresentativo, le sequenze di primer sono le seguenti:
FW1:AAAGCATTTCGCCGAACGCC
RV1: GCGATAAATCCACTCCAGCGCC
FW2: AGTTCGCTTAGGCATGGAAG

Figura 1: Schema del disegno del primer della tripletta sul gene mutS WT e sull'inserzione mutante mutS IS10. Il triangolo nero rappresenta il sito di inserzione IS10 nel gene mutS. Il gene WT è in blu e l'IS10 è in arancione. I primer FW1 e RV1 contrassegnano il sito di inserimento IS10 e producono un amplicone WT da 155 bp. Il primer RV1 e il primer intra-IS10 FW2 producono un secondo amplicone da 226 bp. Fare clic qui per visualizzare una versione ingrandita di questa figura.
{kind=link}
2. Ottimizzazione delle condizioni di PCR
- Coltiva una coltura notturna di WT fisso e cloni allelici mutanti.
- Estrarre il DNA utilizzando qualsiasi kit.
- Quantificare il DNA.
- Preparare il campione di DNA diluendo l'estrazione di WT e DNA mutante a 5 ng/μL. Mescolare i due campioni di DNA in un rapporto 50/50.
- Amplificare 10 ng dei tre campioni di DNA (WT, mutante, mix 50/50) utilizzando una miscela master PCR pronta all'uso 2x, primer FW1 da 0,5 μM, primer RV1 da 1 μM e primer FW2 da 0,5 μM in un volume di reazione di 20 μL. Utilizzare un gel di agarosio al 2% per migrare il prodotto PCR mediante elettroforesi classica e determinare le condizioni ottimali di PCR.
NOTA: I tempi di allungamento devono essere ridotti al minimo per evitare la formazione dell'amplicone FW1 e RV1 sull'allele mutante. La temperatura di ricottura deve essere regolata per ridurre al minimo l'amplificazione distorta degli alleli e l'amplificazione non specifica.- Per seguire il programma in questo esempio, utilizzare le seguenti impostazioni: 98 °C per 10 s, seguito da 25 cicli di 98 °C per 1 s, 58 °C per 15 s, 72 °C per 8 s e una fase di allungamento finale a 72 °C per 1 min.
NOTA: Il tempo di allungamento è stato ridotto a 8 s per evitare l'amplificazione del prodotto da >1.000 bp dal primer anteriore e dal primer RV1 sull'allele mutante (mutS con inserimento IS10).
- Per seguire il programma in questo esempio, utilizzare le seguenti impostazioni: 98 °C per 10 s, seguito da 25 cicli di 98 °C per 1 s, 58 °C per 15 s, 72 °C per 8 s e una fase di allungamento finale a 72 °C per 1 min.
3. Curva di calibrazione
- Mescolare i due campioni di DNA, WT e mutante, nei rapporti 10/90, 25/75, 40/60, 50/50, 60/40, 75/25 e 90/10.
NOTA: Le repliche biologiche sono preparate da colture batteriche notturne indipendenti. - Amplificare utilizzando condizioni PCR ottimizzate (vedere paragrafo 2).
- Quantificare il prodotto dell'amplicone.
- Diluire i prodotti PCR a 0,1 ng/μL.
- Preparare lo strumento di elettroforesi capillare parallela.
- Mescolare gel fresco e colorante (kit di analisi quantitativa NGS (22, 33 o 55); HS NGS frammento 1-6.000 bp per questo esempio).
NOTA: Vedere la guida ai frammenti di elettroforesi capillare parallela HS NGS per istruzioni dettagliate (vedere la tabella dei materiali).
- Mescolare gel fresco e colorante (kit di analisi quantitativa NGS (22, 33 o 55); HS NGS frammento 1-6.000 bp per questo esempio).
- Sostituire la soluzione di stoccaggio capillare e il tampone di ingresso e posizionare la piastra tampone di risciacquo nelle posizioni corrette del cassetto dello strumento di elettroforesi capillare parallela.
- Aggiungere 2 μL di marcatore di diluente HS a 22 μL di ciascun campione diluito in una piastra da 96 pozzetti.
- Aggiungere una scala dimensionale (scala di dimensioni del DNA; intervallo 1-6.000 bp) da un kit di analisi quantitativa HS NGS a un pozzetto della piastra a 96 pozzetti.
- Posizionare la piastra a 96 pozzetti nel cassetto corretto dello strumento di elettroforesi capillare parallela e selezionare Esegui sul software dello strumento di elettroforesi capillare parallela.
- Analizza i risultati utilizzando il software di analisi dei dati, che rileva e identifica ogni picco della scala dimensionale, assegnando ogni picco dei campioni alla loro dimensione effettiva nota.
- Quando si utilizza un kit quantitativo, utilizzare il software per determinare la quantità di DNA di ciascun frammento integrando l'area sotto il picco, come nell'analisi dei dati cromatografici. Ancora una volta, confrontare i campioni con quantità note negli standard per quantificare ogni picco da un campione e calcolare i rapporti tra i diversi picchi rilevati nei campioni.
- Costruire una curva di calibrazione (Figura 2), collegando la proporzione nota dell'allele mutante (mix di DNA) a quella misurata utilizzando lo strumento di elettroforesi capillare parallela. Questa curva di calibrazione consente di valutare e correggere l'affidabilità del metodo per una minore distorsione di amplificazione.
4. Preparazione del campione
- Campioni di punti temporali di crescita durante la notte in condizioni standard.
- Estrarre il DNA.
- Quantificare il DNA.
- Amplificare i campioni utilizzando condizioni PCR ottimizzate (vedere paragrafo 2).
- Eseguire i campioni nello strumento di elettroforesi capillare parallela (vedere i punti 3.5-3.10).
5. Quantificazione allelica
- Estrai quantità di alleli mutanti dai dati dello strumento di elettroforesi capillare parallela con il software e calcola le proporzioni effettive tracciando questi valori sulla curva di calibrazione.
Risultati
Utilizzando il DNA estratto da un clone ancestrale e un clone ipermutatore isolato dalla popolazione S2.11 alla generazione 1.000, abbiamo stabilito la curva di calibrazione mostrata in Figura 2. Le proporzioni effettive dei mutanti provenienti da miscele di DNA preparate in laboratorio e misurate dallo strumento di elettroforesi capillare parallela sono state collegate da una relazione lineare di pendenza 1,0706, con un R2 di 0,9705. Inoltre, c'era un buon accordo tra le repliche...
Discussione
Qui, abbiamo proposto un metodo economico che consente di seguire la dinamica degli alleli SV adattativi emergenti nelle popolazioni evolutive sperimentali. Questo metodo combina le classiche tecniche di PCR e l'elettroforesi capillare parallela automatizzata, consentendo di determinare le quantità relative di due alleli. Una volta impostato, consente la quantificazione delle proporzioni alleliche in molti campioni in parallelo, ed è molto meno costoso del WGS. Questo metodo può essere visto come un equivalente del se...
Divulgazioni
Gli autori non hanno conflitti di interesse da rivelare.
Riconoscimenti
Questo lavoro è stato supportato dall'ERC HGTCODONUSE (ERC-2015-CoG-682819) a S.B. I dati utilizzati in questo lavoro sono stati (parzialmente) prodotti attraverso le strutture tecniche GenSeq dell'Institut des Sciences de l'Evolution de Montpellier con il supporto di LabEx CeMEB, un programma ANR "Investissements d'avenir" (ANR-10-LABX-04-01).
Materiali
| Name | Company | Catalog Number | Comments |
| 96 Well Skirted PCR Plate | 4titude | 4Ti - 0740 | PCR |
| Agarose molecular biology grade | Eurogentec | EP-0010-05 | Agarose gel electrophoresis |
| Agilent DNF-474 HS NGS Fragment Kit Quick Guide for the Fragment Analyzer Systems | Agilent | PDF instruction guide | |
| Buffer TBE | Panreac appliChem | A4228,5000Pc | Agarose gel electrophoresis |
| Calibrated Disposable Inoculating Loops and Needles | LABELIANS | 8175CSR40H | Bacterial culture |
| Dneasy Blood and Tissue Kit | Qiagen | 69506 | DNA extraction |
| Electrophoresis power supply | Amilabo | ST606T | Agarose gel electrophoresis |
| Fragment Analyzer Automated CE System | Agilent | Parallel capillary electrophoresis | |
| Fragment DNA Ladder | Agilent | DNF-396, range 1-6000bp | Parallel capillary electrophoresis |
| GENTAMICIN SULFATE SALT BIOREAGENT | Sigma-Aldrich | G1264-1G | Bacterial culture |
| High Sensitivity diluent marker | Agilent | DNF-373 | Parallel capillary electrophoresis |
| High Sensitivity NGS quantitative analysis kit | Agilent | DNF-474 | Parallel capillary electrophoresis |
| Ladder quick load 1 kb plus DNA ladder | NEB | N0469S | Agarose gel electrophoresis |
| LB Broth, VegitoneNutriSelect Plus | Millipore | 28713 | Bacterial culture |
| Master Mix PCR High Fidelity Phusion Flash | Thermo Fisher Scientific | F548L | PCR |
| Primers | Eurogentec | PCR | |
| Prosize data analysis software v.4 | Agilent | V.4 | Parallel capillary electrophoresis |
| Qubit assays | Invitrogen | MAN0010876 | DNA quantification |
| Qubit dsDNA HS Assay Kit | LIFE TECHNOLOGIES SAS | Q32854 | DNA quantification |
| Thermocycler | Eppendorf | Ep gradients | PCR |
| UVbox, eBOX VX5 | Vilber Lourmat | Agarose gel electrophoresis visualisation | |
| Water for injectable preparation | Aguettant | PROAMP | PCR |
Riferimenti
- Mahmoud, M., et al. Structural variant calling: the long and the short of it. Genome Biology. 20 (1), 246 (2019).
- Bragg, D. C., et al. Disease onset in X-linked dystonia-parkinsonism correlates with expansion of a hexameric repeat within an SVA retrotransposon in TAF1. Proceedings of the National Academy of Sciences. 114 (51), 11020-11028 (2017).
- Stransky, N., Cerami, E., Schalm, S., Kim, J. L., Lengauer, C. The landscape of kinase fusions in cancer. Nature Communications. 5, 4846 (2014).
- Tenaillon, O., et al. The molecular diversity of adaptive convergence. Science. 335 (6067), 457-461 (2012).
- Vandecraen, J., Chandler, M., Aertsen, A., Van Houdt, R. The impact of insertion sequences on bacterial genome plasticity and adaptability. Critical Reviews in Microbiology. 43 (6), 709-730 (2017).
- Andersson, D. I., Gene Hughes, D. amplification and adaptive evolution in bacteria. Annual Review of Genetics. 43, 167-195 (2009).
- Bailey, S. F., Bataillon, T. Can the experimental evolution programme help us elucidate the genetic basis of adaptation in nature. Molecular Ecology. 25 (1), 203-218 (2016).
- Consuegra, J., et al. Insertion-sequence-mediated mutations both promote and constrain evolvability during a long-term experiment with bacteria. Nature Communications. 12 (1), 980 (2021).
- Burch, C. L., Romanchuk, A., Kelly, M., Wu, Y., Jones, C. D. Genome-wide determination of barriers to horizontal gene transfer. bioRxiv. , (2022).
- Slomka, S., et al. Experimental evolution of Bacillus subtilis reveals the evolutionary dynamics of horizontal gene transfer and suggests adaptive and neutral effects. Genetics. 216 (2), 543-558 (2020).
- Choudhury, D., Saini, S. Evolution of Escherichia coli in different carbon environments for 2,000 generations. Journal of Evolutionary Biology. 32 (12), 1331-1341 (2019).
- Tenaillon, O., et al. Tempo and mode of genome evolution in a 50,000-generation experiment. Nature. 536 (7615), 165-170 (2016).
- Behringer, M. G., et al. Escherichiacoli cultures maintain stable subpopulation structure during long-term evolution. Proceedings of the National Academy of Sciences. 115 (20), 4642-4650 (2018).
- Voordeckers, K., et al. Adaptation to high ethanol reveals complex evolutionary pathways. PLoS Genetics. 11 (11), 1005635 (2015).
- Levy, S. F., et al. Quantitative evolutionary dynamics using high-resolution lineage tracking. Nature. 519 (7542), 181-186 (2015).
- Bruger, E. L., Marx, C. J. A decade of genome sequencing has revolutionized studies of experimental evolution. Current Opinion in Microbiology. 45, 149-155 (2018).
- Grubaugh, N. D., et al. An amplicon-based sequencing framework for accurately measuring intrahost virus diversity using PrimalSeq and iVar. Genome Biology. 20 (1), 8 (2019).
- Bedhomme, S., et al. Evolutionary changes after translational challenges imposed by horizontal gene transfer. Genome Biology and Evolution. 11 (3), 814-831 (2019).
- Tenaillon, O., Toupance, B., Le Nagard, H., Taddei, F., Godelle, B. Mutators, population size, adaptive landscape and the adaptation of asexual populations of bacteria. Genetics. 152 (2), 485-493 (1999).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneEsplora altri articoli
This article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati