
Cromatografía de líquidos de alto rendimiento (HPLC)
Visión general
Fuente: Dr. Pablo Bower - Universidad de Purdue
Cromatografía líquida de alto rendimiento (HPLC) es un importante método analítico utilizado para separar y cuantificar los componentes de muestras líquidas. En esta técnica, una solución (primera fase) se bombea a través de una columna que contiene un embalaje de pequeñas partículas porosas con una segunda fase a la superficie. Las diferentes solubilidades de los componentes de la muestra en las dos fases causan los componentes para moverse a través de la columna con diferentes velocidades promedio, creando así una separación de estos componentes. La solución bombeada es llamada la fase móvil, mientras que la fase en la columna se llama la fase estacionaria.
Hay varios modos de cromatografía de líquidos, dependiendo del tipo de fase estacionaria o móvil empleado. Este experimento utiliza cromatografía de fase inversa, donde la fase estacionaria es apolar y la fase móvil es polar. La fase estacionaria a emplear es C18 grupos de hidrocarburos adheridos a partículas de sílice de 3 μm, mientras que la fase móvil es un buffer acuoso con un modificador orgánico polar (acetonitrilo) añadido para variar su fuerza de elución. De esta forma, la silicona puede utilizarse para obtener muestras que son solubles en agua, proporcionando una amplia gama de aplicaciones. En este experimento, se separan las mezclas de tres componentes frecuentes en refrescos de dieta (es decir, cafeína, benzoato y aspartame). Se utilizan siete soluciones preparadas que contengan cantidades conocidas de las tres especies, y luego se registran sus cromatogramas.
Principios
Durante un experimento HPLC, una bomba de alta presión toma la fase móvil de un depósito a través de un inyector. Entonces viaja a través de una columna de fase inversa C18-embalado para la separación de componentes. Por último, la fase móvil se mueve en una célula del detector, donde se mide la absorbancia a 220 nm y termina en una botella de residuos. La cantidad de tiempo que tarda un componente viajar desde el puerto del inyector al detector se llama el tiempo de retención.
Un cromatógrafo líquido es utilizado en este experimento, donde se realiza la separación en una columna de fase inversa. Las dimensiones de la columna son 3 mm (diámetro interno) x 100 mm y la sílice (tamaño de partícula de 3 μm) del embalaje es funcionalizado con C18 octadecylsilane (ODS). Una válvula de inyección rotatoria de 6 puertos Rheodyne se utiliza para almacenar inicialmente la muestra en un pequeño bucle y presenta la muestra a la fase móvil en rotación de la válvula.
La detección es por espectroscopia de absorción en una longitud de onda de 220 nm. Este experimento se puede ejecutar a 254 nm, si un detector no es variable. Los datos del detector tiene una salida de voltaje analógica, que se mide con un multímetro digital (DMM), y leído por un ordenador con un programa de adquisición de datos. El cromatograma resultante tiene un pico para cada componente en la muestra. Para este experimento, los tres componentes responsables dentro de 5 minutos.
Este experimento utiliza una fase móvil y la bomba, que se llama fase móvil isocrática. Para las muestras que son muy difíciles de separar, se puede utilizar un gradiente fase móvil. Esto es cuando la fase móvil inicial es principalmente acuosa, y con el tiempo, una fase móvil segunda orgánica poco a poco se agrega a la fase móvil en general. Este método plantea la polaridad de esta fase con el tiempo, que reduce los tiempos de retención de los componentes y funciona de forma similar a un gradiente de temperatura en un cromatógrafo de gases. Hay algunos casos donde la columna se calienta (generalmente a 40 ° C), que quita cualquier retención errores asociados con un cambio de temperatura.
En HPLC de fase inversa, la fase estacionaria de la columna del embalaje es generalmente un C4, C8 o C18 de embalaje. Las columnas de C4 son principalmente de proteínas de gran peso molecular, mientras que las columnas C18 para péptidos y ejemplos básicos con pesos moleculares más bajos.
Detección por espectroscopia de absorción es mayoritariamente el método de detección de elección, como los espectros de absorción de los componentes están todos disponibles. Algunos sistemas utilizan medidas electroquímicas, como la conductividad o la Amperometría, como métodos de detección.
Para este experimento, la fase móvil es principalmente 20% acetonitrilo y 80% purificada agua desionizada (DI). Una pequeña cantidad de ácido acético se agrega al bajar el pH de la fase móvil, que mantiene el silanol en la fase de embalaje inmóvil en un estado undissociated. Esto reduce el pico de la adsorción de relave, dando picos más estrechos. Luego, se ajusta el pH con hidróxido de sodio 40% para elevar el pH y ayudar a disminuir los tiempos de retención de los componentes.
Cada grupo utiliza un conjunto de 7 frascos que contienen diferentes concentraciones de las soluciones estándar (tabla 1). Los 3 primeros se utilizan para identificar cada pico, y los últimos 4 son para crear un gráfico de calibración para cada componente. Normas 1-3 también se utilizan para el cuadro de calibración.
| Número | Cafeína (mL) | Benzoato (mL) | Aspartamo (mL) |
| 1 | 4 | 0 | 0 |
| 2 | 0 | 4 | 0 |
| 3 | 0 | 0 | 4 |
| 4 | 1 | 1 | 1 |
| 5 | 2 | 2 | 2 |
| 6 | 3 | 3 | 3 |
| 7 | 5 | 5 | 5 |
Tabla 1. Volúmenes de existencias estándares utilizados para preparar los estándares de trabajo proporcionado 7 (volumen total de cada estándar es de 50 mL).
Procedimiento
1. que la fase móvil
- Para preparar la fase móvil, agregar 400 mL de acetonitrilo a aproximadamente 1,5 L de agua purificada de la DI.
- Cuidadosamente añadir 2,4 mL de ácido acético glacial a esta solución.
- Diluir la solución hasta un volumen total de 2.0 L en un matraz aforado con agua purificada de la DI. La solución resultante debe tener un pH entre 2.8 a 3.2.
- Ajustar el pH a 4.2 agregando hidróxido de sodio 40%, mediante goteo con el uso de un medidor de pH digital calibrado. Agregue muy lentamente una vez que el pH llegue a 4.0. Esto debe tomar alrededor de 50 gotas para llevar a cabo.
- Filtrar la fase móvil a través de un filtro de membrana de Nylon 66 de 0.47 μm bajo vacío para desgasificar la solución y eliminar sólidos que podrían conectar la columna cromatográfica. Es importante la fase móvil para evitar que una burbuja, que podría causar un vacío en la fase estacionaria en la entrada de la columna o su forma de trabajo en la célula del detector, causando inestabilidad con la absorbancia de UV de desgasificación.
2. crear las soluciones de componentes
Los tres componentes que deben hacerse son cafeína (0,8 mg/mL), benzoato de potasio (1,4 mg/mL) y aspartamo (éster metílico de L-aspartil-L-fenilalanina) (6,0 mg/mL). Estas concentraciones, una vez diluidas de la misma manera, ponen los estándares en los niveles encontrados en las muestras de soda.
- Añadir 0,40 g de cafeína a un matraz aforado de 500 mL y diluir hasta la marca de 500 mL con agua desionizada.
- Añadir a un matraz aforado de 500 mL 0,70 g de benzoato y luego diluir a la marca de 500 mL con agua desionizada.
- Añadir 0,60 g de aspartamo a un matraz aforado de 100 mL y diluir hasta la marca de 100 mL con agua desionizada. Colocar esta solución en un refrigerador para evitar la descomposición durante el almacenamiento.
3. para las soluciones estándar 7
Los tres componentes todos tienen diferentes coeficientes de distribución, que afecta a cómo cada uno interactúa con los dos de las fases. Cuanto mayor sea el coeficiente de distribución, cuanto más tiempo que pasa el componente en la fase estacionaria, dando por resultado la retención más veces para alcanzar el detector.
- Siguiendo la tabla en el cuadro 1, pipetear la cantidad adecuada de cada componente en un matraz aforado de 50 mL.
- Diluir cada una de las soluciones stock para la marca de 50 mL en matraces aforados con fase móvil.
- Verter cada solución estándar en etiquetado pequeños viales en una gradilla de muestras.
- Almacene los racks de muestras en un refrigerador, junto con las soluciones restantes en los matraces aforados de 50 mL.
4. comprobación de la configuración inicial del sistema HPLC
- Confirman que la línea de residuos en un contenedor de residuos y no es el reciclaje en la fase móvil.
- Verificar que el caudal de la fase móvil se establece en 0, 5 mL/min. Esto es lo suficientemente alto como para permitir que todos los picos eluir dentro de 5 min y frenar lo suficiente para permitir la buena resolución.
- Verificar que la presión mínima y máxima y el caudal se establecen en los valores correctos en el panel frontal del sistema solvente (la bomba).
- Presión mínima: 250 psi (esto es para apagar la bomba, si se produce una fuga).
- Presión máxima: 4.000 psi (esto es para proteger la bomba de última hora, si la forma de un estorbo).
- Pulsa el "cero" en el panel frontal del detector para el espacio en blanco (el blanco es la fase móvil pura).
- Enjuague una jeringa de 100 μl con agua desionizada, luego con varios volúmenes de una de las normas de trabajo para ser analizado y llene la jeringa con la solución. Empezar con las 3 muestras del solo-componente, que permite identificar el pico de cada componente de interés.
5. manualmente inyectar la muestra y recolección de datos
- Con el mango del inyector en la posición de carga, lentamente inyectar 100 μl de solución a través del puerto de tabique.
- Verificar que el programa de recolección de datos está configurado para recopilar datos para 300 s, que permite suficiente tiempo para que todos los 3 picos a fin de eluir a través del detector.
- Cuando esté listo para iniciar la prueba, gire la manija del inyector a la posición de inyectar (que inyecta la muestra en la fase móvil) y haga clic en "Iniciar prueba" en el programa de recolección de datos de computadora inmediatamente. Normas 1-3, sólo uno de los tres picos secuenciales aparecen en la pantalla durante la ejecución (figura 1).
- Una vez 300 s han pasado, la recolección de datos envía un mensaje para guardar el archivo de datos. Guardar los datos bajo un nombre de archivo adecuada (p. ej., enfermedades de transmisión sexual #1).
- Nota el tiempo en segundos para el pico de cada ensayo, que se utiliza en la identificación de ese componente.
- Retire la jeringa del septo y repita el proceso para cada una de las restantes normas de trabajo, utilizando el mismo tiempo por el cromatograma como determinado a partir de la primera carrera.
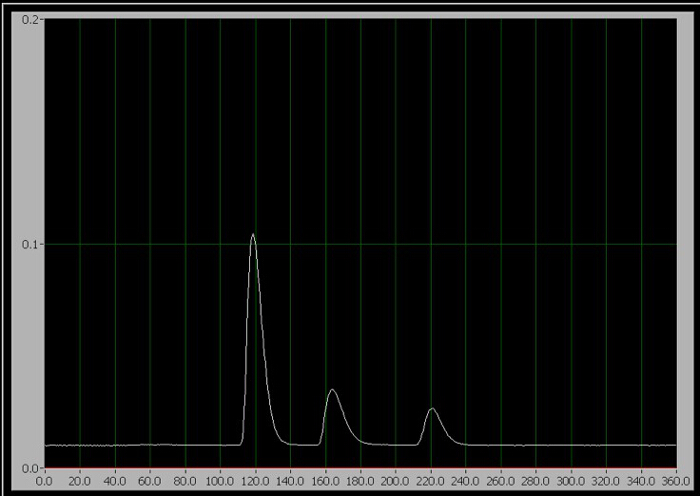
Figura 1. El cromatograma de los 3 componentes. De izquierda a derecha, son cafeína, aspartamo y benzoato.
6. las muestras de refrescos de dieta
Coke Zero, Diet Coke y Pepsi de dieta son las "incógnitas". Se han quedado de envases abiertos durante la noche para deshacerse de la carbonatación, como burbujas no son buenas para el sistema HPLC. Esto suficientemente se deshace de los gases en las muestras.
- Dibujar aproximadamente 2 mL de la soda de dieta en una jeringa de plástico.
- Coloque la punta del filtro a la jeringa a través de Luer-Lok girando en su lugar.
- Empujar el líquido en la jeringa a través del filtro y en un frasco pequeño de vidrio. Esto elimina las partículas no deseadas que potencialmente podrían obstruir la columna de la separación.
- Diluir cada muestra con una cantidad igual de agua DI, por lo que son en el 50% de pureza.
- Inyectar 100 μl de la muestra en el bucle de muestra y realizar ensayos con los mismos parámetros en cuanto a las normas.
7. cálculos
- De las concentraciones de las soluciones de componente, calcular la concentración de todos los componentes en las normas, basadas en las diluciones que se realizaron para las 7 muestras.
- Determinar las áreas de picos en los cromatogramas de cada estándar y muestras desconocidas por el método triangular, que es igual a horas punta de altura el ancho a la altura de ½ (figura 2). Después de determinar que pico corresponde a cada componente basado en el tiempo que tarda cada componente mostrar sus respectivos picos, entrar en estas áreas de pico en una hoja de cálculo de computadora.
- Crear curvas de calibración de área de pico frente a concentración (mg/L) en las normas para los tres componentes.
- Determinar que los mínimos de la cuadrados aptos para cada curva de calibración.
- Calcular la concentración de cada componente en los refrescos de dieta de las áreas de pico que se muestra en los ensayos de HPLC de las muestras. Recuerde que el refresco de dieta se diluyó por un factor de 2 antes de inyectar en el sistema HPLC.
- Calcular la cantidad, en mg/L, de cada componente de los refrescos de dieta.
- Basándose en los resultados, calcular los miligramos de cada componente en una lata de 12 onzas de soda. Asumir 12 oz = 354,9 mL.
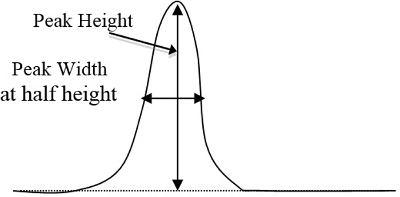
Figura 2. Un ejemplo básico de una curva pico alto y ancho, que se multiplicará (altura del pico veces ancho de altura ½).
Resultados
Los cromatogramas HPLC son capaces de cuantificar cada uno de los 3 componentes para todas las muestras basadas en las curvas de calibración de los estándares (figura 3).
De este conjunto de experimentos, se determinó que una lata de 12 onzas de estas sodas de dieta contiene las siguientes cantidades de cada componente:
Diet Coke: 50,5 mg de cafeína; aspartamo 217,6 mg; benzoato de 83,6 mg.
Coca-Cola Zero: 43,1 mg de cafeína; aspartamo 124,9 mg; benzoato de 85,3 mg.
Dieta Pepsi: cafeína de 34,1 mg; aspartamo 184,7 mg; benzoato de 79,5 mg.
No es de extrañar, los 3 tenían aproximadamente la misma cantidad de benzoato, ya que es sólo un preservativo. Los productos Coca Cola tenían un poco más de cafeína y la Coca Zero tenía mucho menos aspartamo que los otros dos refrescos, ya que también incluye ácido cítrico para algunos saborizantes.
Los siguientes números son las reales cantidades de cafeína y el aspartamo en una lata de 12 oz de las sodas de 3 dieta (el contenido de cafeína se obtuvo de las páginas web de Coca-Cola y Pepsi. El contenido de aspartame se obtuvo de LiveStrong.com y DiabetesSelfManagement.com.):
Diet Coke: 46 mg de cafeína; aspartamo 187,5 mg
Coca-Cola Zero: 34 mg de cafeína; aspartamo 87,0 mg
Dieta Pepsi: de 35 mg de cafeína; aspartamo 177,0 mg
Cálculos de la muestra (tabla 2):
Concentración de cafeína en ETS #1: la solución del componente de cafeína tenía 0,400 g de cafeína diluida en 500 mL = 0,500 L → 0,800 g / L = 0,800 mg / mL.
ETS #1 tenía 1 mL de esta solución diluida a 50,0 mL
0,800 mg/mL * (1,0 mL 50,0 mL) = 0,016 mg / mL = 16,0 mg / L.
STD #2 tenía 2 mL de esta solución diluida a 50,0 mL
0,800 mg/mL * (2,0 mL 50,0 mL) = 0,032 mg / mL = 32,0 mg / L.
Los resultados de las gráficas de calibración de tres (figura 4) rindieron las siguientes ecuaciones:
Área de pico de cafeína = 0.1583* [mg/L de cafeína] - 0.574
Área de pico de aspartamo = 0.02696* [mg/L de Aspartame] - 0.405
Área de pico de benzoato = 0.1363* [benzoato mg/L] - 1.192
Coca-Cola: Área de pico de cafeína = 10.68 = 0.1583* [mg/L de cafeína] - 0.574
[Mg/L de cafeína] = (10.68 + 0.574) / (0.1583) = 71,1 mg/L en la muestra inyectada.
Puesto que la muestra fue diluida en un factor de 2, la Diet Coke tenía cafeína 141,2 mg/L.
El monto por 12 onzas puede = (141,2 mg/L) (mL/12-onzas 0.3549 se puede) = 50,5 mg de cafeína / puede.
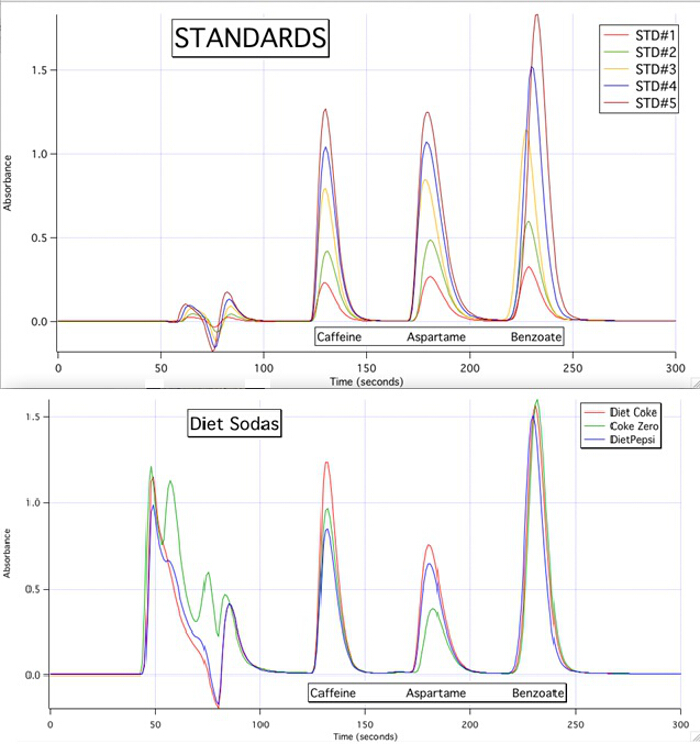
Figura 3. Los cromatogramas HPLC de los 5 estándares y las 3 muestras.
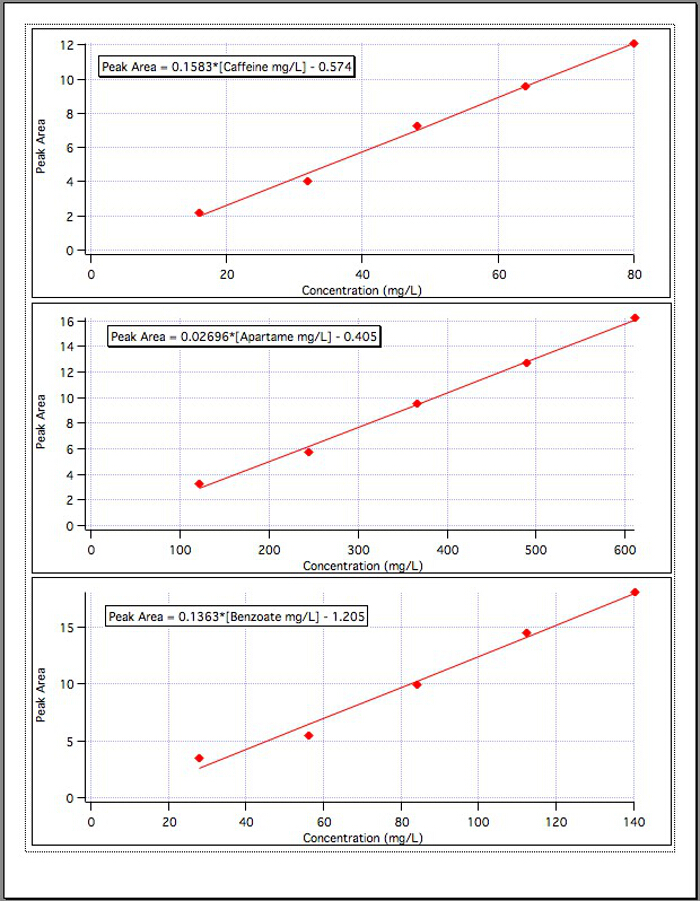
Figura 4. Las curvas de calibración para cada uno de los 3 componentes.
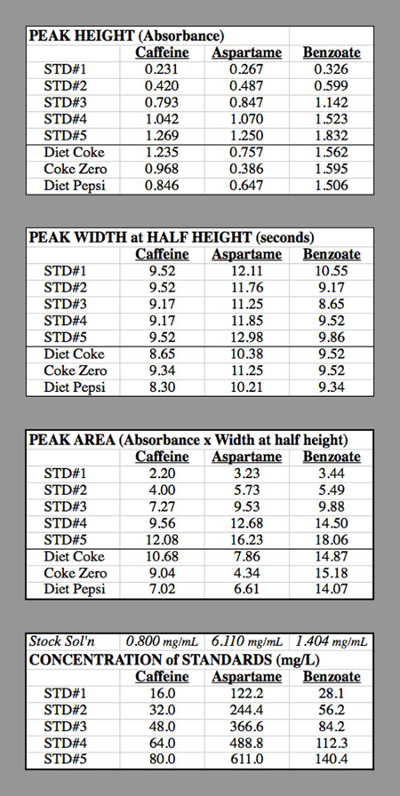
Tabla 2. Las tablas de datos para los ensayos de HPLC utilizados para generar las curvas de calibración.
Aplicación y resumen
HPLC es una técnica ampliamente utilizada en la separación y detección para muchas aplicaciones. Es ideal para compuestos no volátiles, como cromatografía de gases (GC) requiere que las muestras están en su fase de gas. Compuestos no-volátiles incluyen azúcares, vitaminas, drogas y metabolitos. También, es no destructiva, que permite a cada componente a ser recogidos para su posterior análisis (tales como espectrometría de masas). Las fases móviles son prácticamente ilimitadas, que permite cambiar la polaridad del pH para lograr mejor resolución. El uso de fases móviles gradiente permite estos cambios durante los ensayos reales.
Ha habido preocupación sobre los problemas de salud que pueden estar asociados con el aspartame del dulcificante artificial. Etiquetado actual no muestra la cantidad de estos componentes dentro de las bebidas de dieta. Este método permite cuantificar dichas cantidades, junto con la cafeína y benzoato.
Otras aplicaciones incluyen la determinación de las cantidades de plaguicidas en el agua; determinar la cantidad de acetaminofén o ibuprofeno en tabletas analgésicas; determinar si hay fármacos mejoran el rendimiento presentes en la sangre de los atletas; o simplemente determinar la presencia de drogas en un laboratorio de Criminalística. Mientras que las concentraciones de las muestras y a menudo la identidad de los componentes, pueden ser fácilmente determinadas, una limitación es que varias muestras podrían tener cerca de tiempos, dando por resultado Co liberador de retención idéntica.
Tags
Saltar a...
Vídeos de esta colección:

Now Playing
Cromatografía de líquidos de alto rendimiento (HPLC)
Analytical Chemistry
385.0K Vistas

Preparación de muestras para la caracterización analítica
Analytical Chemistry
84.8K Vistas

Estándares internos
Analytical Chemistry
204.9K Vistas

Método de adición estándar
Analytical Chemistry
320.3K Vistas

Curvas de calibración
Analytical Chemistry
797.3K Vistas

Espectroscopía ultravioleta-visible (UV-Vis)
Analytical Chemistry
624.0K Vistas

Espectroscopía de Raman para el análisis químico
Analytical Chemistry
51.2K Vistas

Fluorescencia de rayos x (XRF)
Analytical Chemistry
25.4K Vistas

Cromatografía de gases (CG) con detección de ionización de llama
Analytical Chemistry
282.3K Vistas

Cromatografía de intercambio iónico
Analytical Chemistry
264.7K Vistas

Electroforesis capilar (EC)
Analytical Chemistry
94.0K Vistas

Introducción a la espectrometría de masas
Analytical Chemistry
112.3K Vistas

Microscopía electrónica de barrido (MEB)
Analytical Chemistry
87.3K Vistas

Mediciones electroquímicas de catalizadores soportados utilizando un potenciostato/galvanostato
Analytical Chemistry
51.4K Vistas

Voltametría cíclica (CV)
Analytical Chemistry
125.4K Vistas
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados