Hochleistungs-Flüssigkeitschromatographie (HPLC)
Überblick
Quelle: Dr. Paul Bower - Purdue Universität
Hochleistungs-Flüssigkeitschromatographie (HPLC) ist eine wichtige analytische Methode häufig verwendet, um zu trennen und Komponenten von flüssigen Proben zu quantifizieren. Bei dieser Technik wird eine Lösung (erste Phase) durch eine Spalte gepumpt, die eine Verpackung von kleinen porösen Partikeln mit einer zweiten Phase gebunden an die Oberfläche enthält. Die verschiedenen Solubilities der Probe Komponenten in den beiden Phasen dazu führen, dass die Komponenten durch die Säule mit unterschiedlichen durchschnittlichen Geschwindigkeiten, wodurch eine Trennung dieser Komponenten verschieben. Die gepumpte Lösung heißt der mobilen Phase, während die Phase in der Spalte der stationären Phase genannt wird.
Es gibt verschiedene Modi der Flüssigchromatographie, je nach Art der stationären oder mobilen Phase eingesetzt. Dieses Experiment verwendet umgekehrt-Phase Chromatographie, wo die stationäre Phase unpolar, und die mobile Phase ist polar. Die stationäre Phase eingesetzt werden ist C18 Kohlenwasserstoff-Gruppen auf 3 µm Silica-Partikel gebunden, während die mobile Phase eine wässrige Puffer mit polaren organischen Modifier (Acetonitril ist) hinzugefügt, um seine Medikamentenfreisetzende Stärke variieren. In dieser Form kann die Kieselsäure für Proben verwendet werden, die wasserlöslich ist, bietet ein breites Anwendungsspektrum. In diesem Experiment werden die Mischungen der drei Komponenten häufig in Diät Softdrinks (nämlich Koffein Natriumbenzoat und Aspartam) getrennt. Sieben vorbereitete Lösungen mit bekannten Beträge der drei Arten dienen, und ihre Chromatogramme werden dann aufgezeichnet.
Grundsätze
Während ein HPLC-Experiment nimmt eine Hochdruckpumpe die mobile Phase aus einem Reservoir durch einen Injektor. Er reist dann durch eine Reverse-Phase C18-verpackten Spalte für Trennung der Komponenten. Schließlich zieht die mobile Phase in eine Detektorzelle, wo die Extinktion bei 220 nm und endet in einer Abfallflasche gemessen wird. Die Höhe der Zeitaufwand für eine Komponente vom Injektor Hafen zum Detektor zu reisen ist die Retentionszeit genannt.
Ein liquid Chromatograph ist in diesem Experiment verwendet wo erfolgt die Trennung auf einer Reverse-Phase-Spalte. Die Spalte Abmessungen sind 3 mm (Innendurchmesser) x 100 mm und die Kieselsäure (3 µm Partikelgröße) Verpackung mit C18 Octadecylsilane (ODS) funktionalisiert wird. Eine Rheodyne 6-Port rotary Einspritzventil wird verwendet, um zunächst die Probe in einer kleinen Schleife speichern und führt die Probe in der mobilen Phase beim Drehen des Ventils.
Erkennung ist durch Absorption Spektroskopie bei einer Wellenlänge von 220 nm. Dieses Experiment ausgeführt werden kann, bei 254 nm, wenn ein Melder nicht variabel ist. Daten vom Melder hat einen analogen Spannungsausgang, gemessen mit einem digitalen Multimeter (DMM), und lesen Sie von einem Computer mit einer Daten-Übernahme-Programm geladen. Die daraus resultierende Chromatogramm hat eine Spitze für jede Komponente in der Probe. Für dieses Experiment eluieren aller drei Komponenten innerhalb von 5 Minuten.
Dieses Experiment verwendet eine einzelne mobile Phase und Pumpe, die eine isokratische mobile Phase genannt wird. Für Proben, die schwer zu trennen sind, kann eine Farbverlauf mobile Phase verwendet werden. Dies ist bei die erste mobilen Phase in erster Linie eine wässrige ist und im Laufe der Zeit eine zweite organische mobile Phase allmählich die insgesamt mobile Phase hinzugefügt. Diese Methode löst die Polarität dieser Phase im Laufe der Zeit die senkt der Retentionszeiten der Komponenten und funktioniert ähnlich wie ein Temperaturgradient auf einen Gaschromatographen. Es gibt einige Fälle, wo die Spalte (in der Regel auf 40 ° C) erhitzt wird, die nimmt Aufbewahrung Zeitfehler im Zusammenhang mit einer Änderung der Umgebungstemperatur.
Im Reverse-Phase-HPLC ist die Spalte stationäre Phase Verpackung in der Regel eine C4, C8 und C18 Verpackung. Die C4-Spalten sind in erster Linie für Proteine mit großen Molekulargewichten C18 Spalten für Peptide und einfache Beispiele mit niedrigeren Molmassen sind.
Erkennung durch Absorption Spektroskopie ist mit überwältigender Mehrheit die Methode der Wahl, da die Absorptionsspektren der Komponenten alle leicht zugänglich sind. Einige Systeme verwenden elektrochemische Messungen, wie Leitfähigkeit oder strommesstechnik, als ihre Nachweismethode.
Für dieses Experiment die mobile Phase ist in erster Linie 20 % Acetonitril und 80 % gereinigte deionisiertes Wasser (DI). Eine kleine Menge an Essigsäure wird hinzugefügt, um den pH-Wert der mobilen Phase zu senken, hält die Silanol in der stationären Nachdruckphase in einem undissoziierten Zustand. Dies reduziert die Adsorption Spitze von Tailing, schmalere Spitzen geben. Dann ist der pH-Wert eingestellt, mit 40 % Natriumhydroxid, den pH-Wert zu erhöhen und verringern die Retentionszeiten der Komponenten.
Jede Gruppe nutzt eine Reihe von den 7 Ampullen mit verschiedenen Konzentrationen von Standardlösungen (Tabelle 1). Die ersten 3 werden verwendet, um jede Spitze zu identifizieren, und die letzten 4 sind zur Grafikerstellung Kalibrierung für jede Komponente. Maßstäbe 1 – 3 sind auch für die Kalibrierung-Diagramm verwendet.
| Anzahl | Koffein (mL) | Benzoat (mL) | Aspartam (mL) |
| 1 | 4 | 0 | 0 |
| 2 | 0 | 4 | 0 |
| 3 | 0 | 0 | 4 |
| 4 | 1 | 1 | 1 |
| 5 | 2 | 2 | 2 |
| 6 | 3 | 3 | 3 |
| 7 | 5 | 5 | 5 |
Tabelle 1. Mengen an Lager Standards verwendet, um die 7 bereitgestellten Arbeitsstandards vorbereiten (Gesamtvolumen von jedem Standard ist 50 mL).
Verfahren
(1) macht der mobilen Phase
- Bereiten Sie die mobile Phase von ca. 1,5 L Trinkwasser DI 400 mL Acetonitril hinzufügen.
- 2,4 mL Eisessig sorgfältig diese Projektmappe hinzufügen.
- Verdünnen Sie die Lösung für ein Gesamtvolumen von 2,0 L in einem volumetrischen Kolben mit gereinigtem DI Wasser. Die resultierende Lösung sollte einen pH-Wert zwischen 2,8 bis 3.2 aufweisen.
- Stellen Sie den pH-Wert auf 4.2 durch Zugabe von 40 % Natronlauge tropfenweise mit dem Einsatz von einem kalibrierten digital pH-Meter. Fügen Sie sehr langsam, sobald der pH-Wert 4.0 erreicht. Dies dauert rund 50 Tropfen zu erreichen.
- Filtern Sie die mobile Phase durch ein 0,47-µm-Nylon 66-Membranfilter unter Vakuum um die Lösung zu entgasen und Feststoffe zu entfernen, die die chromatographische Säule stecken könnte. Es ist wichtig zu entgasen die mobile Phase zu vermeiden, dass eine Blase, die entweder eine Lücke in der stationären Phase am Einlass der Spalte verursachen oder Arbeit ihren Weg in der Detektorzelle verursacht Instabilität mit der UV-Absorption.
2. erstellen die Komponentenlösungen
Die drei Komponenten, die getroffen werden müssen sind Koffein (0,8 mg/mL), Kalium-Benzoat (1,4 mg/mL) und Aspartam (L-Aspartyl-L-Phenylalanin Methylester) (6,0 mg/mL). Diese Konzentrationen verdünnt einmal in der gleichen Weise setzen die Standards auf den Ebenen in den Soda-Proben gefunden.
- Eine volumetrische 500-mL-Flasche 0,40 g Koffein hinzufügen, dann auf die 500-mL-Markierung mit VE-Wasser zu verdünnen.
- Eine volumetrische 500-mL-Flasche 0,70 g Natriumbenzoat hinzufügen, dann auf die 500-mL-Markierung mit VE-Wasser zu verdünnen.
- Eine volumetrische 100 mL-Flasche 0,60 g von Aspartam hinzufügen, dann auf den 100-mL-Markierung mit VE-Wasser zu verdünnen. Legen Sie diese Lösung in einem Kühlschrank, Zersetzung während der Lagerung zu vermeiden.
3. machen die 7 Standard-Lösungen
Alle drei Komponenten haben unterschiedliche Verteilung Koeffizienten, die beeinflusst, wie jeder mit den beiden Phasen interagiert. Je größer der Verteilung Koeffizient, desto mehr Zeit verbringt die Komponente in der stationären Phase, was zu längerer Verweildauer in den Detektor erreichen Zeiten.
- Nach der Tabelle in Tabelle 1, pipettieren die korrekte Menge der einzelnen Komponenten in eine volumetrische 50-mL-Flasche.
- Verdünnen Sie die Stammlösungen, die 50-mL-Markierung auf die volumetrische Fläschchen mit mobilen Phase.
- Gießen Sie jede standard-Lösung in beschrifteten kleinen Fläschchen in einem Probenrack.
- Die Regale von Proben im Kühlschrank, zusammen mit den restlichen Lösungen in die volumetrische 50-mL-Fläschchen aufbewahren.
4. überprüfen die Grundeinstellungen der HPLC-Anlage
- Bestätigen Sie, dass die Abwasserlinie in einem Abfallbehälter und ist nicht wieder in der mobilen Phase recycling.
- Stellen Sie sicher, dass die Durchflussmenge der mobilen Phase auf 0,5 mL/min eingestellt ist. Dies ist hoch genug, um ermöglichen allen Gipfeln eluieren innerhalb von 5 min und langsam genug, um schöne Auflösung zu ermöglichen.
- Stellen Sie sicher, dass die minimalen und maximalen Druck und die Durchflussmenge auf die korrekten Werte auf der Vorderseite der Lösungsmittel Delivery Systems (die Pumpe) festgelegt sind.
- Minimale Druckeinstellung: 250 Psi (Dies ist die Pumpe abgeschaltet, wenn ein Leck auftritt).
- Maximale Druckeinstellung: 4.000 Psi (Dies ist zum Schutz der Pumpe vor dem brechen, wenn ein Clog bildet).
- Drücken Sie "Null" auf den Detektor Frontplatte um die Leerzeichen gesetzt (die leere ist die reine mobile Phase).
- Spülen einer 100 µL Spritze mit entionisiertem Wasser, dann mit mehreren Bänden eines Arbeitsstandards werden analysiert, und füllen die Spritze mit der Lösung. Beginnen Sie mit 3 Einkomponenten-Proben, wodurch für die Ermittlung der Höhepunkt jeder Komponente von Interesse.
5. manuell Einspritzen der Probe und Datenerhebung
- Injizieren Sie mit dem Injektor Griff in Ladeposition langsam 100 µL der Lösung durch das Septum Port.
- Überprüfen Sie, ob Datenbeschaffungsprogramm ist für die Datensammlung für 300 s, die genügend für alle 3 Gipfel Zeit, durch den Detektor eluieren.
- Wenn Sie bereit zum Starten der Testversion, drehen Sie den Injektor Griff in die Spritzen-Position (die Probe in der mobilen Phase injiziert) und klicken Sie auf "Test starten" auf dem Computer Datenbeschaffungsprogramm sofort. Erscheinen Sie für Standards 1-3, nur einer der drei sequenziellen Gipfel auf dem Bildschirm während des Laufs (Abbildung 1).
- Einmal 300 s vergangen, die Datenerhebung sendet eine Aufforderung zum Speichern der Datei. Sichern Sie die Daten unter einem passenden Dateinamen (z. B.STD #1).
- Beachten Sie die Zeit in Sekunden für die Peak von jeder Studie, die bei der Ermittlung dieser Komponente verwendet wird.
- Ziehen Sie die Spritze aus dem Septum, und wiederholen Sie den Vorgang für jede der verbleibenden Arbeitsstandards, mit der gleichen Zeit pro Chromatogramm wie aus dem ersten Lauf ermittelt.
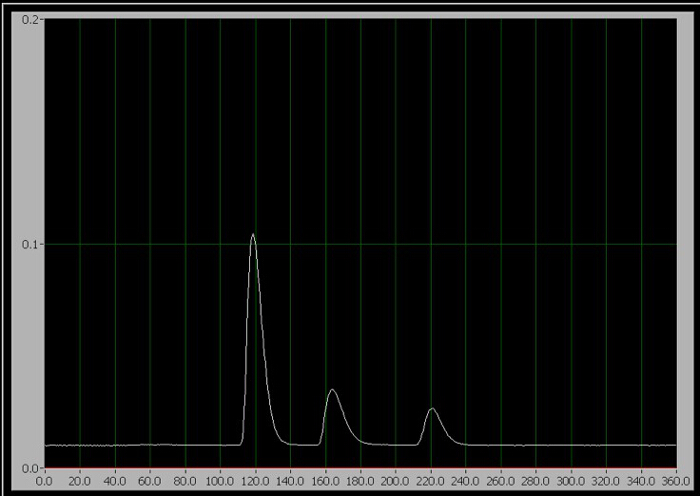
Abbildung 1: Das Chromatogramm der 3 Komponenten. Von links nach rechts sind sie Koffein, Aspartam und Natriumbenzoat.
(6) die Proben der Diät Limonaden
Diet Coke, Diet Pepsi und Coke Zero sind die "unbekannten". Sie haben in offenen Behältern über Nacht get rid of die Kohlensäure ausgelassen worden wie Luftblasen nicht gut für die HPLC-Anlage sind. Dies beseitigt ausreichend keine Gase in den Proben.
- Ziehen Sie etwa 2 mL die Diätsoda in einer Kunststoffspritze.
- Legen Sie die Filterspitze auf die Spritze über Luer-Lok durch Drehen an Ort.
- Drücken Sie die Flüssigkeit in der Spritze durch den Filter und in ein kleines Glas-Fläschchen. Dies beseitigt unerwünschte Partikel, die potenziell die Trennsäule verstopfen könnten.
- Jede Probe mit einer gleichen Menge von VE-Wasser zu verdünnen, so sind sie bei 50 % Reinheit.
- Injizieren Sie 100 µL der Probe in die Probenschleife, und laufen Sie Studien mit den gleichen Parametern wie die Standards.
(7) Berechnungen
- Berechnen Sie von den Konzentrationen der Komponentenlösungen die Konzentration aller Komponenten in den Standards, basierend auf die Verdünnungen, die für die 7 Proben vorgenommen wurden.
- Bestimmen Sie Peakflächen auf die Chromatogramme für jeden Standard und der unbekannten Proben durch die dreieckigen Methode, die Höhe Spitzenzeiten die Breite auf ½ Höhe (Abbildung 2 entspricht). Treten Sie nach der Festlegung, welchen Gipfel entspricht jede Komponente basiert auf den Zeitaufwand für die einzelnen Komponenten zu ihren jeweiligen Höhepunkt zeigen diese Peakflächen in einem Tabellenkalkulationsprogramm auf dem Computer.
- Kalibrierkurven von Peak vs. Konzentration (mg/L) in den Normen für alle drei Komponenten zu erstellen.
- Bestimmen der kleinsten Quadrate für jede Kalibrierungskurve passen.
- Berechnen Sie die Konzentration der einzelnen Komponenten in der Diät-Limonaden aus der Peakflächen der HPLC-Studien für die Beispiele gezeigt. Denken Sie daran, dass die Diät-Cola, um den Faktor 2 vor dem Einspritzen in das HPLC-System verdünnt wurde.
- Berechnen Sie die Menge in mg/L, der einzelnen Komponenten in der Diät-Limonaden.
- Basierend auf den Ergebnissen, berechnen Sie die Milligramm der einzelnen Komponenten in der 12-Unze-Dose Soda gefunden. 12 Unzen = 354,9 mL zu übernehmen.
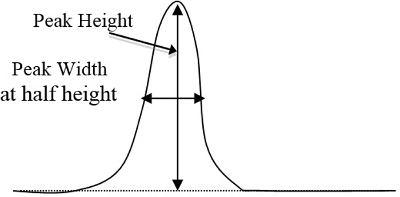
Abbildung 2. Ein einfaches Beispiel für einer Kurve Peakhöhe und Breite, die multipliziert werden sollen (Peakhöhe x Breite in ½ Höhe).
Ergebnisse
Die HPLC-Chromatogramme sind in der Lage, jede der 3 Komponenten für alle Proben, die basierend auf die Kalibrierkurven der Standards (Abbildung 3) zu quantifizieren.
Ab dieser Serie von Experimenten wurde festgestellt, dass eine 12-oz Dose diese Diät-Limonaden die folgenden Beträge der einzelnen Komponenten enthalten:
Diät-Cola: 50,5 mg Koffein; 217,6 mg Aspartam; 83,6 mg Natriumbenzoat.
Coke Zero: 43,1 mg Koffein; 124,9 mg Aspartam; 85,3 mg Natriumbenzoat.
Diät-Pepsi: 34,1 mg Koffein; 184,7 mg Aspartam; 79,5 mg Natriumbenzoat.
Nicht überraschend, hatten alle 3 etwa die gleiche Menge an Natriumbenzoat, da es nur ein Konservierungsmittel ist. Die Cola-Produkte hatte ein bisschen mehr Koffein und Coke Zero hatte viel weniger Aspartam als die anderen beiden Limonaden, wie es auch Zitronensäure für einige Aromastoffe enthält.
Die folgenden Zahlen sind die tatsächlichen Mengen an Koffein und Aspartam in der 12-oz-Dose der 3 Diät-Limonaden (der Koffeingehalt aus den Webseiten von Coca-Cola und Pepsi ermittelt. Die Aspartam-Inhalte wurden LiveStrong.com und DiabetesSelfManagement.com entnommen.):
Diät-Cola: 46 mg Koffein; 187,5 mg Aspartam
Coke Zero: 34 mg Koffein; 87,0 mg Aspartam
Diät-Pepsi: 35 mg Koffein; 177,0 mg Aspartam
Beispielrechnungen (Tabelle 2):
Konzentration von Koffein in STD #1: die Komponentenlösung für Koffein hatte 0,400 g Koffein bis 500 mL = 0,500 L → 0,800 g / L = 0,800 mg / mL verdünnt.
STD #1 hatte 1 mL dieser Lösung bis 50,0 mL verdünnt
0,800 mg/mL * (1,0 mL/50,0 mL) = 0,016 mg / mL = 16,0 mg / L.
#2 STD hatte 2 mL dieser Lösung bis 50,0 mL verdünnt
0,800 mg/mL * (2,0 mL/50,0 mL) = 0,032 mg / mL = 32,0 mg / L.
Die Ergebnisse aus den drei Kalibrierung Grafiken (Abbildung 4) ergab die folgenden Gleichungen:
Koffein Peakfläche = 0.1583* [mg/L Koffein] - 0.574
Aspartam Peakfläche = 0.02696* [Aspartam mg/L] - 0.405
Benzoat Peakfläche = 0.1363* [Benzoat mg/L] - 1,192
Diät-Cola: Koffein Peakfläche = 10,68 = 0.1583* [mg/L Koffein] - 0.574
[Mg/L Koffein] = (10,68 + 0.574) / (0.1583) = 71,1 mg/L in der injizierten Probe.
Da die Probe mit einem Faktor von 2 verdünnt wurde, musste der Diet Coke 141,2 mg/L Koffein.
Der Betrag pro 12-oz = können (141,2 mg/L) (0.3549 mL/12-oz-Can) = 50,5 mg Koffein / können.
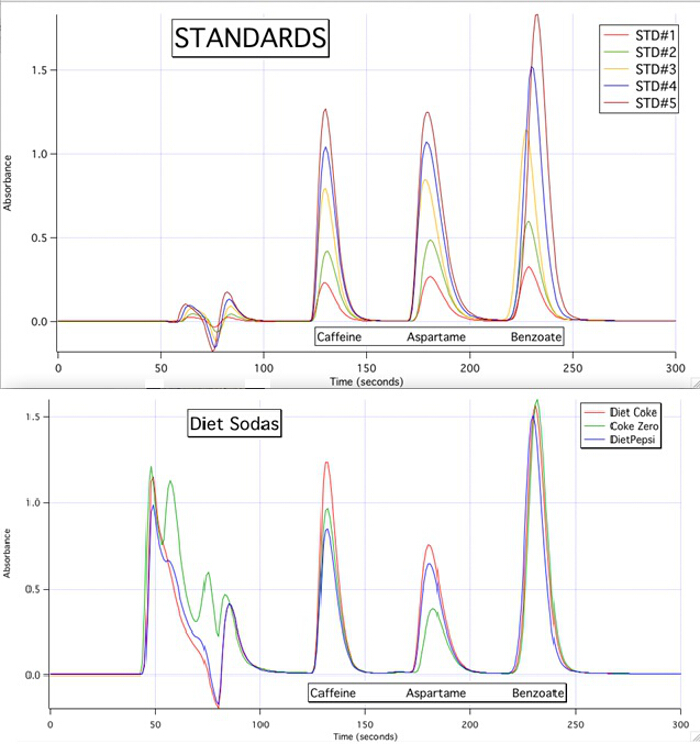
Abbildung 3. Die HPLC-Chromatogramme 5 Standards und die 3 Proben.
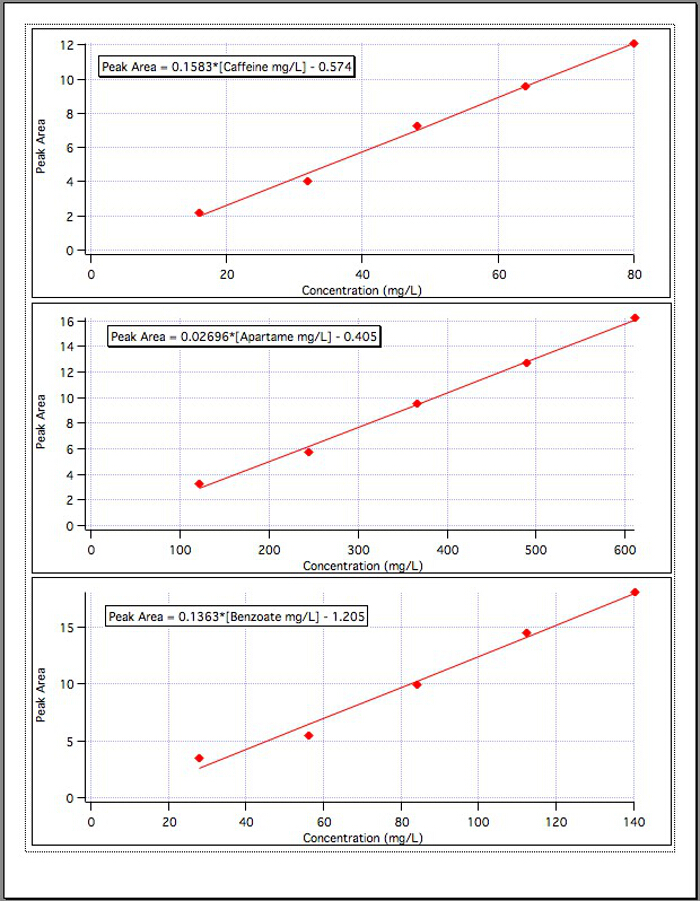
Abbildung 4. Die Kalibrierkurven für jede der 3 Komponenten.
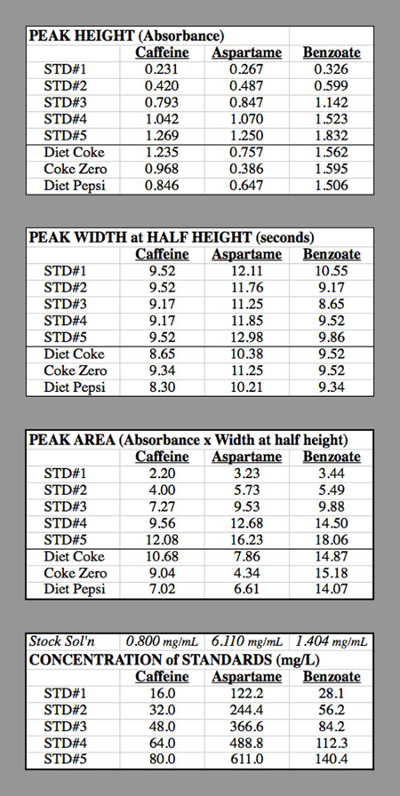
Tabelle 2. Die Tabellen für die HPLC-Studien für die Generierung der Kalibrierkurven verwendet.
Anwendung und Zusammenfassung
HPLC ist eine weit verbreitete Technik in der Trennung und Nachweis für viele Anwendungen. Es ist ideal für nicht-flüchtige Verbindungen, wie Gaschromatographie (GC) setzt voraus, dass die Proben in ihrer Gasphase. Nicht-flüchtiger Verbindungen enthalten Zucker, Vitamine, Medikamente und Metaboliten. Außerdem ist es nicht-destruktiv, wodurch jede Komponente zur weiteren Analyse (z. B. Massenspektrometrie) gesammelt werden. Die mobilen Phasen sind praktisch unbegrenzt, wodurch Änderungen auf die richtige Polung des pH-Wertes, besseren Auflösung zu erreichen. Die Verwendung von gradient mobilen Phasen kann diese Änderungen während der eigentlichen Versuche.
Es wurde Besorgnis über die möglichen gesundheitlichen Probleme, die mit dem Süßstoff Aspartam verbunden sein können. Aktuelle Produkt-Kennzeichnung zeigt nicht die Menge dieser Komponenten im Inneren der Diät-Getränke. Diese Methode ermöglicht diese Beträge zusammen mit dem Koffein und Benzoat zu quantifizieren.
Andere Anwendungen umfassen die Bestimmung der Mengen von Pestiziden in Wasser; Bestimmung der Höhe der Paracetamol oder Ibuprofen in Schmerzen Schmerzmittel Tabletten; bestimmen, ob es leistungssteigernde Mittel in die Blutbahn des Athleten anwesend sind; oder einfach das Vorhandensein von Drogen in eine kriminaltechnische Labor bestimmen. Während die Konzentrationen dieser Proben, und oft die Identität der Komponenten, ohne weiteres ermittelt werden können, ist die eine Einschränkung, dass mehrere Proben in der Nähe von identischen Aufbewahrung Zeiten, wodurch Co eluierenden haben könnte.
Tags
pringen zu...
Videos aus dieser Sammlung:

Now Playing
Hochleistungs-Flüssigkeitschromatographie (HPLC)
Analytical Chemistry
383.1K Ansichten

Probenvorbereitung für die analytische Charakterisierung
Analytical Chemistry
84.2K Ansichten

Interne Standards
Analytical Chemistry
204.3K Ansichten

Standard-Additionsverfahren
Analytical Chemistry
319.4K Ansichten

Kalibrierkurven
Analytical Chemistry
794.7K Ansichten

UV/Vis-Spektroskopie
Analytical Chemistry
621.7K Ansichten

Raman-Spektroskopie für die chemische Analyse
Analytical Chemistry
51.0K Ansichten

Röntgen-Fluoreszenz (XRF)
Analytical Chemistry
25.3K Ansichten

Gaschromatographie (GC) mit Flammen-Ionisations-Detektion
Analytical Chemistry
281.1K Ansichten

Ionenaustausch-Chromatographie
Analytical Chemistry
263.9K Ansichten

Kapillarelektrophorese (CE)
Analytical Chemistry
93.3K Ansichten

Einführung in die Massenspektrometrie
Analytical Chemistry
111.9K Ansichten

Rasterelektronenmikroskopie (SEM)
Analytical Chemistry
86.8K Ansichten

Elektrochemische Messungen von Trägerkatalysatoren mit einem Potentiostat / Galvanostat
Analytical Chemistry
51.3K Ansichten

Zyklische Voltammetrie (CV)
Analytical Chemistry
124.3K Ansichten
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten