
Ensayos ELISA: Indirecto, Sándwich y Competitivo
Visión general
Fuente: Whitney Swanson1,2, Frances V. Sjaastad2,3y Thomas S. Griffith1,2,3,4
1 Departamento de Urología, Universidad de Minnesota, Minneapolis, MN 55455
2 Centro de Inmunología, Universidad de Minnesota, Minneapolis, MN 55455
3 Programa de Posgrado en Microbiología, Inmunología y Biología del Cáncer, Universidad de Minnesota, Minneapolis, MN 55455
4 Centro De Cáncer Masónico, Universidad de Minnesota, Minneapolis, MN 55455
El ensayo inmunoabsorbente ligado a enzimas (ELISA) se utiliza con frecuencia para medir la presencia y/o concentración de un antígeno, anticuerpo, péptido, proteína, hormona u otra biomolécula en una muestra biológica. Es extremadamente sensible, capaz de detectar bajas concentraciones de antígenos. La sensibilidad de ELISA se atribuye a su capacidad para detectar las interacciones entre un único complejo antígeno-anticuerpo (1). Además, la inclusión de un anticuerpo específico de antígeno conjugado en enzimas permite la conversión de un sustrato incoloro en un producto cromogénico o fluorescente que puede ser detectado y fácilmente cuantificado por un lector de placas. En comparación con los valores generados por cantidades valoradas de un antígeno de interés conocido, se puede determinar la concentración del mismo antígeno en las muestras experimentales. Se han adaptado diferentes protocolos ELISA para medir las concentraciones de antígenos en una variedad de muestras experimentales, pero todos tienen el mismo concepto básico (2). La elección del tipo de ELISA para realizar, indirecta, sándwich o competitiva, depende de una serie de factores, incluyendo la complejidad de las muestras a probar y los anticuerpos específicos de antígeno disponibles para su uso. El ELISA indirecto se utiliza con frecuencia para determinar el resultado de una respuesta inmunológica, como la medición de la concentración de un anticuerpo en una muestra. El sándwich ELISA es el más adecuado para analizar muestras complejas, como sobrenatantes de cultivo de tejido o lysates de tejido, donde el analito, o antígeno de interés, es parte de una muestra mixta. Por último, el ELISA competitivo se utiliza con mayor frecuencia cuando sólo hay un anticuerpo disponible para detectar el antígeno de interés. Los ELISA competitivos también son útiles para detectar un pequeño antígeno con un solo epítopo de anticuerpos que no puede acomodar dos anticuerpos diferentes debido a la obstrucción estaica. El protocolo describirá los procedimientos básicos para los ensayos ELISA indirectos, sándwiches y competitivos.
El ensayo elSAY ELISA indirecto se utiliza comúnmente para medir la cantidad de anticuerpos en suero o en el sobrenadante de un cultivo de hibridato. El procedimiento general para el ensayo elAse indirecto el ISA es:
- Capa de pozos con antígenos
- Añadir sobrenadante de cultivo de suero o hibrioma que contenga anticuerpos (primarios o anticuerpos de 1o)
- Incubar y lavar
- Añadir anticuerpos secundarios (o 2o) conjugados enzimáticos
- Incubar y lavar
- Añadir sustrato
El ensayo el sándwich ELISA difiere del ensayo elISA indirecto en que el método no implica el recubrimiento de las placas con un antígeno purificado. En su lugar, se utiliza un anticuerpo "capturar" para recubrir los pocillos de la placa. El antígeno se "se intercala" entre el anticuerpo de captura y un segundo anticuerpo conjugado enzimático de "detección", donde ambos anticuerpos son específicos para el mismo antígeno, pero en diferentes epítopos (3). Al unirse al complejo de anticuerpos/antígenos de captura, el anticuerpo de detección permanece en la placa. Los anticuerpos monoclonales o los antisueros policlonales se pueden utilizar como anticuerpos de captura y detección. La principal ventaja del sándwich ELISA es que la muestra no tiene que ser purificada antes del análisis. Además, el ensayo puede ser bastante sensible (4). Muchos kits ELISA disponibles comercialmente son de la variedad sándwich y utilizan pares de anticuerpos probados y emparejados. El procedimiento general para el ensayo ELISA sándwich es:
- Pozos de abrigo con anticuerpo de captura
- Añadir muestras de prueba que contengan antígeno
- Incubar y lavar
- Añadir anticuerpode detección conjugado enzimático.
- Incubar y lavar
- Añadir sustrato
La mayoría de los kits ELISA sándwiches disponibles comercialmente vienen con anticuerpos de detección conjugados enzimáticos. En los casos en que no se dispone de un anticuerpo de detección conjugado enzimático, se puede utilizar un anticuerpo secundario conjugado enzimático específico para el anticuerpo de detección. La enzima del anticuerpo secundario desempeña la misma función, que consiste en convertir el sustrato incoloro en un producto cromogénico o fluorescente. Al mencionado anticuerpo secundario conjugado enzimática le gustaría más ser utilizado en un sándwich "casero" ELISA desarrollado por un investigador que ha generado sus propios anticuerpos monoclonales, por ejemplo. Un inconveniente del uso de un anticuerpo secundario conjugado enzimático es asegurarse de que sólo se une al anticuerpo de detección, y no al anticuerpo de captura unido a la placa. Esto resultaría en un producto medible en todos los pozos, independientemente de la presencia o ausencia de antígeno o anticuerpo de detección.
Por último, el ensayo ELISA competitivo se utiliza para detectar antígenos solubles. Es fácil de realizar, pero sólo es adecuado cuando el antígeno purificado está disponible en una cantidad relativamente grande. El procedimiento general para el ensayo ELISA competitivo es:
- Capa de pozos con antígeno
- Incubar y lavar
- Muestra de prueba de preincubación con anticuerpos primarios conjugados enzimáticos
- Agregue la mezcla al pozo
- Incubar y lavar cualquier anticuerpo primario conjugado en enzimas sin ataduras
- Añadir sustrato
La "competencia" en este ensayo proviene del hecho de que más antígeno en la muestra de prueba utilizada en el paso 3 resultará en menos anticuerpos disponibles para unir se adhieren al antígeno que recubre el pozo. Por lo tanto, la intensidad del producto cromogénico/fluorogénico en el pozo al final del ensayo está inversamente relacionada con la cantidad de antígeno presente en la muestra de ensayo.
Un componente clave en cualquier tipo de ELISA son los estándares valorados de concentraciones conocidas que permitirán al usuario determinar la concentración de antígeno presente en las muestras de ensayo. Típicamente, una serie de pozos se designan para crear una curva estándar, donde las cantidades conocidas de una proteína recombinante purificada se agregan a los pozos en cantidades decrecientes. Cuando estos pozos se procesan al mismo tiempo que las muestras de prueba, el usuario puede tener un conjunto de referencia de valores de absorbancia obtenidos de un lector de microplacas para que las concentraciones de proteínas conocidas vayan junto con los valores de absorción para las muestras de prueba. A continuación, el usuario puede calcular una curva estándar a la que se pueden comparar las muestras de prueba para determinar la cantidad de proteína de interés presente. La curva estándar también puede determinar el grado de precisión de la fabricación de dilución del usuario.
Por último, el último paso en cada uno de los tipos ELISA enumerados anteriormente requiere la adición de un sustrato. El grado de conversión del sustrato al producto está directamente relacionado con la cantidad de enzima presente en el pozo. La peroxidasa de rábano picante (HRP) y la fosfatasa alcalina (AP) son las enzimas más comunes encontradas conjugadas con los anticuerpos. Como era de esperar, hay una serie de sustratos disponibles específicos para cualquiera de las enzimas que producen un producto cromogénico o fluorescente. Además, los sustratos están disponibles en una gama de sensibilidades que pueden aumentar la sensibilidad general del ensayo. El usuario también debe tener en cuenta el tipo de instrumentación disponible para leer la placa al final del experimento al recoger el tipo de sustrato a utilizar, junto con su correspondiente anticuerpo conjugado enzimático.
Los sustratos cromogénicos de uso común para HRP incluyen 2,2'-Azinobis [3-etilbenzotiazolina-6-ácido sulfónico]-sal de diamonio (ABTS) y 3,3',5,5'-tetrametilbenzidina (TMB), mientras que p-fosfato de nitrofenilo (PNPP) se utiliza para AP. producir productos de reacción de color verde y azul solubles en agua, respectivamente. El producto verde ABTS tiene dos picos de absorbancia principales, 410 y 650 nm, mientras que el producto azul TMB se detecta mejor a 370 y 652 nm. Los colores de ABTS y TMB cambian a amarillo tras la adición de una solución de parada ácida, que se lee mejor a 450 nm. El desarrollo de color para ABTS es lento, mientras que es rápido para TMB. TMB es más sensible que ABTS, y puede producir una señal de fondo más alta si la reacción enzimática continúa demasiado tiempo. PNPP produce un producto amarillo soluble en agua después de la conversión de AP que absorbe la luz a 405 nm.
Procedimiento
1. ELISA indirecto
Un ELISA indirecto es aquel en el que el anticuerpo específico del antígeno primario es reconocido por un anticuerpo conjugado secundario. El siguiente protocolo es un ejemplo de un método ELISA indirecto, donde las muestras séricas de ratones infectados por el virus de la gripe A (IAV) se prueban para detectar la presencia de anticuerpos IgG específicos de IAV. Una fortaleza de este ejemplo es que se pueden utilizar diferentes anticuerpos secundarios que reconocen todos los isotipos de anticuerpos o isotipos específicos (por ejemplo, IgG).
Antígeno de recubrimiento a la microplaca
- Recubrir los pocillos de una placa ELISA de 96 pocillos con antígeno purificado pipeteando 50 l de antígeno purificado (2 mg/ml del virus purificado De/PR/8 influenza A en un tampón Tris-HCl de 0,05M (pH 9,5)) en cada pocal de la placa.
- Cubra la placa con una cubierta adhesiva e incubarla durante la noche a 4oC para permitir que el antígeno se adhiera a la placa.
- Una vez completada la incubación, retire la solución de recubrimiento moviendo la placa sobre un fregadero.
Bloqueo
- Bloquear los sitios de unión a proteínas restantes en los pozos recubiertos mediante la adición de tampón de bloqueo de 200 l, 5% suero de burro en 1X PBS se utiliza aquí, por pozo. Los reactivos de bloqueo alternativos incluyen un 5% de leche seca sin grasa o BSA en PBS o suero normal de un animal en el que se generó el anticuerpo secundario.
- Incubar durante al menos 2 horas a temperatura ambiente o durante la noche a 4oC.
- Después de la incubación, retire el tampón de bloqueo moviendo la placa y luego lave la placa con PBS que contenga 1% de Tween-20.
Incubación con el anticuerpo primario
- Preparar una dilución en serie de la muestra sérica, que contiene el anticuerpo primario, para obtener un rango de dilución de 1 a 204.800, utilizando 1X PBS. Para ello, primero diluir el suero 1:12.5 y luego realizar una dilución 4X (rango de dilución - 1:12.5 a 1:204,800).
- Añadir 100 l de las muestras de suero diluidas en serie a los pozos.
- Placa de cubierta con tapa adhesiva e incubar a temperatura ambiente durante 1-2 h.
- Después de la incubación, mueva la placa sobre un fregadero y lave la placa con PBS que contenga 1% de Tween-20.
Incubación con el anticuerpo secundario
- Añadir 100 l de un anticuerpo secundario conjugado en zimdulación, peroxidasa de rábano picante, anti-ratón conjugado con HRP secundario en este experimento, a cada pocto.
- Incubar la placa durante 1 hora a temperatura ambiente.
- Después de la incubación, mueva la placa sobre un fregadero y luego lave la placa con PBS que contenga 1% de Tween-20.
Detección
- Añadir 100 l del sustrato del indicador (3,3',5,5'-tetrametilbenzidina (TMB)) a una concentración de 1 mg/ml a cada pocal.
- Incubar la placa con el sustrato durante 5-10 min a temperatura ambiente.
- Después de 10 min, detenga la reacción enzimática añadiendo ácido sulfúrico de 100 l 2N (H2SO4).
Dentro de 30 minutos de la adición de la solución de parada, lea la placa utilizando un lector de microplacas a 405 nm para determinar la absorbancia de los pozos.
2. Sandwich ELISA
En esta versión DE ELISA, la muestra experimental se "se intercala" entre un anticuerpo de captura no conjugado y un anticuerpo de detección conjugado, ambos específicos de la misma proteína pero en diferentes epítopos. En el siguiente ejemplo de ELISA sándwich, la concentración de TNF humano se determinó en una muestra desconocida utilizando una curva estándar generada a partir de una dilución en serie 2.5X de un TNF humano recombinante estándar conocido (indicando a una concentración de 75 pg/ml).
Recubrimiento captura de anticuerpos en la microplaca
- Recubrir los pocillos de una placa ELISA de 96 pocillos con anticuerpo de captura purificado añadiendo 100 s de anticuerpo de captura (rango de 1-10 g/ml) a cada pocil de la placa.
- Cubra la placa con una cubierta adhesiva de la placa e incubarla durante la noche a 4oC.
- Después de la incubación, retire la solución de recubrimiento de la placa moviendo la placa sobre un fregadero.
Bloqueo
- Bloquear los sitios de unión a proteínas restantes en los pozos recubiertos de anticuerpos añadiendo una solución de bloqueo de 200 ol, un 5% de leche seca sin grasa que contenga PBS, a los pozos.
- Incubar durante al menos 2 h a temperatura ambiente o durante la noche a 4oC.
- Después de la incubación, retire el tampón de bloqueo moviendo la placa y luego lave la placa con PBS que contenga 1% de Tween-20.
Añadir antígeno que contenga muestras de prueba
- Añadir 100 l de la muestra de prueba a los pozos. Selle la placa con una cubierta adhesiva.
- Incubar durante 1-2 h a temperatura ambiente o durante la noche a 4oC.
- Después de la incubación, retire las muestras moviendo la placa sobre el fregadero y luego lave los pozos con 200 S 1X PBS que contengan 1% De entre 20.
Añadir anticuerpo de detección conjugado enzimático
- Añadir 100 l de anticuerpo de detección conjugado enzimático a los pozos a una concentración preoptimizada.
- Sellar la placa con una cubierta adhesiva e incubar a temperatura ambiente durante 2 h.
- Retire el anticuerpo de detección sin ataduras moviendo la placa sobre un fregadero y lave los pozos con 200 s 1X PBS que contengan 1% de Tween-20.
Detección
- Añadir 100 l del sustrato del indicador a una concentración de 1 mg/ml. Cualquier anticuerpo de detección conjugado enzimático unido convertirá el sustrato en una señal detectable.
- Incubar la placa durante 5-10 min a temperatura ambiente.
- Después de 5-10 min, detenga la reacción enzimática añadiendo 100 s 2N H2SO4 a los pozos. Dentro de los 30 minutos de la adición de la solución de parada, lea la placa utilizando un lector de microplacas para determinar la absorbancia de los pozos.
3. ELISA competitivo
Los pasos de un ELISA competitivo son diferentes de los utilizados en ELISA indirecto y sándwich, siendo la principal diferencia el paso de unión competitivo entre el antígeno de muestra y el antígeno "add-in". El antígeno de la muestra se incuba con el anticuerpo primario sin etiquetar. Estos complejos de anticuerpos y antígenos se añaden a la placa ELISA, que ha sido pre-revestida con el mismo antígeno. Después de un período de incubación, cualquier anticuerpo no unido se lava. Existe una correlación inversa entre la cantidad de anticuerpos libres disponibles para unir el antígeno en el pozo y la cantidad de antígeno en la muestra original. Por ejemplo, una muestra con antígeno abundante tendría más complejos de anticuerpos antigen primarios, dejando poco anticuerpo sin ataduras para unirse a la placa ELISA. Luego se añade a los pozos un anticuerpo secundario conjugado en zimdo específico para el anticuerpo primario, seguido por el sustrato.
Antígeno de recubrimiento a la microplaca
- Recubrir los pocillos de una placa ELISA de 96 pocillos con 100 ml de antígeno purificado a una concentración de 1-10 g/ml.
- Cubra la placa con una cubierta adhesiva de la placa e incubar la placa durante la noche a 4oC.
- Después de la incubación, retire la solución de antígeno sin ataduras de los pozos moviendo la placa sobre un fregadero.
Bloqueo
- Bloquear los sitios de unión a proteínas restantes en los pozos recubiertos añadiendo 200 l de tampón de bloqueo a cada poca, que puede ser un 5% de leche seca no grasa o BSA en PBS.
- Incubar la placa durante al menos 2 h a temperatura ambiente o durante la noche a 4oC.
Muestra de incubación (antígeno) con el anticuerpo primario
- Mientras bloquea los pozos, prepare la mezcla de antígeno-anticuerpo mezclando antígeno de muestra de 150 ml y 150 ml de anticuerpo primario para cada pocaen en el ensayo.
- Incubar esta mezcla durante 1 h a 37oC.
Añadir mezcla antígeno-anticuerpo al pozo
- Ahora, retire el búfer de bloqueo de los pozos moviendo la placa sobre un fregadero.
- Luego, lave los pozos con 1X PBS que contenga Tween-20.
- Añadir 100 l de la mezcla de anticuerpos primarios de antígeno de muestra.
- Incubar la placa a 37oC durante 1 h.
- Retire la mezcla de muestra moviendo la placa sobre un fregadero.
- Luego, lave los pozos con 1X PBS que contenga 1% de Tween-20 para eliminar cualquier anticuerpo no unido.
Añadir el anticuerpo secundario
- Añadir 100 l de un anticuerpo secundario conjugado enzimático, que en este caso es un anticuerpo conjugado AP, a cada poca.
- Incubar la placa durante 1 h a 37oC.
- Después de la incubación, lave la placa con 1X PBS que contenga 1% de Tween-20.
Detección
- Añadir 100 l de la solución de sustrato a cada poca.
- Espere 5-10 min.
- Después de 10 min, detenga la reacción enzimática añadiendo ácido sulfúrico de 100 l 2N a los pozos. A continuación, mida la absorbancia en un lector de microplacas dentro de los 30 minutos de agregar la solución de parada
Resultados
En el siguiente ejemplo de ELISA indirecto, se determinó la presencia de IgG específico del virus de la gripe A (IAV) en el suero de ratones infectados por el IAV. Los ratones C57Bl/6 fueron infectados con el virus de la gripe A (A/PR/8; 105 PFU en 100 l de PBS i.p.) y el suero se recogió 28 días después. Para cuantificar la cantidad de IgG específica de la IAV en el suero, las placas ELISA de 96 pocillos se recubriron con el virus purificado A/PR/8 de la gripe A (50 l/bien de 2 mg/ml de virus PBS) durante la noche a 4 oC. Las placas recubiertas se bloquearon durante 1 hora a temperatura ambiente con un 5% de suero de burro normal en PBS, seguido de la incubación con muestras de suero diluidas de ratones con problemas de IAV durante la noche a 4oC. El suero se diluyó inicialmente 1:12.5, seguido de diluciones 1:4 (rango de dilución - 1:12.5 a 1:204,800). Después del lavado, las placas fueron incubadas con un igG anti-ratón conjugado con fosfatasa alcalina (AP) durante 1 h. Las placas se lavaron, y luego se añadió p-Fosfato de nitrofenilo (PNPP; 1 mg/ml, 100 l/bien). La solución PNPP incolora se convierte en un color amarillo cuando AP está presente. Después de 5-10 min, la reacción enzimática se detuvo añadiendo 100 l/bien 2N H2SO4. La placa se leyó en un lector de microplacas a 405 nm. Los resultados obtenidos se muestran en la Tabla 1 y la Figura 1.
| Muestra | Pozos | OD405 | Decir |
| Suero 1:12.5 | A1 | 2.163 | 2.194 |
| B1 | 2.214 | ||
| C1 | 2.204 | ||
| Suero 1:50 | A1 | 1.712 | 1.894 |
| B1 | 2.345 | ||
| C1 | 1.624 | ||
| Suero 1:200 | A1 | 1.437 | 1.541 |
| B1 | 1.73 | ||
| C1 | 1.456 | ||
| Suero 1:800 | A1 | 1.036 | 0.957 |
| B1 | 0.912 | ||
| C1 | 0.923 | ||
| Suero 1:3200 | A1 | 0.579 | 0.48 |
| B1 | 0.431 | ||
| C1 | 0.429 | ||
| Suero 1:12800 | A1 | 0.296 | 0.281 |
| B1 | 0.312 | ||
| C1 | 0.236 | ||
| Suero 1:51200 | A1 | 0.308 | 0.283 |
| B1 | 0.299 | ||
| C1 | 0.243 | ||
| Suero 1:204800 | A1 | 0.315 | 0.303 |
| B1 | 0.298 | ||
| C1 | 0.297 |
Tabla 1: Datos indirectos del ensayo ELISA. Diluciones séricas (de 1:12.5 a 1:204.800), de ratones infectados por el virus de la gripe A (IAV) que contienen IgG específico según el IAV, valores de densidad óptica (OD) (405 nm) y valores medios de OD405.
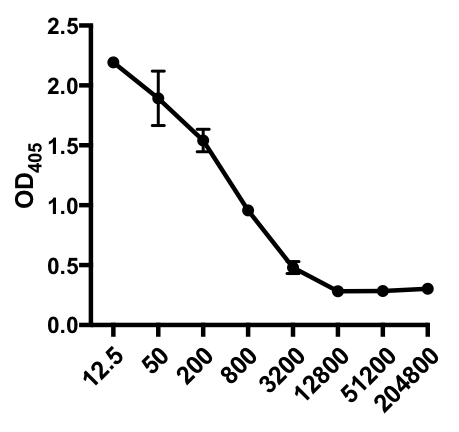
Figura 1: Gráfica indirecta de dispersión del ensayo ELISA de valores medios de OD405 (+ S. D.) y diluciones séricas (de 1:12.5 a 1:204.800), de IgG específica del virus de la gripe A (IAV) en el suero de ratones infectados por el IAV. Los valores de OD405 pueden correlacionarse inversamente con las diluciones séricas.
En el siguiente ejemplo de un sándwich ELISA, se añadió a los pozos indicados una dilución 1:2.5 de las normas de TNF humanas recombinantes (a partir de una concentración de 75 pg/ml) a los pozos indicados de una placa de fondo plano de 96 pocillos. Estas normas dieron lugar a un cambio correspondiente de 2,5 veces en las lecturas de absorbancia.
| Muestra | Concentración (pg/mL) | Pozos | Valores | Valor medio | Cálculo de la concentración posterior | Promedio |
| Estándar 1 | 75 | A1 | 1.187 | 1.169 | 76.376 | 75.01 |
| A2 | 1.152 | 73.644 | ||||
| Estándar 2 | 30 | B1 | 0.534 | 0.52 | 30.827 | 29.962 |
| B2 | 0.506 | 29.098 | ||||
| Estándar 3 | 12 | C1 | 0.23 | 0.217 | 12.838 | 12.105 |
| C2 | 0.204 | 11.372 | ||||
| Estándar 4 | 4.8 | D1 | 0.09 | 0.084 | 5.055 | 4.726 |
| D2 | 0.078 | 4.398 | ||||
| Estándar 5 | 1.92 | E1 | 0.033 | 0.031 | 1.941 | 1.86 |
| E2 | 0.03 | 1.778 | ||||
| Estándar 6 | 0.768 | F1 | 0.009 | 0.011 | 0.626 | 0.764 |
| F2 | 0.014 | 0.901 | ||||
| Estándar 7 | 0.307 | G1 | 0.002 | 0.004 | 0.238 | 0.377 |
| G2 | 0.007 | 0.516 |
Tabla 2: Datos de curva estándar el ELISA del sándwich TNF. Una dilución de 1:2.5 de las normas de TNF humanas recombinantes (75 a 0,3 pg/ml), valores de Do (450 nm), valores medios de450 OD, cálculos de concentración posterior y sus promedios.
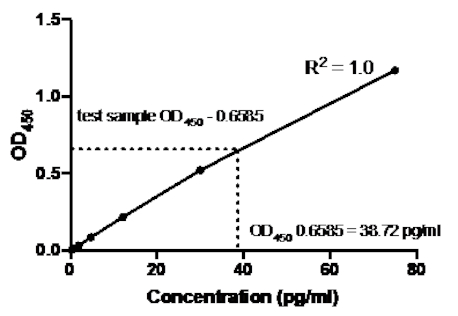
Figura 2: Curva estándar para el sándwich TNF ELISA. Se analizó una dilución de 1:2.5 de las normas de TNF humanas recombinantes (75 a 0,3 pg/ml) utilizando el sándwich ELISA. Los valores de OD450 se pueden correlacionar directamente con las concentraciones de dilución estándar. La cantidad de proteína TNF en la muestra de ensayo se determinó utilizando la curva estándar, que corresponde a una concentración de 38,72 pg/ml.
Una vez generada la curva estándar, se determinó la cantidad de proteína TNF en la muestra de prueba. En este ejemplo de el sándwich ELISA, las muestras de prueba dieron450 lecturas de OD de 0,636 y 0,681, que dan un promedio de 0,6585. Al trazar esta lectura OD450 en el gráfico anterior, esto corresponde a una concentración de TNF de 38,72 pg/ml.
Aplicación y resumen
Como se ha demostrado, una gama de inmunoensayos (con ligera variación en los protocolos) se enmarcan dentro de la familia técnica ELISA. Determinar qué versión de ELISA utilizar depende de una serie de factores, incluyendo qué antígeno se está detectando, el anticuerpo monoclonal disponible para un antígeno en particular y la sensibilidad deseada del ensayo (5). Algunas fortalezas y debilidades de las diferentes ELISA descritas en este documento son:
| Elisa | Fortalezas | Debilidades |
| Indirecta | 1) Alta sensibilidad debido al hecho de que múltiples anticuerpos secundarios conjugados en enzimas pueden unirse al anticuerpo primario | 1) La señal de fondo alta puede ocurrir porque el recubrimiento del antígeno de interés a la placa no es específico (es decir, todas las proteínas de la muestra recubrirán la placa) |
| 2) Muchos anticuerpos primarios diferentes pueden ser reconocidos por un único anticuerpo secundario conjugado en enzimas que da al usuario la flexibilidad de usar el mismo anticuerpo secundario conjugado en enzimas en muchos ELISA diferentes (independientemente del antígeno que se detecte) | ||
| 3) La mejor opción cuando sólo un solo anticuerpo para el antígeno de interés está disponible | ||
| Sándwich | 1) El uso de anticuerpos monoclonales de captura y detección específicos de antígenos aumenta la sensibilidad y especificidad del ensayo (en comparación con el ELISA indirecto) | 1) Optimizar las concentraciones de los anticuerpos monoclonales de captura y detección puede ser difícil (especialmente para kits no comerciales) |
| 2) La mejor opción para detectar una proteína grande con múltiples epítopos (como una citoquina) | ||
| Competitivo | 1) Se pueden utilizar muestras impuras | 1) Requiere una gran cantidad de antígeno altamente puro para ser utilizado para recubrir la placa |
| 2) Menos sensibilidad a los efectos de dilución de reactivos | ||
| 3) Ideal para detectar moléculas pequeñas (como un hapten) |
Tabla 3: Resumen. Un resumen de las fortalezas y debilidades de las diferentes técnicas elISA.
Si bien es una técnica simple y útil, también hay algunos inconvenientes para cualquier ELISA. Una es la incertidumbre de la cantidad de la proteína de interés en las muestras de prueba. Si la cantidad es demasiado alta o demasiado baja, los valores de absorbancia obtenidos por el lector de microplacas pueden caer por encima o por debajo de los límites de la curva estándar, respectivamente. Esto hará que sea difícil determinar con precisión la cantidad de proteína presente en las muestras de prueba. Si los valores son demasiado altos, la muestra de prueba se puede diluir antes de añadir a los pozos de la placa. Los valores finales tendrían que ajustarse de acuerdo con el factor de dilución. Como se mencionó, los kits caseros a menudo requieren una optimización cuidadosa de las concentraciones de anticuerpos utilizados para producir una alta relación señal-ruido.
Referencias
- Porstmann, T. and Kiessig S.T. Enzyme immunoassay techniques. An overview. Journal of Immunological Methods. 150 (1-2), 5-21 (1992).
- Suleyman Aydin. A short history, principles, and types of ELISA, and our laboratory experience with peptide/protein analyses using ELISA. Peptides, 72, 4-15 (2015).
- Gan. S. D. and Patel K. R. Enzyme Immunoassay and Enzyme-Linked Immunosorbent Assay. Journal of Investigative Dermatology, 133 (9), 1-3 (2013).
- Kohl, T. O. and Ascoli C.A. Immunometric Antibody Sandwich Enzyme-Linked Immunosorbent Assay. Cold Spring Harbor Protocols, 1 (6), (2017).
- Sakamoto, S., Putalun, W., Vimolmangkang, S., Phoolcharoen, W., Shoyama, Y., Tanaka, H., and Morimoto S. Enzyme-linked immunosorbent assay for the quantitative/qualitative analysis of plant secondary metabolites. Journal of natural medicines, 72 (1), 32-42 (2018).
Saltar a...
Vídeos de esta colección:

Now Playing
Ensayos ELISA: Indirecto, Sándwich y Competitivo
Immunology
238.8K Vistas

Citometría de flujo y clasificación de células activadas por fluorescencia (FACS): Aislamiento de linfocitos B esplénicos
Immunology
93.1K Vistas

Clasificación celular activada magnéticamente (MACS): Aislamiento de linfocitos T del timo
Immunology
22.9K Vistas

Ensayo ELISPOT: Detección de esplenocitos secretores de IFN-γ
Immunology
28.5K Vistas

Inmunohistoquímica e Inmunocitoquímica: Imágenes de tejidos a través de microscopía óptica
Immunology
79.0K Vistas

Generación de anticuerpos: Producción de anticuerpos monoclonales mediante hibridomas
Immunology
43.6K Vistas

Microscopía de Inmunofluorescencia: Tinción de inmunofluorescencia de secciones de tejido incrustado en parafina
Immunology
53.9K Vistas

Microscopía de Fluorescencia Confocal: Una Técnica para Determinar la Localización de Proteínas en Fibroblastos de Ratón
Immunology
43.2K Vistas

Técnicas basadas en inmunoprecipitación: purificación de proteínas endógenas mediante perlas de agarosa
Immunology
87.8K Vistas

Análisis del ciclo celular: Evaluación de la proliferación de células T CD4 y CD8 después de su estimulación mediante tinción CFSE y citometría de flujo
Immunology
24.3K Vistas

Transferencia celular adoptiva: Introducción de cenocitos de un ratón donante a un ratón huésped y evaluación del éxito a través de FACS
Immunology
22.5K Vistas

Ensayo sobre la muerte celular: Ensayo de liberación de cromo de la capacidad citotóxica
Immunology
151.4K Vistas
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados