
Saggi ELISA: indiretti, sandwich e competitivi
Panoramica
Fonte: Whitney Swanson1,2, Frances V. Sjaastad2,3e Thomas S. Griffith1,2,3,4
1 Dipartimento di Urologia, Università del Minnesota, Minneapolis, MN 55455
2 Centro di Immunologia, Università del Minnesota, Minneapolis, MN 55455
3 Programma di laurea in microbiologia, immunologia e biologia del cancro, Università del Minnesota, Minneapolis, MN 55455
4 Masonic Cancer Center, Università del Minnesota, Minneapolis, MN 55455
Il saggio di immunoassorbimento enzimatico (ELISA) viene spesso utilizzato per misurare la presenza e/o la concentrazione di un antigene, anticorpo, peptide, proteina, ormone o altra biomolecola in un campione biologico. È estremamente sensibile, in grado di rilevare basse concentrazioni di antigene. La sensibilità di ELISA è attribuita alla sua capacità di rilevare le interazioni tra un singolo complesso antigene-anticorpo (1). Inoltre, l'inclusione di un anticorpo antigene-specifico coniugato enzimatico consente la conversione di un substrato incolore in un prodotto cromogenico o fluorescente che può essere rilevato e facilmente quantificato da un lettore di piastre. Se confrontati con i valori generati dalle quantità titolate di un antigene noto di interesse, è possibile determinare la concentrazione dello stesso antigene nei campioni sperimentali. Diversi protocolli ELISA sono stati adattati per misurare le concentrazioni di antigene in una varietà di campioni sperimentali, ma tutti hanno lo stesso concetto di base (2). La scelta del tipo di ELISA da eseguire, indiretto, sandwich o competitivo, dipende da una serie di fattori, tra cui la complessità dei campioni da testare e gli anticorpi antigene-specifici disponibili per l'uso. L'ELISA indiretto viene spesso utilizzato per determinare l'esito di una risposta immunologica, come la misurazione della concentrazione di un anticorpo in un campione. L'ELISA sandwich è più adatto per l'analisi di campioni complessi, come supernatanti di coltura tissutale o lasasità tissutale, in cui l'analita, o antigene di interesse, fa parte di un campione misto. Infine, l'ELISA competitivo viene spesso utilizzato quando è disponibile un solo anticorpo per rilevare l'antigene di interesse. Gli ELISA competitivi sono anche utili per rilevare un piccolo antigene con un solo epitopo anticorpale che non può ospitare due anticorpi diversi a causa dell'ostacolo sterico. Il protocollo descriverà le procedure di base per i test ELISA indiretti, sandwich e competitivi.
Il saggio ELISA indiretto è comunemente usato per misurare la quantità di anticorpi nel siero o nel surnatante di una coltura di ibridoma. La procedura generale per il saggio ELISA indiretto è:
- Rivestire i pozzezze con antigeni
- Aggiungere siero o surnatante contenente surnatante di coltura di siero o ibridoma (anticorpo primario o 1°)
- Incubare e lavare
- Aggiungere un anticorpo secondario (o 2°) coniugato con enzimi
- Incubare e lavare
- Aggiungi substrato
Il test ELISA sandwich differisce dal test ELISA indiretto in quanto il metodo non prevede il rivestimento delle piastre con un antigene purificato. Invece, un anticorpo "cattura" viene utilizzato per rivestire i pozzetti della piastra. L'antigene è "inserito" tra l'anticorpo di cattura e un secondo anticorpo coniugato enzimatico "di rilevamento" - dove entrambi gli anticorpi sono specifici per lo stesso antigene, ma a epitopi diversi (3). Legandosi al complesso anticorpo/antigene di cattura, l'anticorpo di rilevamento rimane nella piastra. Gli anticorpi monoclonali o gli antisieri policlonali possono essere utilizzati come anticorpi di cattura e rilevamento. Il vantaggio principale del sandwich ELISA è che il campione non deve essere purificato prima dell'analisi. Inoltre, il test può essere abbastanza sensibile (4). Molti kit ELISA disponibili in commercio sono della varietà sandwich e utilizzano coppie di anticorpi testati e abbinati. La procedura generale per il test ELISA sandwich è:
- Coat pozzi con anticorpo di cattura
- Aggiungere campioni di prova contenenti antigene
- Incubare e lavare
- Aggiungere anticorpi di rilevamento coniugati con enzimi.
- Incubare e lavare
- Aggiungi substrato
La maggior parte dei kit ELISA sandwich disponibili in commercio sono dotati di anticorpi di rilevamento coniugati con enzimi. Nei casi in cui non è disponibile un anticorpo di rilevazione coniugato con enzima, può essere utilizzato un anticorpo secondario coniugato con enzima specifico per l'anticorpo di rilevamento. L'enzima sull'anticorpo secondario svolge lo stesso ruolo, che è quello di convertire il substrato incolore in un prodotto cromogenico o fluorescente. Il suddetto anticorpo secondario coniugato con enzima vorrebbe più essere utilizzato in un ELISA sandwich "fatto in casa" sviluppato da un investigatore che ha generato i propri anticorpi monoclonali, per esempio. Uno svantaggio dell'utilizzo di un anticorpo secondario coniugato con enzima è quello di essere sicuri che si leghi solo all'anticorpo di rilevamento e non all'anticorpo di cattura legato alla piastra. Ciò si tradurrebbe in un prodotto misurabile in tutti i pozzi, indipendentemente dalla presenza o dall'assenza di antigene o anticorpo di rilevamento.
Infine, il test ELISA competitivo viene utilizzato per rilevare antigeni solubili. È semplice da eseguire, ma è adatto solo quando l'antigene purificato è disponibile in una quantità relativamente grande. La procedura generale per il test ELISA competitivo è:
- Rivestire i pozzezze con antigene
- Incubare e lavare
- Campione di prova preincubato con anticorpi primari coniugati con enzimi
- Aggiungere bene la miscela
- Incubare e lavare via qualsiasi anticorpo primario coniugato con enzimi non legati
- Aggiungi substrato
La "competizione" in questo test deriva dal fatto che più antigene nel campione di prova utilizzato nella fase 3 si tradurrà in meno anticorpi disponibili per legarsi all'antigene che riveste il pozzo. Pertanto, l'intensità del prodotto cromogenico/fluorogenico nel pozzo alla fine del test è inversamente correlata alla quantità di antigene presente nel campione di prova.
Un componente chiave in qualsiasi tipo di ELISA sono gli standard titolati di concentrazioni note che consentiranno all'utente di determinare la concentrazione di antigene presente nei campioni di prova. Tipicamente, una serie di pozzi sono designati per la creazione di una curva standard, in cui quantità note di una proteina ricombinante purificata vengono aggiunte ai pozzetto in quantità decrescenti. Quando questi pozzetto vengono elaborati contemporaneamente ai campioni di prova, l'utente può quindi avere un set di riferimento di valori di assorbanza ottenuti da un lettore di micropiasche per le concentrazioni proteiche note da associare ai valori di assorbanza per i campioni di prova. L'utente può quindi calcolare una curva standard a cui è possibile confrontare i campioni di prova per determinare la quantità di proteina di interesse presente. La curva standard può anche determinare il grado di precisione della diluizione dell'utente.
Infine, l'ultimo passaggio in ciascuno dei tipi ELISA sopra elencati richiede l'aggiunta di un substrato. Il grado di conversione del substrato in prodotto è direttamente correlato alla quantità di enzima presente nel pozzo. La perossidasi di rafano (HRP) e la fosfatasi alcalina (AP) sono gli enzimi più comuni coniugati agli anticorpi. Come previsto, ci sono un certo numero di substrati disponibili specifici per entrambi gli enzimi che producono un prodotto cromogenico o fluorescente. Inoltre, i substrati sono disponibili in una gamma di sensibilità che possono aumentare la sensibilità complessiva del test. L'utente deve anche prendere in considerazione il tipo di strumentazione disponibile per la lettura della piastra alla fine dell'esperimento quando sceglie il tipo di substrato da utilizzare, insieme al corrispondente anticorpo coniugato con enzima.
I substrati cromogenici comunemente usati per l'HRP includono 2,2'-Azinobis [3-ethylbenzothiazoline-6-sulfonic acid]-diammonium salt (ABTS) e 3,3',5,5'-tetramethylbenzidine (TMB), mentre p-Nitrophenyl Phosphate (PNPP) è usato per AP. ABTS e TMB producono rispettivamente prodotti di reazione di colore verde e blu solubili in acqua. Il prodotto ABTS verde ha due picchi di assorbanza principali, 410 e 650 nm, mentre il prodotto TMB blu è meglio rilevato a 370 e 652 nm. I colori di ABTS e TMB cambiano in giallo con l'aggiunta di una soluzione di arresto acido, che è meglio leggere a 450 nm. Lo sviluppo del colore per ABTS è lento, mentre è veloce per TMB. TMB è più sensibile di ABTS e può produrre un segnale di fondo più alto se la reazione enzimatica procede troppo a lungo. PNPP produce un prodotto solubile in acqua giallo dopo conversione AP che assorbe la luce a 405 nm.
Procedura
1. ELISA indiretto
Un ELISA indiretto è quello in cui l'anticorpo antigene antigene-specifico primario è riconosciuto da un anticorpo coniugato secondario. Il seguente protocollo è un esempio di metodo ELISA indiretto, in cui i campioni di siero di topi infetti da virus dell'influenza A (IAV) vengono testati per la presenza di anticorpi IgG specifici per IAV. Un punto di forza di questo esempio è che possono essere utilizzati diversi anticorpi secondari che riconoscono tutti gli isotipi anticorpali o isotipi specifici (ad esempio, IgG).
Antigene di rivestimento alla micropiastra
- Rivestire i pozzetti di una piastra ELISA a 96 pozzetti con antigene purificato pipettando 50 μL di antigene purificato (2 mg/mL di virus A dell'influenza A A/PR/8 purificato in tampone Tris-HCl da 0,05 M (pH 9,5)) in ciascun pozzettino della piastra.
- Coprire la piastra con un coperchio adesivo e incubarla durante la notte a 4°C per consentire all'antigene di legarsi alla piastra.
- Al termine dell'incubazione, rimuovere la soluzione di rivestimento facendo scorrere la piastra su un lavandino.
Inceppamento
- Blocca i restanti siti di legame proteico nei pozzi rivestiti aggiungendo un tampone di blocco da 200 μL, qui viene utilizzato il siero d'asino al 5% in 1X PBS, per pozzo. I reagenti bloccanti alternativi includono il 5% di latte secco non grasso o BSA in PBS o siero normale da un animale in cui è stato generato l'anticorpo secondario.
- Incubare per almeno 2 ore a temperatura ambiente o durante la notte a 4°C.
- Dopo l'incubazione, rimuovere il tampone di blocco facendo scorrere la piastra e quindi lavare la piastra con PBS contenente l'1% di Tween-20.
Incubazione con l'anticorpo primario
- Preparare una diluizione seriale del campione di siero, che contiene l'anticorpo primario, per ottenere un intervallo di diluizione da 1 a 204.800, utilizzando 1X PBS. Per fare ciò, prima diluire il siero 1:12.5 e quindi eseguire una diluizione 4X (intervallo di diluizione - da 1:12,5 a 1:204.800).
- Aggiungere 100 μL dei campioni di siero diluiti in serie ai pozzi.
- Coprire la piastra con coperchio adesivo e incubare a temperatura ambiente per 1-2 ore.
- Dopo l'incubazione, far scorrere la piastra su un lavandino e lavare la piastra con PBS contenente l'1% di Tween-20.
Incubazione con l'anticorpo secondario
- Aggiungere 100 μL di un anticorpo secondario coniugato con enzima, perossidasi di rafano, asino coniugato HRP anti-topo secondario in questo esperimento, a ciascun pozzo.
- Incubare la piastra per 1 ora a temperatura ambiente.
- Dopo l'incubazione, far scorrere la piastra su un lavandino e quindi lavare la piastra con PBS contenente l'1% di Tween-20.
Scoperta
- Aggiungere 100 μL del substrato indicatore (3,3',5,5'-tetrametilbenzidina (TMB)) ad una concentrazione di 1 mg/mL a ciascun pozzettino.
- Incubare la piastra con il substrato per 5-10 minuti a temperatura ambiente.
- Dopo 10 min, interrompere la reazione enzimatica aggiungendo 100 μL 2N acido solforico (H2SO4).
Entro 30 minuti dall'aggiunta della soluzione di arresto, leggere la piastra utilizzando un lettore di micropiasse a 405 nm per determinare l'assorbanza dei pozzetti.
2. Sandwich ELISA
In questa versione ELISA, il campione sperimentale è "inserito" tra un anticorpo di cattura non coniugato e un anticorpo di rilevamento coniugato, entrambi specifici per la stessa proteina ma a epitopi diversi. Nel seguente esempio di ELISA sandwich, la concentrazione di TNFα umano è stata determinata in un campione sconosciuto utilizzando una curva standard generata dalla diluizione seriale 2,5X di un TNFα umano ricombinante standard noto (che indica una concentrazione di 75 pg / mL).
Anticorpo di cattura del rivestimento alla micropiastra
- Rivestire i pozzetti di una piastra ELISA a 96 pozzetti con anticorpo di cattura purificato aggiungendo 100 μL di anticorpo di cattura (intervallo 1-10 μg/mL) a ciascun pozzettino della piastra.
- Coprire la piastra con un coperchio adesivo e incubarla durante la notte a 4°C.
- Dopo l'incubazione, rimuovere la soluzione di rivestimento dalla piastra facendo scorrere la piastra su un lavandino.
Inceppamento
- Bloccare i restanti siti di legame proteico nei pozzi rivestiti di anticorpi aggiungendo 200 μL di soluzione bloccante, 5% di latte secco non grasso contenente PBS, ai pozzi.
- Incubare per almeno 2 ore a temperatura ambiente o durante la notte a 4°C.
- Dopo l'incubazione, rimuovere il tampone di blocco facendo scorrere la piastra e quindi lavare la piastra con PBS contenente l'1% di Tween-20.
Aggiungere campioni di antigene contenenti campioni di prova
- Aggiungere 100 μL del campione di prova ai pozzi. Sigillare la piastra con un coperchio adesivo.
- Incubare per 1-2 ore a temperatura ambiente o durante la notte a 4°C.
- Dopo l'incubazione, rimuovere i campioni facendo scorrere la piastra sul lavandino e quindi lavare i fori con 200 μL 1X PBS contenente l'1% di Tween-20.
Aggiungi anticorpo di rilevamento coniugato con enzima
- Aggiungere 100 μL di anticorpo di rilevamento coniugato con enzimi ai pozzetti a una concentrazione preottimizzata.
- Sigillare la piastra con un coperchio adesivo e incubare a temperatura ambiente per 2 ore.
- Rimuovere l'anticorpo di rilevamento non legato facendo scorrere la piastra su un lavandino e lavare i pozzetti con 200 μL 1X PBS contenente l'1% di Tween-20.
Scoperta
- Aggiungere 100 μL del substrato indicatore ad una concentrazione di 1 mg/mL. Qualsiasi anticorpo di rilevamento coniugato enzimatico legato convertirà il substrato in un segnale rilevabile.
- Incubare la piastra per 5-10 minuti a temperatura ambiente.
- Dopo 5-10 minuti, interrompere la reazione enzimatica aggiungendo 100 μL 2N H2SO4 ai pozzi. Entro 30 minuti dall'aggiunta della soluzione di arresto, leggere la piastra utilizzando un lettore di micropiasse per determinare l'assorbanza dei pozzetti.
3. ELISA competitivo
Le fasi di un ELISA competitivo sono diverse da quelle utilizzate nell'ELISA indiretto e sandwich, con la differenza principale che è la fase di legame competitivo tra l'antigene campione e l'antigene "add-in". L'antigene campione viene incubato con l'anticorpo primario non etichettato. Questi complessi anticorpo-antigene vengono quindi aggiunti alla piastra ELISA, che è stata pre-rivestita con lo stesso antigene. Dopo un periodo di incubazione, qualsiasi anticorpo non legato viene lavato via. Esiste una correlazione inversa tra la quantità di anticorpi liberi disponibili per legare l'antigene nel pozzo e la quantità di antigene nel campione originale. Ad esempio, un campione con antigene abbondante avrebbe più complessi di anticorpi antigene-primari, lasciando pochi anticorpi non legati per legarsi alla piastra ELISA. Un anticorpo secondario coniugato con enzimi specifico per l'anticorpo primario viene quindi aggiunto ai pozzi, seguito dal substrato.
Antigene di rivestimento alla micropiastra
- Rivestire i pozzetti di una piastra ELISA a 96 pozzetti con 100 μL di antigene purificato ad una concentrazione di 1-10 μg/mL.
- Coprire la piastra con un coperchio adesivo e incubare la piastra durante la notte a 4°C.
- Dopo l'incubazione, rimuovere la soluzione di antigene non legato dai pozzetti facendo scorrere la piastra su un lavandino.
Inceppamento
- Bloccare i restanti siti di legame proteico nei pozzi rivestiti aggiungendo 200 μL di tampone bloccante a ciascun pozzo, che può essere il 5% di latte secco non grasso o BSA in PBS.
- Incubare la piastra per almeno 2 ore a temperatura ambiente o durante la notte a 4°C.
Campione di incubazione (antigene) con l'anticorpo primario
- Mentre si bloccano i pozzezze, preparare la miscela antigene-anticorpo mescolando 150 μL di antigene campione e 150 μL di anticorpo primario per ciascun pozzeggio nel test.
- Incubare questa miscela per 1 ora a 37°C.
Aggiungere la miscela antigene-anticorpo al pozzo
- Ora, rimuovi il buffer di blocco dai pozzetti facendo scorrere la piastra su un lavandino.
- Quindi, lavare i pozzi con 1X PBS contenente Tween-20.
- Aggiungere 100 μL della miscela antigene-anticorpo primario campione.
- Incubare la piastra a 37°C per 1 ora.
- Rimuovere la miscela campione facendo scorrere la piastra su un lavandino.
- Quindi, lavare i pozzi con 1X PBS contenente l'1% di Tween-20 per rimuovere eventuali anticorpi non legati.
Aggiungere l'anticorpo secondario
- Aggiungere 100 μL di un anticorpo secondario coniugato enzimatico, che in questo caso è un anticorpo coniugato AP, a ciascun pozzo.
- Incubare la piastra per 1 ora a 37°C.
- Dopo l'incubazione, lavare la piastra con 1X PBS contenente l'1% di Tween-20.
Scoperta
- Aggiungere 100 μL della soluzione di substrato a ciascun pozzo.
- Attendere 5-10 min.
- Dopo 10 minuti, interrompere la reazione enzimatica aggiungendo 100 μL di acido solforico 2N ai pozzi. Quindi, misurare l'assorbanza in un lettore di micropiasse entro 30 minuti dall'aggiunta della soluzione di arresto
Risultati
Nel seguente esempio di ELISA indiretto, è stata determinata la presenza di IgG specifiche del virus dell'influenza A (IAV) nel siero di topi infetti da IAV. I topi C57Bl/6 sono stati infettati dal virus dell'influenza A (A/PR/8;10 5 PFU in 100 μL PBS i.p.) e il siero è stato raccolto 28 giorni dopo. Per quantificare la quantità di IgG IAV-specifico nel siero, le piastre ELISA a 96 pozzetti sono state rivestite con virus A/PR/8 influenzale purificato (50 μL/pozzetti di virus PBS da 2 mg/ml) durante la notte a 4°C. Le piastre rivestite sono state bloccate per 1 ora a temperatura ambiente con il 5% di siero d'asino normale in PBS, seguito da incubazione con campioni di siero diluiti da topi sfidati da IAV durante la notte a 4 ° C. Il siero è stato inizialmente diluito 1:12.5, seguito da diluizioni 1:4 (intervallo di diluizione - da 1:12,5 a 1:204.800). Dopo il lavaggio, le piastre sono state incubate con un asino coniugato con fosfatasi alcalina (AP) anti-topo IgG per 1 ora. Le piastre sono state lavate e quindi è stato aggiunto p-nitrofenil fosfato (PNPP; 1 mg / mL, 100 μL / pozzo). La soluzione PNPP incolore si trasforma in un colore giallo quando è presente AP. Dopo 5-10 min, la reazione enzimatica è stata interrotta aggiungendo 100 μL/well 2N H2SO4. La piastra è stata letta su un lettore di micropiasse a 405 nm. I risultati ottenuti sono riportati nella Tabella 1 e nella Figura 1.
| Campione | Wells | OD405 | Significare |
| Siero 1:12.5 | A1 | 2.163 | 2.194 |
| B1 | 2.214 | ||
| C1 | 2.204 | ||
| Siero 1:50 | A1 | 1.712 | 1.894 |
| B1 | 2.345 | ||
| C1 | 1.624 | ||
| Siero 1:200 | A1 | 1.437 | 1.541 |
| B1 | 1.73 | ||
| C1 | 1.456 | ||
| Siero 1:800 | A1 | 1.036 | 0.957 |
| B1 | 0.912 | ||
| C1 | 0.923 | ||
| Siero 1:3200 | A1 | 0.579 | 0.48 |
| B1 | 0.431 | ||
| C1 | 0.429 | ||
| Siero 1:12800 | A1 | 0.296 | 0.281 |
| B1 | 0.312 | ||
| C1 | 0.236 | ||
| Siero 1:51200 | A1 | 0.308 | 0.283 |
| B1 | 0.299 | ||
| C1 | 0.243 | ||
| Siero 1:204800 | A1 | 0.315 | 0.303 |
| B1 | 0.298 | ||
| C1 | 0.297 |
Tabella 1: Dati del saggio ELISA indiretto. Diluizioni sieriche (da 1:12,5 a 1:204.800), di topi infetti dal virus dell'influenza A (IAV) contenenti IgG specifiche per IAV, valori di densità ottica (OD) (405 nm) e valori medi di OD405.
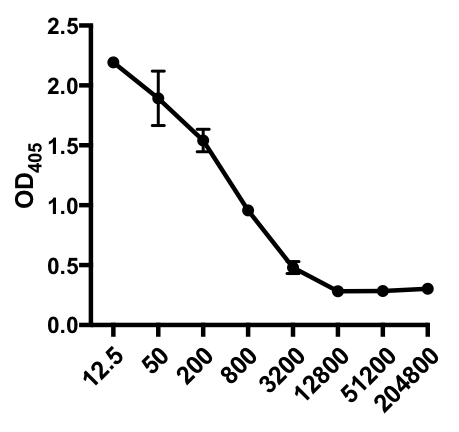
Figura 1: Grafico a dispersione indiretta del saggio ELISA dei valori medi OD405 (+ S. D.) e delle diluizioni sieriche (da 1:12,5 a 1:204.800), delle IgG specifiche del virus dell'influenza A (IAV) nel siero di topi infetti da IAV. I valori di OD405 possono essere inversamente correlati alle diluizioni sieriche.
Nel seguente esempio di un sandwich ELISA, una diluizione 1:2.5 degli standard TNFα umani ricombinanti (a partire da una concentrazione di 75 pg / mL) è stata aggiunta ai pozzetti indicati di una piastra a fondo piatto a 96 pozzetti. Questi standard hanno portato a un corrispondente cambiamento di 2,5 volte nelle letture di assorbanza.
| Campione | Concentrazione (pg/mL) | Wells | Valori | Valore medio | Calcolo della concentrazione posteriore | Nella media |
| Standard 1 | 75 | A1 | 1.187 | 1.169 | 76.376 | 75.01 |
| A2 | 1.152 | 73.644 | ||||
| Standard 2 | 30 | B1 | 0.534 | 0.52 | 30.827 | 29.962 |
| B2 | 0.506 | 29.098 | ||||
| Standard 3 | 12 | C1 | 0.23 | 0.217 | 12.838 | 12.105 |
| C2 | 0.204 | 11.372 | ||||
| Standard 4 | 4.8 | D1 | 0.09 | 0.084 | 5.055 | 4.726 |
| D2 | 0.078 | 4.398 | ||||
| Standard 5 | 1.92 | E1 | 0.033 | 0.031 | 1.941 | 1.86 |
| E2 | 0.03 | 1.778 | ||||
| Standard 6 | 0.768 | F1 | 0.009 | 0.011 | 0.626 | 0.764 |
| F2 | 0.014 | 0.901 | ||||
| Standard 7 | 0.307 | G1 | 0.002 | 0.004 | 0.238 | 0.377 |
| G2 | 0.007 | 0.516 |
Tabella 2: Dati della curva standard ELISA sandwich TNFα. Una diluizione 1:2.5 degli standard TNFα umani ricombinanti (da 75 a 0,3 pg/mL), valori OD (450 nm), valori medi OD450, calcoli di concentrazione posteriore e loro medie.
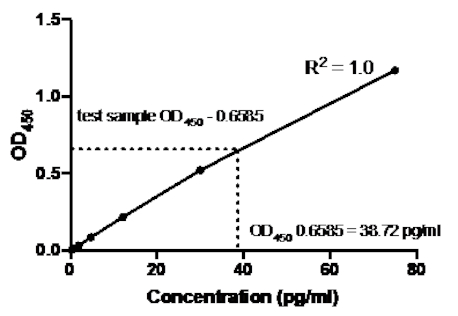
Figura 2: Curva standard per TNFα sandwich ELISA. Una diluizione 1:2.5 degli standard TNFα umani ricombinanti (da 75 a 0,3 pg/mL) è stata analizzata utilizzando ELISA sandwich. I valori OD450 possono essere direttamente correlati alle concentrazioni di diluizione standard. La quantità di proteina TNFα nel campione di prova è stata determinata utilizzando la curva standard, che corrisponde a una concentrazione di 38,72 pg/mL.
Una volta generata la curva standard, è stata determinata la quantità di proteina TNFα nel campione di prova. In questo esempio ELISA sandwich, i campioni di prova hanno fornito letture OD450 di 0,636 e 0,681, che danno una media di 0,6585. Quando si traccia questa lettura di OD450 sul grafico sopra, ciò corrisponde a una concentrazione di TNFα di 38,72 pg / ml.
Applicazione e Riepilogo
Come dimostrato, una serie di saggi immunologici (con lievi variazioni nei protocolli) rientrano nella famiglia delle tecniche ELISA. La determinazione della versione di ELISA da utilizzare dipende da una serie di fattori, tra cui l'antigene rilevato, l'anticorpo monoclonale disponibile per un particolare antigene e la sensibilità desiderata del test (5). Alcuni punti di forza e di debolezza delle diverse ELISA qui descritte sono:
| ELISA | Punti di forza | Debolezze |
| Indiretto | 1) Alta sensibilità dovuta al fatto che più anticorpi secondari coniugati con enzimi possono legarsi all'anticorpo primario | 1) Può verificarsi un segnale di fondo elevato perché il rivestimento dell'antigene di interesse per la piastra non è specifico (cioè, tutte le proteine nel campione rivestiranno la piastra) |
| 2) Molti anticorpi primari diversi possono essere riconosciuti da un singolo anticorpo secondario coniugato con enzima dando all'utente la flessibilità di utilizzare lo stesso anticorpo secondario coniugato con enzima in molti ELISA diversi (indipendentemente dall'antigene rilevato) | ||
| 3) Scelta migliore quando è disponibile un solo anticorpo per l'antigene di interesse | ||
| Sandwich | 1) L'uso di anticorpi monoclonali di cattura e rilevazione antigene-specifici aumenta la sensibilità e la specificità del test (rispetto all'ELISA indiretto) | 1) Ottimizzare le concentrazioni degli anticorpi monoclonali di cattura e rilevazione può essere difficile (soprattutto per i kit non commerciali) |
| 2) La scelta migliore per rilevare una proteina di grandi dimensioni con più epitopi (come una citochina) | ||
| Competitivo | 1) È possibile utilizzare campioni impuri | 1) Richiede una grande quantità di antigene altamente puro da utilizzare per rivestire la piastra |
| 2) Minore sensibilità agli effetti di diluizione dei reagenti | ||
| 3) Ideale per rilevare piccole molecole (come un hapten) |
Tabella 3: Riepilogo. Una sintesi dei punti di forza e di debolezza delle diverse tecniche ELISA.
Mentre una tecnica semplice e utile, ci sono anche alcuni inconvenienti per qualsiasi ELISA. Uno è l'incertezza della quantità di proteine di interesse nei campioni di prova. Se la quantità è troppo alta o troppo bassa, i valori di assorbanza ottenuti dal lettore di micropiasse possono scendere rispettivamente al di sopra o al di sotto dei limiti della curva standard. Ciò renderà difficile determinare con precisione la quantità di proteine presenti nei campioni di prova. Se i valori sono troppo alti, il campione di prova può essere diluito prima di aggiungerlo ai pozzi della piastra. I valori finali dovrebbero quindi essere regolati in base al fattore di diluizione. Come accennato, i kit fatti in casa spesso richiedono un'attenta ottimizzazione delle concentrazioni di anticorpi utilizzate per produrre un elevato rapporto segnale-rumore.
Riferimenti
- Porstmann, T. and Kiessig S.T. Enzyme immunoassay techniques. An overview. Journal of Immunological Methods. 150 (1-2), 5-21 (1992).
- Suleyman Aydin. A short history, principles, and types of ELISA, and our laboratory experience with peptide/protein analyses using ELISA. Peptides, 72, 4-15 (2015).
- Gan. S. D. and Patel K. R. Enzyme Immunoassay and Enzyme-Linked Immunosorbent Assay. Journal of Investigative Dermatology, 133 (9), 1-3 (2013).
- Kohl, T. O. and Ascoli C.A. Immunometric Antibody Sandwich Enzyme-Linked Immunosorbent Assay. Cold Spring Harbor Protocols, 1 (6), (2017).
- Sakamoto, S., Putalun, W., Vimolmangkang, S., Phoolcharoen, W., Shoyama, Y., Tanaka, H., and Morimoto S. Enzyme-linked immunosorbent assay for the quantitative/qualitative analysis of plant secondary metabolites. Journal of natural medicines, 72 (1), 32-42 (2018).
Vai a...
Video da questa raccolta:

Now Playing
Saggi ELISA: indiretti, sandwich e competitivi
Immunology
238.2K Visualizzazioni

Citometria a flusso e selezione cellulare attivata dalla fluorescenza (FACS): isolamento dei linfociti B della milza
Immunology
92.9K Visualizzazioni

Magnetic Activated Cell Sorting (MACS): isolamento dei linfociti T timici
Immunology
22.9K Visualizzazioni

EliSPOT Assay: Rilevamento di splenociti secernenti IFN-γ
Immunology
28.5K Visualizzazioni

Immunoistochimica e immunocitochimica: imaging dei tessuti tramite microscopia ottica
Immunology
78.8K Visualizzazioni

Generazione di anticorpi: produzione di anticorpi monoclonali attraverso l'utilizzo di ibridomi
Immunology
43.5K Visualizzazioni

Microscopia a immunofluorescenza: colorazione a immunofluorescenza di sezioni di tessuto incorporato in paraffina
Immunology
53.8K Visualizzazioni

Microscopia a fluorescenza confocale: una tecnica per determinare la localizzazione delle proteine nei fibroblasti di topo
Immunology
43.1K Visualizzazioni

Tecniche basate sull'immuno-precipitazione: purificazione di proteine endogene con l'impiego di microsfere di agarosio
Immunology
87.7K Visualizzazioni

Analisi del ciclo cellulare: valutazione della proliferazione delle cellule T CD8 e CD4 in seguito a stimolazione tramite colorazione CFSE e citometria a flusso
Immunology
24.2K Visualizzazioni

Trasferimento di cellule adottive: introduzione degli splenociti di topo donatore a un topo ospite e valutazione del successo tramite FACS
Immunology
22.3K Visualizzazioni

Saggio per la morte cellulare: saggio di rilascio di cromo della capacità citotossica
Immunology
151.4K Visualizzazioni
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati