
Ensaios ELISA: Indireto, Sanduíche e Competitivo
Visão Geral
Fonte: Whitney Swanson1,2, Frances V. Sjaastad2,3, e Thomas S. Griffith1,2,3,4
1 Departamento de Urologia, Universidade de Minnesota, Minneapolis, MN 55455
2 Centro de Imunologia, Universidade de Minnesota, Minneapolis, MN 55455
3 Programa de Pós-Graduação em Microbiologia, Imunologia e Biologia do Câncer, Universidade de Minnesota, Minneapolis, MN 55455
4 Centro de Câncer Maçônico, Universidade de Minnesota, Minneapolis, MN 55455
O ensaio imunossorbente ligado à enzima (ELISA) é frequentemente usado para medir a presença e/ou concentração de um antígeno, anticorpo, peptídeo, proteína, hormônio ou outra biomolécula em uma amostra biológica. É extremamente sensível, capaz de detectar baixas concentrações de antígeno. A sensibilidade da ELISA é atribuída à sua capacidade de detectar as interações entre um único complexo de anticorpos de antígeno (1). Além disso, a inclusão de um anticorpo específico de antígeno conjugado por enzimas permite a conversão de um substrato incolor em um produto cromogênico ou fluorescente que pode ser detectado e facilmente quantitado por um leitor de placas. Quando comparada aos valores gerados por quantidades tituladas de um antígeno conhecido de interesse, a concentração do mesmo antígeno nas amostras experimentais pode ser determinada. Diferentes protocolos ELISA foram adaptados para medir concentrações de antígenos em uma variedade de amostras experimentais, mas todos eles têm o mesmo conceito básico (2). A escolha do tipo de ELISA para realizar, indireta, sanduíche ou competitiva, depende de uma série de fatores, incluindo a complexidade das amostras a serem testadas e os anticorpos específicos do antígeno disponíveis para uso. A ELISA indireta é frequentemente utilizada para determinar o resultado de uma resposta imunológica, como medir a concentração de um anticorpo em uma amostra. O sanduíche ELISA é mais adequado para analisar amostras complexas, como supernascedores de cultura tecidual ou lises de tecido, onde o analito, ou antígeno de interesse, faz parte de uma amostra mista. Finalmente, o ELISA competitivo é mais frequentemente utilizado quando há apenas um anticorpo disponível para detectar o antígeno de interesse. EliSAs competitivas também são úteis para detectar um pequeno antígeno com apenas um único epítope de anticorpos que não pode acomodar dois anticorpos diferentes devido à dificultação estérica. O protocolo descreverá os procedimentos básicos para os ensaios ELISA indiretos, sanduíches e competitivos.
O ensaio ELISA indireto é comumente usado para medir a quantidade de anticorpos no soro ou no sobrenatante de uma cultura hibrida. O procedimento geral para o ensaio ELISA indireto é:
- Reveste poços com antígenos
- Adicionar supernanato da cultura sérico ou hibridoma contendo anticorpos (anticorpo primário ou 1°)
- Incubar e lavar
- Adicionar anticorpo secundário (ou 2°) conjugado por enzimas
- Incubar e lavar
- Adicionar substrato
O ensaio ELISA sanduíche difere do ensaio elisa indireto na medida em que o método não envolve revestir as placas com um antígeno purificado. Em vez disso, um anticorpo "captura" é usado para revestir os poços da placa. O antígeno é "sanduíche" entre o anticorpo de captura e um segundo anticorpo conjugado por enzimas de "detecção" - onde ambos os anticorpos são específicos para o mesmo antígeno, mas em epítopos diferentes (3). Ao vincular-se ao complexo de anticorpos/antígenos de captura, o anticorpo de detecção permanece na placa. Anticorpos monoclonais ou antisera policlonal podem ser usados como anticorpos de captura e detecção. A principal vantagem do sanduíche ELISA é que a amostra não precisa ser purificada antes da análise. Além disso, o ensaio pode ser bastante sensível (4). Muitos kits ELISA disponíveis comercialmente são da variedade de sanduíches e usam pares de anticorpos testados e combinados. O procedimento geral para o ensaio elisa sanduíche é:
- Poços de revestimento com anticorpo de captura
- Adicione amostras de teste contendo antígeno
- Incubar e lavar
- Adicione anticorpo de detecção conjugado por enzimas.
- Incubar e lavar
- Adicionar substrato
A maioria dos kits ELISA de sanduíche disponíveis comercialmente vêm com anticorpos de detecção conjugados por enzimas. Nos casos em que um anticorpo de detecção conjugado por enzimas não esteja disponível, um anticorpo coordenado por enzimas secundárias específico para o anticorpo de detecção pode ser usado. A enzima no anticorpo secundário desempenha o mesmo papel, que é converter o substrato incolor em um produto cromogênico ou fluorescente. O anticorpo secundário já mencionado conjugado por enzimas gostaria mais de ser usado em um sanduíche "caseiro" ELISA desenvolvido por um investigador que gerou seus próprios anticorpos monoclonais, por exemplo. Uma desvantagem ao uso de um anticorpo coordenado por enzimas secundárias é ter certeza de que ele se liga apenas ao anticorpo de detecção, e não ao anticorpo de captura ligado à placa. Isso resultaria em um produto mensurável em todos os poços, independentemente da presença ou ausência de antígeno ou anticorpo de detecção.
Finalmente, o ensaio ELISA competitivo é usado para detectar antígenos solúveis. É simples de executar, mas só é adequado quando o antígeno purificado está disponível em uma quantidade relativamente grande. O procedimento geral para o ensaio ELISA competitivo é:
- Poços de casaco com antígeno
- Incubar e lavar
- Amostra de teste pré-incobarto com anticorpos primários conjugados por enzimas
- Adicione a mistura para bem
- Incubar e lavar qualquer anticorpo primário conjugado por enzimas não ligado
- Adicionar substrato
A "competição" neste ensaio vem do fato de que mais antígeno na amostra de ensaio usada na etapa 3 resultará em menos anticorpo disponível para se ligar ao revestimento de antígeno do poço. Assim, a intensidade do produto cromogênico/fluorogênico no poço no final do ensaio está inversamente relacionada à quantidade de antígeno presente na amostra de ensaio.
Um componente-chave em qualquer tipo de ELISA são os padrões titulados de concentrações conhecidas que permitirão ao usuário determinar a concentração de antígeno presente nas amostras de teste. Normalmente, uma série de poços são designados para criar uma curva padrão, onde quantidades conhecidas de uma proteína recombinante purificada são adicionadas aos poços em quantidades decrescentes. Quando esses poços são processados ao mesmo tempo que as amostras de teste, o usuário pode então ter um conjunto de referência de valores de absorção obtidos de um leitor de microplato para concentrações de proteínas conhecidas para acompanhar os valores de absorção para as amostras de teste. O usuário pode então calcular uma curva padrão à qual as amostras de teste podem ser comparadas para determinar a quantidade de proteína de interesse presente. A curva padrão também pode determinar o grau de precisão da produção de diluição do usuário.
Finalmente, a última etapa em cada um dos tipos ELISA listados acima exige a adição de um substrato. O grau de conversão do substrato ao produto está diretamente relacionado com a quantidade de enzima presente no poço. Peroxidase de rabanete (HRP) e fosfatismo alcalino (AP) são as enzimas mais comuns encontradas conjugadas a anticorpos. Como esperado, há uma série de substratos disponíveis específicos para enzimas que produzem um produto cromogênico ou fluorescente. Além disso, substratos estão disponíveis em uma gama de sensibilidades que podem aumentar a sensibilidade geral do ensaio. O usuário também deve levar em consideração o tipo de instrumentação disponível para a leitura da placa no final do experimento ao escolher o tipo de substrato a ser usado, juntamente com seu anticorpo correspondente enzimático conjugado.
Substratos cromogênicos comumente usados para HRP incluem 2,2'-Azinobis [3-ethylbenzothiazoline-6-sulfonic acid]-diammonium salt (ABTS) e 3,3',5,5'-tetrameethylbenzidina (TMB), enquanto p-Nitrofenil Phosphate (PNPP) é usado para AP. ABTS e TMB produzem produtos de reação verde e azul solúveis em água, respectivamente. O produto ABTS verde tem dois picos principais de absorção, 410 e 650 nm, enquanto o produto Azul TMB é melhor detectado em 370 e 652 nm. As cores de ABTS e TMB mudam para amarelo após a adição de uma solução de parada ácida, que é melhor lida em 450 nm. O desenvolvimento de cores para ABTS é lento, enquanto é rápido para TMB. O TMB é mais sensível que o ABTS, e pode produzir um sinal de fundo mais alto se a reação enzimática continuar por muito tempo. A PNPP produz um produto amarelo solúvel em água após a conversão de AP que absorve a luz a 405 nm.
Procedimento
1. Elisa indireta
Um ELISA indireto é aquele em que o anticorpo primário específico do antígeno é reconhecido por um anticorpo conjugado secundário. O protocolo a seguir é um exemplo de um método ELISA indireto, onde as amostras de soro de camundongos infectados pelo vírus influenza A (IAV) são testadas para a presença de anticorpo IgG específico do IAV. Uma força deste exemplo é que diferentes anticorpos secundários podem ser usados que reconhecem todos os isótipos de anticorpos ou isótipos específicos (por exemplo, IgG).
Revestimento de antígeno para a microplacão
- Cubra os poços de uma placa ELISA de 96 poços com antígeno purificado por tubos de 50 μL de antígeno purificado (2 mg/mL de vírus purificado A/PR/8 Influenza A em 0,05M Tampão Tris-HCl (pH 9,5)) em cada poço da placa.
- Cubra a placa com uma tampa adesiva e incuba-a durante a noite a 4°C para permitir que o antígeno se ligue à placa.
- Após a conclusão da incubação, remova a solução de revestimento, jogando a placa sobre uma pia.
Bloqueio
- Bloqueie os locais de ligação de proteína restantes nos poços revestidos adicionando 200 μL de tampão de bloqueio, 5% de soro de burro em 1X PBS é usado aqui, por poço. Os reagentes de bloqueio alternativos incluem 5% de leite seco não gordo ou BSA em PBS ou soro normal de um animal no qual o anticorpo secundário foi gerado.
- Incubar por pelo menos 2 horas em temperatura ambiente ou durante a noite a 4°C.
- Após a incubação, remova o tampão de bloqueio apertando a placa e, em seguida, lave a placa com PBS contendo 1% de Interpol-20.
Incubação com o anticorpo primário
- Prepare uma diluição serial da amostra de soro, que contém o anticorpo primário, para obter uma faixa de diluição de 1 a 204.800, utilizando 1X PBS. Para isso, primeiro dilua o soro 1:12.5 e depois realize uma diluição de 4X (faixa de diluição - 1:12.5 a 1:204.800).
- Adicione 100 μL das amostras de soro diluídas em série aos poços.
- Cubra a placa com tampa adesiva e incubar à temperatura ambiente por 1-2 h.
- Após a incubação, passe a placa sobre uma pia e lave a placa com PBS contendo 1% de Interpol-20.
Incubação com o anticorpo secundário
- Adicione 100 μL de um anticorpo secundário conjugado por enzimas, peroxidase de rabanete, anti-rato de burro conjugado hrp neste experimento, para cada poço.
- Incubar a placa por 1 hora em temperatura ambiente.
- Após a incubação, passe a placa sobre uma pia e depois lave a placa com PBS contendo 1% de Interpol-20.
Detecção
- Adicione 100 μL do substrato indicador (3,3',5,5'-tetramebenzidina (TMB)) a uma concentração de 1 mg/mL para cada poço.
- Incubar a placa com o substrato por 5-10 min em temperatura ambiente.
- Após 10 min, pare a reação enzimática adicionando 100 μL 2N ácido sulfúrico (H2SO4).
Dentro de 30 minutos de adicionar a solução stop, leia a placa usando um leitor de microplacas a 405 nm para determinar a absorvância dos poços.
2. Sanduíche ELISA
Nesta versão ELISA, a amostra experimental é "sanduíche" entre um anticorpo de captura não conjugado e um anticorpo de detecção conjugado, ambos específicos para a mesma proteína, mas em epítopos diferentes. No exemplo elisa sanduíche a seguir, a concentração de TNFα humano foi determinada em amostra desconhecida usando uma curva padrão gerada a partir da diluição serial de 2,5X de um TNFα humano padrão e recombinante conhecido (indicando na concentração de 75 pg/mL).
Revestimento captura anticorpo para a microplacão
- Cubra os poços de uma placa ELISA de 96 poços com anticorpo de captura purificado adicionando 100 μL de anticorpo de captura (1-10 μg/mL de alcance) a cada poço da placa.
- Cubra a placa com uma tampa de placa adesiva e incuba-a durante a noite a 4°C.
- Após a incubação, remova a solução de revestimento da placa, jogando a placa sobre uma pia.
Bloqueio
- Bloqueie os locais de ligação de proteína restantes nos poços revestidos de anticorpos adicionando 200 μL de solução de bloqueio, 5% de leite seco sem gordura contendo PBS, aos poços.
- Incubar por pelo menos 2 h em temperatura ambiente ou durante a noite a 4°C.
- Após a incubação, remova o tampão de bloqueio apertando a placa e, em seguida, lave a placa com PBS contendo 1% de Interpol-20.
Adicionar amostras de teste contendo antígenos
- Adicione 100 μL da amostra de ensaio aos poços. Sele a placa com uma tampa adesiva.
- Incubar por 1-2 h em temperatura ambiente ou durante a noite a 4°C.
- Após a incubação, remova as amostras movendo a placa sobre a pia e, em seguida, lave os poços com 200 μL 1X PBS contendo 1% de Interpol-20.
Adicionar anticorpo de detecção conjugado por enzimas
- Adicione 100 μL de anticorpo de detecção conjugado com enzimas aos poços em uma concentração pré-otimizada.
- Sele a placa com uma tampa adesiva e incubar em temperatura ambiente por 2h.
- Remova o anticorpo de detecção sem saída, movendo a placa sobre uma pia e lave os poços com 200 μL 1X PBS contendo 1% de Interpol-20.
Detecção
- Adicione 100 μL do substrato indicador a uma concentração de 1 mg/mL. Qualquer anticorpo de detecção conjugado por enzimas ligadas converterá o substrato em um sinal detectável.
- Incubar a placa por 5-10 min em temperatura ambiente.
- Após 5-10 min, pare a reação enzimática adicionando 100 μL 2N H2SO4 aos poços. Dentro de 30 minutos de adicionar a solução stop, leia a placa usando um leitor de microplacão para determinar a absorção dos poços.
3. ELISA competitiva
As etapas de um ELISA competitivo são diferentes das utilizadas em ELISA indireta e sanduíche, sendo a principal diferença a etapa de vinculação competitiva entre o antígeno amostral e o antígeno "add-in". O antígeno amostra é incubado com o anticorpo primário não rotulado. Estes complexos anticorpos-antígenos são então adicionados à placa ELISA, que foi pré-revestida com o mesmo antígeno. Após um período de incubação, qualquer anticorpo desvinculado é lavado. Há uma correlação inversa entre a quantidade de anticorpos livres disponíveis para ligar o antígeno no poço e a quantidade de antígeno na amostra original. Por exemplo, uma amostra com antígeno abundante teria mais complexos de anticorpos primários de antígeno, deixando pouco anticorpo desvinculado para se ligar à placa ELISA. Um anticorpo secundário conjugado por enzimas específico para o anticorpo primário é então adicionado aos poços, seguido pelo substrato.
Revestimento de antígeno para a microplacão
- Cubra os poços de uma placa ELISA de 96 poços com 100 μL de antígeno purificado a uma concentração de 1-10 μg/mL.
- Cubra a placa com uma tampa de placa adesiva e incubar a placa durante a noite a 4°C.
- Após a incubação, remova a solução de antígeno desvinculada dos poços, sacudindo a placa sobre uma pia.
Bloqueio
- Bloqueie os locais de ligação de proteína restantes nos poços revestidos adicionando 200 μL de tampão de bloqueio a cada poço, que pode ser 5% de leite seco não gordo ou BSA em PBS.
- Incubar a placa por pelo menos 2 h em temperatura ambiente ou durante a noite a 4°C.
Amostra de incubação (antígeno) com o anticorpo primário
- Ao bloquear os poços, prepare a mistura de anticorpos de antígeno misturando antígeno de amostra de 150 μL e 150 μL de anticorpo primário para cada poço no ensaio.
- Incubar esta mistura por 1h a 37°C.
Adicione mistura de anticorpos de antígeno ao poço
- Agora, remova o tampão de bloqueio dos poços, jogando a placa sobre uma pia.
- Em seguida, lave os poços com 1X PBS contendo Interpol-20.
- Adicione 100 μL da mistura de anticorpos antígenos-primários.
- Incubar a placa a 37°C por 1h.
- Remova a mistura de amostra, movendo a placa sobre uma pia.
- Em seguida, lave os poços com 1X PBS contendo 1% de Interpol-20 para remover qualquer anticorpo desvinculado.
Adicione o anticorpo secundário
- Adicione 100 μL de um anticorpo secundário conjugado enzimático, que neste caso é anticorpo conjugado ap, a cada poço.
- Incubar a placa por 1h a 37°C.
- Após a incubação, lave a placa com 1X PBS contendo 1% de Interpol-20.
Detecção
- Adicione 100 μL da solução do substrato a cada poço.
- Espere 5-10 min.
- Após 10 minutos, pare a reação enzimática adicionando 100 μL 2N de ácido sulfúrico aos poços. Em seguida, meça a absorvância em um leitor de microplacão dentro de 30 minutos de adicionar a solução stop
Resultados
No exemplo a seguir de um ELISA indireto, foi determinada a presença de IgG específico do vírus influenza A (IAV) no soro de camundongos infectados pelo IAV. Os camundongos C57Bl/6 foram infectados pelo vírus influenza A (A/PR/8; 105 PFU em 100 μL PBS i.p.) e o soro foi coletado 28 dias depois. Para quantificar a quantidade de IGG específico do IAV no soro, placas ELISA de 96 poços foram revestidas com vírus purificado de Influenza A (50 μL/well de 2 mg/ml pbs vírus) durante a noite a 4°C. As placas revestidas foram bloqueadas por 1 hora à temperatura ambiente com soro de burro 5% normal na PBS, seguido pela incubação com amostras de soro diluído de camundongos desafiados pelo IAV durante a noite a 4°C. O soro foi inicialmente diluído 1:12.5, seguido por diluições de 1:4 (faixa de diluição - 1:12.5 a 1:204.800). Após a lavagem, as placas foram incubadas com um fosfattase alcalina (AP)-conjugado anti-rato IgG por 1 h. As placas foram lavadas e, em seguida, p-Fósforo nitrofenil (PNPP; 1 mg/mL, 100 μL/bem) foi adicionado. A solução PNPP incolor se transforma em uma cor amarela quando o AP está presente. Após 5-10 min, a reação enzimática foi interrompida adicionando 100 μL/well 2N H2SO4. A placa foi lida em um leitor de microplacas a 405 nm. Os resultados obtidos são mostrados na Tabela 1 e Figura 1.
| Amostra | Wells | OD405 | Significar |
| Soro 1:12.5 | A1 | 2.163 | 2.194 |
| B1 | 2.214 | ||
| C1 | 2.204 | ||
| Soro 1:50 | A1 | 1.712 | 1.894 |
| B1 | 2.345 | ||
| C1 | 1.624 | ||
| Soro 1:200 | A1 | 1.437 | 1.541 |
| B1 | 1.73 | ||
| C1 | 1.456 | ||
| Soro 1:800 | A1 | 1.036 | 0.957 |
| B1 | 0.912 | ||
| C1 | 0.923 | ||
| Soro 1:3200 | A1 | 0.579 | 0.48 |
| B1 | 0.431 | ||
| C1 | 0.429 | ||
| Soro 1:12800 | A1 | 0.296 | 0.281 |
| B1 | 0.312 | ||
| C1 | 0.236 | ||
| Soro 1:51200 | A1 | 0.308 | 0.283 |
| B1 | 0.299 | ||
| C1 | 0.243 | ||
| Soro 1:204800 | A1 | 0.315 | 0.303 |
| B1 | 0.298 | ||
| C1 | 0.297 |
Tabela 1: Dados indiretos de ensaio ELISA. Diluições de soro (de 1:12.5 a 1:204.800), de camundongos infectados pelo vírus influenza A (IAV)contendo IGG específico do IAV, valores de densidade óptica (OD) (405 nm) e valores médios de OD405.
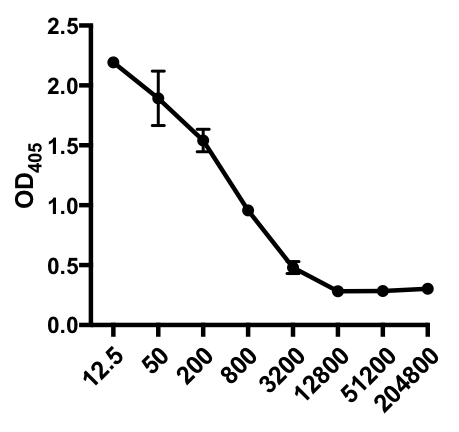
Figura 1: Gráfico de dispersão de ensaios indiretos ELISA de valores médios deOD 405 (+ S. D.) e diluições de soro (de 1:12.5 a 1:204.800), do vírus influenza A (IAV)-specific IgG no soro de camundongos infectados pelo IAV. Os valores OD405 podem ser inversamente correlacionados com as diluições do soro.
No exemplo a seguir de um sanduíche ELISA, uma diluição de 1:2.5 de padrões TNFα humanos recombinantes (a partir de uma concentração de 75 pg/mL) foi adicionada aos poços indicados de uma placa de fundo plana de 96 poços. Esses padrões levaram a uma variação correspondente de 2,5 vezes nas leituras de absorvência.
| Amostra | Concentração (pg/mL) | Wells | Valores | Valor Médio | Cálculo de concentração traseira | Média |
| Padrão 1 | 75 | A1 | 1.187 | 1.169 | 76.376 | 75.01 |
| A2 | 1.152 | 73.644 | ||||
| Padrão 2 | 30 | B1 | 0.534 | 0.52 | 30.827 | 29.962 |
| B2 | 0.506 | 29.098 | ||||
| Padrão 3 | 12 | C1 | 0.23 | 0.217 | 12.838 | 12.105 |
| C2 | 0.204 | 11.372 | ||||
| Padrão 4 | 4.8 | D1 | 0.09 | 0.084 | 5.055 | 4.726 |
| D2 | 0.078 | 4.398 | ||||
| Padrão 5 | 1.92 | E1 | 0.033 | 0.031 | 1.941 | 1.86 |
| E2 | 0.03 | 1.778 | ||||
| Padrão 6 | 0.768 | F1 | 0.009 | 0.011 | 0.626 | 0.764 |
| F2 | 0.014 | 0.901 | ||||
| Padrão 7 | 0.307 | G1 | 0.002 | 0.004 | 0.238 | 0.377 |
| G2 | 0.007 | 0.516 |
Tabela 2: TNFα Sandwich ELISA dados de curva padrão. Uma diluição de 1:2.5 dos padrões humanos recombinantes TNFα (75 a 0,3 pg/mL), valores OD (450 nm), valores médios de OD450, cálculos de concentração de costas e suas médias.
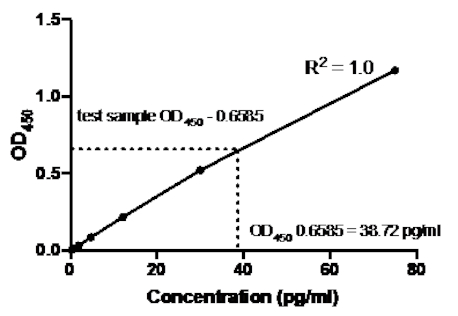
Figura 2: Curva Padrão para sanduíche TNFα ELISA. Foi analisada uma diluição de 1:2.5 das normas TNFα humanas recombinantes (75 a 0,3 pg/mL) utilizando-se sanduíche ELISA. Os valores OD450 podem estar diretamente correlacionados com as concentrações de diluição padrão. A quantidade de proteína TNFα na amostra de ensaio foi determinada utilizando-se a curva padrão, que corresponde a uma concentração de 38,72 pg/mL.
Uma vez gerada a curva padrão, a quantidade de proteína TNFα na amostra de ensaio foi determinada. Neste exemplo elisa sanduíche, as amostras de teste deram OD450 leituras de 0,636 e 0,681, que dão uma média de 0,6585. Ao traçar esta leitura OD450 no gráfico acima, isso corresponde a uma concentração TNFα de 38,72 pg/ml.
Aplicação e Resumo
Como demonstrado, uma gama de imunoensaios (com ligeira variação nos protocolos) se enquadra na família técnica ELISA. Determinar qual versão de ELISA usar depende de uma série de fatores, incluindo o antígeno que está sendo detectado, o anticorpo monoclonal disponível para um determinado antígeno e a sensibilidade desejada do ensaio (5). Alguns pontos fortes e fracos das diferentes ELISAs aqui descritas são:
| ELISA | Pontos fortes | Fraquezas |
| Indireto | 1) Alta sensibilidade devido ao fato de que múltiplos anticorpos secundários conjugados por enzimas podem se ligar ao anticorpo primário | 1) O sinal de fundo alto pode ocorrer porque o revestimento do antígeno de interesse da placa não é específico (ou seja, todas as proteínas da amostra revestirão a placa) |
| 2) Muitos anticorpos primários diferentes podem ser reconhecidos por um único anticorpo secundário conjugado por enzimas, dando ao usuário a flexibilidade de usar o mesmo anticorpo secundário conjugado por enzimas em muitos DIFERENTES ELISA (independentemente do antígeno ser detectado) | ||
| 3) Melhor escolha quando apenas um único anticorpo para o antígeno de interesse está disponível | ||
| Sanduíche | 1) O uso de anticorpo monoclonal de captura e detecção específico de antígeno aumenta a sensibilidade e a especificidade do ensaio (em comparação com a ELISA indireta) | 1) Otimizar as concentrações dos anticorpos monoclonais de captura e detecção pode ser difícil (especialmente para kits não comerciais) |
| 2) Melhor escolha para detectar uma proteína grande com vários epítopos (como uma citocina) | ||
| Competitivo | 1) Podem ser utilizadas amostras impuras | 1) Requer uma grande quantidade de antígeno altamente puro para ser usado para cobrir a placa |
| 2) Menor sensibilidade aos efeitos de diluição do reagente | ||
| 3) Ideal para detectar pequenas moléculas (como um hapten) |
Tabela 3: Resumo. Um resumo dos pontos fortes e fracos das diferentes técnicas ELISA.
Embora seja uma técnica simples e útil, há também algumas desvantagens para qualquer ELISA. Uma delas é a incerteza da quantidade de proteína de interesse nas amostras de teste. Se a quantidade for muito alta ou muito baixa, os valores de absorção obtidos pelo leitor de microplaca podem cair acima ou abaixo dos limites da curva padrão, respectivamente. Isso dificultará a determinação precisa da quantidade de proteína presente nas amostras de teste. Se os valores forem muito altos, a amostra de ensaio pode ser diluída antes de adicionar aos poços da placa. Os valores finais precisariam então ser ajustados de acordo com o fator de diluição. Como mencionado, os kits caseiros muitas vezes requerem uma otimização cuidadosa das concentrações de anticorpos usadas para produzir uma alta relação sinal-ruído.
Referências
- Porstmann, T. and Kiessig S.T. Enzyme immunoassay techniques. An overview. Journal of Immunological Methods. 150 (1-2), 5-21 (1992).
- Suleyman Aydin. A short history, principles, and types of ELISA, and our laboratory experience with peptide/protein analyses using ELISA. Peptides, 72, 4-15 (2015).
- Gan. S. D. and Patel K. R. Enzyme Immunoassay and Enzyme-Linked Immunosorbent Assay. Journal of Investigative Dermatology, 133 (9), 1-3 (2013).
- Kohl, T. O. and Ascoli C.A. Immunometric Antibody Sandwich Enzyme-Linked Immunosorbent Assay. Cold Spring Harbor Protocols, 1 (6), (2017).
- Sakamoto, S., Putalun, W., Vimolmangkang, S., Phoolcharoen, W., Shoyama, Y., Tanaka, H., and Morimoto S. Enzyme-linked immunosorbent assay for the quantitative/qualitative analysis of plant secondary metabolites. Journal of natural medicines, 72 (1), 32-42 (2018).
Pular para...
Vídeos desta coleção:

Now Playing
Ensaios ELISA: Indireto, Sanduíche e Competitivo
Immunology
238.8K Visualizações

Citometria de Fluxo e Separação de Células Ativadas por Fluorescência (FACS): Isolamento de Linfócitos B Esplênicos
Immunology
93.1K Visualizações

Seleção de Células Ativadas por Magnetismo (MACS): Isolamento de Linfócitos T Tímicos
Immunology
22.9K Visualizações

Ensaio ELISPOT: Detecção de Esplenócitos Secretores de IFN-γ
Immunology
28.5K Visualizações

Imunohistoquímica e imunocitoquímica: imageamento de tecidos via microscopia de luz
Immunology
79.0K Visualizações

Geração de anticorpos: produzindo anticorpos monoclonais usando hibridomas
Immunology
43.6K Visualizações

Microscopia de imunofluorescência: Coloração por imunofluorescência de secções de tecido embebidos em parafina
Immunology
53.9K Visualizações

Microscopia confocal de fluorescência: uma técnica para determinar a localização de proteínas em fibroblastos de camundongos
Immunology
43.2K Visualizações

Técnicas Baseadas em Imunoprecipitação: Purificação de Proteínas Endógenas Usando Esferas de Agarose
Immunology
87.8K Visualizações

Análise do Ciclo Celular: Avaliação da Proliferação de Células T CD4 e CD8 Após Estimulação Usando Coloração CFSE e Citometria de Fluxo
Immunology
24.3K Visualizações

Transferência de células adotivas: introduzindo esplenócitos de camundongos doadores para um camundongo hospedeiro e avaliando o sucesso via FACS
Immunology
22.5K Visualizações

Ensaio para Morte Celular: Ensaio de Liberação de Cromo para Avaliar a Capacidade Citotóxica
Immunology
151.4K Visualizações
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados