
ELISA 분석: 간접, 샌드위치 및 길항
Overview
출처: 휘트니 스완슨1,2,프랜시스 V. 자스타드2,3,토마스 에스 그리피스1,2,3,4
1 미네소타 대학교 비뇨기과, 미니애폴리스, MN 55455
2 면역학 센터, 미네소타 대학, 미니애폴리스, MN 55455
3 미생물학, 면역학 및 암 생물학 대학원 프로그램, 미네소타 대학, 미니애폴리스, MN 55455
4 Masonic 암 센터, 미네소타 대학, 미니애폴리스, MN 55455
효소-연결된 면역흡소생식분석(ELISA)은 생물학적 샘플에서 항원, 항체, 펩타이드, 단백질, 호르몬 또는 기타 생체 분자의 존재 및/또는 농도를 측정하는 데 자주 사용된다. 그것은 매우 민감한, 낮은 항 원 농도 감지 할 수. ELISA의 감도는 단일 항원 항체 복합체(1)간의 상호 작용을 검출하는 능력에 기인한다. 더욱이, 효소-공주 항원 특이적 항체의 포함은 무색 기판의 변환을 플레이트 판독기에 의해 검출되고 쉽게 양수할 수 있는 염색체 또는 형광 제품으로 변환할 수 있게 한다. 알려진 항원의 적정량에 의해 생성된 값과 비교하면, 실험 샘플에서 동일한 항원의 농도를 결정할 수 있다. 다른 ELISA 프로토콜은 다양한 실험 샘플에서 항원 농도를 측정하도록 조정되었지만 모두 동일한 기본 개념(2)을 가지고 있습니다. 수행, 간접, 샌드위치, 또는 경쟁력을 수행하기 위해 ELISA의 유형을 선택하는 것은 테스트 할 샘플의 복잡성과 사용할 수있는 항원 특정 항체를 포함하여 여러 가지 요인에 달려 있습니다. 간접 ELISA는 시료에서 항체의 농도를 측정하는 것과 같은 면역 학적 반응의 결과를 결정하는 데 자주 사용됩니다. 샌드위치 ELISA는 분석물 또는 관심있는 항원이 혼합 샘플의 일부인 조직 배양 초월제 또는 조직 용액과 같은 복잡한 샘플을 분석하는 데 가장 적합합니다. 마지막으로, 경쟁적인 ELISA는 관심 있는 항원 검출을 위해 사용할 수 있는 항체가 하나일 때 가장 자주 사용된다. 경쟁적 ELISA는 또한 스테릭 방해로 인해 두 개의 다른 항체를 수용할 수 없는 단일 항체 에피토프만으로 작은 항원검출에 유용하다. 이 프로토콜은 간접, 샌드위치 및 경쟁력있는 ELISA 분석의 기본 절차를 설명합니다.
간접 ELISA 분석체는 일반적으로 혈청 또는 혼종 배양의 상부에서 항체의 양을 측정하는 데 사용됩니다. 간접 ELISA 분석법에 대한 일반적인 절차는 다음과 같이 합니다.
- 항원으로 우물을 코팅
- 항체를 포함하는 혈청 또는 hybridoma 배양 상피제 추가 (1 차 또는 1° 항체)
- 인큐베이션 및 세척
- 이차(또는 2°) 효소 컨쥬게이드 항체 추가
- 인큐베이션 및 세척
- 기판 추가
샌드위치 ELISA 분석법은 정제된 항원으로 플레이트를 코팅하는 것을 포함하지 않는다는 점에서 간접 ELISA 분석과 다릅니다. 대신, "캡처"항체는 플레이트의 우물을 코팅하는 데 사용됩니다. 항원은 포획 항체와 두 번째 "검출" 효소-컨쥬게이트 항체 사이에 "끼어들"이다- 두 항체가 동일한 항원에 특이적이지만, 상이한 에피토프(3)에서. 포획 항체/항원 복합체에 결합함으로써 검출 항체는 플레이트 내에 남아 있다. 단일 클론 항체 또는 폴리클론 항세라는 포획 및 검출 항체로서 사용될 수 있다. 샌드위치 ELISA의 주요 장점은 샘플이 분석하기 전에 정제 될 필요가 없다는 것입니다. 또한, 분석은 매우 민감 할 수있다 (4). 많은 시판되는 ELISA 키트는 샌드위치 품종이며 테스트된 테스트된 항체 쌍을 사용합니다. 샌드위치 ELISA 분석의 일반적인 절차는 다음과 같이 합니다.
- 포획 항체가 있는 코트 웰
- 항원 함유 테스트 샘플 추가
- 인큐베이션 및 세척
- 효소-컨쥬게이드 검출 항체를 추가합니다.
- 인큐베이션 및 세척
- 기판 추가
대부분의 시판 가능한 샌드위치 ELISA 키트는 효소-컨쥬게이트 검출 항체와 함께 제공됩니다. 효소-컨쥬게이트 검출 항체가 제공되지 않는 경우, 검출 항체에 특이적인 이차 효소-컨쥬게이트 항체를 사용할 수 있다. 이차 항체의 효소는 무색 기판을 염색체 또는 형광 제품으로 변환하는 동일한 역할을 수행한다. 상기 언급된 이차 효소-컨쥬게이드 항체는 예를 들어, 자신의 단일클론 항체를 생성한 조사관에 의해 개발된 "수제" 샌드위치 ELISA에 더 많이 사용되고 싶습니다. 이차 효소-컨쥬게이드 항체를 사용하는 한 가지 단점은 검출 항체에만 결합하고, 플레이트에 결합된 항체를 포획하지 않도록 하는 것이다. 이것은 항원 또는 검출 항체의 존재 또는 부재에 관계없이 모든 우물에서 측정 가능한 제품을 초래할 것입니다.
마지막으로, 경쟁적인 ELISA 분석은 수용성 항원검출을 위해 이용됩니다. 수행이 간단하지만 정제 된 항원이 비교적 많은 양으로 사용할 때만 적합합니다. 경쟁력 있는 ELISA 분석법에 대한 일반적인 절차는 다음과 같이 합니다.
- 항원으로 코트 우물
- 인큐베이션 및 세척
- 효소 접합 1차 항체를 갖춘 프리인큐베이트 테스트 샘플
- 잘 혼합물을 추가
- 무한한 효소를 공주한 1차 항체를 배양하고 씻어내다
- 기판 추가
이 분석에서 "경쟁"은 3단계에서 사용되는 시험 샘플에서 더 많은 항원들이 항원을 코팅하여 결합할 수 있는 항체를 적게 초래할 것이라는 사실에서 비롯된다. 따라서, 분석의 끝에 잘 있는 염색체/불소생성 생성물의 강도는 시험 샘플에 존재하는 항원의 양과 반비례한다.
ELISA의 모든 유형의 핵심 성분은 사용자가 시험 샘플에 존재하는 항원 농도를 결정할 수 있도록 알려진 농도의 적정 기준이다. 전형적으로, 일련의 우물은 표준 곡선을 만들기 위해 지정되며, 정제된 재조합 단백질의 알려진 양이 양을 감소시키는 우물에 첨가됩니다. 이러한 우물이 시험 샘플과 동시에 처리될 때, 사용자는 시험 샘플에 대한 흡광도 값과 함께 이동하는 알려진 단백질 농도에 대한 마이크로 플레이트 판독기로부터 얻어진 흡광도 값의 기준 세트를 가질 수 있다. 그런 다음 사용자는 현재 관심 있는 단백질의 양을 결정하기 위해 테스트 샘플을 비교할 수 있는 표준 곡선을 계산할 수 있습니다. 표준 곡선은 또한 사용자의 희석 제조의 정밀도 정도를 결정할 수 있습니다.
마지막으로 위에 나열된 각 ELISA 유형의 마지막 단계는 기판의 추가를 요구합니다. 제품에 기판의 변환 정도는 우물에 존재하는 효소의 양과 직접적으로 관련이 있다. 고추성 과산화구체 (HRP) 및 알칼리인포스파아제 (AP)는 항체에 공주된 가장 흔한 효소이다. 예상대로, 염색체 또는 형광 제품을 생성하는 효소에 대해 특정 한 많은 기판이 있습니다. 더욱이, 기판은 분석의 전반적인 감도를 증가시킬 수 있는 감도의 범위에서 유효하다. 사용자는 또한 해당 효소-컨쥬게이트 항체와 함께 사용할 기판 유형을 선택할 때 실험이 끝날 때 플레이트를 읽을 수 있는 계측의 유형을 고려해야 합니다.
HRP에 일반적으로 사용되는 염색체 기판에는 2,2'-Azinobis [3-ethylbenzothiazoline-6-설포닉 산]-디아MM모늄 염(ABTS) 및 3,3',5'-테트라메틸벤지딘(TMB), p-니트로펜일 포산염(PMB)이 포함됩니다. ABTS와 TMB는 각각 수용성 녹색 및 파란색 색 반응 제품을 생산합니다. 녹색 ABTS 제품에는 두 개의 주요 흡광도 봉우리, 410 및 650 nm가 있으며 파란색 TMB 제품은 370 및 652 nm에서 가장 잘 감지됩니다. 450 nm에서 가장 잘 읽는 산성 정지 용액을 추가하면 ABTS 및 TMB의 색상이 노란색으로 변경됩니다. ABTS의 색상 개발은 느리지만 TMB의 경우 빠릅니다. TMB는 ABTS보다 더 민감하며 효소 반응이 너무 오래 진행되면 더 높은 배경 신호를 생성할 수 있습니다. PNPP는 405 nm에서 빛을 흡수하는 AP 변환 후 노란색 수용성 제품을 생산합니다.
Procedure
1. 간접 ELISA
간접ELISA는 1차 항원 특이적 항체가 이차 공주 항체에 의해 인식되는 곳이다. 다음 프로토콜은 인플루엔자 A 바이러스(IAV)-감염된 마우스의 혈청 샘플이 IAV 특이적 IgG 항체의 존재를 위해 시험되는 간접 ELISA 방법의 예입니다. 이 예의 한 가지 강점은 다른 이차 항체가 모든 항체 등색형 또는 특정 등색형(예를 들어, IgG)을 인식하는 데 사용될 수 있다는 것이다.
마이크로 플레이트에 항원을 코팅
- 정제된 항원의 50 μL(정제된 A/PR/8 인플루엔자 A 바이러스의 2 mg/mL)을 각 플레이트의 각 웰에 코팅하여 정제된 항원을 가진 96웰 ELISA 플레이트의 웰을 코팅한다.
- 접착제 덮개로 접시를 덮고 4°C에서 하룻밤 동안 배양하여 항원이 접시에 결합할 수 있도록 합니다.
- 잠복이 완료되면 플레이트를 싱크대 위로 쓸어 넘으로써 코팅 용액을 제거합니다.
블로킹
- 코팅된 우물에서 남은 단백질 결합 부위를 200 μL 차단 버퍼를 추가하여, 1X PBS에 당나귀 혈청이 5% 잘 사용된다. 대체 차단 시약은 PBS에서 5% 비지방 건조 우유 또는 BSA 또는 이차 항체가 생성된 동물로부터의 정상 혈청을 포함한다.
- 실온에서 최소 2시간 또는 4°C에서 하룻밤 동안 배양하십시오.
- 인큐베이션에 이어, 플레이트를 가볍게 하여 블로킹 버퍼를 제거한 다음 1% Tween-20을 함유한 PBS로 플레이트를 세척합니다.
1차 항체를 가진 배양
- 1X PBS를 사용하여 1-204,800의 희석 범위를 얻기 위해 1차 항체를 포함하는 혈청 시료의 직렬 희석을 준비한다. 이렇게 하려면 먼저 혈청을 1:12.5로 희석한 다음 4X 희석(희석 범위 - 1:12.5 ~ 1:204,800)을 수행합니다.
- 연재된 혈청 샘플 100μL을 우물에 넣습니다.
- 접착제 커버로 플레이트를 덮고 실온에서 1-2 시간 동안 배양하십시오.
- 인큐베이션에 이어 플레이트를 싱크대 위로 쓸어넘기고 1% Tween-20을 함유한 PBS로 플레이트를 세척합니다.
이차 항체를 가진 배양
- 이 실험에서 효소-컨쥬게이드 이차 항체, 고추냉이 과옥시다제, HRP-컨쥬게이드 당나귀 항마우스 보조의 100 μL을 각 웰에 추가합니다.
- 상온에서 1 시간 동안 접시를 배양하십시오.
- 인큐베이션후, 접시를 싱크대 위로 쓸어 넘은 다음 1% Tween-20을 함유한 PBS로 플레이트를 세척합니다.
탐지
- 각 우물에 1 mg/mL의 농도로 표시기 기판(3,3',5,5't'-테트라메틸벤지딘(TMB)의 100 μL을 추가합니다.
- 상온에서 5-10 분 동안 기판으로 접시를 배양하십시오.
- 10 분 후, 100 μL 2N 황산 (H2SO4)를추가하여 효소 반응을 중지하십시오.
정지 용액을 추가한 지 30분 이내에 405nm의 마이크로플레이트 리더기를 사용하여 플레이트를 읽고 우물의 흡광도를 결정합니다.
2. 샌드위치 엘리사
본 ELISA 버전에서, 실험용 샘플은 공주포항체와 공주 검출 항체 사이에 "끼여", 둘 다 동일한 단백질에 특이적이지만 상이한 에피토프에 특이적이다. 다음 샌드위치 ELISA 예에서, 인간 TNFα의 농도는 공지된 표준, 재조합 인간 TNFα(75 pg/mL의 농도에서 진술)의 2.5X 직렬 희석으로부터 생성된 표준 곡선을 사용하여 알 수 없는 시료로 결정되었다.
마이크로 플레이트에 항체를 포획하는 코팅
- 96웰 ELISA 플레이트의 웰을 정제된 포획 항체로 코팅하여 100 μL의 포획 항체(1-10 μg/mL 범위)를 플레이트의 각 웰에 첨가합니다.
- 접착제 플레이트 커버로 플레이트를 덮고 4°C에서 하룻밤 동안 배양합니다.
- 인큐베이션 후 플레이트를 싱크대 위로 쓸어가 서 플레이트에서 코팅 용액을 제거합니다.
블로킹
- 200 μL 차단 용액, PBS를 함유하는 5% 무지방 건조 우유를 우물에 추가하여 항체 코팅 우물에서 남은 단백질 결합 부위를 차단합니다.
- 실온에서 또는 4°C에서 하룻밤 동안 적어도 2 시간 동안 배양하십시오.
- 인큐베이션에 이어, 플레이트를 가볍게 하여 블로킹 버퍼를 제거한 다음 1% Tween-20을 함유한 PBS로 플레이트를 세척합니다.
테스트 샘플을 포함하는 항원 추가
- 테스트 샘플의 100 μL을 우물에 추가합니다. 접착제 덮개로 접시를 밀봉합니다.
- 실온에서 1-2 시간 동안 또는 4 ° C에서 하룻밤 동안 배양하십시오.
- 인큐베이션 후, 싱크대 위로 접시를 쓸어 넘은 다음 1% Tween-20을 포함하는 200 μL 1X PBS로 우물을 세척하여 샘플을 제거합니다.
효소-컨쥬게이드 검출 항체 추가
- 미리 최적화된 농도로 100 μL의 효소 접합 검출 항체를 우물에 첨가합니다.
- 접착제 덮개로 접시를 밀봉하고 실온에서 2 시간 동안 배양하십시오.
- 싱크대 위에 플레이트를 쓸어 넘음으로써 언바운드 검출 항체를 제거하고 1% Tween-20을 함유한 200 μL 1X PBS로 우물을 세척한다.
탐지
- 1 mg/mL의 농도에서 지표 기판의 100 μL을 추가합니다. 모든 바운드 효소-공해 검출 항체는 기판을 검출 가능한 신호로 변환합니다.
- 상온에서 5-10 분 동안 접시를 배양하십시오.
- 5-10 분 후, 우물에 100 μL 2N H2SO4를 추가하여 효소 반응을 중지합니다. 정지 용액을 추가한 지 30분 이내에 마이크로 플레이트 판독기를 사용하여 플레이트를 읽고 우물의 흡광도를 결정합니다.
3. 경쟁 ELISA
경쟁적인 ELISA의 단계는 간접 및 샌드위치 ELISA에 사용되는 것과 다르며, 주요 차이점은 샘플 항원과 "추가 기능"항원 사이의 경쟁 적 바인딩 단계입니다. 시료 항원들은 표지가 없는 1차 항체로 배양된다. 이러한 항체-항원 복합체는 동일한 항원으로 미리 코팅된 ELISA 플레이트에 첨가된다. 잠복기 후에, 어떤 불바운드 항체든지 멀리 씻어내게 됩니다. 원래 시료에서 항원의 우물과 항원 양을 결합하는 데 사용할 수 있는 자유 항체의 양과 역상관관계가 있다. 예를 들어, 항원이 풍부한 샘플은 더 많은 항원 항체 복합체를 가지며, ELISA 플레이트에 결합하기 위해 거의 결합되지 않은 항체를 남겨 둡시다. 1차 항체에 특이적인 효소-컨쥬게이트 이차 항체는 이어서 우물에 첨가되고 기판이 뒤따릅니다.
마이크로 플레이트에 항원을 코팅
- 정제된 항원의 100 μL로 96웰 ELISA 플레이트의 웰을 1-10 μg/mL 농도로 코팅합니다.
- 접착제 플레이트 커버로 플레이트를 덮고 4°C에서 하룻밤 동안 플레이트를 배양합니다.
- 인큐베이션에 따라 싱크대 위로 플레이트를 쓸어넘기면 우물에서 언바운드 항원 용액을 제거합니다.
블로킹
- 코팅된 우물에서 나머지 단백질 결합 부위를 각 웰에 블로킹 버퍼 200 μL을 추가하여 PBS에서 5% 비지방 건조 우유 또는 BSA가 될 수 있습니다.
- 플레이트를 실온에서 2시간 이상 또는 4°C에서 하룻밤 동안 배양합니다.
1차 항체를 가진 배양 샘플(antigen)
- 우물을 차단하는 동안, 분석에서 각각 의 150 μL 샘플 항원 및 1차 항체의 150 μL을 혼합하여 항원 항체 혼합물을 준비한다.
- 이 혼합물을 37°C에서 1시간 동안 배양합니다.
항원-항체 혼합물을 우물에 추가
- 이제 싱크대 위로 플레이트를 가볍게 하여 우물에서 차단 버퍼를 제거합니다.
- 그런 다음, Tween-20을 포함하는 1X PBS로 우물을 씻는다.
- 시료 항원 항체 혼합물의 100 μL을 추가합니다.
- 플레이트를 37°C에서 1시간 동안 배양합니다.
- 싱크대 위에 접시를 쓸어서 샘플 혼합물을 제거합니다.
- 이어서, 1%의 Tween-20을 함유한 1X PBS로 우물을 세척하여 결합되지 않은 항체를 제거한다.
이차 항체 추가
- 이 경우 AP-컨쥬게이드 항체인 효소 컨쥬게이징 이차 항체의 100 μL을 각각 양호하게 넣습니다.
- 플레이트를 37°C에서 1시간 동안 배양합니다.
- 인큐베이션에 이어 1%의 트위엔-20을 함유한 1X PBS로 플레이트를 세척한다.
탐지
- 각 우물에 기판 용액의 100 μL을 추가합니다.
- 5-10 분 기다립니다.
- 10 분 후, 우물에 100 μL 2N 황산을 추가하여 효소 반응을 중지합니다. 그런 다음 스톱 솔루션을 추가한 후 30분 이내에 마이크로 플레이트 판독기에서 흡광도를 측정합니다.
Results
간접 ELISA의 다음 예에서, IAV 감염 마우스의 혈청에서 인플루엔자 A 바이러스(IAV)-특이적 IgG의 존재가 결정되었다. C57Bl/6 마우스는 인플루엔자 A 바이러스(A/PR/8; 100 μL PBS i.p.에서105 PFU)에 감염되었고 28일 후에 혈청을 수집하였다. 혈청에서 IAV 특이적 IgG의 양을 양량화하기 위해 96웰 ELISA 플레이트는 4°C에서 하룻밤 사이에 정제된 A/PR/8 인플루엔자 A 바이러스(50 μL/well 2 mg/ml PBS 바이러스)로 코팅하였다. 코팅 된 플레이트는 PBS에서 5 % 일반 당나귀 세럼으로 실온에서 1 시간 동안 차단되었으며, 4 ° C에서 IAV 도전 마우스의 희석 된 혈청 샘플로 인큐베이션이 뒤따랐습니다. 혈청은 처음에 희석1:12.5, 1:4 희석 (희석 범위 - 1:12.5 받는 것 1:204,800). 세척 후, 플레이트는 알칼리성 인산염(AP)-컨쥬게이드 당나귀 안티마우스 IgG로 1h로 배양되었다. 플레이트를 세척한 다음 p-니트로페닐 인산염(PNPP; 1 mg/mL, 100 μL/웰)이 첨가되었다. AP가 있을 때 무색 PNPP 솔루션은 노란색으로 바뀝니다. 5-10 분 후, 효소 반응은 100 μL / 웰 2N H2SO4를추가하여 중단되었습니다. 플레이트는 405 nm에서 마이크로 플레이트 판독기에서 읽혔습니다. 얻은 결과는 표 1과 도 1에 표시됩니다.
| 견본 | 웰 스 | OD405 | 의미하다 |
| 세럼 1:12.5 | A1 | 2.163 | 2.194 |
| B1 | 2.214 | ||
| C1 | 2.204 | ||
| 세럼 1:50 | A1 | 1.712 | 1.894 |
| B1 | 2.345 | ||
| C1 | 1.624 | ||
| 세럼 1:200 | A1 | 1.437 | 1.541 |
| B1 | 1.73 | ||
| C1 | 1.456 | ||
| 세럼 1:800 | A1 | 1.036 | 0.957 |
| B1 | 0.912 | ||
| C1 | 0.923 | ||
| 세럼 1:3200 | A1 | 0.579 | 0.48 |
| B1 | 0.431 | ||
| C1 | 0.429 | ||
| 세럼 1:12800 | A1 | 0.296 | 0.281 |
| B1 | 0.312 | ||
| C1 | 0.236 | ||
| 세럼 1:51200 | A1 | 0.308 | 0.283 |
| B1 | 0.299 | ||
| C1 | 0.243 | ||
| 세럼 1:204800 | A1 | 0.315 | 0.303 |
| B1 | 0.298 | ||
| C1 | 0.297 |
표 1: 간접 ELISA 분석 데이터. 혈청 희석제(1:12.5 ~1:204,800), 인플루엔자 A 바이러스(IAV)-감염된 마우스는 IAV 특이적 IgG, 광학 밀도(OD) (405nm) 값을 함유하고 평균 OD405 값을 함유하고 있다.
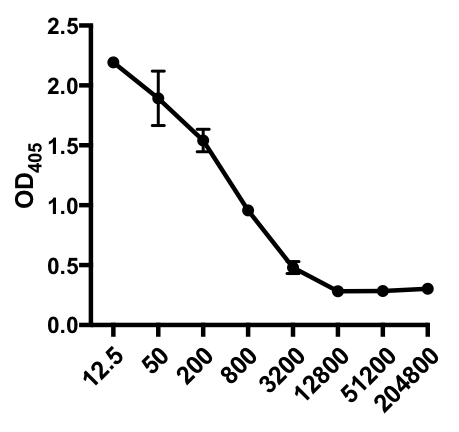
그림 1: IAV 감염 마우스의 혈청에서 인플루엔자 A 바이러스(IAV)-특이적 IgG의 평균 OD405 값(+ S. D.) 및 혈청 희석제(1:12.5에서 1:204,800)의 간접 ELISA 분석산란 플롯. OD405 값은 혈청 희석제와 반비례할 수 있습니다.
샌드위치 ELISA의 다음 예에서, 재조합 인간 TNFα 표준의 1:2.5 희석(75 pg/mL의 농도에서 시작)은 96웰의 평평한 바닥 플레이트의 표시된 우물에 첨가되었다. 이러한 표준은 흡광도 판독값의 2.5배 변화를 주도했습니다.
| 견본 | 농도(pg/mL) | 웰 스 | 값 | 평균 값 | 백 농도 계산 | 평균의 |
| 표준 1 | 75 | A1 | 1.187 | 1.169 | 76.376 | 75.01 |
| A2 | 1.152 | 73.644 | ||||
| 표준 2 | 30 | B1 | 0.534 | 0.52 | 30.827 | 29.962 |
| B2 | 0.506 | 29.098 | ||||
| 표준 3 | 12 | C1 | 0.23 | 0.217 | 12.838 | 12.105 |
| C2 | 0.204 | 11.372 | ||||
| 표준 4 | 4.8 | D1 | 0.09 | 0.084 | 5.055 | 4.726 |
| D2 | 0.078 | 4.398 | ||||
| 표준 5 | 1.92 | E1 | 0.033 | 0.031 | 1.941 | 1.86 |
| E2 | 0.03 | 1.778 | ||||
| 표준 6 | 0.768 | F1 | 0.009 | 0.011 | 0.626 | 0.764 |
| F2 | 0.014 | 0.901 | ||||
| 표준 7 | 0.307 | G1 | 0.002 | 0.004 | 0.238 | 0.377 |
| G2 | 0.007 | 0.516 |
표 2: TNFα 샌드위치 ELISA 표준 곡선 데이터. 재조합 인간 TNFα 표준(75~0.3pg/mL), OD(450nm) 값의 1:2.5 희석은 OD450값, 백 농도 계산 및 평균을 의미한다.
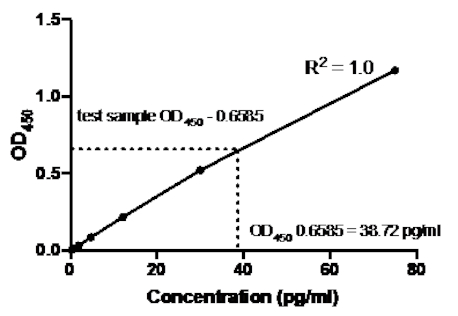
그림 2: TNFα 샌드위치 ELISA에 대한 표준 곡선. 재조합 인간 TNFα 표준(75 ~0.3 pg/mL)의 1:2.5 희석을 샌드위치 ELISA를 사용하여 분석하였다. OD450 값은 표준 희석 농도와 직접 상관관계가 있을 수 있습니다. 시험 샘플에서 TNFα 단백질의 양은 38.72 pg/mL의 농도에 해당하는 표준 곡선을 사용하여 결정되었다.
표준 곡선이 생성되면 시험 샘플에서 TNFα 단백질의 양이 결정되었습니다. 이 샌드위치 ELISA 예제에서 테스트 샘플은 평균0.6585를 제공하는 0.636 및 0.681의 ODI 450 판독값을 제공했습니다. 위의 차트에서 이 OD450 판독값을 플로팅할 때, 이는 38.72 pg/ml의 TNFα 농도에 해당한다.
Application and Summary
입증 된 바와 같이, 면역 분석의 범위 (프로토콜의 약간의 변화와 함께) ELISA 기술 제품군 내에 속한다. 어떤 종류의 ELISA를 사용할지 결정하는 것은 검출되는 항원, 특정 항원용으로 제공되는 단일 클론 항체 및 분석의 원하는 감도(5)를 포함한 여러 가지 요인에 달려 있다. 여기에 설명된 다른 ELISA의 몇 가지 강점과 약점은 다음과 같습니다.
| 엘리사 (주) | 강점 | 약점 |
| 간접적인 | 1) 다중 효소-컨쥬게이드 이차 항체가 1차 항체에 결합할 수 있기 때문에 고감도 | 1) 높은 배경 신호는 플레이트에 대한 관심있는 항원의 코팅이 특이적이지 않기 때문에 발생할 수 있습니다 (즉, 샘플의 모든 단백질은 플레이트를 코팅합니다) |
| 2) 많은 상이한 1차 항체는 단일 효소-컨쥬게이드 이차 항체에 의해 인식될 수 있어 사용자에게 동일한 효소-컨쥬게이드 이차 항체를 사용하는 유연성을 제공하는 데 다수 다른 ELISA(항원 검출에 관계없이) | ||
| 3) 관심있는 항원단일 항체만 사용할 수 있는 최선의 선택 | ||
| 샌드위치 | 1) 항원 특이적 포획 및 검출 단클론 항체의 사용은 분석의 민감도 및 특이성을 증가시킵니다(간접 ELISA에 비해) | 1) 포획 및 검출 단클론 항체의 농도를 최적화하는 것은 어려울 수 있습니다 (특히 비상업적 키트의 경우) |
| 2) 다중 에피토페 (예 : 사이토카인)를 가진 큰 단백질을 검출하기위한 최선의 선택 | ||
| 경쟁적인 | 1) 불순한 샘플을 사용할 수 있습니다. | 1) 플레이트를 코팅하는 데 사용되는 고순수 항원이 다량 필요 |
| 2) 시약 희석 효과에 대한 민감도 가 적다 | ||
| 3) 작은 분자 (예 : 햅텐)를 검출하는 데 이상적입니다. |
표 3: 요약. 다른 ELISA 기술의 강점과 약점에 대한 요약.
간단하고 유용한 기술이지만, 또한 어떤 ELISA에 몇 가지 단점이있다. 하나는 시험 견본에 있는 관심있는 단백질의 양의 불확실성입니다. 양이 너무 높거나 너무 낮은 경우 마이크로 플레이트 판독기에서 얻은 흡광도 값이 각각 표준 곡선의 한계 이상 또는 그 이하로 떨어질 수 있습니다. 이것은 시험 견본에 존재하는 단백질의 양을 정확하게 결정하는 것을 어렵게 할 것입니다. 값이 너무 높으면 플레이트의 우물에 추가하기 전에 테스트 샘플을 희석할 수 있습니다. 그런 다음 희석 계수에 따라 최종 값을 조정해야 합니다. 언급했듯이, 수제 키트는 종종 높은 신호 대 잡음 비율을 산출하는 데 사용되는 항체 농도의 신중한 최적화가 필요합니다.
References
- Porstmann, T. and Kiessig S.T. Enzyme immunoassay techniques. An overview. Journal of Immunological Methods. 150 (1-2), 5-21 (1992).
- Suleyman Aydin. A short history, principles, and types of ELISA, and our laboratory experience with peptide/protein analyses using ELISA. Peptides, 72, 4-15 (2015).
- Gan. S. D. and Patel K. R. Enzyme Immunoassay and Enzyme-Linked Immunosorbent Assay. Journal of Investigative Dermatology, 133 (9), 1-3 (2013).
- Kohl, T. O. and Ascoli C.A. Immunometric Antibody Sandwich Enzyme-Linked Immunosorbent Assay. Cold Spring Harbor Protocols, 1 (6), (2017).
- Sakamoto, S., Putalun, W., Vimolmangkang, S., Phoolcharoen, W., Shoyama, Y., Tanaka, H., and Morimoto S. Enzyme-linked immunosorbent assay for the quantitative/qualitative analysis of plant secondary metabolites. Journal of natural medicines, 72 (1), 32-42 (2018).
Tags
건너뛰기...
이 컬렉션의 비디오:

Now Playing
ELISA 분석: 간접, 샌드위치 및 길항
Immunology
238.8K Views

유세포 분석 및 형광 활성화 세포 분류 (FACS): 비장 B 림프구의 분리
Immunology
93.0K Views

자기 활성화 세포 분류 (MACS): 흉선 T 림프구 분리
Immunology
22.9K Views

ELISPOT 분석: IFN-γ 분비 비장세포 검출
Immunology
28.5K Views

면역 조직 화학 및 면역 세포 화학: 광학 현미경을 통한 조직 이미징
Immunology
79.0K Views

항체 생성: 융합 세포를 사용한 단일 클론 항체 생성
Immunology
43.6K Views

면역 형광 현미경 검사법: 파라핀이 내장 된 조직 절편의 면역 형광 염색
Immunology
53.9K Views

공 초점 형광 현미경: 쥐 섬유 아세포에서 단백질의 국소화를 결정하는 기술
Immunology
43.2K Views

면역침전반응 기반 기술: 아가로스 비즈를 사용한 내인성 단백질의 정제
Immunology
87.8K Views

세포주기 분석: 세포주기 CFSE 염색 및 유세포 분석을 사용한 자극 후 CD4 및 CD8 T 세포 증식 평가
Immunology
24.3K Views

입양 세포 전송: 기증자 쥐 비장 세포를 숙주 쥐에 도입 후 FACS를 통한 성공 평가
Immunology
22.5K Views

세포 사멸 분석: 세포 독성 능력의 크롬 방출 분석
Immunology
151.4K Views
Copyright © 2025 MyJoVE Corporation. 판권 소유