
Method Article
Enrichissement simultané d’affinité de deux modifications post-traductionnelle pour la quantification et la localisation des sites
Dans cet article
Résumé
Ce flux de travail décrit la performance de l’enrichissement rentable et temporel de multiples modifications post-traductionnelle (PTM) de protéines simultanément pour l’analyse protéomique globale quantitative. Le protocole utilise l’enrichissement ptM de niveau peptidique avec de multiples anticorps conjugués, suivi d’une analyse de spectrométrie de masse d’acquisition indépendante des données pour obtenir des connaissances biologiques sur le crosstalk PTM.
Résumé
L’étude de multiples modifications post-traductionnelles (PTM) des protéines est une étape cruciale pour comprendre le travesti PTM et obtenir des informations plus holistiques sur la fonction protéique. Malgré l’importance des études d’enrichissement multi-PTM, peu d’études étudient plus d’un PTM à la fois, en raison en partie des dépenses, du temps, et de grandes quantités de protéines nécessaires pour effectuer l’analyse protéomique globale multiple des PTM. L’enrichissement d’affinité « un pot » détaillé dans ce protocole permet l’identification simultanée et la quantification des peptides avec des résidus de lysine contenant des PTM d’acétylation et de succinylation avec de faibles quantités d’échantillons Entrée. Le protocole comprend la préparation du lysate protéique à partir des foies de souris des souris KNOCK SIRT5, la performance de la digestion de la trypsine, l’enrichissement des PTM et la performance de l’analyse spectrométrique de masse à l’aide d’un flux de travail d’acquisition indépendant de données (DIA). Parce que ce flux de travail permet l’enrichissement de deux PTM à partir d’un même échantillon simultanément, il fournit un outil pratique pour étudier PTM crosstalk sans nécessiter de grandes quantités d’échantillons, et il réduit considérablement le temps nécessaire pour la préparation de l’échantillon, les données l’acquisition et l’analyse. La composante DIA du flux de travail fournit des informations complètes spécifiques au PTM. Ceci est particulièrement important lors de l’étude de la localisation des sites PTM, car LE DIA fournit des ensembles complets d’ions fragmentaires qui peuvent être déchiffrés par calcul pour différencier les différents isoformes de localisation PTM.
Introduction
Une myriade de modifications post-traductionnelle régulent dynamiquement les protéines et les voies par des effets sur l’activité1, signalant2, et le chiffre d’affaires3,4. Par exemple, les kinases protéiques sont activées ou désactivées par l’ajout de groupes de phosphate5, et l’acétylation histone et d’autres modifications fournissent un mécanisme pour changer la structure de la chromatine et servir de mécanismes de régulation transcriptionnel6,7. Ces dernières années, les preuves ont monté que plusieurs PTM travaillent de concert ou en concurrence pour réguler la fonction de protéine ou l’activité8,9,10,11. Par conséquent, la compréhension de ptM crosstalk est un besoin émergent dans la recherche de PTM. Cependant, la plupart des flux de travail protéomiques disponibles pour identifier et quantifier les sites PTM se concentrent sur des modifications uniques, plutôt que sur l’interaction de multiples modifications. Le flux de travail décrit corrèle la modification spécifique des protéines « points chauds » et les résidus de lysine qui sont modifiés par plusieurs PTM différents.
Il y a un besoin croissant dans la communauté scientifique pour des méthodes réalisables pour étudier plusieurs PTM simultanément12. La plupart des méthodes pour identifier et quantifier globalement les sites de plusieurs types de PTM sont difficiles en raison des coûts élevés et de la quantité de tissu requis12,13. Non seulement les expériences d’enrichissement multiPTM prennent beaucoup de temps en termes de préparation d’échantillons, d’acquisition de données et d’analyse de données, mais ces études nécessitent généralement des quantités importantes et souvent prohibitives de protéines11. Décrit ici est un protocole pour l’enrichissement simultané et l’analyse de plusieurs PTM, qui aborde également plusieurs de ces obstacles et permet le profilage à grande échelle PTM et l’évaluation des traques croisées entre les différents PTM14. Ce flux de travail d’un pot décrit une façon pratique pour les chercheurs biomédicaux de profiler à l’échelle mondiale plusieurs PTM, d’identifier les peptides co-modifiés et d’étudier le traqueur PTM d’une manière efficace et rentable14,15.
Ici, cette méthode est présentée en examinant l’acylation des protéines mitochondriales, qui a été étudiée pour la première fois il y a plus de 50 ans16. Il se concentre spécifiquement sur l’acétylation de lysine17 et la succinylation18, y compris la co-occurrence de ces modifications sur les protéines et même la co-modification au niveau peptidique. Étant donné que l’étude utilise un modèle de souris à élimination directe sirtuine 5 (SIRT5), elle a été choisie pour se concentrer sur l’enrichissement des sites d’acétylation et de succinylation. Cette décision a été prise parce que les sites de succinylation sont des cibles de SIRT5 desuccinylase et sont donc censés montrer une upregulation significative chez les souris KO, ce qui en fait les PTM les plus pertinents dans ce cas. Les deux PTM sont biologiquement pertinents comme l’ont récemment résumé Carrico et coll.19. En général, l’acétylation montre des effets importants sur l’expression des gènes et le métabolisme, et la succinylation a été rapportée pour réguler le métabolisme cardiaque et la fonction20.
Le protocole décrit peut être exécuté avec une faible quantité de matière d’entrée de protéine (par exemple, 1 mg de lysate protéique) et réduit la durée totale de l’expérience en réduisant le temps passé pour le traitement de l’échantillon, l’acquisition de la SP et l’analyse des données. Un schéma de flux de travail est fourni à la figure 1. Nous avons également utilisé des quantités encore plus faibles de matière de départ (jusqu’à 100 g de lysate protéique, réduisant les quantités de perles utilisées en conséquence), ce qui, comme prévu, réduit le rendement global des peptides acylés identifiés; cependant, il fournit toujours des résultats très précieux et des peptides acylated quantifiables.
Bien que les workflows dits descendantou intermédiaire n’utilisent généralement pas d’approches de digestion protéolytique (et maintiennent ainsi la connectivité de plusieurs PTM au sein d’une protéine), ce protocole se concentre sur une approche d’enrichissement d’affinité basée sur le peptide afin d’acquérir une profondeur et une sensibilité supplémentaires pour l’identification et la quantification du PTM (figure 1). En outre, ce flux de travail centré sur le peptide utilise des méthodes modernes de spectrométrie de masse, y compris 1) une combinaison d’acquisition dépendante des données (DDA) pour générer des bibliothèques spectrales, et 2) l’acquisition indépendante des données (DIA) pour une quantification précise de PTM dans un flux de travail sans étiquette.
Les flux de travail DIA surmontent la stochasticité de l’échantillonnage des schémas d’analyse typiques du PDD en fragmentant de façon exhaustive tous les signaux peptidiques dans la plage de m/z échantillonnée21. Cette fonctionnalité est également extrêmement bénéfique en termes de localisation du site, car il est plus facile d’obtenir des informations sur les ions fragmentaires spécifiques sont modifiés dans le peptide. En outre, les flux de travail DIA permettent l’identification et la quantification des isoformes PTM-peptide mineurs avec des ions précurseurs identiques. Les méthodes DE DIA peuvent également déterminer une localisation spécifique du site PTM au sein d’un peptide en fonction d’ions fragmentés correspondants spécifiques qui sont mesurés en tout temps de manière exhaustive. Cependant, les approches DDA utilisent souvent des caractéristiques d'« exclusion dynamique » qui excluent l’échantillonnage multiple de la SP/MS pour le même ion précurseur, manquant ainsi des isoformes PTM mineurs.
La stratégie d’enrichissement simultané décrite ici est idéalepour les études qui bénéficieront du profilage et de la quantification globaux de plusieurs PTM, de l’examen du travestissement ptM et de la compréhension des interactions dynamiques des modifications post-traductionnelles. L’identification de multiples PTM enrichis dans un flux de travail combiné a été décrite par : l’enrichissement global, en série ou parallèle de PTM contenant des peptides protéolytiques, ou alternativement par l’analyse des protéines intactes. En comparaison directe avec l’enrichissement en série de l’acétylation et de la succinylation, l’efficacité de la méthodologie d’un pot a été établie comme étant très similaire14. Ces protocoles alternatifs exigent des quantités importantes de matériel de départ et de temps et peuvent être prohibitifs. En revanche, le protocole d’un pot fournit une méthode peu coûteuse et efficace pour l’enrichissement de plus d’un PTM avec l’analyse et l’identification ultérieures.
Les tissus hépatiques de souris sont obtenus à partir de souris KNOCK SIRT5 et sont utilisés ici comme matériau de départ. Ce protocole peut également être réalisé pour les lysates protéiques provenant de différents tissus ou d’expériences de culture cellulaire. Ce protocole peut être appliqué aux lysates protéiques obtenus à partir de tissus ou de granulés de culture cellulaire.
Protocole
Toutes les expériences décrites dans le protocole suivent les lignes directrices du Comité institutionnel de recherche sur les animaux de l’Institut Buck.
1. Extraction de protéines à partir de tissus homogénéisés et digestion à la protéase
- Préparer fraîchement le tampon de lyse à une concentration finale de 8 M d’urée, 50 mM de bicarbonate de triéthylène (TEAB), 1 m de trichostatin A (TSA), 20 mM de nicotinamide, 75 mM de chlorure de sodium (NaCl), 1x cocktail inhibiteur de la protéase/phosphatase (PIC) avec de l’eau HPLC.
- Ajouter une perle d’acier stérile à un tube de 2 ml compatible avec un homogénéisateur de moulin à perles, puis placer le morceau de tissu (de la taille d’un pois) dans le tube. Ajouter brièvement le tampon de lyse (500 l) et le vortex pour s’assurer que le tampon couvre la pièce de tissu (ajouter plus de tampon de lyse, si nécessaire). Placez les tubes sur des ensembles d’adaptateurs préréfrigérés, en veillant à ce que les tubes soient équilibrés entre les deux adaptateurs de l’homogénéisateur.
- Homogénéiser les échantillons 2x à 25 Hz pendant 3 min chacun à 4 oC. Si le tissu n’est pas entièrement homogénéisé, faites tourner brièvement les tubes et transférez le lysate homogénéisé dans un tube de 1,5 ml fraîchement étiqueté. Ensuite, ajoutez un tampon de lyse supplémentaire à la pièce de tissu restante, et répétez l’homogénéisation 2x à 25 Hz pendant 3 min chacun. Deux cycles d’homogénéisation sont généralement suffisants pour briser le tissu.
- Retirer la perle de métal à l’œil d’une pince à épiler. Entre chaque échantillon, rincez la pince à épiler avec de l’eau de qualité HPLC, du méthanol de qualité HPLC et de l’eau de qualité HPLC à nouveau.
- Combinez les lysates homogénéisés si deux cycles d’homogénéisation étaient appliqués et sonicate des échantillons avec un ultrasonateur pendant 10 cycles à 30 s sur/30 s off et 4 oC à haute puissance.
- Centrifuger les lysates homogénéisés à 14 000 x g pendant 10 min à 4 oC pour effacer le lysate. Transférer le supernatant (lysate défriché) dans un nouveau tube microcentrifuge de 1,5 ml, en évitant toute couche de graisse qui peut se siéger sur le supernatant et tout débris au fond du tube.
- Effectuer un test d’acide bicinchoninique (BCA) pour mesurer la concentration protéique du lysate défriché avec une dilution appropriée (par exemple, 1:20 et/ou 1:200). Selon les résultats de BCA, aliquot 1 mg de protéines de chaque échantillon.
- Réduire les protéines en 4,5 mM de dithiothreitol (TNT) à 37 oC pendant 30 min avec une agitation à 1 400 tr/min. Par la suite, alkylate protéines dans 10 mM d’iodoacétamide (IAA) avec incubation dans l’obscurité (place dans le tiroir ou l’armoire) à température ambiante (RT) pendant 30 min.
REMARQUE : Des réactifs alternatifs peuvent être utilisés, comme le N-éthylmaleimide (NEM) au lieu de l’IAA. - Ajouter 50 mM TEAB aux échantillons pour diluer la concentration d’urée à moins de 2 M. Spot 1 'L de l’échantillon sur un papier pH pour s’assurer que le pH de l’échantillon dilué se situe entre 7,0 et 8,5. Ajouter la trypsine à chaque échantillon à un rapport de 1:50 (trypsine-à-protéine, wt/wt) et digérer la protéine avec de l’agitation à 37 oC pendant la nuit (environ 14-16 h).
- Étancher les digestes avec 10% d’acide formique pour atteindre 1% d’acide formique le lendemain. Vortex et tourner brièvement. Spot 1 'L de l’échantillon sur des bandes de pH pour s’assurer que le digest est pH '2-3. Échantillons de centrifugeuse à 1 800 x g pendant 15 min à RT pour granuler tout matériau insoluble.
2. Désalement des peptides protéolytiques non enrichis par l’extraction en phase solide à grande échelle
- Obtenir des cartouches contenant de la résine C18 qui peut lier jusqu’à 10 mg de protéines. Ajustez ces cartouches dans un appareil à vide pour utiliser l’aspiration sous vide qui peut tirer le liquide à travers la cartouche pendant chacune des étapes suivantes. Désignez une cartouche pour chaque échantillon de peptide.
- Ajouter 800 l de 80 % d’acétonitrile (ACN) en acide formique de 0,2 % aux cartouches avec 19,8 % d’eau et utiliser l’aspiration sous vide pour tirer le liquide à travers. Répétez cette étape 1x, en évitant le séchage des cartouches complètement.
- Equilibrate cartouches en ajoutant 800 L de 0,2% d’acide formique dans l’eau avec aspiration sous vide pour tirer tout le volume à travers le filtre. Répétez ce 2x. Chargez les peptides sur les cartouches avec aspiration sous vide. Laver les peptides 2x avec 800 l’acide formique de 0,2 % dans l’eau sous aspiration sous vide.
- Disposer 1,5 mL de tubes microcentrifugesous chaque cartouche pour recueillir l’élutation peptide dans l’étape finale. Sous aspiration sous vide édumise les peptides des cartouches, d’abord avec 800 L de 80% ACN dans 0,2% d’acide formique et 19,8% d’eau, puis avec 400 L de la même solution. Sécher complètement les échantillons de peptide salés dans un concentrateur sous vide (2-3 h).
3. Enrichissement simultané des peptides K-acétylated et K-succinylated avec des perles d’immunoaffinité
- Resuspendre les peptides séchés dans 1,4 ml de tampon froid 1x de purification immuno-affinité (IAP). Vortex pour mélanger et assurer un pH de 7 euros. Échantillons de centrifugeuses à 10 000 x g pendant 10 min à 4 oC. Une petite boulette peut apparaître.
- Mettre les peptides de côté sur la glace tout en préparant des perles d’anticorps. À un tube de K-acétyl et à un tube de lisier de perles d’anticorps K-succinyl (volume : 100 ll de lisier par tube), ajoutez 1 ml de saline à phosphate froid (PBS) et mélangez par pipetting. Transférer toute la solution dans un nouveau tube de 1,5 ml et tourner dans une centrifugeuse miniature pour 30 s à RT.
- Aspirez le PBS, en prenant soin d’éviter d’aspirer les perles. Répétez le lavage 3x en lavant à nouveau avec 1 ml de froid 1x PBS, en centrifuge pour 30 s à RT et en s’apitant hors du PBS.
- Suspendre à nouveau les perles dans environ 440 L de PBS. Un quart du nombre de perles dans chaque tube PTM (100 L, fourni par le fabricant) est utilisé pour l’enrichissement d’un pot (environ 62,5 g de chaque anticorps immobilisé pour les perles d’anticorps K-acétyl et K-succinyl). Pipette les perles plusieurs fois pour bien mélanger. Retirer 100 l de perles d’anticorps acétyl-lysine et les transférer dans un seul tube avec 200 pointes précoupées ll, en veillant à ce que le mélange principal reste constamment bien mélangé.
- Faites de même avec les perles d’anticorps succinyl-lysine en enlevant 100 l de perles d’anticorps succinyl-lysine au tube pour atteindre des parties égales de l’anticorps acétyl-lysine et de l’anticorps succinyl-lysine dans chaque tube. Faites tourner vers le bas pendant 30 s dans une centrifugeuse miniature et aspirez le support avec la pointe de chargeur de gel (les perles deviendront blanches).
REMARQUE: Chaque tube doit avoir une boulette similaire au fond. - Pipette le peptide resuspendu directement sur les perles lavées. Veillez à éviter les granulés (ajouter rapidement pour éviter de sécher les perles). Incuber les peptides avec des perles à 4 oC pendant la nuit avec de l’agitation.
4. Suppression des peptides liés aux perles d’anticorps
- Faire tourner les échantillons de peptides à 2 000 x g pour 30 s à 4 oC. Retirez le supernatant contenant des peptides non liés et économisez pour de futures expériences si désiré. Ajouter 1 ml de tampon IAP froid 1x aux perles pour laver et mélanger en inversant 5x. Faire tourner à 2 000 x g pour 30 s à 4 oC. Aspirez la solution IAP et répétez l’étape de lavage IAP 1x pour un total de deux lavages.
- Ajouter 1 ml d’eau froide HPLC aux perles et mélanger en inversant 5x. Faire tourner à 2 000 x g pour 30 s à 4 oC. Aspirez hors de l’eau. Répétez l’étape de lavage à l’eau deux fois pour un total de trois lavages. Faites tourner un temps supplémentaire à 2 000 x g pour 30 s à 4 oC pour recueillir l’eau restante au fond du tube. Aspirez toute eau supplémentaire qui aurait pu se recueillir avec des pointes de chargement de gel à pointe plate. Veillez à éviter d’aspirater les perles.
- Ajouter 55 ll d’acide trifluoroacetic de 0,15 % dans l’eau aux perles. Incuber les perles pendant 10 min à RT tout en tapant de temps en temps le fond du tube pour mélanger. Faites tourner les perles à RT pendant 30 s dans une centrifugeuse miniature. Retirer les peptides éliqués et réserver à l’aide d’une pointe à bout plat de gel-chargement.
- Ajouter 45 ll d’acide trifluoroacétique de 0,15 % dans l’eau aux perles. Incuber pendant 10 min à RT tout en tapant sur le fond du tube de temps en temps pour mélanger. Faites tourner les perles à RT pendant 30 s dans une centrifugeuse miniature. Retirez la deuxième élution à l’aide d’une pointe à pointe plate à pointe de gel et combinez-la avec la première élution.
- Faites tourner les peptides elutés combinés à 12 000 x g pendant 5 min à RT pour granuler les perles qui se reportent. Transférer la solution d’échantillon de peptides dans un nouveau tube de 500 l.
REMARQUE: Le protocole peut être mis en pause ici.
5. Désalement de peptides enrichis
REMARQUE : Le pH des échantillons doit être inférieur à 4 pour une liaison optimale à la pointe contenant de la résine C18. Utilisez une pointe de pipette de 10 l contenant 0,6 l de résine C18 et une pipette de 10 l pour contrôler la pointe.
- Prémouiller la pointe C18 en pipetting 10 'L d’acetonitrile dans la pointe. Distribuer l’acétonitrile en déchets. Répétez cette étape deux fois de plus.
- Équilibrez la pointe en lavant 3x avec 10 L d’acide formique de 0,2% dans l’eau. Distribuez la solution en déchets après chaque équilibre.
- Chargez les peptides de l’échantillon sur la pointe en continuant à tapoter et à distribuer l’échantillon enrichi de peptide avec la pointe. Pipette de haut en bas à plusieurs reprises au moins 20x et de distribuer la solution restante de la pointe.
- Laver la pointe contenant du peptide lié avec 10 'L de 0,2% d’acide formique dans l’eau. Distribuer la solution dans le gaspillage. Répétez cette étape 9x.
- Élirez les peptides dans un nouveau tube à l’aide de 10 l d’un tampon d’élution contenant 0,2 % d’acide formique, 50 % d’acétonitrile et 49,8 % d’eau. Pipette à plusieurs reprises et distribuer les 10 L de solution dans le nouveau tube 15x. Répétez cette étape avec un tampon d’élution supplémentaire de 10 l, en traînant à plusieurs reprises dans le même tube que l’élution précédente.
- Sécher complètement les peptides dans le concentrateur sous vide (environ 20 min). Resuspendre les peptides séchés en volume approprié de 0,2 % d’acide formique dans l’eau.
REMARQUE: Le protocole peut être mis en pause ici.
6. Acquisition de données à l’aide de DDA et DIA
REMARQUE : Analyser les échantillons avec les méthodes DDA et DIA LC-MS/MS, qui peuvent être ajustées en fonction de l’instrument spectrométrique de masse disponible. Ici, les échantillons ont été analysés à l’aide d’un système nano-LC 2D HPLC couplé à un spectromètre de masse à haute résolution.
- Utilisez un système HPLC combiné à un système HPLC de plate-forme à puce directement relié à un spectromètre de masse (de nombreuses configurations et systèmes LC-MS peuvent également être utilisés).
- Analyse d’échantillons à l’aide de la phase inverse HPLC-ESI-MS/MS
- Après l’injection, transférer les mélanges de peptides sur une puce de pré-colonne C18 et les saler en lavant avec la phase A mobile à 2 l/min pendant 10 min. Ensuite, transférez les peptides dans une colonne analytique et elute à un débit de 300 nL/min avec un gradient de 2-3 h en utilisant les phases mobiles A et B. Plus précisément, utilisez un gradient linéaire de 5% de phase Mobile B à 35% de phase Mobile B de plus de 80 min.
- Par la suite, rampe de la phase B mobile à 80% sur 5 min, puis tenir à 80% B pendant 8 min avant de revenir à 5% B pour une rééquilibrité de 25 min.
- Construire une méthode d’instrument MS pour Le DDA et définir les expériences d’analyse d’instruments suivantes
- Expérience 1 : balayage d’ion précurseur MS1 de m/z 400-1 500 (temps d’accumulation de 250 ms). Définir le seuil d’intensité pour déclencher des scans MS/MS pour les ions de charge indique 2-5 à 200 comptes. Définir l’exclusion dynamique des ions précurseurs à 60 s.
- Expérience 2 : Scan d’ion produit MS/MS avec une plage d’analyse MS2 de m/z 100-1 500 (temps d’accumulation de 100 ms par balayage ion de 30 produits par cycle). Définir la propagation de l’énergie de collision à CES 5, puis sélectionnez le «mode de balayage d’ionde produit à haute sensibilité ».
REMARQUE : La méthode DDA fera l’acquisition des spectres MS/MS pour les 30 ions précurseurs les plus abondants après chaque analyse MS1 par cycle, et le temps de cycle total sera de 3,3 s. Les acquisitions de DDA seront utilisées pour construire des bibliothèques spectrales telles que décrites à la section 7.
- Construire une méthode d’instrument MS pour le DIA et définir les expériences d’analyse d’instruments suivantes.
- Expérience 1 : effectuer un balayage des ions précurseurs MS1 de m/z 400-1 250 (temps d’accumulation de 250 ms).
- Expérience 2 : effectuez des balayages d’ions de produit MS/MS pour 64 segments variables de SWATH avec une gamme de balayage mS2 de m/z 100-1500 (temps d’accumulation de 45 ms par balayage d’ion de 64 produits par cycle). Définir la propagation de l’énergie de collision à CES 10, puis sélectionnez le «mode de balayage d’ionde produit à haute sensibilité ».
- Utilisez la stratégie d’acquisition de 64 fenêtres variables DIA/SWATH décrite par Schilling et coll.22 pour obtenir une quantification sans étiquette avec un temps de cycle total de 3,2 s. En bref, dans cette acquisition, au lieu du quadrupole Q1 transmettant une plage de masse étroite à travers la cellule de collision, une fenêtre plus large de largeur variable de fenêtre (5 -90 m/z) est passée en étapes incrémentielles sur la plage de masse complète (m/z 400 -1 250 avec 64 segments SWATH, chacun avec un temps d’accumulation de 45 ms, donnant un temps de cycle de 3,2 s, qui comprend un scan MS1 avec un temps d’accumulation de 250 ms).
REMARQUE : La largeur variable de la fenêtre est ajustée en fonction de la complexité du courant ionique MS1 typique observé dans une certaine plage de m/z à l’aide d’un algorithme de calculatrice de fenêtre variable22 (des fenêtres plus étroites sont choisies dans des gammes de m/z « occupées », de larges fenêtres dans des gammes m/z avec peu d’ions précurseurs élorants). Sur d’autres plates-formes d’instruments MS, d’autres stratégies de fenêtre DIA peuvent être mises en œuvre.
7. Analyse des données
REMARQUE : Certains paramètres d’analyse de données doivent être modifiés et adaptés à l’expérience spécifique. Par exemple, la base de données de protéines (fichier EXPRES) sélectionnée dépend de l’espèce à partir de laquelle l’échantillon a été préparé (ici, Mus musculus). Ci-dessous, l’analyse de données pour les échantillons de souris enrichis pour les peptides acétylés et succinyés est décrite.
- Utilisez un moteur de recherche de base de données MS pour analyser les acquisitions de PDD. Créez une méthode de moteur de recherche de base de données comme suit :
- Pour les paramètres de description de l’échantillon: sélectionnez "Identification" sous type d’échantillon, sélectionnez "Iodoacetic Acid" sous " Cysteine Alkylation ", sélectionnez "Trypsine" sous " Digestion " (en supposant clivage C-terminal à la lysine et à l’arginine), sélectionnez "TripleTOF 6600" sous Instrument, sous Facteurs spéciaux, contrôlez l’accent sur l’acétylation et l’enrichissement de Succinylation, et sélectionnez Mus musculus sous species.
- Pour les paramètres de traitement spécifiques : sélectionnez «Modifications biologiques» sous ID Focus, sélectionnez «SwissProt» sous Base de données, cochez «ID complet» sous recherche effort, sélectionnez «0,05 (10 %)» sous « Seuil de protéines détectés », et cochez « Exécuter une analyse defaux tauxde découverte » sous « Qualité des résultats ». Enregistrer la méthode du moteur de recherche et soumettre les fichiers bruts spectrométriques de masse pour le traitement par le moteur de recherche de base de données en utilisant la méthode générée.
REMARQUE : Dans un processus itératif, tous les scans MS et MS/MS sont automatiquement recalibrés par le moteur de recherche en fonction des annotations et des résultats initiaux.
- Cliquez sur "Export Peptide Summary" à la fin de la recherche et filtrez tous les résultats d’identification peptide par un " seuil de confiance " de 99 dans un logiciel de tableur (p. ex. Excel; faux taux de découverte [FDR] de 1%).
- Dans le fichier de tableur "Peptide Summary", filtre pour tous les peptides qui contiennent l’annotation PTM "acétylation" et "succinylation" dans la colonne de modification pour générer un rapport de résultats pour présenter exclusivement les peptides acylés et leurs protéines correspondantes.
- Pour construire des bibliothèques spectrales MS/MS pour le traitement ultérieur du fichier brut DIA et une quantification relative ultérieure, ouvrez le logiciel d’analyse quantitative DIA. Sélectionnez l’onglet «Bibliothèque», puis (en bas de la page) cliquez sur «Generate Spectral Library» à partir de « Database SearchEngine» et ouvrez un rapport FDR du moteur de recherche de base de données (le fichier FDR.xlsx), qui a été automatiquement généré dans le cadre du processus de recherche de base de données de fichiers bruts du DDA. Ensuite, cliquez sur "next" et sélectionnez le "LibrarySettings Schema" et "next". Sélectionnez «Uniprot_mouse_proteome« comme base de données, puis cliquez sur «next» et «goa_mouse» comme fichier d’annotation génétique (ontologie). Enfin, cliquez sur "finish", et la bibliothèque spectrale sera générée.
REMARQUE : Les renseignements sur les peptides acétylées et succinylées provenant des fichiers de données brutes, des balayages d’ions précurseurs de MS1 et des scans d’ions fragmentaires de MS2 seront inclus dans les bibliothèques spectrales (ce qui comprend le temps de rétention, le modèle de fragmentation MS/MS, etc.). - Utilisez le logiciel d’analyse quantitative de la protéomique DIA pour effectuer une quantification relative des niveaux d’acétylation et de succinylation et créer des feuilles de calcul de peptides candidats contenant du PTM qui peuvent être utilisés pour une analyse plus approfondie des données.
- Pour analyser et quantifier les peptides contenant des PTM qui ouvrent le logiciel d’analyse quantitative DIA, utilisez le schéma d’analyse du modèle. Le schéma de modèle est disponible dans le logiciel en sélectionnant l’option "Perspective des paramètres" "Analyse DIA" "BGS PTMs" (significatif ou clairsemé, selon l’expérience).
REMARQUE : Au lieu d’utiliser le modèle d’analyse BGS PTM dans le logiciel d’analyse quantitative DIA, tous les paramètres spécifiques au PTM peuvent également être mis en place manuellement en suivant ces instructions : 1) sous «identification», sélectionnez «localisation PTM» (seuil de probabilité de 0,75); 2) en "quantification", sélectionnez "groupement de peptides mineurs" "séquence modifiée"; et 3) sous "post-analyse", sélectionnez "groupement d’abondance différentielle" "groupe mineur" (paramètres de quantification). Ces paramètres activeront la fonction de localisation PTM de sorte que les peptides modifiés sont répertoriés comme des entrées individuelles et l’analyse différentielle sont effectuées au niveau peptidique. - Pour commencer l’analyse de quantification, sélectionnez l’onglet «Pipeline», puis cliquez sur «Configurez une analyse DIA à partir du fichier», ouvrez les fichiers bruts MS DIA d’intérêt pour la quantification relative. Sélectionnez "Assign Spectral Library" et sélectionnez la bibliothèque qui a été construite ci-dessus, cliquez sur "charge" "prochaine". Sélectionnez le "BGS PTMs" schéma d’analyse et cliquez sur "suivant". Sélectionnez la base de données appropriée fichier FASTA "Uniprot_mouse_proteome" "prochaine". Définir la configuration de la condition, qui attribue les différentes conditions aux échantillons, et cliquez sur "suivant". Sélectionnez "goa_mouse" comme fichier d’annotation génique (ontologie) et cliquez sur "Next". Examiner la vue d’ensemble de l’analyse (résumé de la configuration de l’expérience) et sélectionner « répertoire desortie» "finition". Enfin, cliquez sur "Run Pipeline" pour effectuer l’analyse quantitative sans étiquette.
REMARQUE : Les modules statistiques du logiciel d’analyse quantitative DIA effectuent automatiquement l’analyse FDR, génèrent des cartes thermiques et des parcelles volcaniques comparant les différentes conditions, génèrent des listes de peptides et de protéines identifiés et quantifiés, et fournissent Q-values avec des changements relatifs de pli comparant différentes conditions.
- Pour analyser et quantifier les peptides contenant des PTM qui ouvrent le logiciel d’analyse quantitative DIA, utilisez le schéma d’analyse du modèle. Le schéma de modèle est disponible dans le logiciel en sélectionnant l’option "Perspective des paramètres" "Analyse DIA" "BGS PTMs" (significatif ou clairsemé, selon l’expérience).
- Comme alternative, utilisez un logiciel pour la suppression des jeux de données DIA pour le traitement des données et l’exécution d’analyses statistiques après l’exportation des zones de pointe extraites pour les sites d’acétylation et de succinylation.
8. Visualisation des données des peptides modifiés et évaluation de la localisation des sites PTM
- Pour ouvrir une analyse quantitative DIA générée, sélectionnez l’onglet «Analyse» suivi de «charge de l’expérience logicielle d’analyse quantitative» (à partir d’une expérience enregistrée ouvrant un . fichier SNE). Naviguez jusqu’à l’a.. Fichier des résultats SNE et sélectionnez "open".
- Dans le panneau de gauche, cliquez sur la flèche à gauche de la troisième fichier de données brutes de l’ile du haut pour étendre et visualiser les 64 segments SWATH. Cliquez sur la flèche à gauche du segment [428.7-437.3] pour étendre le segment spécifique et afficher les peptides modifiés identifiés pour cette plage de masse. Cliquez sur le peptide triply chargé KQYGEAFEK[Acetyl]R.
REMARQUE : Par défaut, le panneau supérieur droit affiche généralement le MS2 XIC, et le panneau inférieur affiche l’enveloppe iXIque MS1 XIC- Dans le panneau supérieur, sélectionnez "PTM Localization Plot". Dans le panneau inférieur, sélectionnez "PTM Localization Plot".
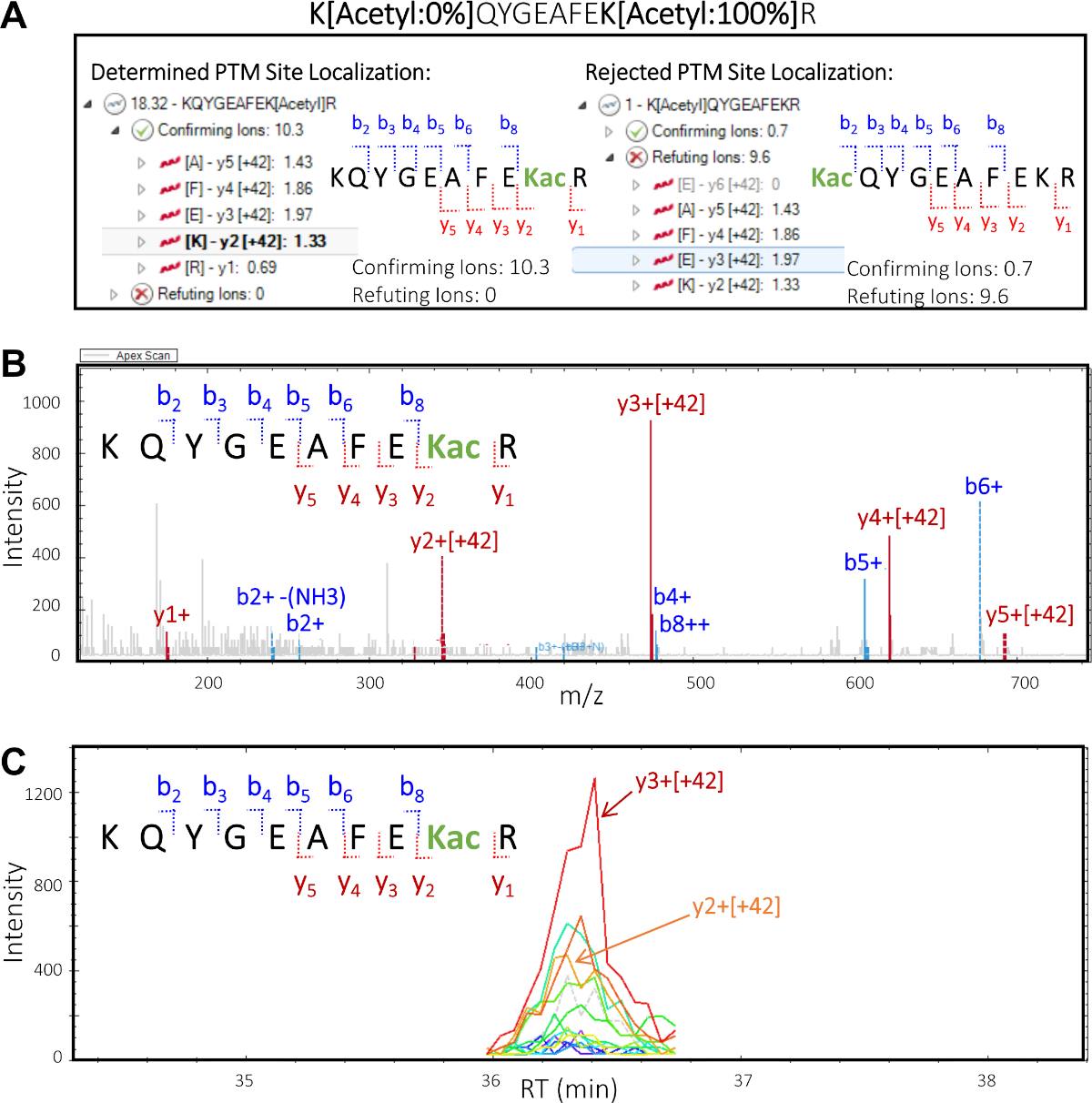
- Dans le terrain de localisation PTM, sélectionnez la séquence peptide supérieure "KQYGEAFEK[Acetyl]R", avec un score total de localisation PTM de 18,32. Cliquez sur la flèche vers la gauche pour élargir la vue et visualiser les ions confirmant et réfutant. Cliquez sur la flèche à côté de la "Confirmation Ions", puis sélectionnez l’ion "y3 [42]" pour mettre en évidence cet ion particulier, ce qui confirme que le site d’acétylation est situé sur K2 de la séquence peptidique.
- Dans le PTM Localization Plot sélectionnez la deuxième séquence peptide "K[Acetyl]QYGEAFEKR" avec un score total (très faible) de localisation PTM de 1. Cliquez sur la flèche vers la gauche pour élargir la vue et visualiser les ions confirmant et réfutant. Cliquez sur la flèche à côté de la "Confirming Ions", puis sur la flèche à côté des "Ions réfutables", puis sélectionnez quelques ions fragmentés pour visualiser et évaluer le chromatogramme d’ions extraits et les spectres MS/MS (Figure 5).
REMARQUE : Le score de localisation ptM assigné et l’inspection visuelle indiquent que l’isosomer PTM correct est KQYGEAFEK[Acetyl]R, alors qu’il n’existe aucune preuve ou minime pour l’autre isomer possible de PTM K[Acetyl]QYGEAFEKR.
Résultats
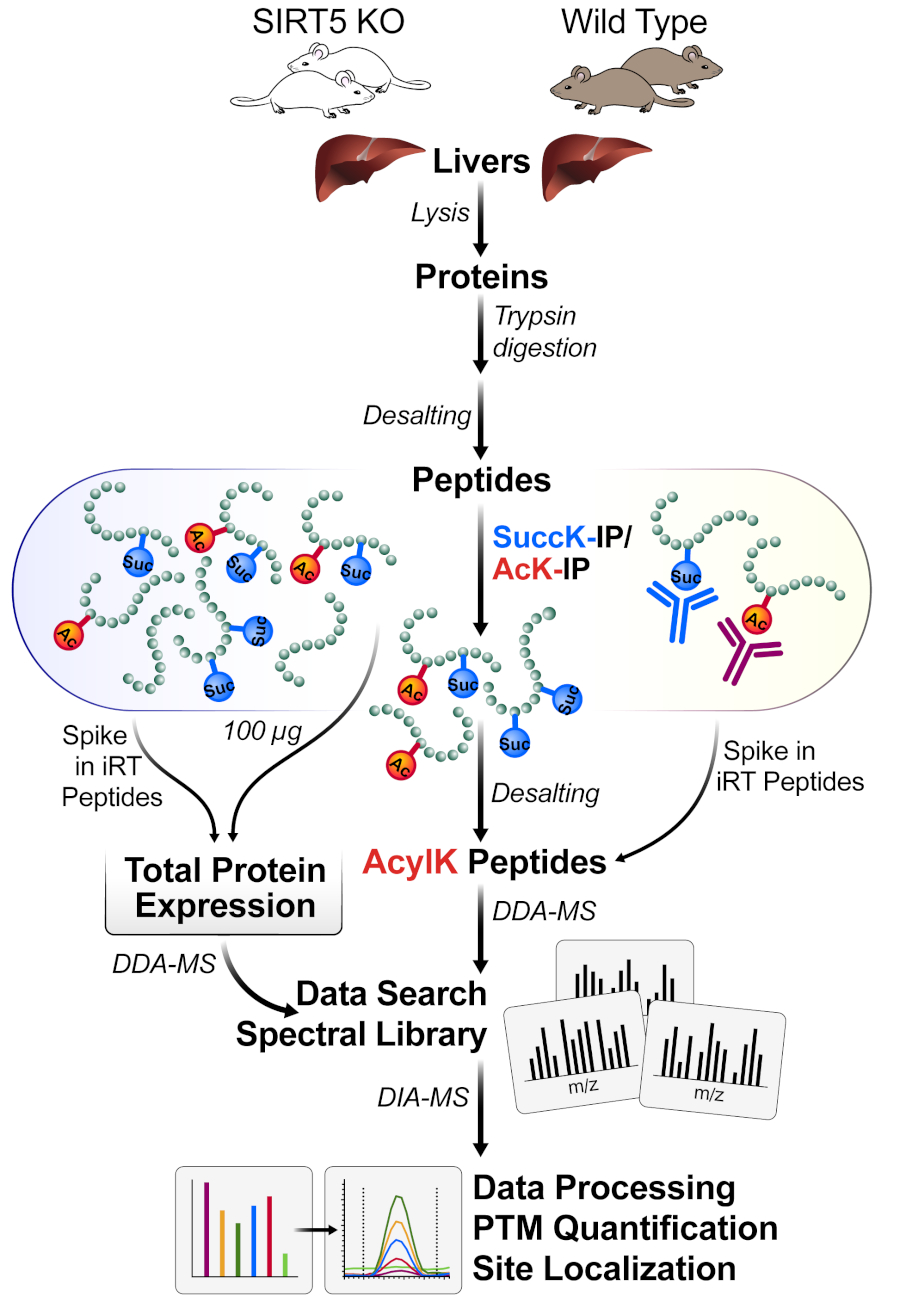
La figure 1 montre un diagramme général du flux de travail, y compris la récolte du tissu à partir du foie de souris, l’utilisation de 1 mg de protéines pour digérer le lysate protéique avec la trypsine, l’incubation de peptides avec des perles conjuguées par anticorps, l’acquisition des échantillons sur la SP, et enfin effectuer l’analyse DIA/SWATH des données à l’aide de divers logiciels quantitatifs protéomiques (académiques et commerciaux).
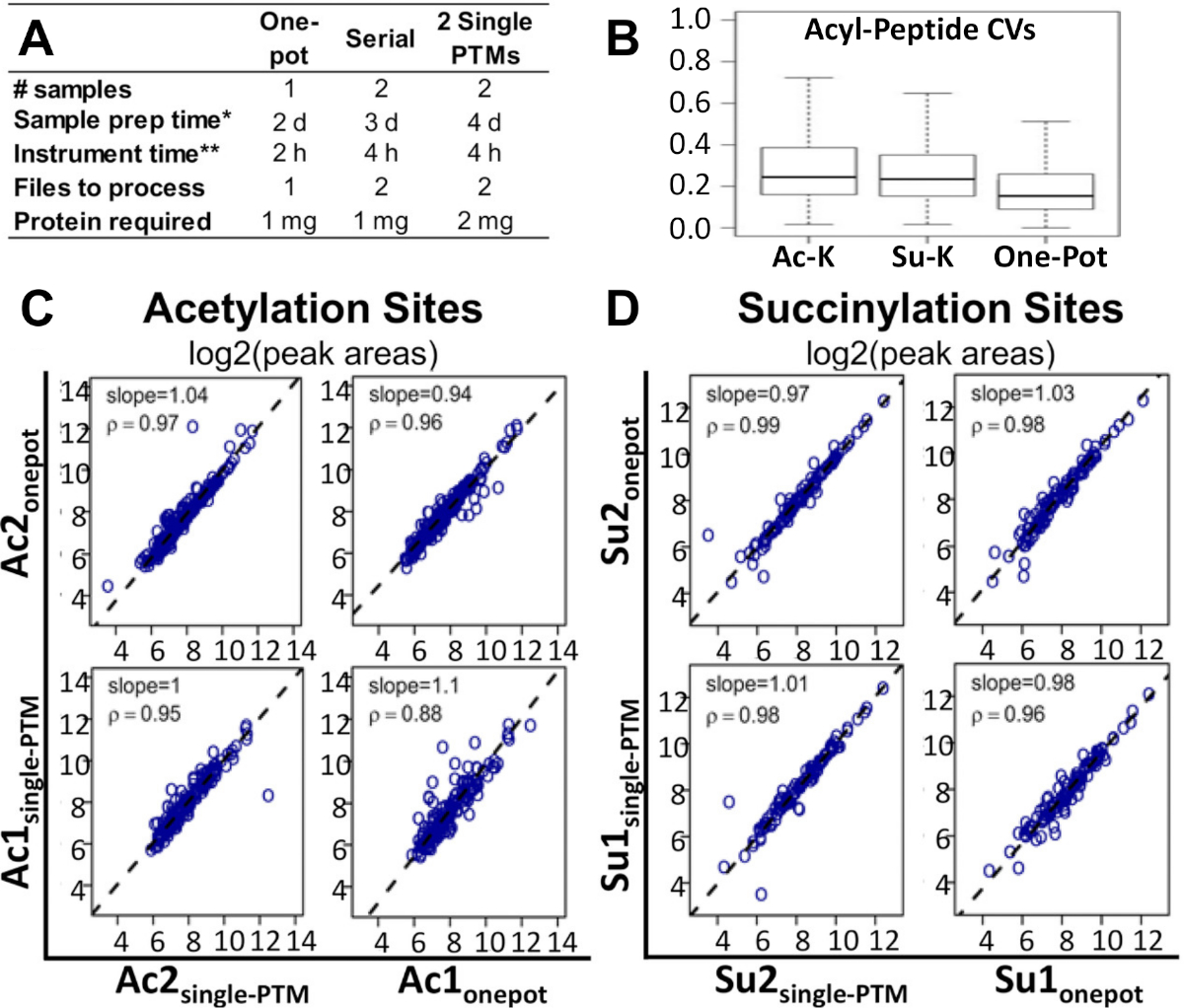
La figure 2A montre comment la chronologie du flux de travail et les quantités d’échantillons et de protéines requises, par rapport aux méthodes alternatives actuellement utilisées pour les études d’enrichissement multi-PTM. La méthode d’un pot peut être effectuée en deux fois moins de temps et avec la moitié du nombre d’échantillons que ces méthodes alternatives. Par rapport à la méthode d’enrichissement à un seul PTM, le protocole d’un pot nécessite également la moitié de la quantité de protéines.
Ce protocole s’est avéré être une solution de rechange réalisable et rentable. La figure 2B montre que le coefficient médian de variation (CV) pour les zones peptidiques modifiées était plus faible dans la méthode d’un pot que dans les enrichissements en un seul PTM et en série-PTM. La figure 2C,D montre que, tout en comparant les méthodes d’enrichissement d’un pot PTM et d’un seul PTM, aucune différence notable n’était apparente entre les corrélations des quantifications au niveau du site pour les deux modifications. Cela était également vrai pour les corrélations au niveau du peptide et au niveau des fragments. La même observation a tenu pour chacun des trois corrélations en comparant les enrichissements d’un pot et de série-PTM. Toutes les données brutes sous-jacentes à la SP et les fiches de résultats Excel traitées associées à un rapport récent de Basisty et coll.14 sont disponibles et peuvent être téléchargées à partir de MassIVE (MSV00081906) et ProteomeXchange (PXD008640).
En général, alors que les stratégies d’enrichissement des anticorps peuvent montrer certaines limitations, telles que l’occlusion épitope potentielle ou une spécificité limitée, les anticorps utilisés dans cette étude sont des mélanges de clones générés indépendamment et fournissent ainsi des gammes plus larges de Spécificités.
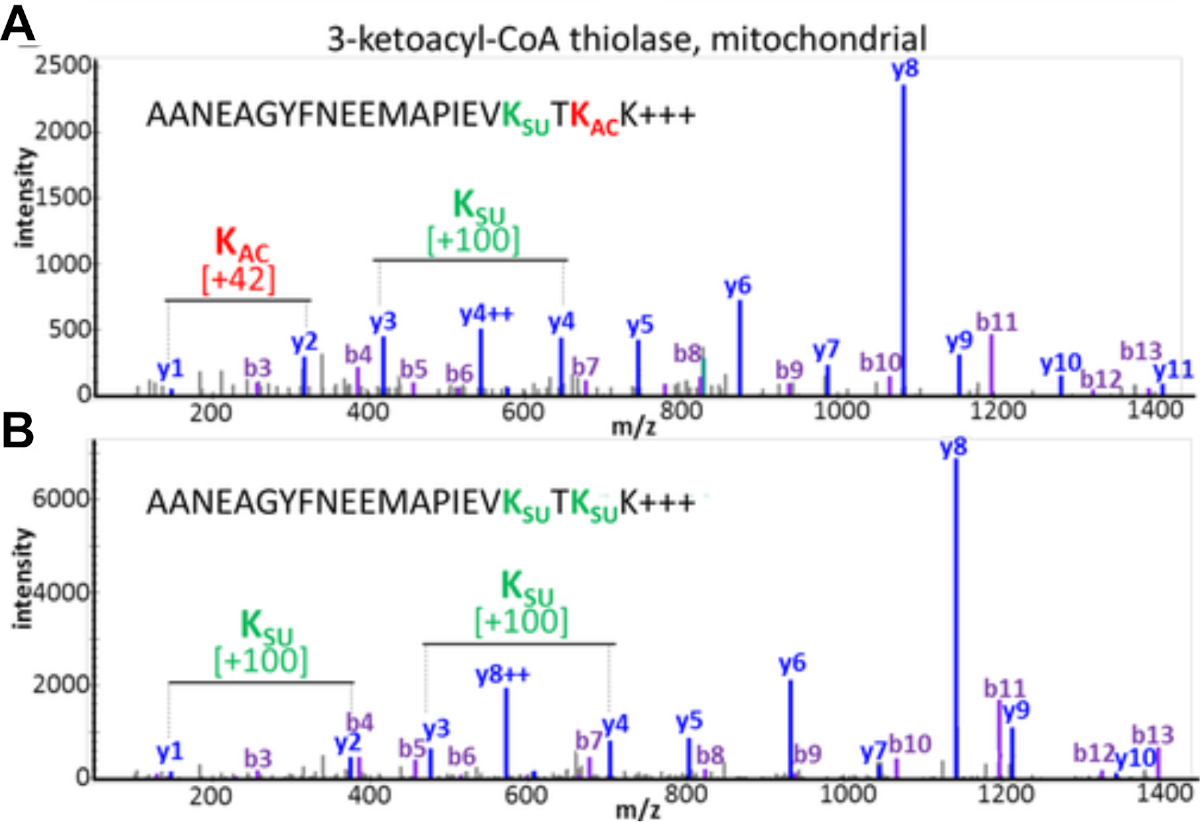
Les résultats expérimentaux documentent la possibilité de détecter et d’évaluer le traqueur ptM. La figure 3 affiche les données d’un enrichissement réussi et illustre un exemple pour un peptide contenant des modifications multiples et différentes d’acyl visualisant le crosstalk de PTM. La figure 3A montre un peptide qui est acétylé sur un résidu de lysine et succinylé sur l’autre, et la figure 3B montre le même peptide succinylé aux deux lysines. Cela démontre que le même résidu de lysine peut être modifié avec les deux groupes d’acylation, et il ya une possibilité de crosstalk se produisant à ce site. La figure 4 montre le nombre de résidus de lysine qui ont été identifiés comme décrits dans les sections 7.1-7.3 pour transporter les deux modifications, pointant également vers un éventuel traqueur ptM.
Comme le montre la figure 5, le traitement des ensembles de données DIA PTM avec un logiciel de protéomique quantitative nous permet de déterminer quels résidus de lysine spécifiques sont modifiés. Il s’agit d’un concept connu sous le nom de localisation du site, qui est une étape essentielle pour toute analyse pour la détermination d’éventuels ptM crosstalk. La figure 5 présente deux isoformes potentiels ainsi que les ions confirmant et réfutant pour chacun d’eux qui pourraient être visualisés et évalués comme décrits dans les sections 8.1-8.3 (en particulier les étapes 8.3.2 et 8.3.3). Sur la base de ces informations, nous avons pu identifier avec confiance lequel des deux isoformes était présent dans l’échantillon original. Le spectre MS/MS de l’isoforme confirmé KQYGEAFEKacR démontre clairement que les ions y (y2-y5) contenant le résidu de lysine acétylé, qui a changé de 42 m/z (la masse d’incrément d’un groupe d’acétyl), ont confirmé le résidu spécifique de lysine dans le peptide qui a été modifié.

Figure 1 : Flux de travail typique pour l’enrichissement d’un pot des PTM. Les tissus (ici, les foies) sont récoltés à partir de SIRT5 KO et de souris de type sauvage (WT), et les protéines sont lysées, trypsines digérées en peptides et dessalées. Les peptides sont ensuite enrichis par l’immunoaffinité avec des combinaisons de perles de succinyl- et d’acétyl-anticorps. Les workflows parallèles de SP mesurent à la fois 1) les petits aliquots des changements entiers d’expression de protéine de lysate (pour la normalisation de protéine) et 2) les peptides acyl-contenants enrichis pour l’identification de site d’acylation (DDA-MS) et la localisation de site, suivis de quantification (DIA-MS). Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 2 : Comparaison du flux de travail d’un pot avec d’autres méthodes. (A) Comparaison du temps, des coûts et des matériaux requis pour le flux de travail d’un pot, l’enrichissement en série-PTM, et deux enrichissements d’un seul PTM. (B) Comparaison des CV entre le flux de travail d’un pot, l’enrichissement simple de PTM d’acétyl-lysine, et l’enrichissement simple de PTM de succinyl-lysine. Analyse de corrélation de spearman comparant les zones de pic de peptide d’acyl obtenues à partir du flux de travail d’un pot, et les enrichissements d’un seul PTM : parcelles correspondantes des résultats de la zone de crête de log2 pour (C) sites d’acétylation et (D) sites de succinylation. Les pentes de régression et les facteurs de corrélation sont indiqués dans les panneaux individuels14. Deux répliques biologiques indépendantes ont été traitées pour chacune des conditions. Ce chiffre a été modifié à partir de Basisty et coll.14. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 3 : Crosstalk entre l’acétylation et les modifications de succinylation des résidus de lysine. Les spectres de SP/MS des peptides tryptiques de la thiolase mitochondriale 3-ketoacyl-CoA qui montrent la même séquence d’acide aminé mais ont été modifiés à deux résidus de lysine avec différents PTMs. (A) MS/MS de peptide AANEAGYFNEEMAPIEVKsuccTKacK et (B) MS/MS de peptide AANEAGYFNEEMAPIEVKsuccTKsuccK. Ce chiffre a été modifié à partir de Basisty et coll.14. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 4 : Overlap et crosstalk entre les résidus de lysine acétylées et succinyées, exemples spécifiques dans les complexes protéiques. (A) Diagramme de Venn affichant un chevauchement entre 2 235 acétylation et 2 173 sites de succinylation. De ce nombre, 943 sites ont été actylés et succinylés. Le foie d’une souris knock-out SIRT5 (de-succinylase) a été analysé, et de nombreux sites de succinylation ont été identifiés. En fait, ils étaient plus abondants que normalement observés dans le foie de souris (les peptides modifiés ont été filtrés à une valeur Q de 'lt;0.05). (B) Complexes protéiques montrant le pourcentage de leurs sous-unités contenant à la fois des sites acrasés et succinylés (la ligne rouge audacieuse représente l’importance déterminée par le test exact de Fisher). (C) Diagramme du complexe de synthase ATP : les sous-unités protéiques en rouge représentent les sous-unités qui contiennent à la fois des sites acétylés et succinylés. Ce chiffre a été modifié à partir de Basisty et coll.14. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 5 : Le logiciel de protéomique quantitatif déchiffre la localisation des PTM sur le site peptidique. Sur la base de la fragmentation mS/MS du peptide, il est possible de fournir des informations sur les résidus spécifiques de lysine que le groupe d’acétyl modifie. Cela met en valeur la capacité du logiciel à offrir des informations précieuses sur la localisation du site des PTM. (A) Deux possibilités de modification des résidus de lysine et de localisation du site PTM : KQYGEAFEKacR (à gauche) et KacQYGEAFEKR (à droite). Des ions fragmentés « confirmant » et « réfutant » sont montrés pour chacun des isoformes potentiels de localisation de site du peptide. Sur la base de ces informations, les scores de confirmation et de réfutation sont attribués, confirmant la présence de l’isoforme KQYGEAFEKacR dans l’échantillon. (B) Spectre MS/MS correspondant à l’isoforme confirmé KQYGEAFEKacR indiquant que tous les ions y, y compris les résidus de lysine acétylé (y2 et plus) portent une masse d’incrément de 42 m/z, ce qui correspond à la modification de l’acétyl. Les ions b observés ne contiennent pas la modification. (C) Chromatogramme d’ions extraits (XIC) avec des zones de pointe abondantes résultant de y2 et y3 ions, qui constituent tous deux le site d’acétylation dans l’isoforme confirmée KQYGEAFEKacR. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.
{kind=link}
Discussion
Ce protocole décrit une technique nouvelle pour l’enrichissement simultané multiple de PTM pour mieux comprendre le crosstalk de PTM. Les méthodes alternatives pour atteindre cet objectif ont tendance à être excessivement longues et coûteuses, et elles nécessitent de grandes quantités de protéines pour réussir11,13. Ce protocole présente un flux de travail d’enrichissement multi-PTM qui implique l’incubation dans des perles conjuguées par anticorps pour deux PTM à la fois pour améliorer l’efficacité globale de l’expérience. Cette méthode implique également l’utilisation du PDD pour la génération spectrale de bibliothèques et les acquisitions de MS DIA pour détecter et quantifier les peptides présents avec une interférence réduite des ionsfragment2,23. Les logiciels, tels que les moteurs de recherche de base de données MS, sont utilisés pour analyser et quantifier les données des acquisitions de PDD, tandis que Quantitative Proteomics Software24 et DIA Quantitative Analysis Software25 sont nécessaires pour interpréter les spectres complexes produits par les acquisitions de DIA.
Il y a plusieurs étapes critiques dans ce protocole qui doivent être suivies avec soin. Comme l’objectif principal du protocole est d’enrichir simultanément plusieurs PTM, l’étape d’enrichissement de l’affinité anticorps (section 2) est essentielle au succès de l’expérience. Lors de l’exécution des lavages sur les perles, il est nécessaire de s’assurer qu’aucune des perles sont aspirées accidentellement. Il est également nécessaire de s’assurer que la concentration d’urée a été diluée à 1 M avant la digestion avec la trypsine (étape 1.8). Bien que l’urée de 8 M soit nécessaire plus tôt dans le protocole pour la solubilité des protéines, les concentrations d’urée supérieures à 1 M inhiberont l’activité enzymatique de la trypsine. En outre, il est important de vérifier systématiquement le pH de l’échantillon tout au long du protocole. Ceci est particulièrement important avant la digestion. Si le pH de l’échantillon et la solution de trypsine ne sont pas neutralisés de manière appropriée avant l’incubation, il peut entraîner une digestion inefficace dans laquelle de nombreux sites de clivage peuvent être manqués, ce qui entraîne moins d’identifications peptidiques.
Quelques modifications au protocole peuvent être utiles lors de la préparation d’échantillons. Pour un lysate protéique acheté à partir de 1 mg de matériau de départ, un quart des perles d’anticorps fournies dans chaque tube de balayage PTM peut être utilisé comme une alternative rentable. Une plus grande quantité de matériel de départ peut être utilisée pour de meilleurs résultats, tant que la quantité de perles d’anticorps utilisées sont augmentées proportionnellement. Une autre modification qui peut améliorer le flux de travail est de digérer les échantillons avec une autre protéase en plus de la trypsine. Cette modification entraînerait une plus grande variabilité dans les peptides clivés, offrant une couverture accrue des résidus de protéines. Bien que non nécessaire, il est recommandé que la trypsine soit l’une des enzymes utilisées pour l’analyse PTM en raison de sa spécificité de clivage élevé26.
Une limitation de ce protocole est que les PTM à l’étude doivent avoir des chimies similaires afin d’être enrichis simultanément14. Les procédures pour les différentes perles conjuguées par anticorps doivent être similaires, en utilisant des solvants et des solutions similaires, y compris des conditions d’élution similaires, et de préférence du même fournisseur. Pour cette raison, la méthode décrite ici utilise systématiquement l’acétylation et la succinylation comme exemple, qui utilisent tous deux des perles conjuguées par anticorps (Cell Signaling Technology, Inc). Bien que cette méthode puisse théoriquement être appliquée à n’importe quel nombre de PTM, des études supplémentaires seraient nécessaires pour évaluer la limitation exacte du protocole à cet égard. En outre, comme il s’agit d’une méthode d’enrichissement à base d’anticorps, la méthode ne peut fournir qu’une quantification relative des sites PTM.
En comparaison avec les méthodes d’enrichissement multi-PTM existantes, ce flux de travail est une alternative plus faisable et plus rentable. De cette expérience, il a été observé que l’efficacité de cette méthode se compare extrêmement bien avec des méthodes alternatives, telles que les enrichissements individuels ou en série. La figure 2B montre que le CV médian pour les zones de pic de peptides modifiés a en fait été diminué dans la méthode d’un pot par rapport aux enrichissements à un seul PTM et aux enrichissements en série-PTM13. Nous avons analysé en outre les résultats expérimentaux évaluant les quantifications au niveau du site pour l’acétylation ou la succinylation. De plus, l’analyse de corrélation de Spearman (figure 2C,D) a démontré que l’enrichissement en PTM d’un seul pot a été exécuté de la même façon que les workflows d’enrichissement d’un seul PTM. Cela était également vrai pour les corrélations peptides et fragment-niveau. La même observation a tenu pour chacun des trois corrélations en comparant un pot avec l’enrichissement de série-PTM.
Ce protocole permet aux chercheurs de faire des connaissances biologiques fascinantes sur les traques ptM d’une manière rapide et rentable. La composante DIA du flux de travail permet aux chercheurs de mieux comprendre les PTM, car il fournit des informations sur la localisation des sites et surmonte les défis tels que le faible taux d’occupation des PTM. Les ions précurseurs ont tendance à être exclus du PDD, ce qui est particulièrement important lorsqu’ils étudient les PTM, car l’occupation du site est souvent faible au point que ces peptides ne sont pas sélectionnés pour la SP/MS, mais contiennent toujours des informations cruciales. Des expériences de suivi pourraient être effectuées pour évaluer la limite supérieure du nombre de PTM pouvant être enrichis simultanément à l’aide de cette méthode. Une amélioration future de ce flux de travail pourrait inclure le développement de plates-formes logicielles plus avancées pour automatiser davantage l’analyse de la localisation du site et de l’occupation du site PTM.
Déclarations de divulgation
Les auteurs n’ont rien à révéler.
Remerciements
Nous reconnaissons le soutien de la subvention d’instrumentation partagée des NIH pour le système TripleTOF à l’Institut Buck (1S10 OD016281). Ce travail a également été soutenu par le National Institute of Allergy and Infectious Disease (R01 AI108255 à B.S.) et l’Institut national du diabète et des maladies digestives et rénales (R24 DK085610 à Eric Verdin; R01 DK090242 à Eric Goetzman). X.X. a reçu l’appui d’une subvention des National Institutes of Health (subvention des NIH T32GM8806, à Judith Campisi et Lisa Ellerby), au Nouveau-B.), grâce à une bourse postdoctorale de la Glenn Foundation for Medical Research.
matériels
| Name | Company | Catalog Number | Comments |
| 1 M Triethylammonium biocarbonate buffer (TEAB) | Sigma Aldrich, St. Louis, MO, USA | T7408 | |
| Acetonitrile, Burdick and Jackson LC-MS grade | Burdick and Jackson, Muskegon, MI, USA | 36XL66 | |
| Bioruptor sonicator | Diagenode, Denville, NJ, USA | B01020001 | |
| C18 pre-column chip (200 µm x 6 mm ChromXP C18-CL chip, 3 um, 120 A) | SCIEX, Framingham, MA, USA | 5015841 | |
| C18-CL chip (75 µm x 15 cm ChromXP, 3 µm, 300 Å) | SCIEX, Framingham, MA, USA | 804-00001 | |
| Dithiothreitol (DTT) | Sigma Aldrich, St. Louis, MO, USA | D9779-5G | |
| Eppendorf Thermomixer Compact | Eppendorf AG, Hamburg, Germany | T1317-1EA | |
| Eppendorf Tube (2.0 mL Safelock) | Eppendorf AG, Hamburg, Germany | 22363352 | |
| Formic acid | Sigma Aldrich, St. Louis, MO, USA | F0507-500ML | |
| Indexed retention time (iRT) normalization peptide standard | Biognosys AG, Schlieren, Zurich, Switzerland | Ki-3002-2 | |
| Iodoacetamide (IAA) | Sigma Aldrich, St. Louis, MO, USA | I1149-25G | |
| mapDIA | web link | software for interference removal of DIA datasets | |
| Methanol, Burdick and Jackson LC-MS grade | Burdick and Jackson, Muskegon, MI, USA | BJLC230-4 | |
| PURELAB flex 1 ultrapure water dispenser | VWR International, Radnor, PA, USA | 89204-088 | |
| mProphet in Skyline | incorporated in Skyline | integrated statistical algorithms for FDR assessments | |
| Oasis HLB SPE cartridges | Waters Corp., Milford, MA, USA | WAT094225 | cartridges for desalting protein lysates, up to 50 mg material |
| Phosphate buffered saline solution | Life Technologies | 10010023 | |
| Pierce BCA Assay | Thermo Fisher Scientific, Waltham, MA, USA | 23225 | |
| ProteinPilot 5.0 - 'MS database search engine' | SCIEX, Framingham, MA, USA | software download SCIEX | MS database search engine |
| PTMScan Succinyl-Lysine Motif [Succ-K] Kit #13764 | Cell Signaling Technology | 13764 | antibody beads for affinity enrichment |
| PTMScan Acetyl-Lysine Motif [Ac-K] Kit #13416 | Cell Signaling Technology | 13416 | antibody beads for affinity enrichment |
| Sequencing-grade lyophilized trypsin | Life Technologies | 23225 | |
| Skyline - 'Quantitative Proteomics Software' | MacCoss lab (academic) | open source software | Quantitative Proteomics Software (academic) |
| Spectronaut - 'DIA Quantitative Analysis Software' | Biognosys AG, Schlieren, Zurich, Switzerland | Sw-3001 | DIA Quantitative Analysis Software / PTM site localization |
| Thermo Scientific Savant SPD131DDA Speedvac Concentrator | Thermo Fisher Scientific, Waltham, MA, USA | SPD131DDA-115 | instrument to concentrate liquid volume of samples |
| TissueLyser II | Qiagen, Hilden, Germany | 85300 | instrument for efficient lysis of tissue |
| Trifluoroacetic acid (TFA) | Sigma Aldrich, St. Louis, MO, USA | T6508-1L | |
| TripleTOF 6600: orthoganol quadrupole time-of-flight (QqTOF)mass spectrometer | SCIEX, Framingham, MA, USA | Per quote | high resolution mass spectrometer |
| Ultra Plus nano-LC 2D HPLC system | SCIEX, Eksigent Division, Framingham, MA, USA | Model #845 | chromatographic separation system |
| Urea | Thermo Fisher Scientific, Waltham, MA, USA | PI29700 | |
| Water, Burdick and Jackson LC-MS | Burdick and Jackson, Muskegon, MI, USA | 600-30-76 | |
| ZipTip C18 Pipette Tips, P10 | Merck Millipore Ltd, Tullagreen, Carrigtwohill, Co. Cork, IRL | ZTC18S096 | C-18 resin loaded tips for desalting of peptide mixtures |
Références
- Christensen, D. G., et al. Post-translational Protein Acetylation: An Elegant Mechanism for Bacteria to Dynamically Regulate Metabolic Functions. Frontiers in Microbiology. 10, 1604 (2019).
- Deribe, Y. L., Pawson, T., Dikic, I. Post-translational modifications in signal integration. Nature Structural Molecular Biology. 17 (6), 666-672 (2010).
- Sadoul, K., Boyault, C., Pabion, M., Khochbin, S. Regulation of protein turnover by acetyltransferases and deacetylases. Biochimie. 90 (2), 306-312 (2008).
- Swaney, D. L., et al. Global analysis of phosphorylation and ubiquitylation cross-talk in protein degradation. Nature Methods. 10 (7), 676-682 (2013).
- Cohen, P. The regulation of protein function by multisite phosphorylation--a 25 year update. Trends in Biochemical Sciences. 25 (12), 596-601 (2000).
- Grunstein, M. Histone acetylation in chromatin structure and transcription. Nature. 389 (6649), 349-352 (1997).
- Struhl, K. Histone acetylation and transcriptional regulatory mechanisms. Genes and Development. 12 (5), 599-606 (1998).
- Mocciaro, A., Rape, M. Emerging regulatory mechanisms in ubiquitin-dependent cell cycle control. Journal of Cell Sciences. 125 (Pt 2), 255-263 (2012).
- Lopez-Otin, C., Hunter, T. The regulatory crosstalk between kinases and proteases in cancer. Nature Reviews in Cancer. 10 (4), 278-292 (2010).
- Du, Z., et al. DNMT1 stability is regulated by proteins coordinating deubiquitination and acetylation-driven ubiquitination. Science Signalling. 3 (146), (2010).
- McManus, F. P., Lamoliatte, F., Thibault, P. Identification of cross talk between SUMOylation and ubiquitylation using a sequential peptide immunopurification approach. Nature Protocols. 12 (11), 2342-2358 (2017).
- Venne, A. S., Kollipara, L., Zahedi, R. P. The next level of complexity: crosstalk of posttranslational modifications. Proteomics. 14 (4-5), 513-524 (2014).
- Mertins, P., et al. Integrated proteomic analysis of post-translational modifications by serial enrichment. Nature Methods. 10 (7), 634-637 (2013).
- Basisty, N., Meyer, J. G., Wei, L., Gibson, B. W., Schilling, B. Simultaneous Quantification of the Acetylome and Succinylome by 'One-Pot' Affinity Enrichment. Proteomics. 18 (17), e1800123 (2018).
- Wang, G., et al. Regulation of UCP1 and Mitochondrial Metabolism in Brown Adipose Tissue by Reversible Succinylation. Molecular Cell. 74 (4), 844-857 (2019).
- Verdin, E., Ott, M. 50 years of protein acetylation: from gene regulation to epigenetics, metabolism and beyond. Nature Reviews Molecular Cell Biology. 16 (4), 258-264 (2015).
- Rardin, M. J., et al. Label-free quantitative proteomics of the lysine acetylome in mitochondria identifies substrates of SIRT3 in metabolic pathways. Proceedings of the National Academy of Science U. S. A. 110 (16), 6601-6606 (2013).
- Rardin, M. J., et al. SIRT5 regulates the mitochondrial lysine succinylome and metabolic networks. Cell Metabolism. 18 (6), 920-933 (2013).
- Carrico, C., Meyer, J. G., He, W., Gibson, B. W., Verdin, E. The Mitochondrial Acylome Emerges: Proteomics, Regulation by Sirtuins, and Metabolic and Disease Implications. Cell Metabolism. 27 (3), 497-512 (2018).
- Sadhukhan, S., et al. Metabolomics-assisted proteomics identifies succinylation and SIRT5 as important regulators of cardiac function. Proceedings of the National Academy of Science U. S. A. 113 (16), 4320-4325 (2016).
- Collins, B. C., et al. Multi-laboratory assessment of reproducibility, qualitative and quantitative performance of SWATH-mass spectrometry. Nature Communications. 8 (1), 291 (2017).
- Schilling, B., Gibson, B. W., Hunter, C. L. Generation of High-Quality SWATH((R)) Acquisition Data for Label-free Quantitative Proteomics Studies Using TripleTOF((R)) Mass Spectrometers. Methods in Molecular Biology. 1550, 223-233 (2017).
- Meyer, J. G., Schilling, B. Clinical applications of quantitative proteomics using targeted and untargeted data-independent acquisition techniques. Expert Reviews in Proteomics. 14 (5), 419-429 (2017).
- MacLean, B., et al. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 26 (7), 966-968 (2010).
- Sticker, A., Martens, L., Clement, L. Mass spectrometrists should search for all peptides, but assess only the ones they care about. Nature Methods. 14 (7), 643-644 (2017).
- Olsen, J. V., Ong, S. E., Mann, M. Trypsin cleaves exclusively C-terminal to arginine and lysine residues. Molecular Cell Proteomics. 3 (6), 608-614 (2004).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.