Method Article
Analyse de l’hypermutation somatique dans l’intron JH4 des cellules B du Centre Germinal à partir des patchs de Mouse Peyer
Dans cet article
Résumé
Présenté ici est un essai pour quantifier l’hypermutation somatique dans le locus de gène de chaîne lourde d’immunoglobuline utilisant les cellules b germinales de centre des corrections de Peyer de souris.
Résumé
Dans les centres germinaux des organes lymphoïdes, les cellules B matures modifient leur immunoglobuline exprimée (Ig) en introduisant des mutations non tempérées dans les exons codant variables du gène Ig heavy and light chain loci. Ce processus d’hypermutation somatique (SHM) nécessite l’enzyme induite par l’activation cytidine deaminase (AID), qui convertit les désoxycytidines (C), en désoxyuridines (U). Le traitement des inadéquations U:G générées par l’AID en mutations par les voies d’excision de base et de réparation des inadéquations introduit de nouvelles séquences de codage Ig qui peuvent produire une affinité plus élevée Ig. Les mutations dans les gènes aid ou de réparation de l’ADN peuvent bloquer ou modifier de manière significative les types de mutations observées dans l’Ig loci. Nous décrivons un protocole pour quantifier les mutations introniques de JH4 qui emploie le tri cellulaire activé par fluorescence (FACS), PCR, et séquençage de Sanger. Bien que cet essai ne mesure pas directement la maturation de l’affinité Ig, il est indicatif de mutations dans les séquences de codage variable Ig. En outre, ces méthodes utilisent des techniques courantes de biologie moléculaire qui analysent les mutations dans les séquences Ig de plusieurs clones de cellules B. Ainsi, cet essai est un outil précieux dans l’étude de la diversification SHM et Ig.
Introduction
Les cellules B, membres du système immunitaire adaptatif, reconnaissent et éliminent les antigènes en produisant des anticorps, également connus sous le nom d’immunoglobulines (Ig). Chaque Ig est composé de deux polypeptides à chaîne lourde (IgH) et deux polypeptides à chaîne légère (IgL), qui sont maintenus ensemble par des liaisons de disulfide pour former la structure caractéristique de forme « Y » de l’Ig1. Les N-termini d’IgH et d’IgL comprennent la région variable (V) de chaque polypeptide et forment ensemble le site de liaison antigène de l’Ig, tandis que la région constante d’IgH confère la fonction effectrice de l’Ig. Le développement de cellules B dans la moelle osseuse réarrange les exons codants V d’IgH et d’IgL dans un processus connu sous le nom de recombinaison V(D)J2,3,4. La transcription des exons V recombinés, couplée aux exons constants respectifs de la région, forme l’ARNm qui est traduit dans l’Ig.
Les cellules B matures exprimant une membrane liée Ig, également connue sous le nom de récepteur de cellules B (BCR), circulent aux organes lymphoïdes secondaires, tels que la rate, le ganglion lymphatique, ou les corrections de Peyer, où elles enquêtent sur l’environnement pour trouver des antigènes et interagissent avec d’autres cellules du systèmeimmunitaire 1. Dans les centres germinaux (GC) des organes lymphoïdes secondaires, les cellules B qui reconnaissent l’antigène par le BCR deviennent activées. Aidées par des cellules folliculaires dendritiques et des lymphocytes T folliculaires, les cellules B activées peuvent alors proliférer et se différencier en cellules plasmatiques et mémoire, qui sont des effecteurs importants d’une réponse immunitairerobuste 5,6,7,8,9. En outre, ces cellules B activées peuvent subir des processus secondaires de diversification des gènes Ig - recombinaison des commutateurs de classe (RSE) et hypermutation somatique (SHM). Pendant la RSE, les cellules B échangent la μ région constante par défaut du polypeptide IgH avec une autre région constante (γ, α, ε) par une réaction de suppression de l’ADN -recombinaison (figure 1). Cela permet l’expression d’un exon constant différent et la traduction d’un nouvel Ig. La cellule B passera de l’expression d’IgM à un autre isotype (IgG, IgA, IgE). La RSE modifie la fonction effectrice de l’Ig sans altérer sa spécificité antigène10,11,12. Toutefois, au cours de SHM, les cellules B mutent les régions codantes V d’IgH et d’IgL pour permettre la production et la sélection d’Igs d’affinité plus élevée, ce qui peut éliminer plus efficacement unantigène 13,14,15 ( Figure1). Fait important, la RSE et le SHM dépendent tous deux de la fonction d’une enzyme : cytidine deaminase induite par activation (AID)16,17,18. Les humains et les souris déficients en AID ne peuvent pas compléter la RSE ou le SHM et se présentent avec des titres de sérum IgM élevés ou Hyper-IgM17,19.
Dans la RSE, AID déaminate les désoxycytidines (C) dans les régions de commutation répétitives qui précèdent chaque exons codant constant, les convertissant en désoxyuridines (U)20,21, ce qui crée un appariement de base inégalé entre les désoxyuridines et les désoxyguanosines (U:G). Ces inadéquations U:G sont converties en ruptures d’ADN à double brin, qui sont nécessaires pour la recombinaison de l’ADN, soit par la réparation de l’excision de base (BER) ou la réparation de décalage (MMR)voie 22,23,24,25,26,27,28,29. Dans SHM, AID déaminateS C dans les exons codants V. La réplication à travers l’inadéquation U:G génère des mutations de transition C:G à T:A, tandis que l’élimination de la base uracil par la protéine BER, uracil DNA glycosylase (UNG), avant la réplication de l’ADN produit à la fois des mutations de transition et de transversion16. Les mutations nulles de l’UNG augmentent significativement les mutations de transition C:G à T:A21,22. Tout comme la RSE, SHM requiert les rôles complémentaires de MMR et BER. Pendant SHM, MMR génère des mutations aux paires de base A:T. L’activation de mutations dans l’homologie MutS 2 (MSH2) ou la polymésase de l’ADN η (Polν) réduit considérablement les mutations aux bases A:T et les mutations composées dans MSH2 et Polţ abolit pratiquement les mutations aux bases A:T21,30,31. Conformément au rôle crucial de BER et de MMR dans la conversion des U générés par l’AID en mutations de transition ou de transversion, les souris déficientes pour MSH2 et UNG (MSH2-/-UNG-/-)n’affichent que des mutations de transition C:G à T:A résultant de la réplication à travers l’inadéquation U:G21.
L’analyse du SHM dans les régions codantes en V reste compliquée car les cellules B en développement peuvent recombiner n’importe quel exons codant V(D)J dans les exons codants IgH et IgL loci1,2,4. L’analyse précise de ces régions V recombinées et somatiquement mutées exige l’identification et l’isolement des clones des cellules B ou de l’ARNmIg 11,13. L’intron JH4, qui est 3' du dernier exon codant J dans le locus IgH, abrite des mutations somatiques dues à la propagation des mutations 3' du promoteur V32,33,34 et est donc fréquemment utilisé comme marqueur de substitution pour SHM dans les régions V31,35 ( Figure1). Pour élucider expérimentalement comment des gènes spécifiques ou des mutations génétiques modifient les modèles ou les taux de SHM, l’intron JH4 peut être séquencé à partir des cellules B germinales (GCBCs) du centre germinal (PP) de Peyer, qui subissent des taux élevés de SHM36,37,38. Les CCG peuvent être facilement identifiés et isolés avec des anticorps conjugués fluorescents contre les marqueurs de surface cellulaire (B220+PNAHI)17,39.
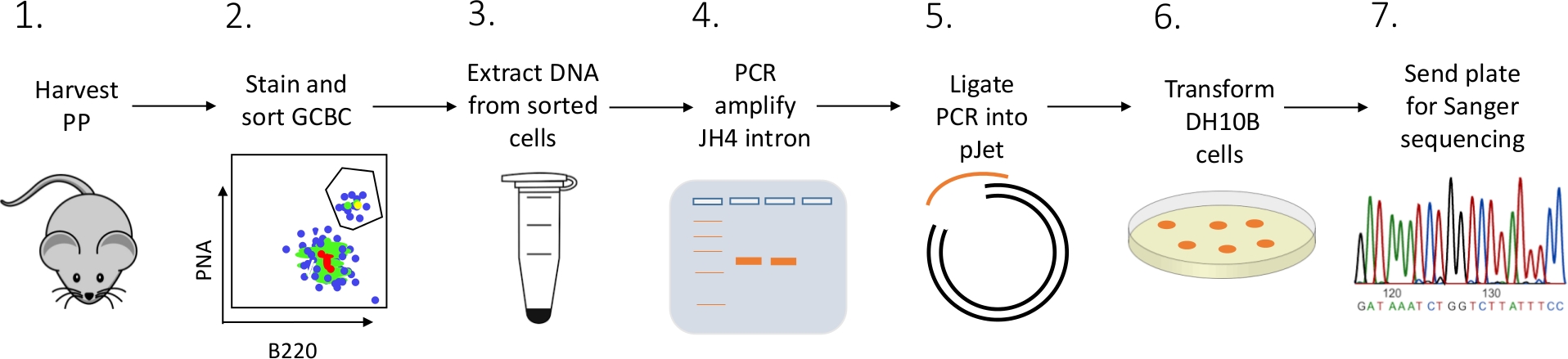
Un protocole détaillé est présenté pour caractériser les mutations introniques JH4 chez les GCBC PP à partir de souris utilisant une combinaison de FACS (fluorescence activée tri cellulaire), PCR, et sanger séquençage (Figure 2).
Protocole
Toutes les souris mutantes ont été maintenues sur un fond C57BL/6. Des souris mâles et femelles appariées dans l’âge (2-5 mois) ont été employées pour toutes les expériences. L’élevage et les expériences avec des souris ont été menés selon les protocoles approuvés par le City College of New York Institutional Animal Care and Use Committee.
1. Dissection des patchs de Peyer
- Euthanasier la souris avec 100% de CO2 à 3 L/min pendant 5 min suivie d’une dislocation cervicale pour confirmer la mort. Stériliser les outils de dissection (ciseaux, forceps, forceps fins) et les mains gantées avec 70% d’éthanol.
- Menter la souris sur le tampon de dissection avec l’abdomen exposé. Vaporiser généreusement le corps de la souris avec 70% d’éthanol avant de faire des incisions pour stériliser la zone de dissection.
- Faire une incision dans la peau à travers l’abdomen et enlever la peau de l’abdomen en tirant simultanément des deux côtés de l’incision vers la tête et la queue à l’aide de forceps (ou stérilisés, mains gantées).
- Épinglez les membres avant et arrière de la souris.
- Couper la cavité péritonéale avec des ciseaux pour exposer les organes internes.
- Localiser l’intestin grêle entre l’estomac et le caecum (structure en forme de J près du côlon). Retirer l’intestin grêle en coupant sous l’estomac et au-dessus du caecum.
- Enlever tout tissu conjonctif et la graisse reliant les plis de l’intestin grêle ensemble.
REMARQUE : La graisse aura une couleur blanche distinctive, contrairement à la couleur rose de l’intestin grêle. - Examinez la surface externe de l’intestin grêle pour les taches du Peyer (PPs), qui sont petites (~1 mm), structures ovales qui apparaissent blanches sous une fine couche de cellules épithéliales translucides.
- Excisez soigneusement tous les PP visibles avec des ciseaux.
REMARQUE : Une souris de type sauvage C57BL/6 (WT) peut produire de 4 à 8 PPP, tandis qu’une souris AIDaura de 6 à 10 PP. - Recueillir les PPP dans un tube de microcentrifugeuse de 1,5 mL contenant 1 mL de tampon FACS sur la glace.
REMARQUE : Le PP doit couler, tandis que la graisse flottera à la surface et pourra être enlevée.
2. Isolement cellulaire pour FACS
- Placer un filtre de 40 μm dans un plat de 6 puits avec 1 mL de tampon FACS froid (4 °C).
- Verser les PPs du tube de 1,5 mL sur le filtre.
- Lavez les PPP avec 1 mL de tampon FACS froid, en vous assurant qu’ils sont toujours dans le liquide et sur la glace.
- Utilisez l’extrémité plate du piston à partir d’une seringue de 1 mL comme pilon pour écraser les PPs sur le filtre jusqu’à ce que seul le tissu conjonctif reste sur le filtre.
- Lavez le filtre et le piston avec 1 mL de tampon FACS froid pour libérer les cellules dans le plat de 6 puits.
- Recueillir les ~4 mL de cellules dans le tampon FACS froid et les filtrer à travers un tube FACS cap passoire de 40 μm.
- Laver le bouchon de la passoire avec 1 mL de tampon FACS froid.
- Pelleter les cellules à 600 x g à 4 °C pendant 5 min dans une centrifugeuse balançante.
- Décanter le surnatant.
- Resuspendez les cellules dans 0,4 mL de tampon FACS froid.
- Retirez 10 μL pour le comptage cellulaire pour vérifier le rendement (attendez-vous à ~5 x10 6 cellules/souris, voir figure 3A)
- Filtrer les cellules restantes à travers un bouchon de passoire de 40 μm dans un tube FACS et passer à la coloration pour FACS.
3. Coloration des CCG pour FACS
- Ajouter 1 bloc μL Fc (cd16/CD32 anti-souris non étiqueté) à la suspension cellulaire de 400 μL et placer les cellules sur la glace pendant 15 min.
- Ajouter 2 mL de tampon FACS froid pour laver les cellules.
- Pelleter les cellules à 600 x g à 4 °C pendant 5 min et jeter le supernatant.
- Resuspendez les cellules dans 80 μL de tampon FACS froid.
- Retirez 10 μL de cellules du WT PP pour chaque contrôle de coloration (4 au total, y compris 3 commandes de taches simples et 1 contrôle non taché). Laissez 40 μL du WT PP pour l’étape suivante. Alternativement, utilisez des perles de compensation pour les commandes de coloration.
- Tacher chacun des échantillons expérimentaux (p. ex., WT et AID-/-) dans 500 μL de tampon facs froid avec 2,5 μL d’agglutinine d’arachide (PNA)-biotine pendant 15 min sur la glace.
- Ajouter 2 mL de tampon FACS froid pour laver les cellules.
- Pelleter les cellules à 600 x g à 4 °C pendant 5 min et jeter le supernatant.
- Tacher chaque échantillon expérimental avec 500 μL du cocktail dans l’obscurité, sur glace, pendant 15 min (tableau 1). Assurez-vous que les cellules sont entièrement résuspendées dans le cocktail de coloration.
- Préparez des commandes de taches simples pour la matrice de compensation.
- Tacher les cellules dans 500 μL de tampon FACS froid à l’aide des dilutions spécifiées dans le tableau 2.
- Incuber les commandes de coloration dans l’obscurité, sur la glace, pendant 15 min.
- Ajouter 2 mL de tampon FACS froid à tous les tubes dans les étapes 3.7 et 3.8, pelleter les cellules, et jeter le supernatant pour laver les anticorps non liés ou DAPI.
- Resuspendez les cellules dans 500 μL de tampon FACS froid et placez-les sur la glace.
- À l’aide d’un trieur cellulaire, recueillir B220+PNAHI à partir de chaque échantillon expérimental taché. La figure 3B montre les pourcentages typiques de B220+ PNAHI obtenus à partir de WT et AID-/- PPs. La figure 3C affiche la stratégie de gating facs.
4. Extraction de l’ADN des CCG
- Les granulés ont trié les cellules à 600 x g à 4 °C pendant 5 minutes et jettent le supernatant.
- Resuspendez les cellules en 1 mL de tampon FACS froid et transférez les cellules dans un tube de microcentrifugeuse de 1,5 mL.
- Pelleter les cellules à 600 x g à 4 °C pendant 5 min et jeter le supernatant.
- Resuspendez les cellules dans 500 μL de tampon d’extraction d’ADN et 5 μL de 20 mg/mL Proteinase K.
- Incuber à 56 °C pendant la nuit.
- Précipiter l’ADN avec 500 μL d’isopropanol et 1 μL de 20 mg/mL de glycogène. Bien mélanger le tube en inversant 5-6x.
- Incuber à température ambiante pendant 10 min.
- Centrifugeuse dans une microcentrifugeuse pendant 15 min à 25 °C à 21 000 x g.
- Jetez le supernatant et conservez la pastille, qui contient l’ADN précipité et le glycogène.
- Laver la pastille d’ADN avec 1 mL d’éthanol à 70 %.
- Pelleter l’ADN dans une microcentrifugeuse pendant 10 min à 25 °C à 21 000 x g.
- Retirer l’éthanol à 70 % et sécher à l’air le granulé d’ADN pendant 5 à 10 min.
REMARQUE : Évitez le sursèchement car l’ADN peut ne pas se réhydrater complètement.
- Resuspendez l’ADN dans un tampon te de 30 μL et incubez pendant la nuit à 56 °C.
5. Amplification et analyse de la séquence intron JH4
- Quantifier l’ADN en mesurant l’absorption à une longueur d’onde de 260 nm (A260).
REMARQUE : La concentration typique d’ADN récupérée à partir de GCBCs TRIÉS B220+PNAHI d’une souris C57BL/6 est de 20-40 ng/μL. - Effectuez le PCR imbriqué pour l’intron JH4 (Tableau 3, Tableau 4). Normaliser la quantité totale d’ADN génomique utilisée dans le premier PCR à l’échantillon le moins concentré. (p. ex., si l’échantillon le moins concentré est de 5 ng/μL, utilisez 58,75 ng d’ADN pour tous les échantillons dans le volume maximal d’eau (11,75 μL) dans les #1 PCR).
- Résoudre le produit PCR sur un gel agarose de 1,5% à 200 V pendant 20 min. La taille amplicon attendue est de 580 bp.
- Exciser l’amplicon du gel et extraire l’ADN à l’aide d’un kit d’extraction de gel selon les instructions du fabricant (voir figure supplémentaire 1).
- Elute l’ADN avec 30 μL d’eau et quantifier la quantité d’ADN en mesurant l’A260.
REMARQUE : La concentration typique du produit PCR purifié est de 3 à 10 ng/μL.
- Elute l’ADN avec 30 μL d’eau et quantifier la quantité d’ADN en mesurant l’A260.
- Ligate le produit PCR purifié dans un plasmide avec des extrémités émoussées. Normaliser la quantité totale de produits PCR utilisés dans chaque réaction de ligature (tableau 5).
- Incuber la réaction de ligature à température ambiante pendant 5 minutes ou toute la nuit à 16 °C.
- Transformer les cellules bactériennes électrocompétentes avec 2 μL de la réaction de ligature.
- Électroporate à 1.65 kV.
- Sauvetage en 600 μL de soc média pendant 1 h à 37 °C dans un incubateur secouant à 225 rpm.
- Plaque 100 μL de bactéries transformées sur LB complétée par des plaques d’agar ampicilline (100 μg/mL) et incuber toute la nuit à 37 °C.
- Soumettez la plaque des colonies bactériennes pour le séquençage Sanger à l’aide de l’amorce avant T7. Alternativement, cultiver des cultures du jour au lendemain de chaque colonie bactérienne et effectuer une purification plasmide.
- Si nécessaire, répétez la PCR, la ligature et/ou la transformation pour optimiser le rendement des colonies bactériennes
REMARQUE : Un minimum de 30 colonies doivent être cueillies dans chaque assiette.
- Si nécessaire, répétez la PCR, la ligature et/ou la transformation pour optimiser le rendement des colonies bactériennes
- Normaliser les données de séquence dans les .txt fichiers
- Supprimer la séquence plasmide.
- Assurez-vous que chaque séquence est orientée de 5' à 3' selon la séquence de référence JH4 intron (NG_005838). Générer le complément inverse de n’importe quelle séquence, si nécessaire.
- Aligner les séquences obtenues pour chaque PCR sur la séquence de référence intron JH4 (NG_005838) à l’aide d’un logiciel Clustal Omega (Figure 4A).
- Identifier les différences par rapport à la séquence de référence sous forme de mutations
- Vérifiez que toutes les mutations sont de véritables mutations ponctuelles en examinant l’électrophérogramme du séquençage Sanger. Répétez le séquençage si nécessaire. (Figure 4B,C).
- Compiler et quantifier les mutations uniques dans l’intron JH4 pour chaque génotype (Figure 5).
- Comptez les séquences avec des mutations identiques une seule fois
REMARQUE : Il n’est pas possible de déterminer si les séquences identiques ont été générées pendant le PCR ou des événements identiques de SHM dans différentes cellules B. - Comptez chaque cas de séquences d’intron de germline WT JH4 (c.-à-d. celles qui n’ont pas de mutations) comme une séquence unique.
- Comptez les séquences avec des mutations identiques une seule fois
Résultats
Cytométrie d’écoulement
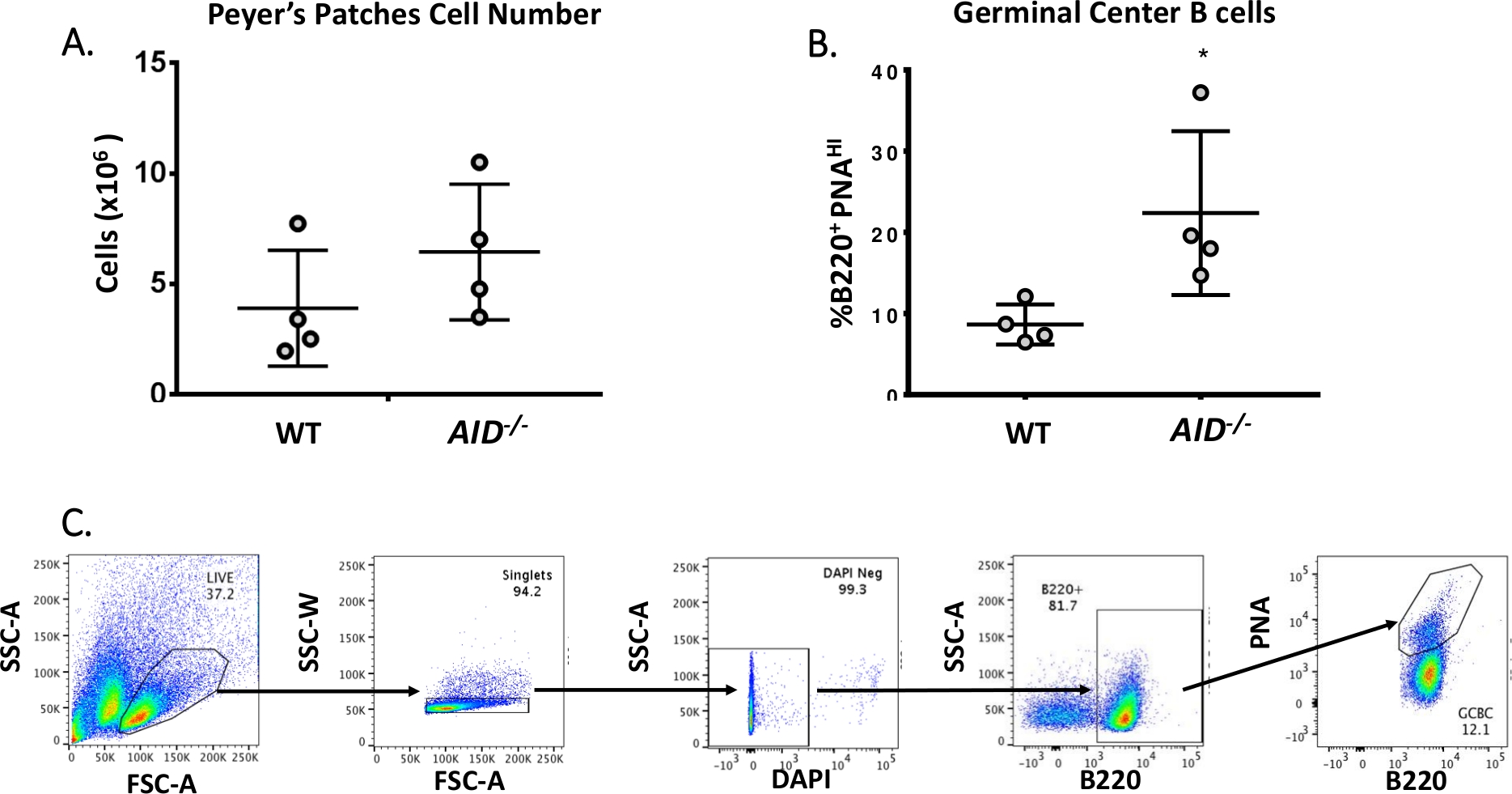
Les cellules B matures circulent vers des centres germinaux où elles subissent une maturation par affinité, une expansion clonale et une différenciation en cellules plasmatiquesou de mémoire 40,41,42,43,44. Ces GCBC peuvent être identifiés par de nombreux marqueurs de surface cellulaire, y compris l’expression élevée du récepteur CD45R/B220 et la liaison de l’agglutinine d’arachide (PNA)45,46. Pour isoler les CCG activés, les cellules PP ont été tachées d’anticorps anti-B220 conjugués à la phycoerythrine (PE) et à la biotinylated-PNA, suivis de streptavidine conjuguée à APC-eFluor780. Les cellules mortes ont été éliminées à l’aide du colorant fluorescent 4',6-Diamidino-2-Phenylindole (DAPI), qui tache l’acide nucléique des cellules mourantesou mortes 47,48. Les cellules tachées ont ensuite été analysées et triées par cytométrie d’écoulement. Les PPP se composaient de ~80% B220+ cellules49,50. Les PP WT contiennent en moyenne 4 x 106 cellules par souris (figure 3A). Environ 8% des cellules WT PP étaient B220+PNAHI, soit la moitié du nombre observé dans AID-/ - ( Figure3B). Ainsi, 0.3-0.6 x 106 B220+PNAHI GCBCs ont été obtenus après tri, qui étaient suffisants pour analyser des mutations dans l’intron de JH4.
Analyse des séquences JH4
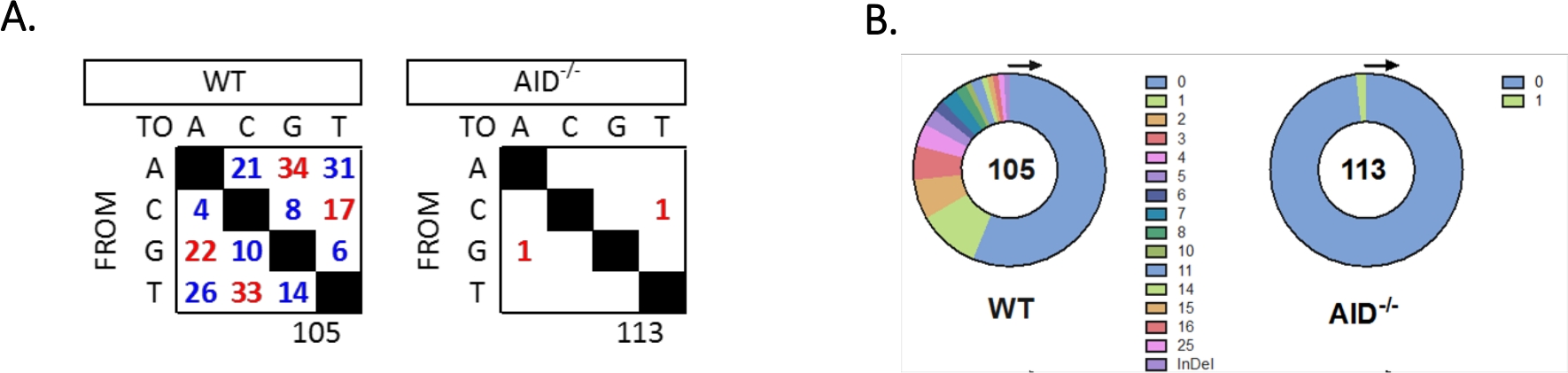
L’intron JH4 a été amplifié par un PCR imbriqué à l’aide d’amorces familiales VHJ558 communes (J558FR3Fw et VHJ558.2) suivies de JH4 intron couvrant les amorces VHJ558.3 et VHJ558.435,37. Sur les 105 séquences uniques obtenues à partir de WT GCBCs, un total de 226 mutations ont été trouvées (Figure 5A). L’analyse du spectre de mutation de GCBC chez les souris de WT a montré une gamme de transitions et de transversions à un taux de 4 x 10-3 mutations/bp, qui a été calculée en divisant le nombre total de bases mutées par le nombre total de bases qui ont été séquencées32,36,37,38. En outre, chaque produit JH4 PCR de WT GCBCs contenait 1-25 mutations (Figure 5B), où de multiples mutations ont été fréquemment trouvées sur une séquence33,36. Seulement deux mutations ont été identifiées dans 113 séquences AID-/- (figure 5A). AID-/- Les cellules B présentaient 1,66 x 10-5 mutations/bp, ce qui était significativement inférieur aux cellules B WT (p <0,05)36 et se compare au taux d’erreur de la polymése haute fidélité (5,3 x 10-7 sous/base/doublement)51,52. Ainsi, aid-/- Les cellules B ont servi de contrôle négatif utile pour cet essai.

Figure 1 : Schéma du locus génétique IgH et des régions ciblées par l’AID pendant la RSE et le SHM. La barre rouge indique l’intron JH4 de 580 bp qui est de 3' de réarrangements VDJH4 et est analysé dans ce protocole. Dans la RSE, la déamination dépendante de l’AID des régions introniques de commutateur (Sμ et Sε) favorise la formation d’ORD qui permet la recombinaison de la suppression et l’expression d’un nouvel isotype d’anticorps (IgM à IgE). Au cours de SHM, les régions V (boîtes grises) accumulent des mutations (lignes bleues) qui peuvent conduire à une affinité plus élevée Ig. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 2 : Flux de travail pour analyser le SHM de l’intron JH4 dans les CCG isolés des PPP. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 3 : Caractérisation des CCG PP. (A) Nombre total de cellules PP de WT et AID-/- souris (n = 4 par génotype). Les barres d’erreur représentent un écart type par rapport à la moyenne. (B) Pourcentage de B220+PNAHI GCBCs obtenus à partir de PPP de WT et AID-/- souris (n = 4 par génotype)36. Les barres d’erreur représentent un écart type par rapport à la moyenne, *p<0.05 en utilisant le t-test de l’élève. (C) Parcelles représentatives FACS pour trier B220+PNAHI GCBCs à partir de PPP. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 4 : Analyse des données de la séquence JH4 Sanger. (A) Alignements de séquences d’échantillons des données de séquence Sanger du produit PCR JH4 de WT (en haut) et d’AID-/- (en bas) GCBCs à la séquence génomique de référence (NG_005838), qui est la séquence immédiatement en dessous des marques de tiques numérotées. Des alignements ont été générés à l’aide de Clustal Omega. (B) Électrophéogramme de données de séquence Sanger de haute qualité, qui affichaient des pics distincts pour chaque base. (C) Électrophéogramme de données de séquence de mauvaise qualité, qui montraient des pics ambigus et des bases non spécifiées (N). Le nucléotide indiqué en rouge doit être annoté manuellement dans le fichier texte séquence. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 5 : Analyse des mutations dans l’intron JH4 dans les WT et aid-/- GCBCs. (A) Le nombre total de mutations de transition (rouge) et de transversion (bleu) aux bases A, C, G et T pour chaque génotype est résumé dans les tableaux. Le nombre total de séquences analysées est indiqué sous le tableau. (B) Le nombre de mutations par amplicon PCR pour chaque génotype est représenté dans les diagrammes à secteurs. Ce chiffre a été modifié à partir de Choi et coll.36 Copyright 2020. L’American Association of Immunologists, Inc. Veuillez cliquer ici pour voir une version plus large de ce chiffre.
{kind=link}
| Cocktail de coloration pour les CCG | Volume: 500 μL | ||
| Anticorps ou colorant | Fluorophore | dilution | μL (μL) |
| B220 (en) | Pe | 1000 | 0.5 |
| Streptavidine ( Streptavidin ) | APC-eFluor780 APC-eFluor780 | 500 | 1 |
| DAPI (DAPI) | n/a | 500 | 1 |
Tableau 1 : Coloration des cocktails pour les CCG. Cocktail des anticorps ou colorants indiqués (indiqués en italique) aux dilutions spécifiées ont été employés pour tacher des cellules de PP dans 500 μL pour la cytométrie d’écoulement.
| Taches simples pour compensation | Volume: 500 μL | ||
| Anticorps ou colorant | Fluorophore | dilution | μL (μL) |
| B220 (en) | Pe | 1000 | 0.5 |
| B220 (en) | APC-eFluor780 APC-eFluor780 | 750 | 0.67 |
| DAPI (DAPI) | n/a | 500 | 1 |
Tableau 2 : Contrôle unique des taches pour l’indemnisation. Des anticorps B220 conjugués aux fluorophores indiqués ont été utilisés pour des contrôles de taches simples afin de compenser le chevauchement spectral.
| PCR #1 | ||||
| Réactif | volume | Thermocycler Conditions | ||
| Tampon 5x | 4 μL | 1 | 95 °C | 3 min |
| 10 mM dNTP | 2 μL | 2 | 94 °C | 30 sec |
| 10 μM J558FR3Fw | 1 μL | 3 | 55 °C | 30 sec |
| 10 μM VHJ558.2 | 1 μL | 4 | 72 °C | 1 h 30 min |
| Polymése d’ADN haute fidélité | 0,25 μL | Cycle 2-4 9x | ||
| ADN | x (normaliser à l’échantillon le moins concentré) | |||
| H2O | à 20 μL | 5 | 72 °C | 5 min |
| Diluer le produit PCR 1:5 en H2O avant de passer à pcr #2 | ||||
Tableau 3 : PCR imbriqué de l’intron JH4. Composants PCR et conditions thermocycler pour la première réaction d’amplification. Diluer le premier produit PCR 1:5 avec de l’eau et utiliser 1 μL de cette dilution pour le deuxième PCR.
| PCR #2 | ||||
| Réactif | volume | Conditions thermocycler #2 | ||
| Tampon 5x | 4 μL | 1 | 94 °C | 3 min |
| 10 mM dNTP | 2 μL | 2 | 94 °C | 30 sec |
| 10 μM VHJ558.3 | 1 μL | 3 | 55 °C | 30 sec |
| 10 μM VHJ558.4 | 1 μL | 4 | 72 °C | 30 sec |
| Polymése d’ADN haute fidélité | 0,25 μL | Cycle 2-4 21x | ||
| PCR dilué#1 | 1 μL | |||
| H2O | à 20 μL | 5 | 72 °C | 5 min |
Tableau 4 : Composants PCR et conditions thermocycler pour le deuxième PCR.
| Réactif | volume |
| Tampon 2x | 10 μL |
| PCR purifié | x (normaliser à l’échantillon le moins concentré) |
| Plasmide aux extrémités émoussées | 1 μL |
| T4 ADN Ligase | 1 μL |
| H2O | à 20 μL |
| Incuber à température ambiante pendant 5 min ou toute la nuit à 16ºC | |
Tableau 5 : Réaction de ligature. Composants pour la ligature du produit purifié JH4 intron PCR dans le plasmide.
| Tampon FACS |
| Chauffer l’inactivation du FBS à 56 °C pendant une heure avant l’utilisation. Supplément PBS, pH 7.4 (Gibco, #10010049) avec 2,5% (v/v) de FBS inactivé par la chaleur. Conserver à 4°C. |
| Tampon d’extraction d’ADN (100 mM Tris pH 8,0, 0,1 M EDTA, 0,5% (w/v) SDS) |
| Ajouter 50 mL de 1 M Tris pH 8.0, 100mL de 0.5 M EDTA, et 12.5 mL de 20% SDS. Ajouter l’eau distillée à 500 mL. Conserver à température ambiante. |
| Tampon TE (10 mM Tris pH 8.0, 1 mM EDTA) |
| Ajouter 2,5 mL de 1 M Tris pH 8,0 et 500 mL de 0,5 M EDTA. Ajouter l’eau distillée à 250 mL. Conserver à température ambiante. |
Tableau 6 : Recettes tampons.
| Liste oligonucléotides | ||
| J558FR3Fw J558FR3Fw | 5'-GCCTGACATCTGAGGACTCTGC-3' | |
| VHJ558.2 | 5'-CTGGACTTTCGGTTTGGTG-3' | |
| VHJ558.3 | 5'-GGTCAAGGAACCTCAGTCA-3' | |
| VHJ558.4 | 5'-TCTCTAGACAGCAACTAC-3' | |
Tableau 7 : Oligonucléotides utilisés dans l’analyse.
Figure supplémentaire 1 : Image représentative du gel agarose après l’achèvement de l’étape 5.4. Le produit PCR imbriqué JH4 intron a été résolu sur un gel d’agarose de 1,5 % et l’amplicon de 580 pb a été excisé. WT PP indique que l’ADN génomique WT PP GCBC a été utilisé comme modèle pour le premier PCR et AID PP indique que l’AID-/- PP GCBC génomique ADN a été utilisé comme un modèle pour le premier PCR. ɸ indique le contrôle pcr sans modèle et - indique que rien n’a été chargé dans le puits du gel agarose. La dernière voie montre une échelle d’ADN de 100 bp. S’il vous plaît cliquez ici pour télécharger ce chiffre.
{kind=link}
Discussion
Caractériser SHM dans les séquences de codage IgH et IgL V d’une population hétérogène de cellules B présente un défi, étant donné que chaque cellule B réorganise de façon unique les segments de codage V pendant la recombinaison V(D)J34. Dans cet article, nous décrivons une méthode pour identifier des mutations dans l’intron JH4 des CCG. L’intron JH4, qui est situé à 3' du dernier segment de codage J dans le locus IgH, est utilisé comme substitut pour SHM des régions V (Figure 1)31,33,34,35. Pour cataloguer ces mutations introniques JH4 et évaluer comment des gènes spécifiques affectent la production ou le modèle des mutations, les GCBC PP sont spécifiquement analysés. Ces cellules accumulent des mutations introniques JH4 à la suite d’une stimulation chronique par microbiote intestinal53. En outre, les GCBCs B220+PNAHI des PPP de souris non immunisées ont un spectre de mutation qui se compare aux CCG spléniques des animaux vaccinés54,55. Cependant, les mutations dans l’intron JH4 ne peuvent pas être corrélées à la maturation d’affinité d’Ig parce que ces mutations sont non-codant.
Pour déterminer si SHM altère l’affinité d’Ig, les souris devraient être immunisées intraperitoneally avec un antigène, tel que NP (4-hydroxy-3-nitrophenylacetyl) conjugué à CGG (globulin gamma de poulet) ou KLH (hémocyanine de limpet de trou de serrure)56. Par la suite, l’ARNm peut être purifié à partir de Plénique B220+PNAHI GCBCs pour examiner SHM dans VH186.2, l’exon codant V qui reconnaît le plus fréquemment NP et est muté après NP-CGG ou NP-KLH vaccination31,57,58,59,60. Mutation du tryptophane-33 à une leucine en VH186.2 a été caractérisée pour augmenter l’affinité Ig jusqu’à 10 fois59,60 et est, par conséquent, un indicateur que SHM et la sélection clonale a généré une affinité élevée Ig. La mesure des titres Ig sériques spécifiques au NP7 et au NP20 par ELISA et le calcul du rapport NP7/NP20 spécifique à Ig au cours de la vaccination documentent également la maturation par affinité ig résultant de la SHM des régions V17,21,36. Ces deux analyses peuvent être utilisées pour corréler le SHM dans les séquences de codage VH186.2 avec des changements dans la maturation d’affinité ig spécifique à NP.
Qu’il s’agisse d’animaux vaccinés ou non vaccinés utilisés pour analyser le SHM de VH186.2 ou l’intron JH4, les CCG doivent être identifiés avec précision. Nous présentons une approche basée sur facs pour isoler B220+PNAHI GCBCs. Alternativement, fas et antigène α2-6-sialyl-LacNAc non sulfated, qui est reconnu par l’anticorps GL761,62,63,64, peut également être utilisé pour isoler les GCBCs, qui sont identifiés comme B220+Fas+GL7+65 ou CD19+Fas+GL7+37. L’expression GL7 reflète étroitement pna dans les GCBCs activés des ganglions lymphatiques64,65,66. En plus d’utiliser des marqueurs anticorps spécifiques aux CCG, les cocktails contaminés devraient maximiser l’excitation d’un fluorophore et la détection d’un biomarqueur tout en minimisant le chevauchement spectral des émissions de fluorescence. Les antigènes exprimés à de faibles niveaux doivent être détectés avec un anticorps qui est conjugué à un fluorophore avec une fluorescence d’émissionrobuste 67. Le protocole de coloration recommandé a été optimisé pour l’analyse sur un trieur cellulaire équipé de quatre lasers (405nm, 488nm, 561nm, 633nm) et 12 filtres; cependant, les configurations des filtres et la disponibilité du laser varient d’un cytomètre à l’autre. Pour modifier le protocole en fonction de la disponibilité du reagent et de l’équipement, le lecteur est référé à des ressources supplémentaires, les téléspectateurs du spectre en ligne et la littératurepubliée 67,68,69,70,71,72,73. Le protocole de coloration multicolore décrit ci-après exige une compensation du chevauchement spectral pour s’assurer que les populations de cellules triées sont des CCG plutôt qu’une détection inexacte des émissions de fluorescence. B220 sert de contrôle utile de la coloration pour le FACS décrit (tableau 1B) parce que les PPP auront des populations négatives et positives distinctes B220 (figure 3C), ce qui permet une compensation appropriée du chevauchement spectral. La stratégie de gating présentée à la figure 3C devrait servir de ligne directrice. Les parcelles de cytométrie d’écoulement peuvent varier en fonction des conditions de coloration et des paramètres du cytomètre. Néanmoins, 4-10% des cellules vivantes devraient être B220+PNAHI 35, 52.
Toutes les mutations dans l’intron JH4 des GCBC PP doivent être validées pour s’assurer que les mutations observées reflètent vraiment le SHM et non un artefact de PCR ou de séquençage. AID-/- Les cellules B peuvent servir de contrôle négatif utile lors de l’examen du phénotype SHM dans d’autres modèles de souris mutantes parce que ces cellules ne peuvent pas compléter SHM17,19. Le taux de mutation intron JH4 dans l’AID-/- GCBCs (1.66x10-5 mutations/bp)20,21,36,37,38,5 0,74 est comparable au taux d’erreur de la polymése haute fidélité (5,3x10-7 sous/base/doublement)51,52 qui est utilisé pour amplifier l’ADN dans le PCR imbriqué. Si aid-/- les souris ne sont pas disponibles, comparez le modèle et la fréquence observés de mutation à la littérature éditée. Les régions d’Ig V accumulent 10-3-10-4 mutations par division de paire de base, qui est approximativement10 6fois plus élevée que le taux de mutation d’autres locide gène 73,75. Les résultats peuvent varier avec l’âge de l’animal76. Alternativement, les cellules B220+PNALO, qui marquent les non-GCBCs, peuvent être utilisées comme un contrôle négatif en l’absence d’AID-/- souris52. Si la fréquence de mutation dans les WT GCBCs est plus faible que prévu, la séquence intronic de germline de WT JH4 peut être représentée de manière disproportionnée. Dans ce cas, assurez-vous que les CCG ont été tachés et triés de façon appropriée et que les RPC sont exempts de contamination par la germline JH4 intron du WT. En outre, les données brutes de séquençage dans les électrophéogrammes doivent être analysées en profondeur pour s’assurer que les mutations dans les données de texte séquence ne sont pas des artefacts d’erreurs de séquençage. Par exemple, de mauvais résultats de séquençage Sanger peuvent réduire la fiabilité des données de séquence (figure 4). Ce contrôle de qualité des données de la séquence Sanger augmentera la précision et la reproductibilité de l’analyse de la mutation intron JH4.
Déclarations de divulgation
Les auteurs n’ont rien à divulguer.
Remerciements
Nous remercions Tasuku Honjo pour l’AID-/- souris. Ces travaux ont été soutenus par le National Institute on Minority Health and Health Disparities (5G12MD007603), l’Institut national du cancer (2U54CA132378) et l’Institut national des sciences médicales générales (1SC1GM132035-01).
matériels
| Name | Company | Catalog Number | Comments |
| 0.2 ml PCR 8-tube FLEX-FREE strip, attached clear flat caps, mixed | USA Scientific | 1402-4708 | |
| Ampicillin sodium salt | Fisher | BP1760-5 | |
| APC-eFluor780 anti-CD45R/B220 | eBioscience | 47-0452-80 | clone RA3-6B2 |
| BD FACSAria II | BD | 643186 | four lasers (405nm, 488nm, 561nm, 633nm) and 12 filters (PacBlue (450/50), AmCyan (502LP; 530/30), SSC (488/10), FITC (502LP; 530/30), PerCP-Cy5.5 (655LP; 695/40), PE (585/15), PE-Texas Red (600LP; 610/20), PE-Cy5 (630LP; 670/14), PE-Cy7 (735LP; 780/60), APC (660/20), Alexa700 (710LP; 730/45), APC-Cy7 (755LP; 780/60)) |
| BD slip tip 1mL syringe | Fisher | 14-823-434 | sterile |
| Biotinylated peanut agglutinin (PNA) | Vector Labs | B-1075-5 | |
| C57BL/6J mice | Jackson Laboratories | 664 | |
| Corning Falcon test tube with cell strainer snap cap | Fisher | 08-771-23 | |
| DAPI (4',6-Diamidino-2-Phenylindole, dihydrochloride) | Fisher | D1306 | 0.5 mg/ml |
| dNTP | NEB | N0447L | 10 mM |
| ElectroMAX DH10B competent cells | Fisher | 18-290-015 | |
| Falcon cell strainer 40mm | Fisher | 08-771-1 | |
| Falcon round-bottom polystyrene tubes (FACS tubes) | Fisher | 14-959-5 | |
| Falcon round-bottom polystyrene tubes (capped) | Fisher | 149591A | |
| Fetal bovine serum | R&D Systems (Atlanta Biologicals) | S11150 | |
| Gibco phosphate buffered saline PBS pH 7.4 | Fisher | 10-010-049 | |
| Glycogen | Sigma | 10901393001 | |
| Lasergene Molecular Biology (MegAlign Pro) | DNA Star | version 15 | |
| PE anti-CD45R/B220 | BD | 553090 | clone RA3-6B2 |
| Proteinase K | Fisher | BP1700-100 | |
| Q5 High-Fidelity DNA Polymerase | NEB | M0491L | |
| QIAquick Gel Extraction Kit | Qiagen | 28706 | |
| Seal-Rite 1.5mL microcentrifuge tubes | USA Scientific | 1615-5500 | |
| Streptavidin APC-eFluor 780 Conjugate | eBioscience | 47-4317-82 | |
| T4 DNA ligase | NEB | M020L | |
| Thermo Scientific CloneJET PCR Cloning Kit | ThermoFisher | FERK1231 | |
| Tissue culture plate 6 well | Fisher | 08-772-1B | sterile |
| Unlabeled anti-mouse CD16/CD32 (Fc block), BD | Fisher | BDB553142 | Clone 2.4G2 |
Références
- Murphy, K., Weaver, C. . Janeyway's Immunobiology. , (2016).
- Alt, F. W., et al. VDJ recombination. Immunology Today. 13 (8), 306-314 (1992).
- Schatz, D. G., Ji, Y. Recombination centres and the orchestration of V (D) J recombination. Nature Reviews Immunology. 11 (4), 251-263 (2011).
- Oettinger, M. A., Schatz, D. G., Gorka, C., Baltimore, D. RAG-1 and RAG-2, adjacent genes that synergistically activate V (D) J recombination. Science. 248 (4962), 1517-1523 (1990).
- Berek, C., Berger, A., Apel, M. Maturation of the immune response in germinal centers. Cell. 67 (6), 1121-1129 (1991).
- Linterman, M. A., et al. Foxp3+ follicular regulatory T cells control the germinal center response. Nature Medicine. 17 (8), 975 (2011).
- Shulman, Z., et al. T follicular helper cell dynamics in germinal centers. Science. 341 (6146), 673-677 (2013).
- Good-Jacobson, K. L., et al. PD-1 regulates germinal center B cell survival and the formation and affinity of long-lived plasma cells. Nature Immunology. 11 (6), 535 (2010).
- Kerfoot, S. M., et al. Germinal center B cell and T follicular helper cell development initiates in the interfollicular zone. Immunity. 34 (6), 947-960 (2011).
- Chaudhuri, J., Alt, F. W. Class-switch recombination: interplay of transcription, DNA deamination and DNA repair. Nature Reviews Immunology. 4 (7), 541-552 (2004).
- Alt, F. W., Zhang, Y., Meng, F. L., Guo, C., Schwer, B. Mechanisms of programmed DNA lesions and genomic instability in the immune system. Cell. 152 (3), 417-429 (2013).
- Xu, Z., Zan, H., Pone, E. J., Mai, T., Casali, P. Immunoglobulin class-switch DNA recombination: induction, targeting and beyond. Nature Reviews Immunology. 12 (7), 517-531 (2012).
- Di Noia, J. M., Neuberger, M. S. Molecular mechanisms of antibody somatic hypermutation. Annual Reviews of Biochemistry. 76, 1-22 (2007).
- Peled, J. U., et al. The biochemistry of somatic hypermutation. Annual Review of Immunology. 26, 481-511 (2008).
- Liu, M., Schatz, D. G. Balancing AID and DNA repair during somatic hypermutation. Trends in Immunology. 30 (4), 173-181 (2009).
- Methot, S., Di Noia, J. Molecular Mechanisms of Somatic Hypermutation and Class Switch Recombination. Advances in Immunology. 133, 37-87 (2017).
- Muramatsu, M., et al. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 102 (5), 553-563 (2000).
- Petersen-Mahrt, S. K., Harris, R. S., Neuberger, M. S. AID mutates E. coli suggesting a DNA deamination mechanism for antibody diversification. Nature. 418 (6893), 99 (2002).
- Revy, P., et al. Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the Hyper-IgM syndrome (HIGM2). Cell. 102 (5), 565-575 (2000).
- Petersen-Mahrt, S. DNA deamination in immunity. Immunological Reviews. 203 (1), 80-97 (2005).
- Rada, C., Di Noia, J. M., Neuberger, M. S. Mismatch recognition and uracil excision provide complementary paths to both Ig switching and the A/T-focused phase of somatic mutation. Molecular Cell. 16 (2), 163-171 (2004).
- Rada, C., et al. Immunoglobulin isotype switching is inhibited and somatic hypermutation perturbed in UNG-deficient mice. Current Biology. 12 (20), 1748-1755 (2002).
- Schrader, C. E., Vardo, J., Stavnezer, J. Role for mismatch repair proteins Msh2, Mlh1, and Pms2 in immunoglobulin class switching shown by sequence analysis of recombination junctions. The Journal of Experimental Medicine. 195 (3), 367-373 (2002).
- Martin, A., et al. Msh2 ATPase activity is essential for somatic hypermutation at AT basepairs and for efficient class switch recombination. The Journal of Experimental Medicine. 198 (8), 1171-1178 (2003).
- Imai, K., et al. Human uracil-DNA glycosylase deficiency associated with profoundly impaired immunoglobulin class-switch recombination. Nature Immunology. 4 (10), 1023-1028 (2003).
- Masani, S., Han, L., Yu, K. Apurinic/apyrimidinic endonuclease 1 is the essential nuclease during immunoglobulin class switch recombination. Molecular and Cellular Biology. 33 (7), 1468-1473 (2013).
- Guikema, J. E., et al. APE1-and APE2-dependent DNA breaks in immunoglobulin class switch recombination. The Journal of Experimental Medicine. 204 (12), 3017-3026 (2007).
- Schrader, C. E., Guikema, J. E., Wu, X., Stavnezer, J. The roles of APE1, APE2, DNA polymerase β and mismatch repair in creating S region DNA breaks during antibody class switch. Philosophical Transactions of the Royal Society B: Biological Sciences. 364 (1517), 645-652 (2009).
- Roa, S., et al. MSH2/MSH6 complex promotes error-free repair of AID-induced dU: G mispairs as well as error-prone hypermutation of A: T sites. PLoS One. 5 (6), 11182 (2010).
- Delbos, F., Aoufouchi, S., Faili, A., Weill, J. C., Reynaud, C. A. DNA polymerase η is the sole contributor of A/T modifications during immunoglobulin gene hypermutation in the mouse. The Journal of Experimental Medicine. 204 (1), 17-23 (2007).
- Maul, R. W., Gearhart, P. J. AID and somatic hypermutation. Advances in Immunology. 105, 159-191 (2010).
- Shen, H. M., Tanaka, A., Bozek, G., Nicolae, D., Storb, U. Somatic hypermutation and class switch recombination in Msh6-/- Ung-/- double-knockout mice. The Journal of Immunology. 177 (8), 5386-5392 (2006).
- Cheng, H. L., et al. Integrity of the AID serine-38 phosphorylation site is critical for class switch recombination and somatic hypermutation in mice. Proceedings of the National Academy of Sciences. 106 (8), 2717-2722 (2009).
- Lebecque, S. G., Gearhart, P. J. Boundaries of somatic mutation in rearranged immunoglobulin genes: 5'boundary is near the promoter, and 3'boundary is approximately 1 kb from V (D) J gene. The Journal of Experimental Medicine. 172 (6), 1717-1727 (1990).
- Jolly, C. J., Klix, N., Neuberger, M. S. Rapid methods for the analysis of immunoglobulin gene hypermutation: application to transgenic and gene targeted mice. Nucleic Acids Research. 25 (10), 1913-1919 (1997).
- Choi, J. E., Matthews, A. J., Michel, G., Vuong, B. Q. AID Phosphorylation Regulates Mismatch Repair-Dependent Class Switch Recombination and Affinity Maturation. The Journal of Immunology. 204 (1), 13-22 (2020).
- McBride, K. M., et al. Regulation of class switch recombination and somatic mutation by AID phosphorylation. The Journal of Experimental Medicine. 205 (11), 2585-2594 (2008).
- Liu, M., et al. Two levels of protection for the B cell genome during somatic hypermutation. Nature. 451 (7180), 841-845 (2008).
- Ross, M., Birbeck, M., Wills, V., Forrester, J., Davis, A. Peanut lectin binding properties of germinal centers of mouse lymphoid tissues. Nature. 284, 364-366 (1980).
- Zhang, J., MacLennan, I. C., Liu, Y. J., Lane, P. J. Is rapid proliferation in B centroblasts linked to somatic mutation in memory B cell clones. Immunology Letters. 18 (4), 297-299 (1988).
- Nieuwenhuis, P., Opstelten, D. Functional anatomy of germinal centers. American Journal of Anatomy. 170 (3), 421-435 (1984).
- Lau, A. W., Brink, R. Selection in the germinal center. Current Opinion in Immunology. 63, 29-34 (2020).
- Victora, G. D., Nussenzweig, M. C. Germinal centers. Annual Review of Immunology. 30, 429-457 (2012).
- Mesin, L., Ersching, J., Victora, G. D. Germinal center B cell dynamics. Immunity. 45 (3), 471-482 (2016).
- Reichert, R. A., Gallatin, W. M., Weissman, I. L., Butcher, E. C. Germinal center B cells lack homing receptors necessary for normal lymphocyte recirculation. The Journal of Experimental Medicine. 157 (3), 813-827 (1983).
- Rose, M., Birbeck, M., Wills, V., Forrester, J., Davis, A. Peanut lectin binding properties of germinal centers of mouse lymphoid tissues. Nature. 284, 364-366 (1980).
- Hamada, S., Fujita, S. DAPI staining improved for quantitative cytofluorometry. Histochemistry. 79 (2), 219-226 (1983).
- Otto, F. DAPI staining of fixed cells for high-resolution flow cytometry of nuclear DNA. Methods in Cell Biology. 33, 105-110 (1990).
- Butcher, E., et al. Surface phenotype of Peyer's patch germinal center cells: implications for the role of germinal centers in B cell differentiation. The Journal of Immunology. 129 (6), 2698-2707 (1982).
- Rogerson, B. J., Harris, D. P., Swain, S. L., Burgess, D. O. Germinal center B cells in Peyer's patches of aged mice exhibit a normal activation phenotype and highly mutated IgM genes. Mechanisms of Ageing and Development. 124 (2), 155-165 (2003).
- Potapov, V., Ong, J. L. Examining sources of error in PCR by single-molecule sequencing. PloS One. 12 (1), 0169774 (2017).
- Gonzalez-Fernandez, A., Milstein, C. Analysis of somatic hypermutation in mouse Peyer's patches using immunoglobulin kappa light-chain transgenes. Proceedings of the National Academy of Sciences. 90 (21), 9862-9866 (1993).
- Reboldi, A., Cyster, J. G. Peyer's patches: organizing B-cell responses at the intestinal frontier. Immunological Reviews. 271 (1), 230-245 (2016).
- Betz, A. G., Rada, C., Pannell, R., Milstein, C., Neuberger, M. S. Passenger transgenes reveal intrinsic specificity of the antibody hypermutation mechanism: clustering, polarity, and specific hot spots. Proceedings of the National Academy of Sciences. 90 (6), 2385-2388 (1993).
- Rada, C., Gupta, S. K., Gherardi, E., Milstein, C. Mutation and selection during the secondary response to 2-phenyloxazolone. Proceedings of the National Academy of Sciences. 88 (13), 5508-5512 (1991).
- Heise, N., Klein, U. Somatic Hypermutation and Affinity Maturation Analysis Using the 4-Hydroxy-3-Nitrophenyl-Acetyl (NP) System. Methods in Molecular Biology. 1623, 191-208 (2017).
- Smith, F., Cumano, A., Licht, A., Pecht, I., Rajewsky, K. Low affinity of kappa chain bearing (4-hydroxy-3-nitrophenyl) acetyl (NP)-specific antibodies in the primary antibody repertoire of C57BL/6 mice may explain lambda chain dominance in primary anti-NP responses. Molecular Immunology. 22 (10), 1209-1216 (1985).
- Takahashi, Y., Dutta, P. R., Cerasoli, D. M., Kelsoe, G. In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl) acetyl. V. Affinity maturation develops in two stages of clonal selection. The Journal of Experimental Medicine. 187 (6), 885-895 (1998).
- Allen, D., Simon, T., Sablitzky, F., Rajewsky, K., Cumano, A. Antibody engineering for the analysis of affinity maturation of an anti-hapten response. The EMBO Journal. 7 (7), 1995-2001 (1988).
- Cumano, A., Rajewsky, K. Clonal recruitment and somatic mutation in the generation of immunological memory to the hapten NP. The EMBO Journal. 5 (10), 2459-2468 (1986).
- Smith, K., Nossal, G., Tarlinton, D. M. FAS is highly expressed in the germinal center but is not required for regulation of the B-cell response to antigen. Proceedings of the National Academy of Sciences. 92 (25), 11628-11632 (1995).
- Hao, Z., et al. Fas receptor expression in germinal-center B cells is essential for T and B lymphocyte homeostasis. Immunity. 29 (4), 615-627 (2008).
- Cervenak, L., Magyar, A., Boja, R., László, G. Differential expression of GL7 activation antigen on bone marrow B cell subpopulations and peripheral B cells. Immunology Letters. 78 (2), 89-96 (2001).
- Naito, Y., et al. Germinal center marker GL7 probes activation-dependent repression of N-glycolylneuraminic acid, a sialic acid species involved in the negative modulation of B-cell activation. Molecular and Cellular Biology. 27 (8), 3008-3022 (2007).
- Olson, W. J., et al. Orphan Nuclear Receptor NR2F6 Suppresses T Follicular Helper Cell Accumulation through Regulation of IL-21. Cell Reports. 28 (11), 2878-2891 (2019).
- Dorsett, Y., et al. MicroRNA-155 suppresses activation-induced cytidine deaminase-mediated Myc-Igh translocation. Immunity. 28 (5), 630-638 (2008).
- Goetz, C., Hammerbeck, C., Bonnevier, J. . Flow Cytometry Basics for the Non-Expert. , (2018).
- Hawley, T. S., Herbert, D. J., Eaker, S. S., Hawley, R. G. . Flow Cytometry Protocols. , (2004).
- Costa, E., et al. A new automated flow cytometry data analysis approach for the diagnostic screening of neoplastic B-cell disorders in peripheral blood samples with absolute lymphocytosis. Leukemia. 20 (7), 1221-1230 (2006).
- McKinnon, K. M. Flow cytometry: An overview. Current Protocols in Immunology. 120 (1), 1-11 (2018).
- McKinnon, K. M. Multiparameter Conventional Flow Cytometry. Methods in Molecular Biology. , 139-150 (2018).
- Lucchesi, S., et al. Computational Analysis of Multiparametric Flow Cytometric Data to Dissect B Cell Subsets in Vaccine Studies. Cytometry Part A. 97, 259-267 (2019).
- Longerich, S., Tanaka, A., Bozek, G., Nicolae, D., Storb, U. The very 5' end and the constant region of Ig genes are spared from somatic mutation because AID does not access these regions. The Journal of Experimental Medicine. 202 (10), 1443-1454 (2005).
- Retter, I., et al. Sequence and characterization of the Ig heavy chain constant and partial variable region of the mouse strain 129S1. The Journal of Immunology. 179 (4), 2419-2427 (2007).
- Shen, H. M., Peters, A., Baron, B., Zhu, X., Storb, U. Mutation of BCL-6 gene in normal B cells by the process of somatic hypermutation of Ig genes. Science. 280 (5370), 1750-1752 (1998).
- Richter, K., et al. Altered pattern of immunoglobulin hypermutation in mice deficient in Slip-GC protein. Journal of Biological Chemistry. 287 (38), 31856-31865 (2012).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.