
Method Article
Une procédure ChIP-Seq semi-automatisée pour les études épigénétiques à grande échelle
Dans cet article
Erratum Notice
Résumé
Cet article décrit un protocole ChIP-Seq semi-automatisé et micro-mis à l’échelle des régions d’ADN associées à une modification spécifique des histones (H3K27ac) à l’aide d’une plate-forme de manipulation de liquide ChIP, suivi d’une préparation de bibliothèque à l’aide de la marquage. La procédure comprend une évaluation du contrôle à des fins de qualité et de quantité et peut être adoptée pour d’autres modifications d’histones ou facteurs de transcription.
Résumé
L’immunoprécipitation de la chromatine suivie d’un séquençage (ChIP-Seq) est une approche puissante et largement utilisée pour profiler l’ADN de la chromatine associé à des modifications spécifiques des histones, telles que H3K27ac, afin d’aider à identifier les éléments de l’ADN cis-régulateurs. Le processus manuel pour compléter un ChIP-Seq est laborieux, techniquement difficile et nécessite souvent un grand nombre de cellules (>100 000 cellules). La méthode décrite ici aide à surmonter ces défis. Une procédure H3K27ac ChIP-Seq semi-automatisée et micro-automatisée complète comprenant la fixation cellulaire, le cisaillement de la chromatine, l’immunoprécipitation et la préparation de la bibliothèque de séquençage, pour un lot de 48 échantillons pour des entrées de nombre de cellules inférieures à 100 000 cellules est décrite en détail. La plate-forme semi-autonome réduit la variabilité technique, améliore les rapports signal/bruit et réduit considérablement la main-d’œuvre. Le système peut ainsi réduire les coûts en permettant de réduire les volumes de réaction, limitant ainsi le nombre de réactifs coûteux tels que les enzymes, les billes magnétiques, les anticorps et le temps pratique requis. Ces améliorations apportées à la méthode ChIP-Seq conviennent parfaitement aux études épigénétiques à grande échelle d’échantillons cliniques avec un nombre limité de cellules de manière hautement reproductible.
Introduction
L’utilisation généralisée des tests ChIP-Seq pour déterminer des fragments d’ADN associés à des modifications spécifiques des histones est en partie due à sa capacité à identifier les éléments d’ADN cis-régulateurs, y compris les activateurs actifs, les promoteurs, les silencieux, l’hétérochromatine et d’autres1,2,3,4. L’identification des régions régulatrices non codantes à travers le génome a montré des informations précieuses pour mieux comprendre la régulation des gènes dans la santé et les maladies4. Des travaux antérieurs du laboratoire ont utilisé ChIP-Seq pour montrer que les élémentsrégulateurscis peuvent jouer un rôle important dans différents types de cellules5. Les tests ChIP du facteur de transcription (TF) ont été utilisés pour montrer le risque associé à la maladie des polymorphismes mononucléotidiques6.
L’utilisation de ChIP-Seq avec des échantillons cliniques humains est difficile, principalement en raison de la limitation du nombre de cellules ou de l’échantillon de tissu souhaité. En conséquence, il y a eu un effort concerté sur le terrain pour améliorer et micro-étendre ces techniques et, par conséquent, plusieurs essais ont émergé, tels que CUT & TAG5,7,8,9,10,11,12. Ce test utilise une transposase pour marquer et isoler les régions génomiques liées par un anticorps spécifique9. Cette technique a été en mesure de réduire le nombre de cellules à 1 000 et dans certains cas à une seule cellule, cependant, l’utilisation de cette technique dans la recherche translationnelle et la configuration clinique a montré des limites en raison des exigences d’utilisation de cellules vivantes pourcetteméthode 9,12. L’exigence de cellules vivantes rend les échantillons cliniques logistiquement difficiles à manipuler et peut introduire des effets de lot si les échantillons ne sont pas traités en même temps. D’autres ont optimisé les techniques microscales pour les cellules fixées au formaldéhyde, notamment le développement de ChIPmentation11, qui est adapté ici de manière à haut débit. L’utilisation de cellules fixes permet de stocker les échantillons jusqu’au prélèvement et au traitement ultérieur de tous les échantillons ensemble afin de minimiser les effets des lots.
Ici, un test ChIP-Seq micro-automatisé est décrit, ce qui réduit le temps pratique expérimental pour profiler les modifications des histones10. La méthode semi-automatisée permet des tests ChIP-Seq à haut débit, permettant à jusqu’à 48 échantillons d’être entièrement traités et prêts pour le séquençage en aussi peu que 5 jours, pour aussi peu que 10 000 cellules par échantillon à l’aide d’un manipulateur de liquide ChIP. Le manipulateur effectue l’immunoprécipitation (IP) et les lavages ultérieurs de manière autonome, ce qui contribue à réduire la variabilité entre les échantillons. La méthode semi-automatisée réduit à la fois le temps de mise en pratique de plus de 15 h pour 48 échantillons et la variabilité technique, ce qui permet de mener des études épigénétiques à grande échelle de manière reproductible et rapide pour les cellules primaires ou cultivées. Le protocole explique le processus du début à la fin pour un ChIP-Seq de haute qualité. Si les machines spécifiques ne sont pas disponibles, le protocole sera toujours une ressource utile pour configurer et dépanner manuellement les expériences ChIP-Seq.
Le test a été effectué avec trois types de cellules immunitaires humaines primaires différentes et une lignée cellulaire cultivée (HUT78 – ATCC: TIB-161). Pour plus de clarté, le protocole a été divisé en sept sections: fixation cellulaire, cisaillement de la chromatine par sonication, immunoprécipitation automatisée de la chromatine, préparation de la bibliothèque par marquage de fragments d’ADN, amplification de la bibliothèque, purification de la bibliothèque, suivie de la quantification de l’ADN. Pour les recettes tampons, veuillez consulter le tableau supplémentaire 1.
Protocole
Le Conseil d’examen institutionnel (CISR) de l’Institut d’allergie et d’immunologie de La Jolla (LJI; Protocole IRB n° SGE-121-0714) a approuvé l’étude. Des volontaires sains ont été recrutés et ont fourni des échantillons de leucaphérèse après consentement éclairé écrit.
1. Fixation des cellules
- Porter la concentration de la suspension cellulaire à 1 à 2 x 106 cellules/mL avec un milieu de culture cellulaire complet dans un tube de 15 mL (<10 mL de suspension) ou un tube de 50 mL (10-30 mL de cellules). Si <1 x 106 cellules, utilisez 0,5 mL du milieu dans un tube de 1,5 mL.
- Vortexez doucement la suspension de la cellule, ajoutez 10x tampon de fixation de cellule goutte à goutte (1:10; vol:vol) et faites pivoter à basse vitesse pendant 10 min à température ambiante (RT).
- Arrêtez la réaction en tourbillonnant doucement et ajoutez 2,5 M de glycine dans un rapport de 1:20 (vol:vol). Inverser les tubes plusieurs fois et incuber sur de la glace pendant au moins 5 min.
- Effectuez les étapes restantes à 4 °C ou sur de la glace. Faire tourner les tubes à 800 x g pendant 5 min à 4 °C et jeter le surnageant.
- Remettre en suspension la pastille doucement avec 5 mL de PBS glacé et incuber pendant 2 min sur de la glace.
- Répétez 1,4 et 1,5 avec 1 mL de PBS glacé et transférez l’échantillon dans un tube prérefroidi de 1,5 mL (étiqueté pour un stockage à long terme). Le cas échéant, la préparation d’aliquotes est recommandée.
- Faire tourner les tubes à 1 200 x g à 4 °C et retirer autant de surnageant que possible sans perturber la pastille cellulaire. Congelez la pastille dans de l’azote liquide. Conserver à -80 °C.
ATTENTION : Prenez une protection appropriée lors de la manipulation de l’azote liquide.
2. Cisaillement de la chromatine
REMARQUE: Ce protocole est optimisé pour le cisaillement de la chromatine de granulés avec 0,3 à 3 x 106 cellules dans des tubes à faible liaison de 0,65 mL.
- Retirer le tube d’échantillons avec des granulés de cellules congelés à partir de -80 °C et les conserver sur de la glace carbonique pour éviter toute décongélation des granulés avant d’ajouter le tampon de lyse. Cette étape est essentielle.
- Ajouter 70 μL de tampon de lyse complet RT frais à la pastille et conserver à RT pendant 1 min.
- Remettre la pastille pendant 1 min sans introduire de bulles puis incuber la suspension cellulaire à RT pendant 1 min avant de mettre l’échantillon sur glace.
- Transférer la pastille remise en suspension dans un tube à faible liaison de 0,65 mL et la garder sur la glace.
REMARQUE: Pour obtenir une sonication reproductible, préchauffez le sonicateur en le faisant fonctionner avec seulement des tubes vierges pendant 3 à 6 cycles avant de soniquer les échantillons. - Placez les échantillons dans le porte-tube du sonicator et remplissez les espaces avec des tubes d’équilibrage remplis de 70 μL d’eau. Laissez les échantillons au bain-marie pendant environ 1 min avant de commencer la sonication.
- Effectuer une sonication pour x cycles (selon le type de cellule) avec 16 s ON / 32 s OFF par cycle.
REMARQUE: Cette étape nécessitera des expériences de validation pour déterminer le nombre optimal de cycles pour une sonication efficace. - Après tous les 3 cycles, retirez les échantillons du sonicateur, vortexz doucement et faites tourner les tubes par impulsion avant de les remettre dans le support. Assurez-vous qu’il n’y a pas de petites gouttelettes à l’extérieur du tube, car cela peut provoquer la formation de bulles.
- Après avoir terminé les cycles nécessaires, filer les échantillons à >14 000 x g pendant 15 min à 4 °C. Transférer le surnageant dans un nouveau tube à faible liaison de 0,65 mL pré-refroidi et à faible liaison et maintenir sur la glace.
- Déterrer une fraction des échantillons soniqués pour vérifier l’efficacité de la sonication.
- Transférer 1 à 7 μL du surnageant (équivalent à environ 250 ng de chromatine cisaillée) dans un tube PCR de 0,2 mL et porter le volume à 10 μL avec un tampon de lyse à court terme à RT.
- Ajouter 1 μL de RNase A et incuber pendant 30 min à 37 °C à 800 tr/min, puis ajouter 1 μL de protéinase K.
- Incuber pendant 2 h à 55 °C en agitant à 1 000 tr/min.
- Retirer 2 μL de l’échantillon réticulé pour la quantification de l’ADN à l’aide du test de quantification fluorescente10 (un spectrophotomètre n’est pas recommandé car le savon et les protéines dégradées peuvent produire un biais dans les résultats).
- Faire couler l’échantillon restant sur un gel d’agarose à 1,2 % pendant 1 h à 70 V. Tacher avec un colorant d’acide nucléique (1:20 000) et lire le gel à l’aide d’un transilluminateur UV.
- Préparer les aliquotes de chromatine pour le stockage (diluer l’échantillon à 25 ng/μL dans 20 μL avec un tampon de lyse complet). Conserver toute la chromatine cisaillée à -80 °C.
3. ChIP-Seq automatisé pour la modification des histones
REMARQUE : Ce protocole est conçu pour s’exécuter sur un gestionnaire de liquide ChIP. Bien que le système puisse utiliser des tampons personnalisés, tous les tampons sont fournis avec le kit ChIP. Les bandes ChIP avec 8 tubes utilisées dans cette section sont spécifiques au manipulateur de liquides ChIP.
- Transférer 16 aliquotes d’échantillon avec 500 ng de chromatine cisaillée dans 20 μL à partir de -80 °C et les placer sur de la glace pour décongeler lentement la chromatine. Une fois complètement décongelé, vortex brièvement et pulse-spin.
- Préparation de la chromatine
- Pipette 100 μL de tampon tC1 complété par 1x inhibiteur de protéase et 20 mM de butyrate de sodium (tampon tC1 complet) en deux bandes ChIP à 8 tubes.
- Transférer 20 μL de chaque échantillon de chromatine dans un tube approprié des bandes ChIP à 8 tubes contenant les 100 μL de tampon tC1 complet. Lavez les tubes de chromatine en ajoutant un tampon tC1 complet de 80 μL aux tubes de chromatine, puis transférez-les dans le tube approprié des bandes à 8 tubes ChIP pour un volume final de 200 μL.
- Préparation de l’anticorps
- Calculer le volume d’anticorps de telle sorte que 0,5 μg d’anticorps se trouve dans chaque tube.
Volume d’anticorps = (nombre d’échantillons x anticorps par réaction) / concentration d’anticorps - Ajouter la quantité calculée d’anticorps dans 500 μL de tampon tBW1. Vortex rapide et pulse-spin.
- Pipette 70 μL de tBW1 dans chacune des deux bandes ChIP à 8 tubes et ajouter 30 μL de l’anticorps + tBW1 à chacun des tubes. Cela portera le volume total dans chacun des tubes à 100 μL.
- Calculer le volume d’anticorps de telle sorte que 0,5 μg d’anticorps se trouve dans chaque tube.
- Préparation de la bille magnétique
- Vortex la protéine A solution de perle à fond. Pour 0,5 μg d’anticorps, pipettez 5 μL de billes dans un nouvel ensemble de bandes ChIP à 8 tubes et de spin d’impulsion.
- Remplissez la dernière rangée du manipulateur de liquides ChIP avec des bandes à 8 tubes ChIP vides et étiquetées.
- Suivez les spécifications du programme ChIP-16-IPure-200D pour le placement de toutes les bandes dans la machine de traitement des liquides ChIP. Ajoutez les tampons dans la bonne position, mais utilisez tW4 au lieu de tE1.
REMARQUE: Organisez la journée de manière à ce que le manipulateur de liquide ChIP effectue le ChIP pendant la nuit. Le programme durera environ 16 h pour 16 échantillons. Cela marque la fin du Jour 1.
4. Intégration transposase d’adaptateurs de bibliothèque pour la préparation de la bibliothèque
- Prérégrez un thermomixeur à 37 °C et 500 tr/min. Refroidissez un aimant pour des bandes de tube de 0,2 mL sur de la glace.
- Pour 16 échantillons, préparer 440 μL de tampon de marquage sur de la glace. Pipette 53 μL dans une seule nouvelle bande de 8 tubes et garder sur la glace.
- Dans une nouvelle bande de 8 tubes de 0,2 mL, ajoutez 220 μL de tampon tC1 froid et conservez sur la glace. Les 8 tubes à bandes peuvent contenir ce volume tout en étant bouchés.
- Retirez le tube à bande « IP samples » de la machine de manutention des liquides ChIP (rangée 12) et bouchez les tubes avant la rotation des impulsions. Capturez les perles à l’aide de l’aimant pour bandes à 8 tubes pendant 2 minutes et retirez soigneusement le surnageant.
- Transférer 25 μL du tampon de marquage sur les billes avec un multicanal, retirer de l’aimant et mélanger doucement jusqu’à ce que les billes soient homogènes (environ 5 fois de haut en bas avec la pipette réglée à 20 μL).
- Boucher les tubes et placer dans le thermomixeur préchauffé et incuber pendant 3 min. L’augmentation du temps diminuera l’efficacité de la préparation de la bibliothèque.
- Transférer les tubes dans une grille métallique réfrigérée et ajouter 100 μL de tampon tC1 réfrigéré à chaque échantillon. Réglez une pipette multicanal à 80 μL et mélangez l’échantillon jusqu’à ce que les billes soient homogènes, arrêtant ainsi la réaction de marquage.
- Replacez les échantillons dans le manipulateur de liquide ChIP et procédez à la procédure de lavage Washing_for_IP-reacts_16_Ipure. Assurez-vous que le lavage est effectué deux fois avec le tampon tC1 et deux fois avec tW4. L’élution doit être complétée comme indiqué par la disposition du programme, avec le tampon tE1.
- Réticulation de l’ADN
- Retirez les bandes ChIP à 8 tubes dans la dernière rangée du manipulateur de liquide ChIP et ajoutez 2 μL de RNase A à chaque échantillon.
- Boucher les tubes, pulse-spin, mélanger doucement les billes avec une pipette multicanal jusqu’à ce que le mélange soit homogène, et reboucher les tubes.
- Incuber les échantillons dans un thermomixeur pendant 30 minutes à 37 °C et 900 tr/min.
- Retirer les échantillons du thermomélangeur, ajouter 2 μL de protéinase K. Suivez la même procédure que 4.9.2 après l’ajout.
- Incuber les échantillons dans un thermomixeur pendant 4 h à 55 °C et 1 250 tr/min, puis 65 °C à 1 000 tr/min pendant la nuit.
REMARQUE: C’est la fin du Jour 2.
5. Purification des fragments d’ADN marqués
- Étiquetez seize tubes de 1,5 mL avec le numéro d’échantillon approprié et ajoutez 400 μL de tampon de liaison à l’ADN du kit de nettoyage de l’ADN à chacun.
- Retirez les bandes à 8 tubes du thermomélangeur et faites tourner les bandes par impulsions pour vous assurer que tout produit évaporé est retenu. Placez des bandes sur un aimant à 8 bandes pour capturer les perles.
- Transférer 100 μL d’ADN réticulé dans chacun des tubes de 1,5 mL. Ajouter 100 μL du tampon de liaison à l’ADN aux bandes à 8 tubes pour laver les billes, puis transférer dans le tube approprié de 1,5 mL.
- Vortex pendant environ 10 s et spin d’impulsion des tubes de 1,5 mL.
- Chargez les colonnes avec les 600 μL contenant le tampon de liaison à l’ADN et l’échantillon ChIP.
- Faites tourner les échantillons pendant 20 s à 10 000 x g et rechargez la colonne avec le flow-through. Tournez à nouveau dans les mêmes conditions et jetez le flux.
- Lavez les colonnes deux fois avec un tampon de lavage de 200 μL (même centrifugation que l’étape précédente) et jetez le débit.
- Sécher les colonnes en centrifugant pendant 2 min à 12 000 x g.
- Transférer la colonne dans un nouveau tube collecteur de 1,5 mL et ajouter un tampon TE chaud de 9 μL (préchauffé à 55 °C) directement à la matrice de colonne. Laisser la colonne incuber pendant 1 min avant la centrifugation pendant 1 min à 10 000 x g.
- Transférer les 9 μL de l’éluat dans un nouvel ensemble approprié de bandes à 8 tubes.
- Complétez à nouveau l’élution avec un tampon TE de 8 μL comme auparavant. Transférer l’éluat dans les bandes appropriées à 8 tubes (volume final 17 μL par échantillon) et conserver sur la glace.
6. Amplification et sélection de la taille des échantillons purifiés
- Les étapes suivantes utilisent la qPCR pour déterminer le nombre de cycles requis pour une amplification optimale (CtD – détermination Ct)
- Préparez le mélange CtD pour tous les échantillons en multipliant le contenu du tampon CtD Mix par le nombre d’échantillons.
- Distribuer 3,6 μL de mélange CtD dans une plaque de qPCR et ajouter 1,4 μL d’échantillons d’ADN marqués (~10 % du volume total). Effectuez la qPCR suivante : 98 °C pendant 3 min, 72 °C pendant 5 min, 98 °C pendant 30 s, 26 cycles de 98 °C pendant 10 s, 63 °C pendant 30 s et 72 °C pendant 30 s.
- Préparez le mélange d’amplis pour tous les échantillons en multipliant le contenu du tampon de mélange AMP par le nombre d’échantillons. Distribuer 14 μL d’ADN marqué dans des puits séparés d’une plaque de PCR, puis ajouter 2,5 μL de deux amorces d’indice de séquençage (25 μM) à chaque échantillon (le volume de réaction final est de 50 μL).
- Mélangez les échantillons par pipette multicanal et effectuez le programme d’amplification utilisé dans le CtD avec le nombre approprié de cycles.
REMARQUE: C’est un bon point d’arrêt car les échantillons amplifiés peuvent être conservés à -20 ° C pendant quelques semaines. Cependant, la purification peut être terminée le même jour. Pour 48 échantillons, les étapes 3 à 6.5 ont été complétées avec deux autres lots distincts, puis amplifiées en un seul lot comme décrit ci-dessous. - Effectuer la post-amplification, la sélection de la taille et la quantification de l’ADN marqué comme décrit ci-dessous. Cela peut généralement être complété avec 48 échantillons (peut être complété avec moins d’échantillons que vous le souhaitez).
- Ajouter 90 μL de perles paramagnétiques (rapport 1:1,8) dans chaque puits, mélanger et laisser incuber à RT pendant 2 min.
- Capturez les perles à l’aide d’un aimant à plaque et jetez le surnageant. Lavez les billes 3 fois avec 200 μL d’éthanol frais à 80% sans perturber la pastille de perle.
- Retirez tout excès d’éthanol avec des pointes de 20 μL après le lavage final et laissez sécher les billes pendant 10 minutes ou jusqu’à ce que des fissures apparaissent dans les granulés de perles.
- Avec la plaque toujours sur l’aimant, ajoutez 40 μL d’eau préchauffée à chaque puits. Scellez soigneusement la plaque, vortexez et faites tourner brièvement la plaque par impulsions.
- Capturez les billes en replaçant la plaque sur l’aimant et transférez l’élute de 40 μL sur une nouvelle plaque « échantillon ». Les échantillons sont maintenant purifiés et les étapes suivantes enrichissent des fragments allant de 200 à 1 000 pb.
- Étape de contrôle de la qualité en option : Retirez 4 μL des échantillons et transférez-la sur une plaque de contrôle qualité. Ajouter 4 μL d’eau aux échantillons. Cela détermine le pourcentage de gros fragments.
- Ajouter 22 μL de billes paramagnétiques (rapport 1:0,55) aux échantillons, mélanger soigneusement et incuber à RT pendant 2 min.
- Placer sur l’aimant pour capturer les perles pendant 5 min et transférer les surnageants dans les colonnes 7 à 12 de la plaque « échantillon ». Retirez la plaque de l’aimant et ajoutez 30 μL de billes (rapport final de 1:1,3). Mélanger soigneusement et laisser reposer à RT pendant 2 min.
- Capturez les perles pendant 5 min, puis jetez le surnageant.
- Laver toutes les billes 3 fois avec 200 μL d’éthanol frais à 80 % comme décrit précédemment (étapes 6.6.2 – 6.6.3).
- Une fois les granulés secs, éluez l’ADN avec un tampon TE préchauffé de 8 μL à chaque puits, tout en restant sur l’aimant.
- Retirez soigneusement la plaque de l’aimant, du joint et du vortex. Laissez la plaque incuber pendant 2 min à TA, pulse-spin, et replacez la plaque sur l’aimant pendant 2 min. Transférer le surnageant sur une nouvelle plaque (planche 2).
- Pour une récupération maximale, répétez l’élution avec 8 μL supplémentaires de tampon TE préchauffé. Placer les échantillons dans les puits appropriés de manière à ce que chaque échantillon ait 16 μL de bibliothèque finale.
REMARQUE: À la fin de cette étape, il devrait y avoir deux plaques (une si aucune plaque QC n’a été complétée). La plaque QC aura les fragments présélectionnés et la deuxième plaque devra avoir 48 puits de bibliothèque finale (16 μL au total).
7. Quantifier les bibliothèques finales et les échantillons de contrôle qualité à l’aide d’un test de fluorescence
- Quantification complète de l’ADN à l’aide d’un test de quantification de fluorescence ou d’une méthode similaire.
- Si la quantification du CQ a été effectuée, déterminez le pourcentage de perte d’échantillon qui a été < 1 000 pb. Il ne devrait pas y avoir plus de 20% de perte – s’il y en avait plus, il pourrait y avoir un problème avec les ratios de perles appliqués.
- Déterminer la taille des fragments de chaque échantillon, de préférence à l’aide d’un appareil d’électrophorèse capillaire. Pour calculer la concentration molaire, utilisez l’équation suivante : [Concentration de bibliothèque (ng/μL) * 106]/[660 * Taille médiane du fragment (pb)]).
REMARQUE : Les bibliothèques sont prêtes à être regroupées (quantités équimolaires) et séquencées selon les procédures de séquençage standard de nouvelle génération.
Résultats
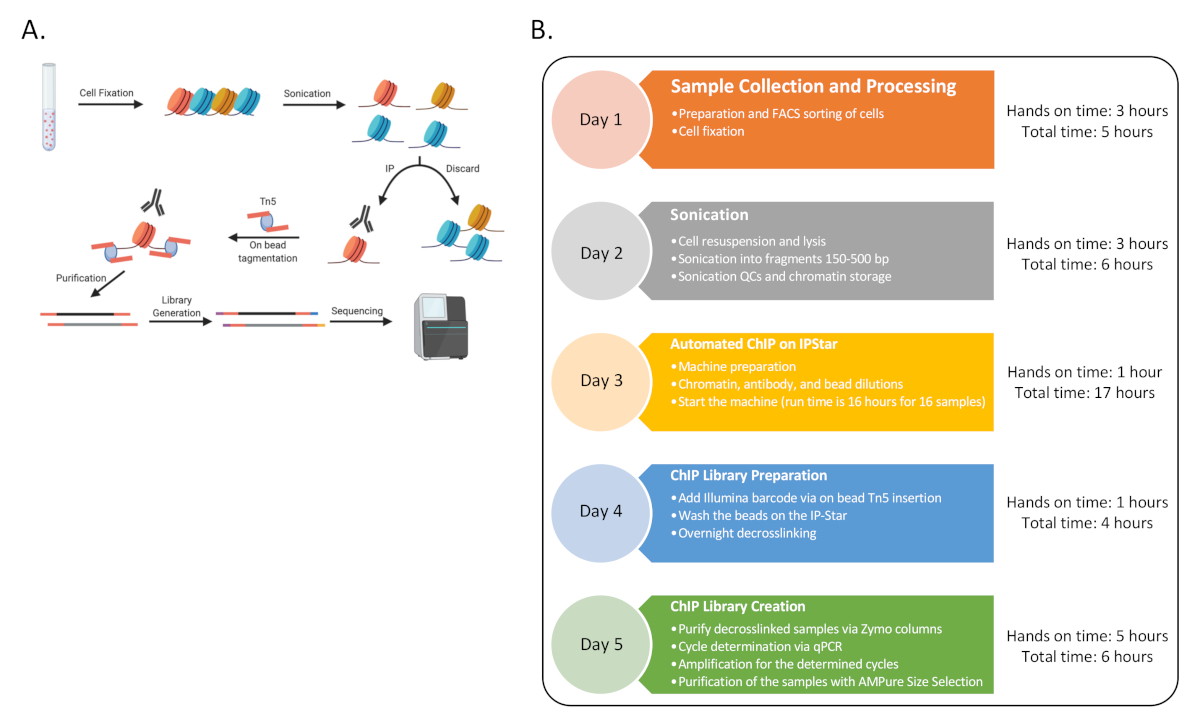
Comme preuve de concept, ChIP-Seq a été complété pour six donneurs humains avec trois ensembles de types de cellules immunitaires: les cellules T CD4 naïves (CD4), les monocytes classiques (MO) et les cellules tueuses naturelles (NK), enrichies par le tri FACS décrit avant13. La procédure soulignée se compose de neuf procédures distinctes telles que représentées à la figure 1.

Figure 1 : Organigramme général de la procédure. (A) Dessin animé de l’ensemble de la procédure (généré dans BioRender). (B) Organigramme pour toutes les principales étapes du protocole et le temps pratique et total estimé associé à chaque jour. Le séquençage peut avoir lieu à la fin du jour 5 ou plus tard avec plusieurs tours. La chronologie peut également être échelonnée tout au long de la semaine, où les jours séquentiels 3 à 4 peuvent être complétés plusieurs fois par semaine pour générer 48 échantillons ChIP. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
Après l’isolement cellulaire par cytométrie en flux13,les cellules triées ont été centrifugées et les cellules fixées et stockées comme décrit ci-dessus. Une fois tous les échantillons prélevés, les échantillons ont été lysés et préparés pour le cisaillement chromatique par lots de 12 comme décrit ci-dessus. Pour chaque échantillon, le nombre de cycles pour atteindre la sonication optimale a été complété10. Les mesures quantitatives, ainsi que les mesures de la taille des fragments de chromatine cisaillés ont montré une grande reproductibilité de notre méthode sur les trois ensembles de cellules immunitaires(Figure 2A). Les différentes cellules immunitaires humaines ont été soniquées en lots distincts et ont donné un rendement très constant avec > 70% de l’échantillon entre 100 et 500 bp pendant 14 cycles (16 s ON, 32 s OFF par cycle). À ce stade, les échantillons avec de gros fragments après sonication (< 70% de l’échantillon entre 100 et 500 pb) ont été considérés comme ayant échoué. Ces échantillons pouvaient soit soniquer pendant 1 à 2 cycles supplémentaires, soit être jetés et remplacés plus tard par des cellules provenant d’une autre pastille. Notre méthode a montré qu’aucun des échantillons ne nécessitait plus de sonication ou n’était éliminé, ce qui suggère une robustesse absolue de la procédure.

Figure 2 : Exemples de QC pré-séquençage. (A) Les gels d’agarose à 1,2 % montrent la reproductibilité de la sonication. Échantillons de sonication pour 6 donneurs dans trois types de cellules : les lymphocytes T CD4 naïfs (CD4), les monocytes classiques (MO) et les cellules tueuses naturelles (NK). Les échantillons ont été soniqués pendant 14 cycles (16 s ON, 32 s OFF par cycle). Pour chaque échantillon, environ 200 ng de chromatine réticulée ont été chargés sur un gel d’agarose à 1%. Les échantillons ont été considérés comme bons si plus de 70 % des fragments se trouvent entre 100 et 500 pb. (B) Haut - Analyse des courbes d’amplification qPCR pour déterminer le nombre optimal de cycles d’amplification (Ct où il y a 1/2 de l’intensité maximale). Les échantillons idéaux ont un Ct d’environ 15 et l’amplification peut être complétée jusqu’à 2 cycles de plus du Ct mesuré. La flèche est un exemple d’un mauvais exemple où le Ct est supérieur à 18. Bas - Un exemple d’un mauvais ensemble d’échantillons est montré qui ont un Ct supérieur à 18. Ces échantillons ont également montré une intensité de fluorescence plus faible. (C) Gauche - Les traces d’électrophorèse de l’analyseur de fragments ont montré la distribution des bibliothèques finales étiquetées après amplification et sélection de la taille. Les échantillons contenant plus de 85 % de la bibliothèque de fragments se situant entre 200 et 1 000 pb ont été considérés comme de bons échantillons. La mesure de l’intensité maximale de fluorescence est également considérée comme un paramètre important du CQ, en effet si le signal est faible, il est peu probable que l’échantillon s’enchaîne bien. Droite - Des exemples d’échantillons positifs dans CD4, MO et NK sont présentés. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
Après quantification, les échantillons ont été exécutés sur un manipulateur de liquide ChIP avec des anticorps H3K27ac, suivi d’un marquage avec l’enzyme transposase Tn5. Pour déterminer le nombre approprié de cycles d’amplification par qPCR, 10 % des échantillons marqués ont été utilisés. Pour la détermination du nombre de cycles pour l’amplification des échantillons, nous trouvons le cycle auquel l’intensité de l’échantillon est la moitié du maximum moyen pour la détermination du cycle (Figure 2B). Les échantillons avec des valeurs Ct supérieures à 18 n’ont pas bien fonctionné après le séquençage et leur valeur Ct était donc révélatrice d’un échantillon ChIP échoué. Ces échantillons ont généralement également produit une quantité plus faible d’ADN après amplification. Les échantillons (100 000 cellules entrées) avec une valeur Ct égale ou inférieure à 15 étaient idéaux et les échantillons entre 15 et 18 étaient acceptables mais moins cohérents après le séquençage. Pour les échantillons avec moins de 100 000 cellules d’entrée, les valeurs Ct étaient généralement trouvées entre 15 et 18, mais n’avaient pas besoin de plus de 18 cycles pour produire suffisamment de produit pour le séquençage.
Après amplification marquée par l’ADN, les bibliothèques ont été purifiées et sélectionnées en taille pour obtenir une distribution de taille idéale, allant de 200 à 1 000 pb, pour le séquençage NextGen. L’évaluation de la distribution de la taille sur chacune des bibliothèques a été effectuée parce que les meilleures données de séquençage ont été obtenues lorsque plus de 85 % des fragments d’ADN variaient entre 200 et 1 000 pb(figure 2C). Notamment, comme la même quantité d’ADN (mesurée par quantification de fluorescence) était chargée, on a remarqué que les échantillons avec une intensité de fluorescence plus faible séquencaient généralement mal(Figure 2C).
Après le séquençage, des contrôles de qualité standard basés sur les directives ENCODE ChIP-Seq ont été appliqués5,14,15.

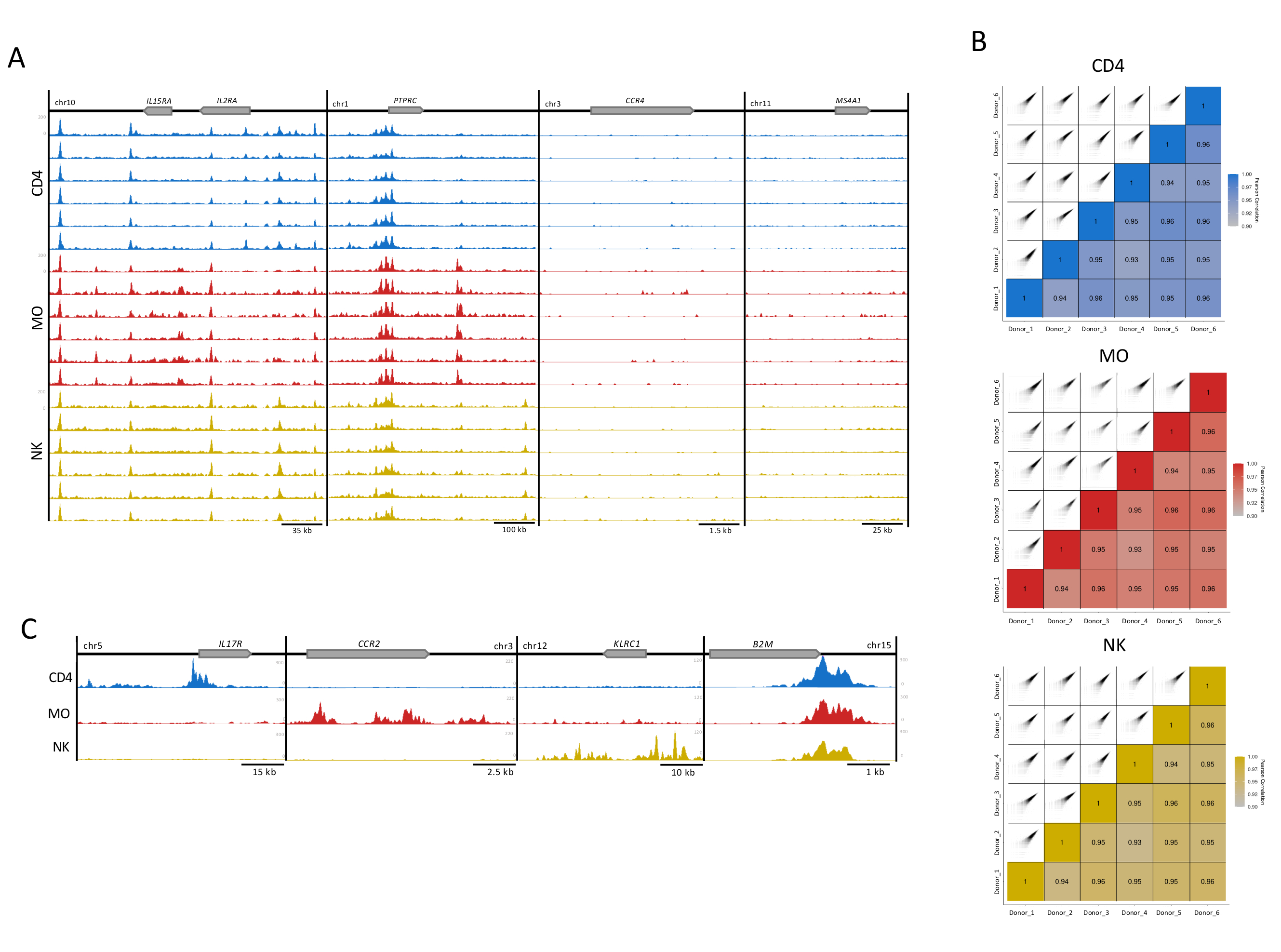
Figure 3 : Reproductibilité des échantillons de cellules immunitaires. (A) Pistes H3K27ac (UCSC Genome Browser, intensité maximale, fonction de lissage de 4, le tout avec un axe Y à échelle égale) pour 6 donneurs (100 000 cellules par réplique) dans chaque type de cellule (CD4, MO et NK). Quatre loci exemplaires sont montrés, deux avec (il2RA locus et PTPRC) et deux sans enrichissement pour H3K27ac (CCR4 et MS4A1). (B) La corrélation de Pearson entre les donneurs et les diagrammes de corrélation correspondants générés à l’aide d’une extension de 300 pb et d’une fenêtre de 500 pb dans le package MEDIPS pour chacun des types de cellules réplique16. (C) Fichiers de donneurs fusionnés pour chaque type de cellule montrant des pistes H3K27ac (intensité maximale du navigateur de génome UCSC, fonction de lissage de 4) dans des régions spécifiques au type de cellule (IL17R pour CD4, CCR2 pour MO et KLRC1 pour NK) et le gène de ménage B2M, présent dans tous les types de cellules. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
Pour le contrôle visuel de la qualité, des pistes d’enrichissement H3K27ac pour affichage dans le navigateur de génome UCSC ont été préparées. Pour quatre loci de gènes, des pistes individuelles pour chaque échantillon ont montré une qualité de cartographie élevée et un rapport signal/bruit reflétant la grande cohérence et la robustesse de notre essai(Figure 3A). Les deux loci à gauche abritent des gènes bien exprimés dans ces types de cellules, tandis que les gènes des deux loci à droite ne sont pas exprimés et ont servi de témoins de fond13 (Figure 3A). En outre, le progiciel d’analyse MEDIPS a été utilisé comme variable post-séquençage pour évaluer l’indice de corrélation entre les réplications techniques(Figure 3B)5,16,17, établissant le degré de corrélation pour le niveau d’enrichissement des lectures pour les bacs de 500 bp16. Pour la majorité des comparaisons par paires, les indices de corrélations de Pearson ont montré une corrélation de plus de 90 %, ce qui suggère un niveau élevé de cohérence entre les réplicats biologiques(figure 3B). Les répliques avec une corrélation acceptable ont été fusionnées pour augmenter le rapport signal/bruit. Alors que les loci spécifiques au type cellulaire ont montré un enrichissement élevé dans les cellules appropriées, un gène de ménage (B2M) a montré une modification très cohérente des histones(Figure 3C). Pour l’analyse, la fusion des pistes des réplicats augmentera l’enrichissement, renforcera le signal spécifique, y compris pour les amplificateurs importants spécifiques au type de cellule, et réduira la variabilité interindividuelle inhérente aux échantillons humains5.
Bien que 100 000 cellules aient été utilisées pour cette étude, il y avait une reproductibilité élevée pour aussi peu que 10 000 cellules dans une lignée de cellules T cultivées chez l’homme (HUT78). L’analyse de corrélation entre l’ensemble de données ChIP-Seq effectuée à partir d’échantillons de moins de 100 000 cellules a montré une reproductibilité élevée et une corrélation allant jusqu’à 10 000 cellules (Figure 4A).

Figure 4 : Reproductibilité des échantillons à faible entrée. (A) Exemples de la consistance de H3K27ac ChIP-Seq pour les cellules 100 000 à 10 000 dans les cellules HUT-78 (une lignée cellulaire de lymphome T). Les pistes (UCSC Genome Browser, intensité maximale, fonction de lissage de 4, toutes avec un axe Y à échelle égale) montrent le locus IL4. (B) Corrélations de Pearson des réplications à l’aide d’une extension de 300 pb et d’une fenêtre de 500 pb dans le package MEDIPS16. (C) Corrélations de Pearson entre les différents groupes de numéros de cellules (100 000, 50 000 et 10 000 cellules) en utilisant les mêmes paramètres MEDIPS que dans (B)16. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
L’analyse de corrélation de Pearson a montré un indice de corrélation élevé (83% à 92%), suggérant le maintien du signal dans les échantillons à faible nombre de cellules. Cependant, il y a eu une augmentation du fond à mesure que le nombre de cellules a été réduit ainsi qu’une baisse des coefficients de corrélation(figure 4B). Pour maintenir des signaux de fond faibles, les doublons techniques ont été fusionnés et la corrélation a été testée entre les groupes(figure 4C).
| Tampon de fixation de cellule 10X | |
| Composé | Concentration finale |
| Solution de formaldéhyde | 11% |
| NaCl | 100 mM |
| EDTA, pH 8,0 | 1 mM |
| EGTA, pH 8,0 | 0,5 mM |
| HEPES, pH 7,5 | 50 mM |
| Tampon de lyse complet | |
| Composé | Concentration finale |
| Tris-HCI, pH 8,0 | 50 mM |
| EDTA, pH 8,0 | 10 mM |
| SDS | 0.25% |
| Sodium Butyrate | 20 mM |
| Cocktail d’inhibiteurs de protéase | 1X |
| Tampon de lyse à court terme | |
| Composé | Concentration finale |
| Tris-HCI, pH 8,0 | 50 mM |
| EDTA, pH 8,0 | 10 mM |
| SDS | 0.25% |
| Mélange de tagmentation | |
| Composé | Concentration finale |
| Tris-HCI, pH 8,0 | 10 mM |
| MgCl2 | 5 mM |
| N,N-diméthylformamide | 10% |
| Enzyme de marquage illumina | 1:24 vol:vol |
| CtD Mix | |
| Composé | Par échantillon (μL) |
| SuivantA Index Primer A (25 μM) | 0.275 |
| NextEra Index Primer B (25 μM) | 0.275 |
| 2X KAPA HiFi HotStart Ready Mix | 2.75 |
| 1:1000 SYBR Colorant vert | 0.11 |
| Colorant passif ROX | 0.11 |
| Eau | Remplir à 4 μL |
| Mélange AMP | |
| Composé | Par échantillon (μL) |
| 2X KAPA HiFi HotStart Ready Mix | 27.5 |
| Eau | Remplir à 31 μL |
Tableau supplémentaire 1 : Recettes tampons.
Tableau supplémentaire 2 : Corrélations des échantillons spearman et Pearson pour les 6 donneurs et chaque type de cellule. Veuillez cliquer ici pour télécharger ce tableau.
Discussion
La méthode décrite ici développe la procédure ChIPmentation11, qui implémente un protocole de préparation de bibliothèque de tagmentation avant la purification de l’ADN, en automatisant et en micro-mettant à l’échelle le protocole. Depuis le début de ChIP-Seq, le nombre de cellules requis a été considérablement réduit, passant d’environ 20 millions de cellules pour les histones à des centaines et même à des cellules uniques1,7,10,12,18,19,20,21. Ces méthodes nouvellement développées ont permis de mieux comprendre comment les mécanismes de régulation cisfonctionnent dans les cellules en augmentant la sensibilité et en permettant de tester des populations de cellules cliniques rares5,6,12,17. Par exemple, l’une des procédures les plus récentes et les plus populaires, appelée CUT & TAG, comme alternative ChIP-Seq robuste et sensible9. Il produit un excellent rapport signal/bruit car l’enzyme Tn5 est liée de manière covalente à la protéine A et reconnaît la chaîne Fc de l’anticorps ChIP avec une spécificité élevée9. L’activité de fond de l’enzyme Tn5 est réduite car l’enzyme n’est pas fonctionnelle avant de se lier à l’anticorps cible9. Cependant, la mise en œuvre de cette méthode dans un contexte clinique est limitée car elle nécessite des cellules vivantes non fixes. En outre, l’élimination des fragments d’ADN du noyau hypotonique pourrait avoir des effets négatifs sur la chromatine car elle est retirée pendant le test. L’exigence nécessaire pour travailler avec des cellules fraîches et vivantes est une source de problèmes pour les échantillons cliniques rares et pour les grandes cohortes d’échantillons, car les grandes cohortes peuvent prendre de nombreuses années à collecter5. Un autre type de méthode, drop-ChIP, utilise élégamment un dispositif microfluidique pour générer un marquage à base de gouttelettes avant de traiter le ChIP19. Cependant, il utilise un dispositif microfluidique hautement spécialisé et, bien qu’il soit possible de compléter ChIP-Seq à cellule unique, il est également limité à l’utilisation de cellules vivantes7,8,9,18,19. Des méthodes plus récentes s’appuyant sur ChIP-Seq telles que PLAC-Seq ou HiChIP, tentent de comprendre les interactions en 3 dimensions (3D) entre les pics ChIP-Seq22,23. Ces méthodes 3D sont passionnantes car elles identifient les interactions cis-régulatricesou médiées par TF à travers le génome et améliorent la compréhension de la régulation de l’expression des gènes dans les types de cellules d’intérêt, dans les tissus sains et dans le contexte de la maladie.
Il y a quelques étapes critiques à considérer pour que le protocole soit efficace, telles que la qualité de la chromatine sonique et la qualité de l’anticorps. L’efficacité de cisaillement est critique, si la chromatine n’est pas bien soniquée, l’efficacité du test diminue drastiquement24. La sonication est un aspect difficile de ChIP-Seq en raison du nombre de cellules requis. Sur le sonicateur utilisé dans le protocole, l’efficacité a été considérablement réduite en dessous de 300 000 cellules. C’est un aspect difficile dans ChIP-Seq car soniquer sous ce niveau nécessiterait souvent une fragmentation enzymatique, qui est moins impartiale. En conséquence, la sonication est un facteur limité majeur pour le véritable ChIP-Seq micro-mis à l’échelle. D’autres plates-formes de sonication et des kits disponibles dans le commerce ont été testés pour la sonication de la chromatine, mais le sonicateur utilisé ici a eu les résultats les plus robustes et reproductibles. Un autre avantage du sonicateur est de ne pas avoir à acheter des tubes spécialisés pour exécuter la sonication, ce qui réduit les coûts lors du traitement d’un grand nombre d’échantillons. Pour une sonication optimale, tout d’abord, il est important de préchauffer le sonicator comme décrit ci-dessus. Deuxièmement, pour lyser la pastille, il est recommandé que la pointe de la pipette touche le fond du tube tout en lysant pour briser les cellules avec plus de contraintes physiques. Troisièmement, toute formation de bulles avant la sonication entrave la capacité de l’échantillon à être soniquer uniformément. S’il y a des bulles formées pendant la lyse, il est important de les enlever avec une pipette. Cela peut être difficile sans enlever beaucoup d’échantillon, mais si la pointe est légèrement pressée contre la bulle, elle peut être lentement dessinée sans perte de beaucoup d’échantillon. Enfin, lors de la détermination du nombre de cycles, effectuez un cours de temps où tous les trois cycles, l’échantillon est retiré, purifié et exécuté sur un gel d’agarose. Évitez la sur/sous-sonication des échantillons car cela diminue l’efficacité du ChIP. Si l’échantillon est sous-sonique, les gros fragments peuvent avoir un effet négatif sur la qualité ChIP-Seq24. D’autre part, si l’échantillon est sursexué, il y a un risque que l’épitope cible se perde dans le processus.
Une autre partie essentielle de ChIP-Seq est la qualité de l’anticorps. Avant de mener toute étude à grande échelle, il est nécessaire d’optimiser l’anticorps qui sera utilisé. L’objectif est d’obtenir un rapport signal/bruit significativement élevé des régions connues du génome et un autre est la reproductibilité. Si l’anticorps tire beaucoup de signal de fond, il peut être recommandé d’utiliser une entrée plus grande ou d’essayer un autre lot / fournisseur. Cela ajoutera du temps avant de commencer une expérience à grande échelle, mais c’est une étape essentielle. Pour tester le signal au bruit, il est recommandé d’utiliser la qPCR avec des régions connues pour être une cible de votre anticorps et une autre région connue pour être absente. Il a été remarqué que les modifications d’histones sont plus robustes et plus faciles à optimiser que les TF.
Le protocole décrit ci-dessus fournit une méthode robuste pour la modification des histones à haut débit ChIP-Seq d’une manière semi-autonome et micro-échelle. La méthode limite le temps de travail pratique et augmente la reproductibilité par rapport au ChIP-Seq manuel. Des études antérieures réalisées en laboratoire utilisaient le ChIP manuel sur des répliques techniques et obtenaient une moyenne de corrélation de Spearman de 0,505, cependant, avec le système semi-automatisé, la corrélation de Spearman entre différents donneurs avec une moyenne de cellules NK de 0,66 (Tableau supplémentaire 2). Cela a également été complété avec environ 40% de temps pratique en moins. La méthode décrite ici a été optimisée pour les modifications d’histones (H3K27ac montré ici, mais le protocole ne devrait pas nécessiter de modification pour d’autres) et ne nécessiterait que des modifications mineures pour être implémentée pour TF ChIP-Seq. Malgré la qualité de l’anticorps, la principale modification serait pour le temps de sonication et potentiellement les tampons utilisés pendant l’IP. Habituellement, pour les tests TF ChIP, la méthode peut mieux fonctionner avec des fragments de chromatine légèrement plus longs (avec une plage d’environ 350-800 bp) car les complexes TF:ADN sont probablement moins capables d’être maintenus par une sonication rigoureuse6. Les tampons peuvent également avoir besoin de passer à un mélange personnalisé ou à d’autres kits disponibles dans l’industrie, car les TF peuvent se comporter différemment des modifications d’histones.
Bien que le manipulateur automatique de liquide ChIP ait été testé pour aussi peu que 10 000 cellules, il y a eu une diminution notable de la reproductibilité à des concentrations de chromatine plus faibles. Pour cette raison, le protocole n’a pas été recommandé à moins de 10 000 cellules, 100 000 cellules étant les conditions optimales. Le protocole a également été complété à l’aide de tampons ChIP de l’industrie, ce qui représentait une dépense supplémentaire, mais fournissait des données de meilleure qualité. Le protocole pourrait être modifié en ce qui concerne les conditions de sonication (tant que la chromatine cisaillée est maintenue dans la même plage), les tampons pourraient être personnalisés pour l’immunoprécipitation (IP; une optimisation peut être nécessaire), ou le manipulateur de liquide ChIP ne peut pas être utilisé. Une limitation du protocole est l’utilisation du manipulateur de liquide ChIP, qui peut être un investissement coûteux et ne peut exécuter que 16 échantillons à la fois. Le manipulateur de liquide ChIP est limité aux réactions à petite échelle et le nombre de cellules supérieur à un million n’est pas recommandé. Cependant, le protocole pourrait être complété sans lui, en complétant les étapes IP et de lavage manuellement. Si l’IP et les lavages ont été effectués à la main, le temps nécessaire pour terminer le test augmentera et la reproductibilité peut diminuer, mais ce guide sera toujours utile pour mener une expérience ChIP-Seq de haute qualité. Il est à noter que d’autres manipulateurs de liquides pourraient être adaptés pour exécuter des réactions ChIP semi-automatisées.
Pour résumer, les principaux avantages de ce système sont la nature à haut débit, car les étapes IP et de lavage sont effectuées de manière autonome. En tant que tel, des cycles séquentiels d’expériences ChIP peuvent être complétés, permettant à jusqu’à 48 échantillons d’être entièrement traités et prêts pour le séquençage en 5 jours, avec un temps pratique limité par rapport aux expériences ChIP-Seq manuelles. Un autre avantage est la reproductibilité accrue puisque ChIP-Seq peut être difficile à obtenir des résultats hautement reproductibles. D’autres méthodes nécessitent soit des cellules vivantes, des systèmes complexes de micro-pipetage, soit le travail à effectuer à la main. Ce système devra être optimisé pour les échantillons à faible entrée (10 000 cellules <), permettant ainsi des réactions ChIP unicellulaires. Le système peut également être adapté aux nouvelles méthodes ChIP, telles que PLAC-Seq et HiChIP22,23.
Déclarations de divulgation
Les auteurs n’ont rien à divulguer.
Remerciements
Nous remercions les membres du laboratoire Vijayanand pour leur aide technique et leurs discussions constructives, ainsi que le Dr Sharron Squazzo et M. Geoffrey Berguet de Diagenode pour leur assistance technique avec le sonicator et la machine de traitement des liquides ChIP. Ce travail a été soutenu par les subventions des NIH AI108564, R01HL114093, S10RR027366 (BD FACSAria II) et S10OD016262 (Illumina HiSeq 2500).
matériels
| Name | Company | Catalog Number | Comments |
| 200 µl tube strips (8 tubes/strip) + cap strips | Diagenode | C30020002 | Strip tubes for use on the IP Star; ChIP 8-tube strip |
| AMPure XP for PCR Purification | Beckman Coulter | A63880 | SPRI beads |
| Axygen 0.6 mL MaxyClear Snaplock Microcentrifuge Tube | Corning | MCT-060-C | 0.65 mL low binding tube |
| Bioruptor Pico Sonicator | Diagenode | B01060010 | Sonicator used in the lab but others can be used |
| ChIP DNA Clean & Concentrator (Capped Columns) | Zymo Research | D5205 | DNA clean-up kit |
| Dynabeads Protein A for Immunoprecipitation | ThermoFisher | 10001D | |
| EDTA (0.5 M), pH 8.0, RNase-free | ThermoFisher | AM9260G | |
| EGTA pH 8.0 | Millipore Sigma | E3889-25G | |
| Eppendorf ThermoMixer C | Eppendorf | 2231000667 | |
| Formaldehyde solution | Millipore Sigma | 252549-1L | |
| Glycine | Millipore Sigma | 50046-250G | |
| H3K27ac polyclonal antibody - Premium | Diagenode | C15410196 | |
| HEPES (1 M) pH 7.5 | ThermoFisher | 15630080 | |
| IDT for Illumina Nextera DNA Unique Dual Indexes | Illumina | 20027213 | |
| Illumina Tagment DNA Enzyme and Buffer Small Kit | Illumina | 20034197 | |
| IP-Star Compact Automated System | Diagenode | B03000002 | Automated system for ChIP-Seq studies; ChIP liquid handler |
| KAPA HiFi HotStart ReadyMix | Roche | KK2601 | PCR mix |
| Medium reagent container for SX-8G IP-Star Compact | Diagenode | C30020003 | |
| MgCl2 (magnesium chloride) (25 mM) | ThermoFisher | R0971 | |
| N,N-Dimethylformamide | Millipore Sigma | D4551-250ML | CAUTION - low flash point |
| NaCl (5 M), RNase-free | ThermoFisher | AM9760G | |
| PBS (10X), pH 7.4 | ThermoFisher | 70011044 | |
| PCR Flex-free 8-tube stripes, attached individual optical caps | USA Scientific | 1402-4700 | 8 strip tubes, 0.2 mL 8-tube strip |
| Proteinase Inhibitor Cocktail | Millipore Sigma | P8340 | |
| Proteinase K Solution (20 mg/mL), RNA grade | ThermoFisher | 25530049 | |
| PureLink RNase A (20 mg/mL) | ThermoFisher | 12091021 | |
| Quant-iT PicoGreen dsDNA Reagent | ThermoFisher | P7581 | Used in the flourescence quantification |
| QuantStudio 6 Flex Real-Time PCR System | ThermoFisher | 4485699 | qPCR |
| ROX Reference Dye | ThermoFisher | 12223012 | |
| Sodium butyrate | Millipore Sigma | 303410-100G | |
| SYBR Gold Nucleic Acid Gel Stain (10,000X Concentrate in DMSO) | ThermoFisher | S11494 | nucleic acid dye |
| SYBR Green I Nucleic Acid Gel Stain - 10,000X concentrate in DMSO | ThermoFisher | S7563 | |
| TE Buffer | ThermoFisher | 12090015 | |
| Tips (bulk) | Diagenode | C30040020 | Tips for the IP Star |
| True MicroChIP Kit | Diagenode | C01010130 | Contains all the buffers for the IP; ChIP kit |
| UltraPure 1M Tris-HCI, pH 8.0 | ThermoFisher | 15568025 | |
| UltraPure SDS Solution, 10% | ThermoFisher | 24730020 |
Références
- Barski, A., et al. High-resolution profiling of histone methylations in the human genome. Cell. 129 (4), 823-837 (2007).
- Johnson, D., Mortazavi, A., Myers, R., Wold, B. Genome-Wide Mapping of in Vivo Protein-DNA Interactions. Science. 316, 1497-1502 (2007).
- Mikkelsen, T. S., et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 448 (7153), 553-560 (2007).
- Furey, T. S. ChIP-seq and beyond: new and improved methodologies to detect and characterize protein-DNA interactions. Nature Reviews Genetics. 13 (12), 840-852 (2012).
- Seumois, G., et al. Epigenomic analysis of primary human T cells reveals enhancers associated with TH2 memory cell differentiation and asthma susceptibility. Nature Immunology. 15 (8), 777-788 (2014).
- Schmiedel, B. J., et al. 17q21 asthma-risk variants switch CTCF binding and regulate IL-2 production by T cells. Nature Communication. 7, 13426(2016).
- Ai, S., et al. Profiling chromatin states using single-cell itChIP-seq. Nature Cell Biology. 21 (9), 1164-1172 (2019).
- Brind'Amour, J., et al. An ultra-low-input native ChIP-seq protocol for genome-wide profiling of rare cell populations. Nature Communication. 6, 6033(2015).
- Kaya-Okur, H. S., et al. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nature Communication. 10 (1), 1930(1930).
- Youhanna Jankeel, D., Cayford, J., Schmiedel, B. J., Vijayanand, P., Seumois, G. An Integrated and Semiautomated Microscaled Approach to Profile Cis-Regulatory Elements by Histone Modification ChIP-Seq for Large-Scale Epigenetic Studies. Methods Molecular Biology. 1799, 303-326 (2018).
- Schmidl, C., Rendeiro, A. F., Sheffield, N. C., Bock, C. ChIPmentation: fast, robust, low-input ChIP-seq for histones and transcription factors. Nature Methods. 12 (10), 963-965 (2015).
- Skene, P. J., Henikoff, S. An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. Elife. 6, 21856(2017).
- Schmiedel, B. J., et al. Impact of Genetic Polymorphisms on Human Immune Cell Gene Expression. Cell. 175 (6), 1701-1715 (2018).
- Landt, S. G., et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Research. 22 (9), 1813-1831 (2012).
- Chen, L., et al. Genetic Drivers of Epigenetic and Transcriptional Variation in Human Immune Cells. Cell. 167 (5), 1398-1414 (2016).
- Lienhard, M., Grimm, C., Morkel, M., Herwig, R., Chavez, L. MEDIPS: genome-wide differential coverage analysis of sequencing data derived from DNA enrichment experiments. Bioinformatics. 30 (2), 284-286 (2014).
- Engel, I., et al. Innate-like functions of natural killer T cell subsets result from highly divergent gene programs. Nature Immunology. 17 (6), 728-739 (2016).
- Grosselin, K., et al. High-throughput single-cell ChIP-seq identifies heterogeneity of chromatin states in breast cancer. Nature Genetics. 51 (6), 1060-1066 (2019).
- Rotem, A., et al. Single-cell ChIP-seq reveals cell subpopulations defined by chromatin state. Nature Biotechnology. 33 (11), 1165-1172 (2015).
- Wang, Q., et al. CoBATCH for High-Throughput Single-Cell Epigenomic Profiling. Molecular Cell. 76 (1), 206-216 (2019).
- Consortium, E. P. An integrated encyclopedia of DNA elements in the human genome. Nature. 489 (7414), 57-74 (2012).
- Fang, R., et al. Mapping of long-range chromatin interactions by proximity ligation-assisted ChIP-seq. Cell Research. 26 (12), 1345-1348 (2016).
- Mumbach, M. R., et al. HiChIP: efficient and sensitive analysis of protein-directed genome architecture. Nature Methods. 13 (11), 919-922 (2016).
- Diagenode. The Ultimate Guide for Chromatin Shearing Optimization with Bioruptor Standard and Plus. Diagenode. , (2012).
Erratum
Formal Correction: Erratum: A Semiautomated ChIP-Seq Procedure for Large-scale Epigenetic Studies
Posted by JoVE Editors on 9/14/2020. Citeable Link.
An erratum was issued for: A Semiautomated ChIP-Seq Procedure for Large-scale Epigenetic Studies. An author's name was updated.
The name was corrected from:
Pandurangan Vijayanad
to:
Pandurangan Vijayanand
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.