
Method Article
Perles chargeant des protéines et des acides nucléiques dans des cellules humaines adhérentes
Dans cet article
Résumé
La charge de billes introduit des protéines, des plasmides et des particules dans les cellules adhérentes des mammifères. Cette technique de chargement cellulaire est peu coûteuse, rapide et n’affecte pas considérablement la santé cellulaire. Il est le mieux adapté à l’imagerie de cellules vivantes.
Résumé
De nombreuses expériences d’imagerie de cellules vivantes utilisent des particules exogènes (p. ex., peptides, anticorps, billes) pour étiqueter ou fonctionner dans les cellules. Cependant, l’introduction de protéines dans une cellule à travers sa membrane est difficile. La sélection limitée de méthodes actuelles est difficile avec une faible efficacité, nécessite des équipements coûteux et techniquement exigeants, ou fonctionne dans des paramètres étroits. Ici, nous décrivons une technique relativement simple et rentable pour charger l’ADN, l’ARN et les protéines dans des cellules humaines vivantes. La charge de billes induit une perturbation mécanique temporaire de la membrane cellulaire, permettant aux macromolécules de pénétrer dans les cellules adhérentes et vivantes des mammifères. À moins de 0,01 USD par expérience, le chargement des billes est la méthode de chargement de cellules la moins coûteuse disponible. De plus, la charge en billes ne stresse pas considérablement les cellules et n’a pas d’impact sur leur viabilité ou leur prolifération. Ce manuscrit décrit les étapes de la procédure de chargement des perles, les adaptations, les variations et les limitations techniques. Cette méthodologie est particulièrement adaptée à l’imagerie des cellules vivantes, mais fournit une solution pratique pour d’autres applications nécessitant l’introduction de protéines, de perles, d’ARN ou de plasmides dans des cellules de mammifères vivantes et adhérentes.
Introduction
Le chargement de macromolécules dans des cellules de mammifères nécessite une méthodologie qui leur permet de traverser la membrane plasmique de la cellule1. Plusieurs méthodes peuvent introduire des plasmides dans les cellules de mammifères par transfection, y compris la transfection liposomale2 et la transfection diéthylaminoéthyl-dextran3. Cependant, les méthodes de chargement de protéines ou de particules imperméables à la membrane dans les cellules sont plus limitées.
Plusieurs techniques ont contourné cet obstacle difficile en utilisant diverses stratégies. Tout d’abord, la microinjection délivre des particules à travers une micropipette dans des cellules vivantes au microscope4. Bien qu’elle soit sans doute la méthode la plus contrôlée et la moins invasive, cette technique est relativement peu débitée car les cellules doivent être chargées une par une. De plus, la microinjection nécessite un équipement spécialisé et est techniquement exigeante.
Deuxièmement, l’électroporation est un moyen d’électro-injecter des protéines dans les cellules via une perturbation membranaire induite par la tension5,6,7. Cependant, cette méthode nécessite à nouveau un équipement spécialisé et coûteux, et le choc peut causer un stress cellulaire et la mortalité. De plus, les cellules doivent être trypsinisées avant l’électroporation, puis replaquées, ce qui limite le délai auquel les cellules peuvent être étudiées après l’électroporation.
Troisièmement, les membranes cellulaires peuvent être chimiquement modifiées pour une perméabilisation temporaire et réversible8,9. La charge de streptolysine-O insère une endotoxine dans les membranes cellulaires, qui forme des pores temporaires, permettant aux particules exogènes imperméables à la membrane, y compris les protéines et les plasmides d’ADN, d’entrer dans les cellules10. Après une récupération de 2 heures, environ la moitié des cellules réparent ces pores et arrêtent l’internalisation des particules de la solution. Cependant, cette technique nécessite un long temps de récupération et est incompatible avec les types de cellules qui ne peuvent pas tolérer les endotoxines.
Quatrièmement, la perturbation mécanique charge les particules dans les cellules par perturbation physique de la membrane cellulaire11. Cela peut être fait de plusieurs façons, y compris le grattage, le grattage et le roulement des perles au sommet des cellules12,13. Dès 1987, des billes ont été utilisées pour charger mécaniquement des protéines dans les cellules14. Plus récemment, la technique de chargement des billes a été optimisée et adaptée au-delà des protéines pour inclure la charge en plasmides et en ARN, comme décrit ici.
Le chargement de billes est une méthode facile, peu coûteuse et rapide pour charger des protéines et des plasmides dans des cellules humaines adhérentes. Les perles de verre sont brièvement roulées sur les cellules, perturbant temporairement leur membrane cellulaire. Cela permet aux particules en solution d’entrer. Comme la charge de billes a une faible efficacité, elle convient mieux aux expériences de microscopie à molécule unique ou à cellule unique. La charge de billes peut introduire une grande variété de protéines, y compris des anticorps fragmentés (Fab),15,16 protéines purifiées comme les scFvs,17 intracorps,18, 19ou des protéines de couche d’ARNm, par exemple, la protéine de couche MS2 (MCP)20,21. Les vecteurs d’expression plasmidique peuvent également être ajoutés à la solution protéique et chargés simultanémentde billes 22,23,24,25.
Au-delà des protéines et des plasmides, des molécules aussi grosses que des billes de polystyrène de 250 nm ont été introduites dans les cellules via la charge de billes (communication personnelle). Le chargement des perles est incroyablement peu coûteux, coûtant moins de 0,01 USD par expérience dans les matériaux et ne nécessitant aucun équipement coûteux supplémentaire. Le coût est encore réduit en minimisant la quantité de sondes utilisées par expérience, car seules les cellules du micropuit central de 14 mm de diamètre d’une chambre d’imagerie sont chargées. Il convient de noter que la zone de chargement limitée signifie que le chargement des billes n’est pas idéal pour le chargement des cellules en vrac.
Ce manuscrit présente le processus de chargement des perles, y compris la façon de construire l’appareil de chargement des perles et d’effectuer une expérience. Il montre que les protéines, l’ARN et l’ADN peuvent être chargés dans différents types de cellules et que deux protéines différentes, chargées simultanément de billes, ont des concentrations cellulaires fortement corrélées et une variance relativement faible. Les variations du protocole en fonction du type de cellule et de la charge en protéines, plasmides ou ARN sont également discutées. Bien que l’on pense que les perles perforent et perturbent la membrane cellulaire, lorsqu’elles sont effectuées de manière appropriée, le processus de chargement des billes ne déloge qu’un petit nombre de cellules du fond de la chambre d’imagerie. Après une courte période de récupération, les cellules continuent de croître et de se diviser. Cette méthodologie est idéale pour les expériences de microscopie de cellules vivantes, y compris le suivi des protéines et de l’ARN à molécule unique, la détection de modifications post-traductionnelles, l’observation de mécanismes cellulaires dynamiques ou la surveillance de la localisation subcellulaire15,16,22,26,27.
Protocole
1. Nettoyez, stérilisez et séchez les billes de verre pour éviter l’agglutination et assurer même la propagation sur les cellules.
- Stériliser environ 5 mL de billes de verre dans de l’hydroxyde de sodium (NaOH). Mesurez les billes dans un tube conique de 50 mL. Ajouter 25 mL de 2 M naOH et mélanger doucement à l’aide d’un agitateur ou d’un rotateur pendant 2 h.
- Décantez le NaOH, en conservant autant de perles que possible. Si les billes sont en suspension, faites tourner brièvement le tube de perles dans une centrifugeuse (1 min à ~1000 × g,température ambiante).
- Lavez soigneusement les billes avec de l’eau de qualité culture cellulaire jusqu’à ce que le pH soit neutre (utilisez une bandelette de test de pH sur l’éluant pour confirmer un pH neutre). Décantez l’eau de lavage à chaque fois, comme auparavant.
- Lavez soigneusement les perles avec 100% d’éthanol 2-3x. Décantez l’éthanol à chaque fois, comme auparavant.
- Séchez les perles. Saupoudrer les perles pour former une fine couche à l’intérieur d’un récipient stérile (comme une boîte de Petri de 10 cm). En laissant le récipient ouvert, laissez les perles sécher à l’air libre dans une armoire de biosécurité pendant la nuit. Assurez-vous que les perles sont complètement sèches en tapotant ou en secouant doucement le récipient et en vérifiant que les perles ont une texture sablonneuse sans agglutination ni desquamation.
- Stériliser les billes sèches aux UV pendant 15 min.
2. Assemblez l’appareil de chargement de billes.
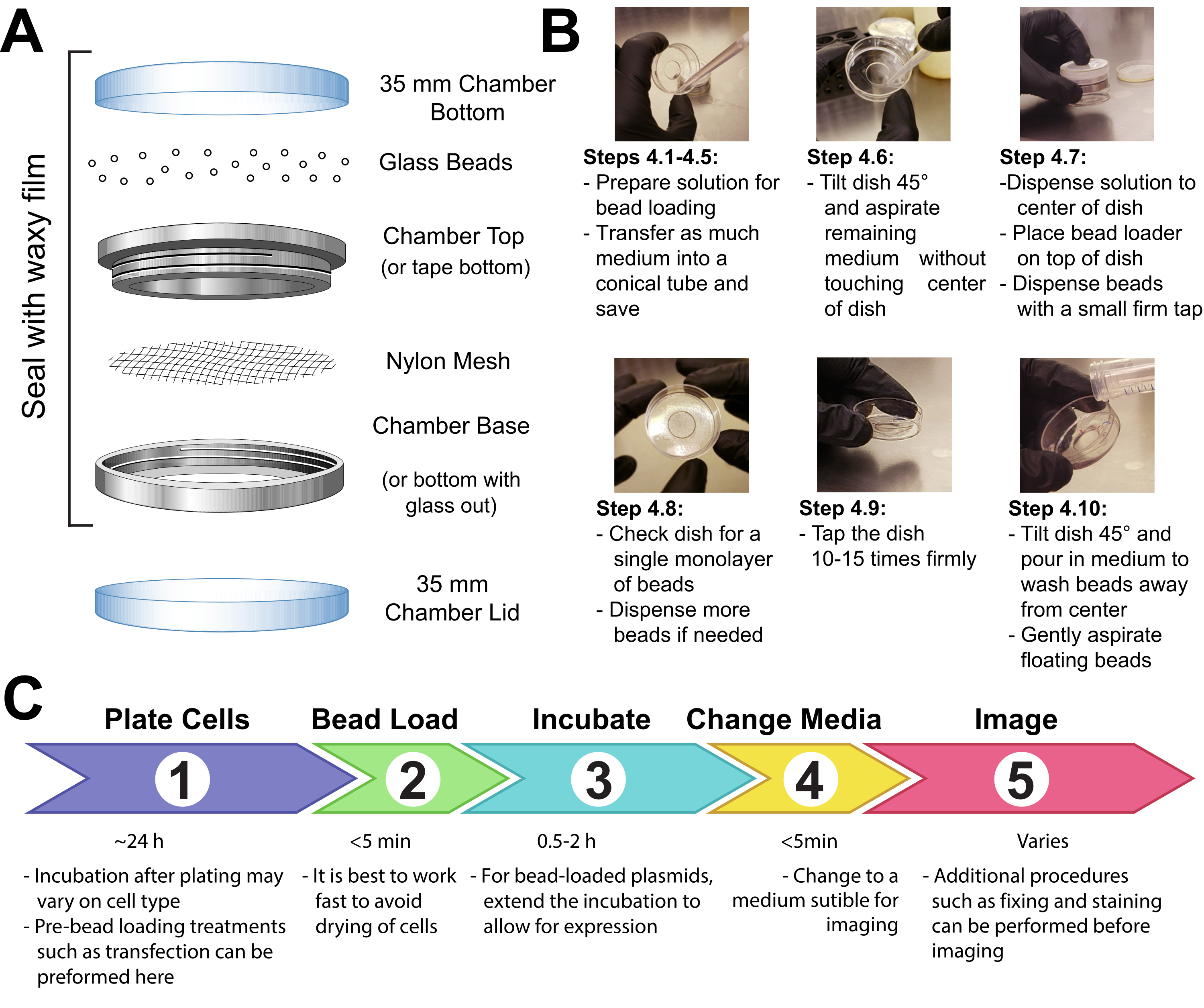
- Fixer une tache de maille (polypropylène ou matériau équivalent, ouvertures de 105 μm pour permettre aux perles de passer) pour couvrir toute l’ouverture de la chambre de maintien des perles avec du ruban adhésif ou en serrant la maille entre les extrémités mâle et femelle d’une chambre d’imagerie métallique réutilisable (Figure 1A).
- Stériliser l’appareil par UV pendant 15 min. Ajoutez les perles à l’appareil et scellez-le hermétiquement avec un film cireux.
REMARQUE: Il est essentiel que les perles soient complètement propres et sèches à cette étape. Ils doivent être lâches et avoir l’air sablonneux sans touffes. S’ils n’apparaissent pas, lavez à nouveau et séchez complètement les perles. - Conservez l’appareil dans un récipient scellé et sec desséché par du gel de silice ou un autre milieu dessicant. Si les perles deviennent humides, ce qui sera apparent par l’agglutination des perles, séchez soigneusement et stérilisez le chargeur de perles et remplacez-le par des perles fraîches.
REMARQUE: Toutes ces précautions empêcheront toute moisissure ou bactérie de se développer sur ou autour des perles dans le chargeur de perles. L’appareil de chargement de billes peut être fabriqué de différentes manières. Voir les détails dans la discussion.
3. Préparez des chambres à fond de verre de cellules adhérentes.
- Ensemencez des cellules de mammifères adhérentes sur une chambre à fond de verre de 35 mm. Assurez-vous que les cellules sont confluentes à environ 80 % au moment du chargement des billes. (Voir le tableau 1 pour plus d’informations sur les différents types de cellules et des notes sur l’efficacité de la charge de billes dans différents types de cellules.)
REMARQUE: Les cellules ne peuvent être ensemencées que dans le micropuit au centre de la chambre pour conserver le nombre de cellules utilisées. - Incuber les cellules dans des conditions normales jusqu’à ce qu’elles adhèrent complètement au verre.
REMARQUE: Il est essentiel que la densité cellulaire soit suffisamment élevée et que les cellules adhèrent solidement au verre. Si ces exigences ne sont pas respectées, les cellules se décolleront probablement pendant le chargement des billes. Le délai entre l’ensemencement cellulaire et le chargement des billes peut être allongé pour assurer une adhésion et une fluidité cellulaires appropriées.
4. Cellules de chargement de perles
REMARQUE: Si nécessaire, lavez brièvement les cellules avec une solution saline tamponnée au phosphate (PBS), puis ajoutez 2 mL du milieu optimal. Incuber pendant au moins 30 min.
- Faire une solution de 3-8 μL contenant les plasmides, protéines et/ou particules désirés. Utilisez ~1 μg (0,1-1 pmol) de chaque type de plasmide et ~0,5 μg (0,01 nmol) de protéine, selon les besoins expérimentaux. Utilisez un tube à faible rétention pour les protéines afin qu’elles ne soient pas laissées sur les parois du tube. Porter la solution à un minimum de 3 μL avec pbS et ajuster le volume de la solution pour recouvrir toute la surface des cellules à charger (c.-à-d. le micropuit de la chambre, figure 1B).
- Mélangez soigneusement la solution en pipetant de haut en bas et/ou en agitant le tube. Faites tourner brièvement la solution jusqu’au fond du tube dans une microfuges de table.
- Transférer la solution de chargement de billes et la chambre des cellules dans une hotte de culture tissulaire. Effectuez les étapes restantes dans la hotte de culture tissulaire en utilisant une technique stérile.
- Retirez le milieu des cellules et stockez-le temporairement dans un tube stérile. Aspirez doucement tout le milieu autour des bords de la chambre, inclinez la chambre à un angle d’environ 45 ° et retirez la goutte restante de média dans le micropuit central. Lors de l’enlèvement du milieu, assurez-vous d’éviter de laisser la pointe de la pipette toucher le verre, ce qui peut entraîner un pelage et une perte de cellules. Passez rapidement à l’étape suivante afin que les cellules ne soient pas sèches longtemps.
- Pipettez doucement la solution de chargement de billes sur le micropuit en verre au centre de la chambre. En option: Incuber avec un balancement doux pendant environ 30 s sans laisser la chambre sécher complètement.
- Disperser doucement une monocouche de billes de verre sur le dessus des cellules, de préférence à l’aide d’un appareil de chargement de billes(Figure 1A). Assurez-vous que les perles recouvrent complètement les cellules du micropuit à fond de verre.
- Pincez la chambre avec deux doigts, tapotez-la contre la surface du capot en la soulevant d’environ 2 pouces et en la faisant descendre fermement. Utilisez une force approximativement équivalente à la chute du plat de cette hauteur. Répétez l’opération pour un total de ~10 taps.
REMARQUE: Assurez-vous que les robinets ne pelent pas substantiellement les cellules. Le tapotement peut être optimisé pour le type de cellule. Si les cellules se chargent mal, appuyez plus fort; cependant, si de nombreuses cellules se détachent, appuyez plus légèrement. - Ajoutez doucement du milieu dans la chambre en pipetant lentement sur le côté en plastique de la chambre. Essayez d’aspirer les perles flottantes sans perturber les cellules. Ajoutez plus de médias préchauffés à cette étape si trop de médias ont été supprimés. Incuber les cellules pendant 0,5-2 h dans l’incubateur.
- Stérilisez les UV du chargeur de billes pendant 15 minutes avant de le remettre en stockage dans des conditions de desséchéité.
- Ajouter un colorant (p. ex., colorant de ligand DAPI ou HaloTag, si l’expérience l’exige) aux cellules conformément au protocole recommandé par le fabricant.
- Lavez les cellules 3x avec du milieu avant l’imagerie pour enlever les billes et les composants de charge excédentaire en solution. Évitez de pipeter directement sur les cellules pour les empêcher de peler.
5. Imagerie des cellules chargées de perles
- Imagez les cellules immédiatement ou lorsque l’expérience l’exige. Utilisez un microscope capable de capturer la fluorescence (lasers ou source de lumière monochromatique). S’assurer que les longueurs d’onde d’excitation sont appropriées pour le fluorophore ou le colorant choisi (p. ex., lumière de longueur d’onde de 488 nm pour la protéine de fluorescence verte (GFP)).
REMARQUE: Les protéines chargées de billes peuvent être imagées une fois que les cellules se sont rétablies (dès 30 minutes après le chargement pour les lignées cellulaires décrites ici). L’expression plasmidique prend ≥2 h en fonction des éléments du vecteur d’expression(Figure 1C, et explication plus détaillée dans la discussion). L’imagerie des cellules chargées de billes peut être réalisée sur n’importe quel microscope équipé des sources fluorescentes appropriées associées à des sondes chargées, une caméra capable de capturer des images de fluorescence, telle qu’un dispositif à couplage de charge multiplicateur d’électrons (EMCCD) ou une caméra scientifique à semi-conducteurs à oxyde métallique complémentaire (sCMOS), et un incubateur pour contrôler la température, l’humidité et le dioxyde de carbone. Pour un guide sur la microscopie à fluorescence, reportez-vous à la section 27.
Résultats
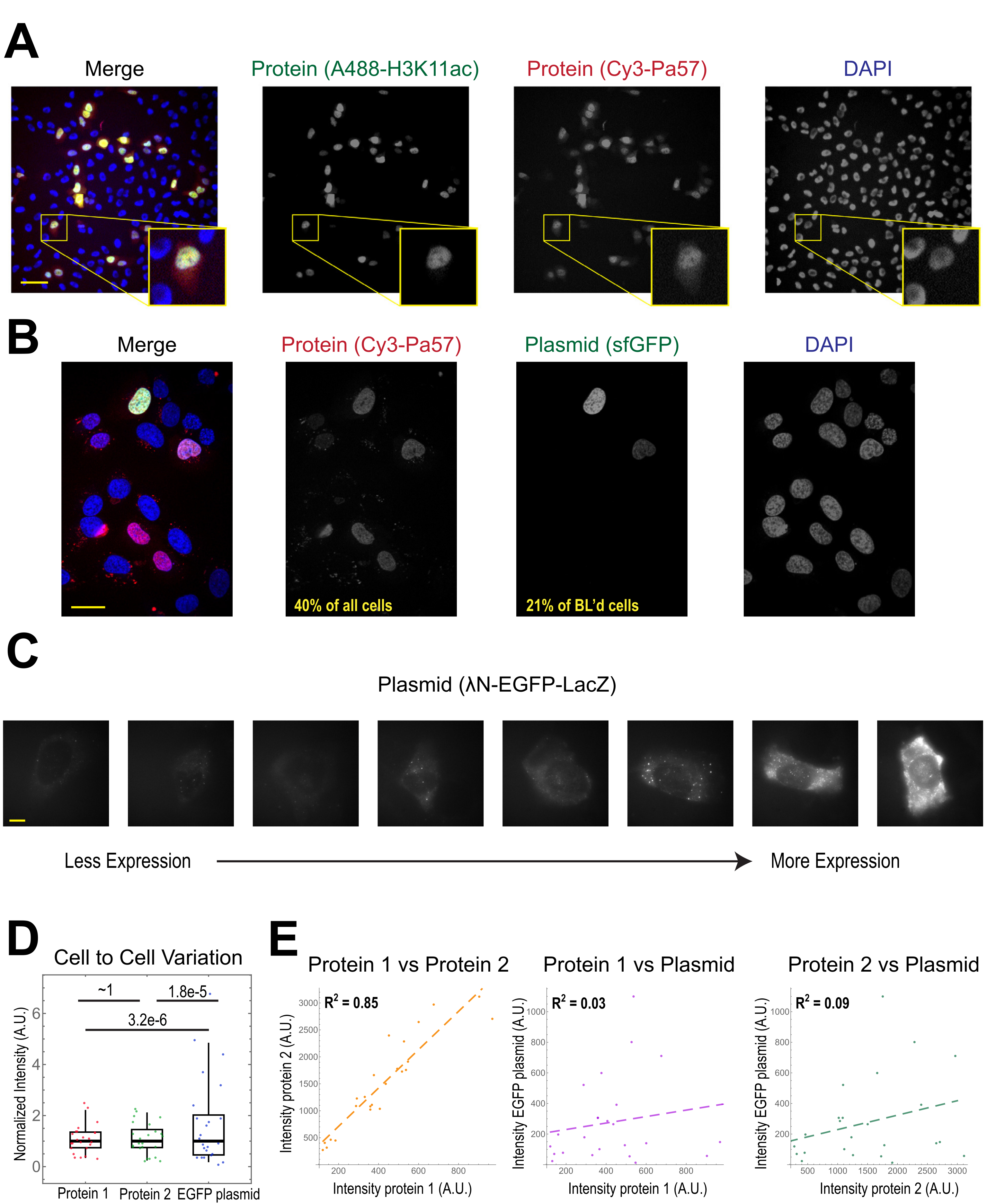
L’application la plus courante de la charge de billes est d’introduire un ou plusieurs types de protéines dans les cellules humaines adhérentes. Pour illustrer cela, les cellules ont été chargées de billes avec une solution d’une protéine Fab conjuguée Cy3 et Alexa488. Bien que toutes les cellules du micropuit n’aient pas été chargées de billes, les cellules chargées avaient presque toujours des protéines étiquetées Cy3 et Alexa488 ensemble(Figure 2A). Selon une estimation antérieure, lorsque 0,5 microgramme de Fab dilué dans 4 microlitres est chargé de billes29, comme dans la figure 2A, chaque cellule est chargée d’environ 106 molécules Fab.
L’ADN plasmidique codant pour le GFP (1 μg d’ADN plasmidique, 1,8 μL d’une solution de 557 ng/μL) et 0,5 μg de Fab marqué Cy3 a également été introduit dans les cellules par chargement de billes, puis exprimé et visualisé(Figure 2B). La fluorescence GFP indiquait que le plasmide coté GFP était non seulement chargé dans les cellules, mais également exprimé. Ainsi, dans la même cellule, la charge de billes peut introduire une sonde protéique (par exemple, Fab marqué Cy3) et un plasmide rapporteur (par exemple, GFP), comme effectué dans ce laboratoire précédemment22,23,24. Nous avons déterminé que 40 % des cellules étaient chargées de billes de protéine Fab et que 21 % des cellules chargées de billes exprimaient le plasmide co-chargé, comme le montrent les champs de vision représentatifs de la figure 2B. Typiquement, chaque chambre est chargée de 1-2 μg de plasmide, à peu près la même quantité que la lipofection.
Les cellules chargées de billes expriment des niveaux très variables de plasmides (Figure 2C, D). Pour mesurer spécifiquement cela, nous avons utilisé le test du rapport de Fisher pour comparer les distributions des données d’intensité des protéines et des plasmides. Les résultats ont montré que bien que les protéines 1 et 2 aient des distributions d’intensité similaires (p = ~1), chaque protéine avait une distribution significativement plus petite que le plasmide (p = 3,2e-6 et 1,8e-5). Bien que cela puisse être dû à la variabilité du nombre de plasmides chargés par cellule, la plus grande source de variabilité peut provenir des nombreuses étapes requises pour l’expression du plasmide qui sont susceptibles de varier considérablement d’une cellule à l’autre, y compris l’importation dans le noyau cellulaire, la transcription et la traduction. En revanche, les niveaux de protéines chargées de billes avaient une légère variance de cellule à cellule, et les niveaux de deux protéines chargées simultanément étaient fortement corrélés l’un à l’autre (Figure 2D, E).
L’expression des plasmides peut être observée dès 2 à 4 heures après la charge des billes, mais peut se produire plus tard en fonction du moment où l’expression optimale des plasmides est obtenue. Nous vous recommandons d’effectuer un cours temporel pour déterminer la meilleure fenêtre d’expression pour un plasmide spécifique couvrant 2 à 24 heures après le chargement des billes. Cela peut être fait dans une chambre avec une imagerie à longue période ou en chargeant des billes et en échelonnant plusieurs chambres. Les cellules chargées de perles restent adhérentes et sont suffisamment saines pour croître et se diviser. Les cellules U2OS humaines chargées de perles ont été imagées directement avant, directement après et 24 heures après le chargement des perles. Une charge correcte des billes n’a eu presque aucun effet notable sur le nombre de cellules ou leur morphologie, comme le montre la figure 3A (à gauche, au milieu).
En revanche, une faible charge de perles avec trop de perles et une force de taraudage excessive sont représentées à la figure 3B. Cela a causé beaucoup de perte de cellules (grandes plaques de verre de couverture sans cellules et cellules détachées, flottantes, floues), une mauvaise morphologie cellulaire (cellules apparaissant arrondies vers le haut et mal adhérentes) et des grappes de perles restant sur le verre de couverture après le chargement des perles. Bien que l’on pense que les cellules subissent des dommages mécaniques lors du chargement des billes, les cellules se sont développées et ont proliféré dans la chambre correctement chargée de perles, comme en témoigne l’augmentation du nombre de cellules 24 h après le chargement des billes (Figure 3A, à droite). L’effet sur la viabilité cellulaire peut être évalué par divers essais, tels qu’un essai de bromure de 3-(4,5-diméthylthiazol-2-yl)-2,5-diphényltétrazolium (MTT), pour comparer les cellules chargées de billes aux cellules chargées de simulacres30. En outre, ce travail et les travaux antérieurs montrent que les cellules chargées de billes subissent une division cellulaire(Figure 3C et vidéo supplémentaire 1),et le moment de la mitose n’est pas affecté par la charge de perles31, ce qui constitue une preuve supplémentaire de la santé cellulaire soutenue après la charge de perles.
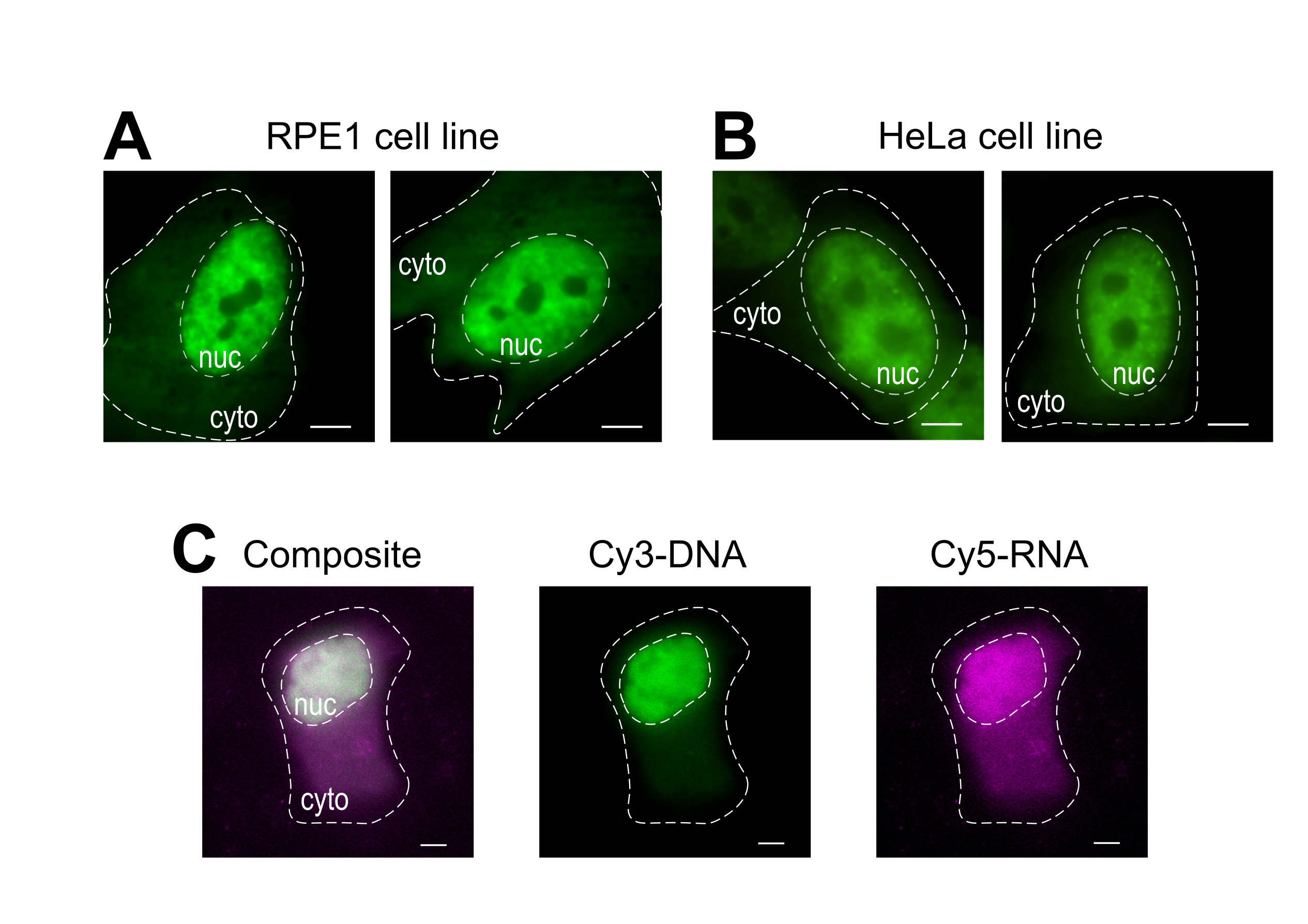
Le chargement de billes est une technique polyvalente, accueillant plusieurs lignées cellulaires adhérentes et diverses macromolécules. Ici, cette variété a été démontrée en chargeant les lignées cellulaires RPE1 et HeLa avec Fab (Figure 4A, B). Le tableau 1 fournit d’autres exemples de chargement de billes dans différentes lignées cellulaires, dans ce laboratoire et au-delà, et souligne certaines des différences nuancées entre les protocoles de chargement de perles d’autres laboratoires. Il convient de noter que le diamètre des billes de verre utilisées pour le chargement varie considérablement d’un laboratoire à l’autre, bien que la charge la plus efficace ait été trouvée pour les petites billes de 75 μm de diamètre dans plusieurs lignées cellulaires14. De plus, ce laboratoire a également commencé à charger de l’ARN par perles (données non présentées). La figure 4C montre une perle cellulaire U2OS représentative chargée d’un Cy5-ARN 9mer et d’un Cy3-ADN 28mer ensemble.

Figure 1: Appareil de chargement des billes, technique et chronologie Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Graphique 2. La charge en billes introduit une faible variabilité dans la concentration en protéines, mais une grande variabilité dans l’expression des plasmides. (A) Les cellules étaient chargées de billes avec 0,5 μg de chacune des Fab anti-H3K27 anti-H3K27 conjuguées à Alexa488 (vert) et de la Fab anti-RNAPII-Sérine 5-phosphorylé conjuguée à Cy3 (rouge) dans 4 μL de solution de chargement de billes. Les cellules ont été colorées en DAPI (bleu), puis immédiatement imagées en direct. Barres d’échelle = 20 μm. (B) Les cellules étaient chargées de billes avec 0,5 μg de protéine Fab (protéine 5-phosphorylée anti-RNAPII-sérine conjuguée À Cy3, rouge) et 1 μg de plasmide encodant le superdossier GFP-H2B (vert) dans 4 μL de solution de chargement de billes. Après 24 heures, les cellules ont été colorées en DAPI (bleu) et imagées en direct. Les barres d’échelle = 30 μm. (C-E) La protéine 1 (JF646-HaloLigand-marqué HaloTag-MCP), la protéine 2 (Cy3-conjugué anti-FLAG Fab) et un plasmide encodant EGFP (λN-EGFP-LacZ) ont été chargés ensemble dans des cellules. L’intensité totale dans chaque canal fluorescent a été mesurée dans un patch de 1,3 x 1,3 μm dans le cytoplasme de chaque cellule. N = 25 cellules. (C) Cellules représentatives exprimant le plasmide chargé de billes, λN-EGFP-LacZ. Les mêmes conditions et intensités d’imagerie ont été utilisées pour toutes les cellules. Les taches sont des agrégats de la protéine exprimée. Barres d’échelle = 10 μm. (D) Le graphique montre l’intensité totale de chaque cellule de la protéine 1, de la protéine 2 ou de l’EGFP exprimée à partir du plasmide. Chaque canal a été normalisé à la médiane. Les valeurs de P corrigées de Bonferroni ont été calculées par le test du rapport de Fisher pour déterminer si la distribution des données sur l’intensité des protéines ou des plasmides présente la même variabilité. Chaque point représente une cellule. (E) Les intensités totales des deux protéines, de la protéine 1 et du plasmide, ou de la protéine 2 et du plasmide, sont tracées l’une par rapport à l’autre. Les valeurs R2 calculées sont affichées. Chaque point représente une cellule. Abréviations : DAPI = 4′,6-diamidino-2-phénylindole ; EGFP = protéine fluorescente verte améliorée; A.U. = unités arbitraires; MCP = protéine de la couche MS2; RNAPII = ARN polymérase II. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 3: Les cellules chargées de billes restent adhérentes et sont suffisamment saines pour croître et se diviser. (A) Les cellules U2OS ont été chargées de perles avec 0,5 μg d’anti-FLAG Fab conjugué cy3 dans 4 μL de solution de chargement de billes. Les cellules ont été imagées directement avant, directement après le chargement des billes et 24 heures après le chargement des perles. Les flèches orange identifient les zones où les cellules se sont décollées pendant le chargement des perles. Barres d’échelle = 2 mm. (B) Image représentative des cellules U2OS chargées de composants de (A) mais avec un taraudage dur et trop de perles. La flèche rouge identifie les perles de verre supplémentaires. Barre d’échelle = 2 mm. (C) Les cellules U2OS ont été chargées avec 1,5 μg du plasmide smFLAG-KDM5B-15xBoxB-24xMS2 de 14,4 kbp, 0,5 μg de plasmide anti-FLAG Fab conjugué Cy3 (vert), 130 ng de HaloTag-MCP (magenta) dans 8 μL de solution de chargement de billes. Juste avant l’imagerie, le HaloTag a été coloré avec JF646-HaloLigand. Les boucles de tige MS2 de l’ARNm transcrites à partir du plasmide rapporteur sont marquées par MCP (taches magenta), et la protéine rapporteur traduite marquée FLAG est marquée par anti-FLAG Fab (colocalisation verte en ARNm). La protéine mature de marque Fab se localise dans le noyau. Cette cellule a été imagée 4-15 h après le chargement des perles. Les flèches jaunes identifient le noyau cellulaire avant et les noyaux après la division cellulaire. Barres d’échelle = 5 μm. Abréviation : MCP = protéine de la couche MS2. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 4: Variations du matériau de chargement de type de cellule du protocole de chargement de billes. (A-B) Les cellules RPE1(A)et HeLa(B)ont été chargées en billes de 1,5 μg d’une protéine Fab nucléaire (anti-RNAPII-Sérine 5-phosphorylation) dans 4 μL de solution de charge. Le noyau (nuc) et le cytoplasme (cyto) de chaque cellule sont marqués. Les cellules ont été imagées 6 h après avoir été chargées de perles. Barres d’échelle = 5 μm. (C) Les cellules U2OS humaines étaient chargées de billes avec des oligos Cy5-ARN 9mer (magenta) et Cy3-ADN 28mer (vert), 10 picomoles de chacun, dans 4 μL de solution de chargement de billes. Les cellules ont été imagées 4 h après avoir été chargées de perles. Tous les noyaux cellulaires sont mis en évidence par une ligne pointillée. Barres d’échelle = 5 μm. Abréviations : RNAPII = ARN polymérase II. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
| Lignée cellulaire | Type de cellule | Efficacité du chargement des billes | Notes/référence |
| Cellules souches (humaines) | Cellules souches embryonnaires | Difficile | * De nombreuses cellules se détachent pendant le chargement des billes si elles sont plaquées sur des plaques recouvertes de gélatine |
| 293 HEK | Cellules rénales embryonnaires humaines | Difficile | * Besoin de déposer une matrice de gel à la surface de la chambre d’imagerie avant le chargement des billes. Appuyez doucement lors du chargement des perles au début. |
| Neurones (rat) | Neurones embryonnaires primaires (e-18), dissociés | Très inefficace | *La charge efficace des billes des neurones n’a pas été observée à l’aide de ce protocole standard de chargement des billes. Cela pourrait être dû à la nature non adhérente des neurones ou à des dommages conséquents aux processus neuronaux. |
| MDCK (canin) | Cellules rénales canines de Madin-Darby | Voir McNeil et Warder (1987)14 | * Chargement de billes à faible efficacité14 |
| U2OS (humain) | Ostéosarcome | Protocole de chargement de billes standard | |
| HeLa (humain) | Cancer cervical | Protocole de chargement de billes standard | |
| RPE1 (humain) | Cellules épithéliales immortalisées avec hTERT | Protocole de chargement de billes standard | |
| HFF (humain) | Fibroblastes primaires du prépuce | Voir Besteiro et coll. (2009)31 | *Protocole modifié d’inclinaison au lieu de tapoter31 |
| BALB/c 3T3, NIH 3T3 et Swiss 3T3 (souris) | Fibroblastes embryonnaires | Voir Gilmore et Romer (1996)32, Emerson et al. (2014)33 et McNeil et Warder (1987)14 | *425–600 μm de billes de verre signalées32 |
| *Perles de verre usagées de 200 à 300 μm33 | |||
| *Des billes de verre de 75 μm ont donné de meilleurs résultats que 400 μm14 | |||
| DM (muntjac indien) | Fibroblastes cutanés | Voir Manders, Kimura et Cook (1999)34 | |
| CHO (hamster) | Cellules ovaraires de type épithélial | Voir Memedula et Belmont (2003)35 | *Perles de verre usagées de 425 à 600 μm35 |
| BAE (bovin) | Cellules endothéliales aortiques bovines (BAEC-11) | Voir McNeil et Warder (1987)14 | *Des billes de verre de 75 μm ont donné de meilleurs résultats que 400 μm14 |
| PtK-2 (Potomus tridaclylis) | Cellules rénales épithéliales | Voir McNeil et Warder (1987)14 | *Des billes de verre de 75 μm ont donné de meilleurs résultats que 400 μm14 |
| HUVEC (humain) | Cellules endothéliales de la veine ombilicale | Voir Gilmore et Romer (1996)32 | *Perles de verre usagées de 425 à 600 μm32 |
| J774 et J774.2 (souris) | cellules macrophages monocytaires | Voir Becker et coll. (2005)36 et McNeil et Warder (1987)14 | * Agitation douce (au lieu de tapoter) et perles de verre de 425 à 600 μm36 |
| MS-5 (souris) | cellules stromales de la moelle osseuse | Voir Molenaar et al. (2003)37 | |
| WPE1-NB11 (humain) | cellules épithéliales de la prostate | Voir Gilmore et Romer (1996)32 et | *Perles de verre usagées de 425 à 600 μm32 |
| Emerson et coll. (2014)33 | *Perles de verre usagées de 200 à 300 μm33 |
Tableau 1 : Chargement des billes dans différentes lignées cellulaires. Pour les lignées cellulaires qui n’ont pas encore été chargées de perles dans ce laboratoire, des références et des notes sur les variations du protocole sont fournies.
Vidéo supplémentaire 1 : Exemple d’une cellule chargée de perles subissant une division cellulaire. Les cellules U2OS ont été chargées avec 1,5 μg du plasmide smFLAG-KDM5B-15xBoxB-24xMS2 de 14,4 kbp, 0,5 μg de fab anti-FLAG conjugué Cy3 (vert), 130 ng de HaloTag-MCP (magenta) dans 8 μL de solution de chargement de billes. Juste avant l’imagerie, le HaloTag a été coloré avec JF646-HaloLigand. Les boucles de tige MS2 de l’ARNm transcrites à partir du plasmide rapporteur sont marquées par MCP (taches magenta), et la protéine rapporteure traduite marquée FLAG est marquée via anti-FLAG Fab (colocalisation verte en ARNm). La protéine mature de marque Fab se localise dans le noyau. Cette cellule a été imagée 4-15 h après le chargement des perles. Barre d’échelle = 10 μm. Abréviation : MCP = protéine de la couche MS2. Veuillez cliquer ici pour télécharger cette vidéo.
Discussion
La technique de chargement des billes décrite ici est une méthode rentable et rapide pour introduire des macromolécules et d’autres particules dans les cellules adhérentes. Ce procédé polyvalent peut charger des protéines (Figure 2A)15,16,26,27, une combinaison de protéines et de plasmides ( Figure2B,C)22,25, ARN ( Figure4C), 100 et 250 nm perles de polystyrène (correspondance personnelle), colorants synthétiques39 ou points quantiques34,40 . La charge de billes peut également avoir la capacité de charger d’autres types de particules imperméables à la membrane. Son application la plus fréquemment utilisée est le chargement d’anticorps ou de Fabs pour cibler les épitopes endogènes, tels que les modifications post-traductionnelles (PTM), dans les cellules vivantes. Les cibles, telles que les PTM, sont souvent difficiles à étiqueter dans les cellules vivantes sans sondes établies spécifiques au PTM, génétiquement codées41,42. En revanche, le chargement de billes peut introduire plusieurs types de sondes, de rapporteurs ou d’autres outils moléculaires ensemble dans la même cellule pour surveiller plusieurs lectures simultanément. Nous prévoyons que le chargement de billes sera une technique utile pour charger une variété de macromolécules ou de particules.
Un avantage majeur du chargement de perles est le faible coût: chaque expérience coûte moins de 0,01 USD. Un appareil de chargement de billes peut être fabriqué facilement en utilisant des matériaux peu coûteux coûtant au total ~ 150 $, ce qui est nettement moins cher que toute autre méthode de chargement de cellules. Le coût d’un appareil de chargement de billes peut être réduit à moins de 10 $ en remplaçant la chambre métallique réutilisable par une chambre en plastique. Pour cela, percez un trou dans une chambre de 35 mm ou retirez le verre d’une chambre à fond de verre de 35 mm, puis fixez solidement le maillage en place avec du ruban adhésif. Au lieu d’un appareil, le chargement des billes peut même être effectué à l’aide d’une pointe de pipette de 1000 μL à large alésage pour prévenir et saupoudrer des billes sur les cellules, bien que cette variation rende difficile l’aspersion d’une monocouche de billes sur les cellules (étape 4.6).
Un autre avantage de la charge en billes est que les cellules peuvent conserver une morphologie globale normale, se rétablir rapidement et continuer à croître et à se diviser, au moins pour les cellules U2OS, RPE1 et HeLa étudiées ici et pour les autres lignées cellulaires étudiées ailleurs (Figure 3; Figure 4A,B; Vidéo supplémentaire 1; et Tableau 1)31. Pendant le chargement des billes, les cellules subissent un stress physique et parfois se délogent et se pelent (~ 5% des cellules pelent dans des conditions optimales, mais une perte cellulaire plus importante peut se produire si la charge des billes est effectuée avec trop de force ou si trop de perles de verre sont chargées sur les cellules, comme illustré à la figure 3B). Cependant, les cellules chargées de billes qui restent attachées au couvercle semblent généralement saines et peuvent être imagées dès 30 minutes après le chargement des billes(Figure 3A). Nous accordons généralement aux cellules une période de récupération de 30 minutes, mais nous prévoyons que l’imagerie plus tôt après le chargement des billes est possible.
Un inconvénient majeur de cette technique est que les cellules doivent être capables de résister à un stress physique mineur pendant le chargement et rester solidement adhérentes au couvercle. Les lignées cellulaires ou les cellules mal ou non adhérentes cultivées sur des plaques revêtues (p. ex., HEK et cellules souches) se détachent souvent en tapotant doucement pendant le chargement des billes. De plus, l’expérience a montré que les neurones primaires sont trop sensibles pour la charge des billes.
La charge de billes est la mieux adaptée aux expériences à cellule unique ou à molécule unique. D’après notre expérience, la charge en billes a une efficacité de charge en protéines d’environ 20 à 40%, et ~ 20% des cellules chargées en perles ont également exprimé un plasmide co-chargé(Figure 2A,B). Ainsi, les plasmides à charge de billes peuvent être moins efficaces pour l’expression des protéines que les protéines purifiées à charge de billes, car les plasmides doivent non seulement pénétrer dans les cellules, mais aussi être exprimés (ce qui implique, entre autres, l’importation nucléaire, la transcription et la traduction, dont chacun peut réduire l’efficacité d’expression). La faible efficacité de l’expression plasmidique chargée de billes peut être contournée en utilisant des protocoles de transfection alternatifs, tels que la lipofection, avant les protéines de charge de billes ou les sondes16,27. De plus, l’incubation de cellules dans un milieu optimal pendant 30 minutes avant le chargement des billes peut aider à l’expression des plasmides. En raison de la faible expression plasmidique, la charge en billes n’a pas souvent été utilisée comme alternative à la transfection à base de lipofection dans ce laboratoire. La seule exception est lorsqu’une protéine purifiée, telle que Fab, doit être co-chargée, auquel cas il est très pratique de charger la protéine et le plasmide en même temps. De plus, pour les cellules qui ne répondent pas ou qui sont intolérantes à la lipofection, la charge en billes peut fournir une autre méthode, bien que peu efficace, pour l’expression transitoire des plasmides.
Déclarations de divulgation
Les auteurs n’ont aucun conflit d’intérêts à divulguer.
Remerciements
Nous sommes reconnaissants aux membres du laboratoire Stasevich pour les innombrables conversations qui ont contribué à améliorer et à développer ce protocole. Plus précisément, la Dre Linda Forero et le Dr Phil Fox pour obtenir des conseils sur le chargement de perles de différents types de cellules. Nous tenons à remercier sincèrement la Dre Yoko Hayashi-Takanaka, la Dre Yuko Sato et le Dr Hiroshi Kimura d’avoir partagé leur protocole de chargement des billes de verre. Nous sommes très reconnaissants au Dr Ashok Prasad et au Dr Diego Krapf d’avoir généreusement partagé leurs protocoles de chargement de billes pour introduire des particules inorganiques dans les cellules. Nous sommes reconnaissants au Dr Travis Sanders, Craig Marshall et au Dr Thomas Santangelo d’avoir généreusement partagé leur réactif d’ARN étiqueté. ALK, MNS, CAC, GG et TJS ont été soutenus par la subvention R35GM119728 des National Institutes of Health (NIH) et la subvention CAREER MCB-1845761 de la National Science Foundation (NSF), toutes deux à TJS. CAC a également été soutenu par le prix NSF NRT DGE-1450032.
matériels
| Name | Company | Catalog Number | Comments |
| 10 cm cell culture dishes | VWR | 82050-916 | Use to culture cells |
| 35 mm cell culture dishes | Falcon | 353001 | Use to construct bead loader |
| Attofluor Cell Chamber | Thermo Fisher Scientific | A7816 | Use to construct the custom bead loader |
| DMEM, high glucose, no glutamine | Thermo Fisher Scientific | 11960069 | Use in general cell culture |
| Drierite Indicating Absorbents | Thermo Fisher Scientific | 07-578-3B | Store the bead loader in a desiccator with these absorbent pellets |
| Fetal Bovine Serum | Atlas Biologics | F-0050-A | Use in general cell culture and as a supplement before bead loading |
| Glass beads, acid washed, ≤106 µm* | Millipore Sigma | G4649 | Sprinkle on cells to bead load plasmid DNA and proteins |
| Glass bottom dishes, 35 mm, #1.5, 14 mm glass | MatTek Corporation | P35G-1.5-14-C | Seed cells onto these chambers for imaging |
| L-Glutamine-200 mM | Thermo Fisher Scientific | 25030081 | Use to make DMEM + media |
| Opti-MEM, Reduced Serum Medium | Thermo Fisher Scientific | 31985070 | Optimal media for incubating cells before bead loading (optional step) |
| Parafilm | VWR | 52858-032 | waxy film used to construct bead loader |
| Penicillin-Streptomycin | Thermo Fisher Scientific | 15140-122 | Use to make DMEM + media |
| Phenol-free DMEM | Thermo Fisher Scientific | 31053036 | Use on cells before imaging |
| Phosphate Buffered Saline (PBS) | Thermo Fisher Scientific | AM9625 | Working stock of sterile 1X PBS |
| Sodium Hydroxide (NaOH) | Thermo Fisher Scientific | S318-500 | Use 2 M solution to wash glass beads |
| Spectra Mesh Woven Filters, Polypropylene, 105 µm opening | Spectrum Labs | 148496 | Use to construct bead loader |
| Trypsin | Thermo Fisher Scientific | 25300062 | Use in general cell culture |
Références
- Stewart, M. P., et al. In vitro and ex vivo strategies for intracellular delivery. Nature. 538 (7624), 183-192 (2016).
- Felgner, P. L., et al. Lipofection: a highly efficient, lipid-mediated DNA-transfection procedure. Proceedings of the National Academy of Sciences of the United States of America. 84 (21), 7413-7417 (1987).
- Schenborn, E. T., Goiffon, V. DEAE-dextran tansfection of mammalian cultured cells. Methods in Molecular Biology. 130, 147-153 (2000).
- Celis, J. E. Microinjection of somatic cells with micropipettes: comparison with other transfer techniques. Biochemical Journal. 223 (2), 281-291 (1984).
- Chakrabarti, R., Wylie, D. E., Schuster, S. M. Transfer of monoclonal antibodies into mammalian cells by electroporation. Journal of Biological Chemistry. 264 (26), 15494-15500 (1989).
- Wilson, A. K., Horwitz, J., De Lanerolle, P. Evaluation of the electroinjection method for introducing proteins into living cells. American Journal of Physiology. 260 (2), 355-363 (1991).
- Potter, H. Transfection by electroporation. Current Protocols in Molecular Biology. 62 (1), 1-6 (2003).
- Fawell, S., et al. Tat-mediated delivery of heterologous proteins into cells. Proceedings of the National Academy of Sciences of the United States of America. 91 (2), 664-668 (1994).
- Prior, T. I., FitzGerald, D. J., Pastan, I. Translocation mediated by domain II of Pseudomonas exotoxin A: transport of barnase into the cytosol. Biochemistry. 31 (14), 3555-3559 (1992).
- Walev, I., et al. Delivery of proteins into living cells by reversible membrane permeabilization with streptolysin-O. Proceedings of the National Academy of Sciences of the United States of America. 98 (6), 3185-3190 (2001).
- Pitchiaya, S., Androsavich, J. R., Walter, N. G. Intracellular single molecule microscopy reveals two kinetically distinct pathways for microRNA assembly. EMBO Reports. 13 (8), 709-715 (2012).
- McNeil, P. L., Murphy, R. F., Lanni, F., Taylor, D. L. A method for incorporating macromolecules into adherent cells. The Journal of Cell Biology. 98 (4), 1556-1564 (1984).
- Ortiz, D., Baldwin, M. M., Lucas, J. J. Transient correction of genetic defects in cultured animal cells by introduction of functional proteins. Molecular and Cellular Biology. 7 (8), 3012-3017 (1987).
- McNeil, P. L., Warder, E. Glass beads load macromolecules into living cells. Journal of Cell Science. 88 (5), 669-678 (1987).
- Hayashi-Takanaka, Y., et al. Tracking epigenetic histone modifications in single cells using Fab-based live endogenous modification labeling. Nucleic Acids Research. 39 (15), 6475-6488 (2011).
- Morisaki, T., et al. Real-time quantification of single RNA translation dynamics in living cells. Science. 352 (6292), 1425-1429 (2016).
- Tanenbaum, M. E., Gilbert, L. A., Qi, L. S., Weissman, J. S., Vale, R. D. A protein-tagging system for signal amplification in gene expression and fluorescence imaging. Cell. 159 (3), 635-646 (2014).
- Zhao, N., et al. A genetically encoded probe for imaging nascent and mature HA-tagged proteins in vivo. Nature Communications. 10 (1), 2947(2019).
- Jedlitzke, B., Mootz, H. D. Photocaged nanobodies delivered into cells for light activation of biological processes. ChemPhotoChem. 5 (1), 22-25 (2021).
- Coulon, A., et al. Kinetic competition during the transcription cycle results in stochastic RNA processing. eLife. 3, 03939(2014).
- Pichon, X., Robert, M. -C., Bertrand, E., Singer, R. H., Tutucci, E. New generations of MS2 variants and MCP fusions to detect single mRNAs in living eukaryotic cells. Methods in Molecular Biology. 2166, 121-144 (2020).
- Koch, A., et al. Quantifying the dynamics of IRES and cap translation with single-molecule resolution in live cells. Nature Structural & Molecular Biology. 27, 1095-1104 (2020).
- Moon, S. L., et al. Multicolor single-molecule tracking of mRNA interactions with RNP granules. Nature cell biology. 21 (2), 162-168 (2019).
- Moon, S. L. Coupling of translation quality control and mRNA targeting to stress granules. Journal of Cell Biology. 219 (8), 202004120(2020).
- Cialek, C. A., et al. Imaging translational control by Argonaute with single-molecule resolution in live cells. bioRxiv. , (2021).
- Forero-Quintero, L. S., et al. Live-cell imaging reveals the spatiotemporal organization of endogenous RNA polymerase II phosphorylation at a single gene. bioRxiv. , (2020).
- Lyon, K., Aguilera, L. U., Morisaki, T., Munsky, B., Stasevich, T. J. Live-cell single RNA imaging reveals bursts of translational frameshifting. Molecular Cell. 75 (1), 172-183 (2019).
- JoVE. Introduction to Fluorescence Microscopy. General Laboratory Techniques. JoVE Science Education Database. , JoVE. Cambridge, MA. (2021).
- Hayashi-Takanaka, Y., Yamagata, K., Nozaki, N., Kimura, H. Visualizing histone modifications in living cells: spatiotemporal dynamics of H3 phosphorylation during interphase. Journal of Cell Biology. 187 (6), 781-790 (2009).
- Kumar, P., Nagarajan, A., Uchil, P. D. Analysis of cell viability by the MTT assay. Cold Spring Harbor Protocols. 2018 (6), (2018).
- Stasevich, T. J., et al. Regulation of RNA polymerase II activation by histone acetylation in single living cells. Nature. 516 (7530), 272-275 (2014).
- Besteiro, S., Michelin, A., Poncet, J., Dubremetz, J. -F., Lebrun, M. Export of a Toxoplasma gondii rhoptry neck protein complex at the host cell membrane to form the moving junction during invasion. PLOS Pathogens. 5 (2), 1000309(2009).
- Gilmore, A. P., Romer, L. H. Inhibition of focal adhesion kinase (FAK) signaling in focal adhesions decreases cell motility and proliferation. Molecular Biology of the Cell. 7 (8), 1209-1224 (1996).
- Emerson, N. T., Hsia, C. -H., Rafalska-Metcalf, I. U., Yang, H. Mechanodelivery of nanoparticles to the cytoplasm of living cells. Nanoscale. 6 (9), 4538-4543 (2014).
- Manders, E. M. M., Kimura, H., Cook, P. R. Direct imaging of DNA in living cells reveals the dynamics of chromosome formation. Journal of Cell Biology. 144 (5), 813-822 (1999).
- Memedula, S., Belmont, A. S. Sequential recruitment of HAT and SWI/SNF components to condensed chromatin by VP16. Current Biology. 13 (3), 241-246 (2003).
- Becker, T., Volchuk, A., Rothman, J. E. Differential use of endoplasmic reticulum membrane for phagocytosis in J774 macrophages. Proceedings of the National Academy of Sciences of the United States of America. 102 (11), 4022-4026 (2005).
- Molenaar, C., et al. Visualizing telomere dynamics in living mammalian cells using PNA probes. The EMBO Journal. 22 (24), 6631-6641 (2003).
- Jones, S. A., Shim, S. -H., He, J., Zhuang, X. Fast, three-dimensional super-resolution imaging of live cells. Nature Methods. 8 (6), 499-505 (2011).
- Sabri, A., Xu, X., Krapf, W. M. Elucidating the origin of heterogeneous anomalous diffusion in the cytoplasm of mammalian cells. Physical Review Letters. 125 (5), 053901(2020).
- Sato, Y., et al. Genetically encoded system to track histone modification in vivo. Scientific Reports. 3, 2436(2013).
- Sato, Y., Stasevich, T. J., Kimura, H. Visualizing the dynamics of inactive X chromosomes in living cells using antibody-based fluorescent probes. X-Chromosome Inactivation. Methods in Molecular Biology. 1861, Humana. New York, NY, USA. 91-102 (2018).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationExplorer plus d’articles
This article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.