
Method Article
Bead Loading Proteins and Nucleic Acids into Adherent Human Cells
In This Article
Summary
Bead loading introduces proteins, plasmids, and particles into adherent mammalian cells. This cell loading technique is inexpensive, rapid, and does not substantially affect cell health. It is best suited for live-cell imaging.
Abstract
Many live-cell imaging experiments use exogenous particles (e.g., peptides, antibodies, beads) to label or function within cells. However, introducing proteins into a cell across its membrane is difficult. The limited selection of current methods struggles with low efficiency, requires expensive and technically demanding equipment, or functions within narrow parameters. Here, we describe a relatively simple and cost-effective technique for loading DNA, RNA, and proteins into live human cells. Bead loading induces a temporary mechanical disruption to the cell membrane, allowing macromolecules to enter adherent, live mammalian cells. At less than 0.01 USD per experiment, bead loading is the least expensive cell loading method available. Moreover, bead loading does not substantially stress cells or impact their viability or proliferation. This manuscript describes the steps of the bead loading procedure, adaptations, variations, and technical limitations. This methodology is especially suited for live-cell imaging but provides a practical solution for other applications requiring the introduction of proteins, beads, RNA, or plasmids into living, adherent mammalian cells.
Introduction
Loading macromolecules into mammalian cells necessitates methodology that allows them to cross the cell's plasma membrane1. Several methods can introduce plasmids into mammalian cells through transfection, including liposomal transfection2 and diethylaminoethyl-dextran transfection3. However, methods for loading proteins or membrane-impermeable particles into cells are more limited.
Several techniques have bypassed this difficult hurdle using various strategies. First, microinjection delivers particles through a micropipette into live cells under a microscope4. While arguably the most controlled and least invasive method, this technique is relatively low-throughput because cells must be loaded one by one. Further, microinjection requires specialized equipment and is technically demanding.
Second, electroporation is a way to electro-inject proteins into cells via voltage-induced membrane disruption5,6,7. However, this method again requires specialized, expensive equipment, and the shock can cause cell stress and mortality. Further, cells must be trypsinized before electroporation and subsequently replated, limiting the timeframe at which cells can be investigated post-electroporation.
Third, cell membranes may be chemically modified for temporary, reversible permeabilization8,9. Streptolysin-O loading inserts an endotoxin into cell membranes, which forms temporary pores, allowing exogenous membrane-impermeable particles, including proteins and DNA plasmids, to enter cells10. After a 2 h recovery, about half the cells repair these pores and halt internalizing particles from the solution. However, this technique requires a long recovery time and is incompatible with cell types that cannot tolerate endotoxins.
Fourth, mechanical disruption loads particles into cells through physical perturbation of the cell membrane11. This can be done in multiple ways, including scratching, scraping, and rolling beads atop cells12,13. As early as 1987, beads have been used to load proteins into cells mechanically14. More recently, the bead loading technique has been optimized and adapted beyond proteins to include the loading of plasmids and RNA, as described here.
Bead loading is an easy, inexpensive, and fast method for loading protein and plasmids into adherent human cells. Glass beads are briefly rolled atop cells, temporarily disrupting their cellular membrane. This allows particles in solution to enter. As bead loading has low efficiency, it is best suited for single-molecule or single-cell microscopy experiments. Bead loading can introduce a wide variety of proteins, including fragmented antibodies (Fab),15,16 purified proteins like scFvs,17 intrabodies,18,19, or mRNA coat proteins, e.g., MS2 coat protein (MCP)20,21. Plasmid expression vectors can also be added to the protein solution and bead-loaded simultaneously22,23,24,25.
Beyond proteins and plasmids, molecules as large as 250 nm polystyrene beads have been introduced into cells via bead loading (personal communication). Bead loading is incredibly inexpensive, costing less than 0.01 USD per experiment in materials and requiring no additional expensive equipment. The cost is further reduced by minimizing the amount of probes used per experiment because only the cells in the central 14 mm-diameter microwell of an imaging chamber are loaded. It should be noted that the limited loading area means that bead loading is not ideal for bulk-cell loading.
This manuscript presents the bead loading process, including how to construct the bead loading apparatus and perform an experiment. It shows that proteins, RNA, and DNA can be loaded into various cell types and that two different, simultaneously bead-loaded proteins have highly correlated cellular concentrations and relatively low variance. Also discussed are variations in the protocol based on cell type and loading of protein, plasmid, or RNA. Although beads are thought to perforate and disrupt the cell membrane, when appropriately performed, the bead loading process dislodges only a small number of cells from the bottom of the imaging chamber. After a short recovery period, cells continue to grow and divide. This methodology is ideal for live-cell microscopy experiments, including single-molecule protein and RNA tracking, post-translational modification detection, observation of dynamic cellular mechanisms, or subcellular localization monitoring15,16,22,26,27.
Protocol
1. Clean, sterilize, and dry glass beads to avoid clumping and ensure even spreading atop the cells.
- Sterilize approximately 5 mL of glass beads in sodium hydroxide (NaOH). Measure the beads in a 50 mL conical tube. Add 25 mL of 2 M NaOH and mix gently using a shaker or rotator for 2 h.
- Decant the NaOH, retaining as many beads as possible. If the beads are in suspension, spin down the tube of beads briefly in a centrifuge (1 min at ~1000 × g, room temperature).
- Wash the beads thoroughly with cell culture-grade water until the pH is neutral (use a pH test strip on the eluent to confirm a neutral pH). Decant the wash water each time, as before.
- Wash the beads thoroughly with 100% ethanol 2-3x. Decant the ethanol each time, as before.
- Dry the beads. Sprinkle the beads to form a thin layer inside a sterile container (such as a 10 cm Petri dish). Leaving the container open, let the beads air dry in a biosafety cabinet overnight. Ensure that the beads are completely dry by tapping or gently shaking the container and checking that the beads have a sandy texture with no clumping or flaking.
- UV-sterilize the dry beads for 15 min.
2. Assemble the bead loader apparatus.
- Fasten a patch of mesh (polypropylene or equivalent material, 105 µm openings to allow the beads to pass through) to cover the entire opening of the beads holding chamber with either tape or clamping the mesh between the male and female ends of a metal reusable imaging chamber (Figure 1A).
- UV-sterilize the apparatus for 15 min. Add the beads to the apparatus and seal it tightly with waxy film.
NOTE: It is essential that the beads are completely clean and dry at this step. They should be loose and look sandy with no clumps. If they do not appear so, re-wash and completely dry the beads. - Store the apparatus in a sealed, dry container desiccated by silica gel or other desiccant medium. If the beads become damp, which will be apparent by bead clumping, thoroughly dry and sterilize the bead loader and replace with fresh beads.
NOTE: All these precautions will prevent any mold or bacteria from growing on or around the beads within the bead loader. The bead loader apparatus can be made in different ways. See the details in the discussion.
3. Prepare glass-bottom chambers of adherent cells.
- Seed adherent mammalian cells onto a 35 mm glass-bottom chamber. Ensure that the cells are approximately 80% confluent at the time of bead loading. (See Table 1 for more information on various cell types and notes on the effectiveness of bead loading in different cell types.)
NOTE: Cells can be seeded in only the microwell in the center of the chamber to conserve how many cells are used. - Incubate the cells under normal conditions until they are completely adherent to the glass.
NOTE: It is essential that the cell density is high enough and that the cells are securely adhered to the glass. If these requirements are not met, cells will likely peel off during bead loading. The timeline between cell seeding and bead loading can be lengthened to ensure proper cell adhesion and confluency.
4. Bead loading cells
NOTE: If required, wash the cells briefly with phosphate-buffered saline (PBS) and then add 2 mL of the optimal medium. Incubate for at least 30 min.
- Make a solution of 3-8 µL containing the desired plasmids, protein, and/or particles. Use ~1 µg (0.1-1 pmol) of each type of plasmid and ~0.5 µg (0.01 nmol) of protein, depending on experimental requirements. Use a low-retention tube for proteins so that they are not left behind on the tube walls. Bring the solution up to a minimum of 3 µL with PBS, and adjust the solution volume to coat the entire area of cells to be loaded (i.e., the chamber's microwell, Figure 1B).
- Mix the solution thoroughly by pipetting up and down and/or flicking the tube. Briefly spin the solution down to the bottom of the tube in a tabletop microfuge.
- Transfer the bead loading solution and the chamber of cells into a tissue culture hood. Perform the remaining steps in the tissue culture hood using sterile technique.
- Remove the medium from the cells and temporarily store it in a sterile tube. Gently aspirate all medium from around the edges of the chamber, and tilt the chamber at approximately a 45° angle and remove the remaining drop of media in the center microwell. During medium removal, make sure to avoid letting the pipette tip touch the glass, which may result in cell peeling and loss. Move quickly to the next step so that the cells are not dry for long.
- Gently pipette the bead loading solution onto the glass microwell in the center of the chamber. Optional: Incubate with gentle rocking for ~30 s without allowing the chamber to dry up completely.
- Gently disperse a monolayer of glass beads on top of the cells, preferably using a bead loading apparatus (Figure 1A). Ensure that the beads cover the cells in the glass-bottom microwell completely.
- Pinching the chamber with two fingers, tap it against the hood surface by lifting it ~2 inches and bringing it down firmly. Use a force approximately equivalent to dropping the dish from that height. Repeat for a total of ~10 taps.
NOTE: Ensure that the taps do not substantially peel the cells. Tapping can be optimized for the cell type. If cells load poorly, tap harder; however, if many cells peel off, tap more lightly. - Gently add medium back into the chamber by pipetting slowly onto the plastic side of the chamber. Try to aspirate any floating beads without disturbing the cells. Add more pre-warmed media at this step if too much was removed. Incubate the cells for 0.5-2 h in the incubator.
- UV-sterilize the bead loader for 15 min before returning it to storage under desiccating conditions.
- Add dye (e.g., DAPI or HaloTag ligand stain, if required by the experiment) to the cells as per the manufacturer's recommended protocol.
- Wash the cells 3x with medium before imaging to remove the beads and excess loading components in solution. Avoid pipetting directly onto cells to keep them from peeling.
5. Imaging the bead-loaded cells
- Image the cells immediately or when required by the experiment. Use a microscope capable of capturing fluorescence (lasers or monochromatic light source). Ensure that the excitation wavelengths are appropriate for the chosen fluorophore or dye (e.g., 488 nm wavelength light for green fluorescence protein (GFP)).
NOTE: Bead-loaded proteins may be imaged once the cells have recovered (as soon as 30 min post loading for the cell lines described here). Plasmid expression takes ≥2 h depending on expression vector elements (Figure 1C, and further explanation in the discussion). Imaging of bead-loaded cells can be performed on any microscope equipped with the appropriate fluorescent sources associated with loaded probes, a camera capable of capturing fluorescence images, such as an electron-multiplying charge-coupled device (EMCCD) or scientific complementary metal-oxide semiconductor (sCMOS) camera, and an incubator to control temperature, humidity, and carbon dioxide. For a guide to fluorescence microscopy, refer to 27.
Results
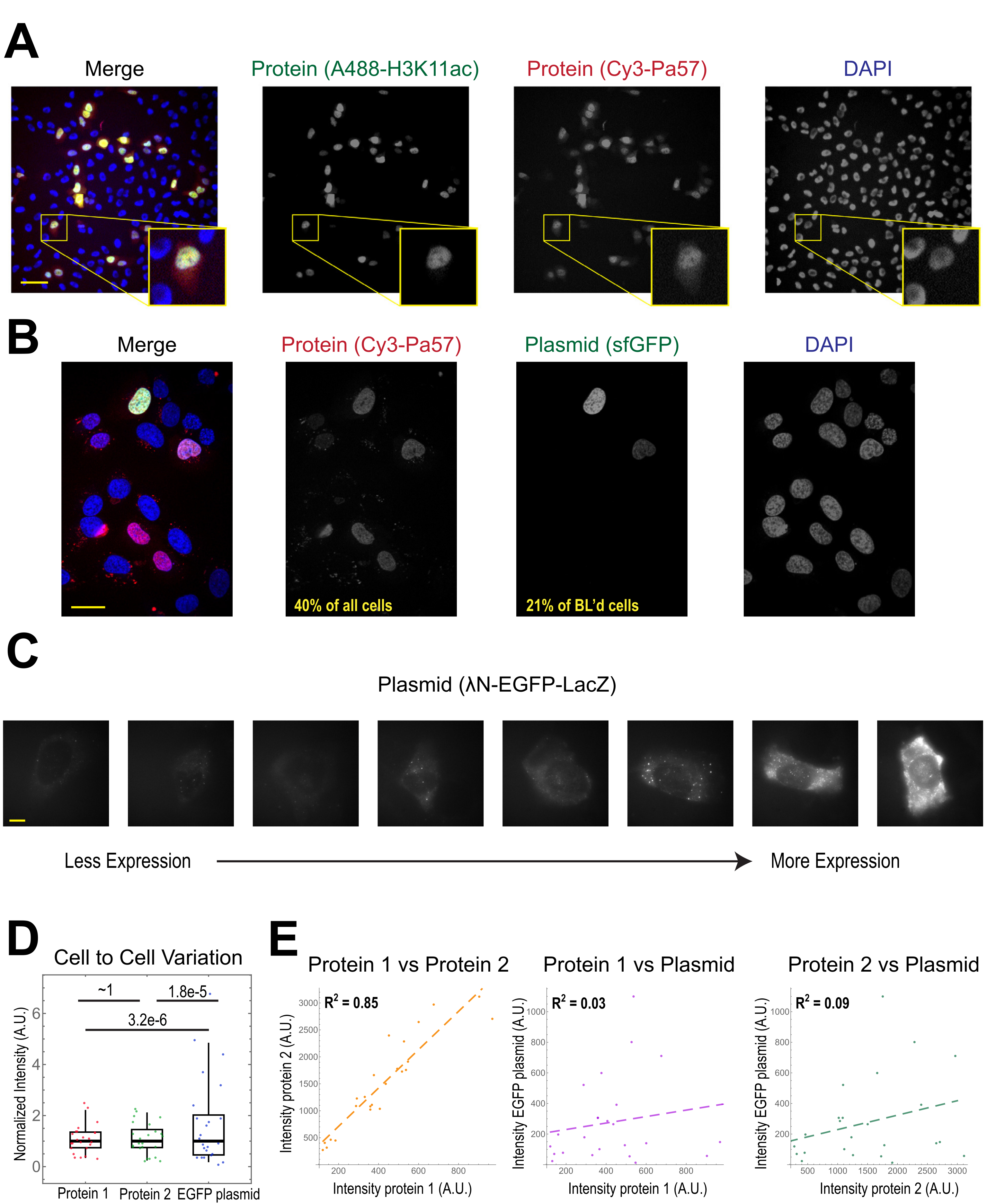
The most common application of bead loading is to introduce one or more types of protein into adherent human cells. To illustrate this, cells were bead-loaded with a solution of a Cy3- and Alexa488-conjugated Fab protein. Although not every cell in the microwell was bead-loaded, the cells that were loaded almost always had both Cy3- and Alexa488-labeled proteins together (Figure 2A). According to an earlier estimate, when 0.5 microgram of Fab diluted in 4 microliters is bead-loaded29, as in Figure 2A, each cell is loaded with roughly 106 Fab molecules.
Plasmid DNA encoding GFP (1 µg of plasmid DNA, 1.8 µL of a 557 ng/µL solution) and 0.5 µg of Cy3-labeled Fab was also introduced into cells via bead loading and subsequently expressed and visualized (Figure 2B). The GFP fluorescence indicated that the GFP-encoding plasmid was not only loaded into cells but also expressed. Thus, in the same cell, bead loading can introduce a protein probe (e.g., Cy3-labeled Fab) and reporter plasmid (e.g., GFP), as performed in this laboratory previously22,23,24. We determined that 40% of the cells were bead-loaded with Fab protein and 21% of the bead-loaded cells expressed the co-loaded plasmid, as shown in the representative fields-of-view in Figure 2B. Typically, each chamber is loaded with 1-2 µg of plasmid, approximately the same amount as lipofection.
Bead-loaded cells express widely varying levels of plasmids (Figure 2C,D). To specifically measure this, we used the Fisher Ratio test to compare the distributions of protein and plasmid intensity data. The results showed that although proteins 1 and 2 had similar intensity distributions (p = ~1), each protein had a significantly smaller distribution than the plasmid (p = 3.2e-6 and 1.8e-5). Although this could be due to variability in how many plasmids are loaded per cell, the greater source of variability may arise from the many steps required for plasmid expression that are likely to vary greatly between cells, including being imported into the cell nucleus, transcription, and translation. In contrast, the levels of bead-loaded proteins had slight cell-to-cell variance, and the levels of two simultaneously loaded proteins were highly correlated with each other (Figure 2D,E).
Plasmid expression can be seen as early as 2-4 h post bead loading but may occur later depending on when optimal plasmid expression is obtained. We recommend performing a time course to determine the best window of expression for a specific plasmid spanning 2-24 h post bead loading. This can be done in one chamber with long timeframe imaging or by bead loading and staggering multiple chambers. Bead-loaded cells remain adherent and are healthy enough to grow and divide. Bead-loaded human U2OS cells were imaged directly before, directly after, and 24 h after bead loading. Proper bead loading had almost no noticeable effect on the number of cells or their morphology, as shown in Figure 3A (left, middle).
In contrast, poor bead loading with too many beads and excessive tapping force is depicted in Figure 3B. This caused much cell loss (large patches of the coverglass without cells and detached, floating, out-of-focus cells), poor cell morphology (cells appearing rounded up and poorly adhered), and clusters of beads remaining on the coverglass after bead loading. Though cells are thought to undergo mechanical damage during bead loading, cells grew and proliferated in the properly bead-loaded chamber, as evidenced by the increased number of cells 24 h after bead loading (Figure 3A, right). The effect on cell viability can be assessed through a variety of assays, such as a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay, to compare bead-loaded to mock-loaded cells30. Further, this and previous work show that the bead-loaded cells undergo cell division (Figure 3C and Supplemental Video 1), and the timing of mitosis is not affected by bead loading31, which serves as further evidence for sustained cell health after bead loading.
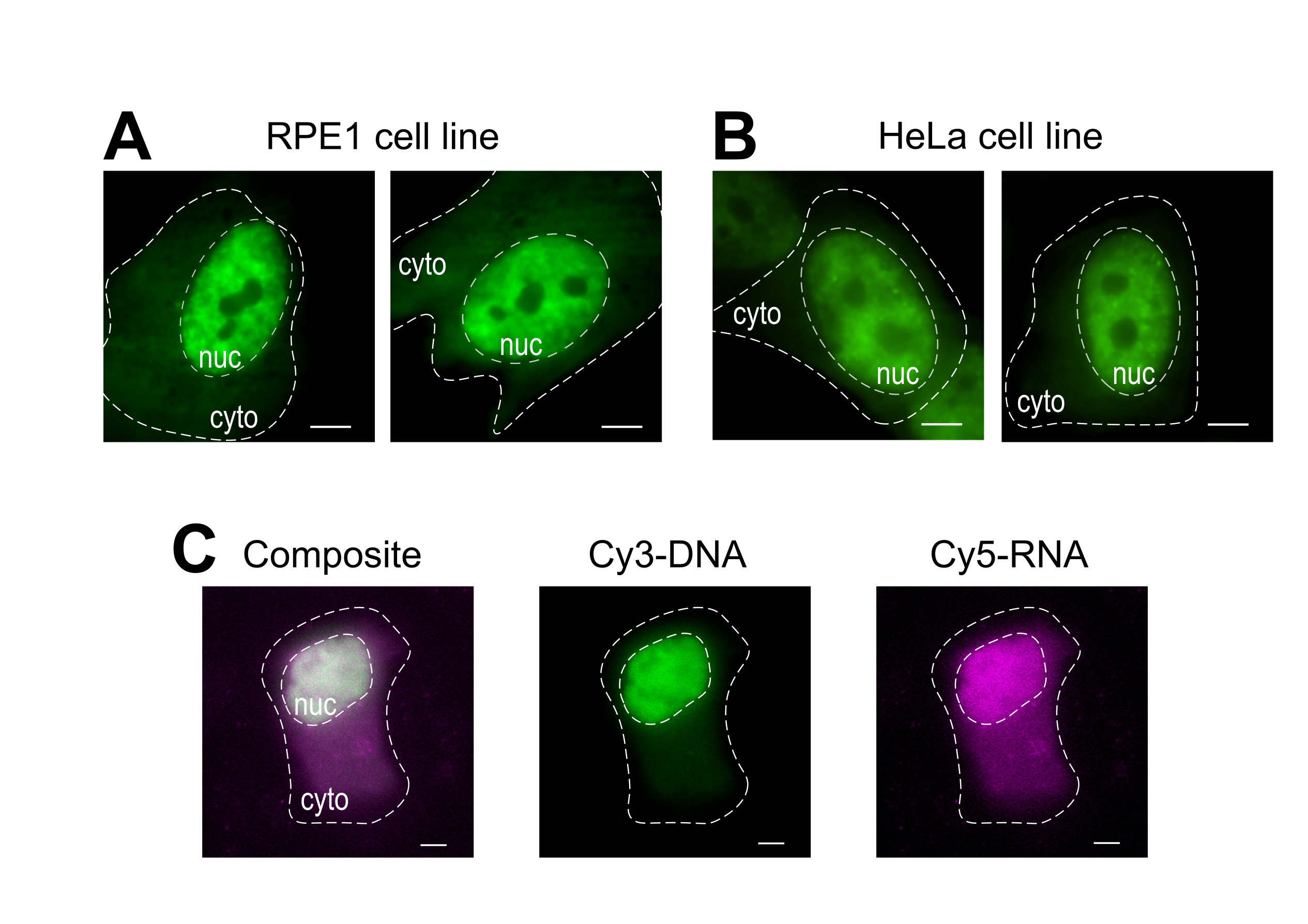
Bead loading is a versatile technique, accommodating several adherent cell lines and various macromolecules. Here, this variety has been demonstrated by loading RPE1 and HeLa cell lines with Fab (Figure 4A,B). Table 1 provides further examples of bead loading in different cell lines, in this laboratory and beyond, and points out some of the nuanced differences between bead loading protocols from other labs. Of note, the diameter of glass beads used for loading varies greatly between laboratories, though the most efficient loading was found for small, 75 µm diameter beads in several cell lines14. Further, this laboratory has begun bead loading RNA as well (data not shown). Figure 4C displays a representative U2OS cell bead-loaded with a Cy5-RNA 9mer and Cy3-DNA 28mer together.

Figure 1: Bead loading apparatus, technique, and timeline Please click here to view a larger version of this figure.
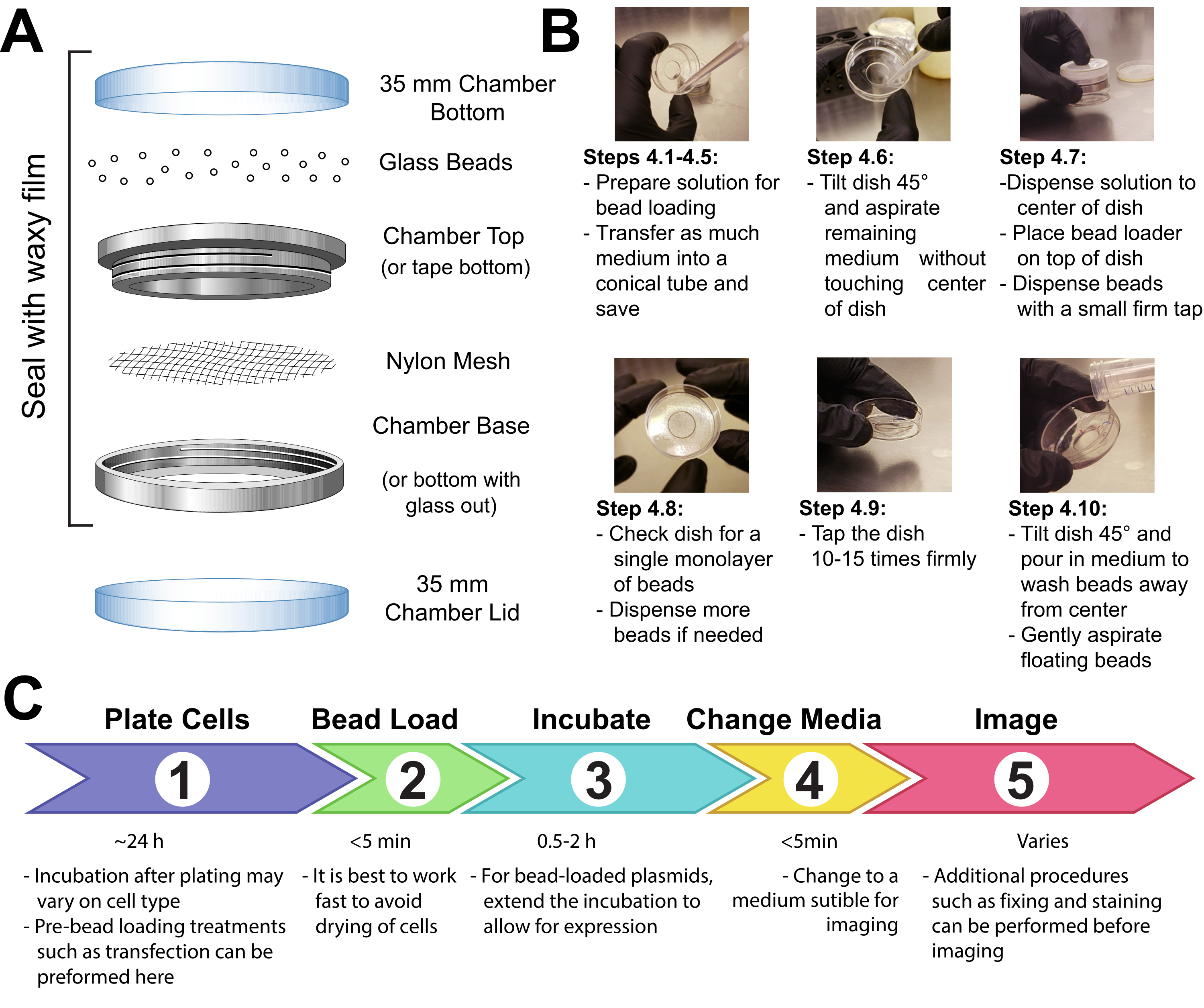{kind=link}

Figure 2. Bead loading introduces low variability in protein concentration but high variability in plasmid expression. (A) Cells were bead-loaded with 0.5 µg of each of Alexa488-conjugated anti-H3K27 acetyl Fab (green) and Cy3-conjugated anti-RNAPII-Serine 5-phosphorylated Fab (red) in 4 µL of bead loading solution. Cells were DAPI-stained (blue) and then live-imaged immediately. Scale bars = 20 µm. (B) Cells were bead-loaded with 0.5 µg of Fab protein (Cy3-conjugated anti-RNAPII-Serine 5-phosphorylated protein, red) and 1 µg of plasmid encoding superfolder GFP-H2B (green) in 4 µL of bead loading solution. After 24 h, cells were DAPI-stained (blue) and imaged live. Scale bars = 30 µm. (C-E) Protein 1 (JF646-HaloLigand-labeled HaloTag-MCP), protein 2 (Cy3-conjugated anti-FLAG Fab), and a plasmid encoding EGFP (λN-EGFP-LacZ) were bead-loaded together into cells. The total intensity in each fluorescent channel was measured in a 1.3 x 1.3 µm patch in the cytoplasm of each cell. N = 25 cells. (C) Representative cells expressing the bead-loaded plasmid, λN-EGFP-LacZ. The same imaging conditions and intensities were used for all cells. Spots are aggregates of the expressed protein. Scale bars = 10 µm. (D) The chart shows each cell's total intensity of either protein 1, protein 2, or EGFP expressed from the plasmid. Each channel was normalized to the median. Bonferroni-corrected P values were calculated by the Fisher Ratio test to determine whether the distribution of protein or plasmid intensity data has the same variability. Each point represents a cell. (E) The total intensities for either both proteins, protein 1 and the plasmid, or protein 2 and the plasmid, are plotted against each other. Calculated R2 values are displayed. Each point represents a cell. Abbreviations: DAPI = 4′,6-diamidino-2-phenylindole; EGFP = enhanced green fluorescent protein; A.U. = arbitrary units; MCP = MS2 coat protein; RNAPII = RNA polymerase II. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Bead-loaded cells remain adherent and are healthy enough to grow and divide. (A) U2OS cells were bead-loaded with 0.5 µg of Cy3-conjugated anti-FLAG Fab in 4 µL of bead loading solution. The cells were imaged directly before, directly after bead loading, and 24 h after bead loading. Orange arrows identify areas where cells peeled off during bead loading. Scale bars = 2 mm. (B) Representative image of U2OS cells bead-loaded with components from (A) but with harsh tapping and too many beads. The red arrow identifies extra glass beads. Scale bar = 2 mm. (C) U2OS cells were loaded with 1.5 µg of the 14.4 kbp plasmid smFLAG-KDM5B-15xBoxB-24xMS2, 0.5 µg of Cy3-conjugated anti-FLAG Fab (green), 130 ng of HaloTag-MCP (magenta) in 8 µL of bead loading solution. Directly before imaging, the HaloTag was stained with JF646-HaloLigand. The MS2 stem-loops of the mRNA transcribed from the reporter plasmid are labeled by MCP (magenta spots), and FLAG-tagged translated reporter protein is labeled by anti-FLAG Fab (green colocalization to mRNA). Mature Fab-labeled protein localizes to the nucleus. This cell was imaged 4-15 h after bead loading. Yellow arrows identify the cell nucleus before and nuclei after cell division. Scale bars = 5 µm. Abbreviation: MCP = MS2 coat protein. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Variations in cell type loading material of the bead loading protocol. (A-B) RPE1 (A) and HeLa (B) cells were bead-loaded with 1.5 µg of a nuclear Fab protein (anti-RNAPII-Serine 5-phosphorylation) in 4 µL of loading solution. Each cell's nucleus (nuc) and cytoplasm (cyto) are marked. Cells were imaged 6 h after being bead-loaded. Scale bars = 5 µm. (C) Human U2OS cells were bead-loaded with both Cy5-RNA 9mer (magenta) and Cy3-DNA 28mer (green) oligos, 10 picomoles of each, in 4 µL of bead loading solution. Cells were imaged 4 h after being bead-loaded. All cell nuclei are highlighted by a dashed line. Scale bars = 5 µm. Abbreviations: RNAPII = RNA polymerase II. Please click here to view a larger version of this figure.
{kind=link}
| Cell line | Cell type | Bead loading effectiveness | Notes/reference |
| Stem cells (human) | Embryonic stem cells | Difficult | *Many cells peel off during bead loading if plated on gelatin-coated plates |
| HEK 293 | Human embryonic kidney cells | Difficult | *Need to lay down a gel matrix to imaging chamber surface before bead loading. Tap gently when bead loading at first. |
| Neurons (rat) | Primary embryonic neurons (e-18), dissociated | Very inefficient | *Efficient bead loading of neurons was not observed using this standard bead loading protocol. This could be due to the non-adherent nature of neurons or from consequent damage to neural processes. |
| MDCK (canine) | Madin-Darby canine kidney cells | See McNeil and Warder (1987)14 | *Low-efficiency bead loading14 |
| U2OS (human) | Osteosarcoma | Standard bead loading protocol | |
| HeLa (human) | Cervical cancer | Standard bead loading protocol | |
| RPE1 (human) | Epithelial cells immortalized with hTERT | Standard bead loading protocol | |
| HFF (human) | Primary foreskin fibroblasts | See Besteiro et al. (2009)31 | *Modified protocol of tilting instead of tapping31 |
| BALB/c 3T3, NIH 3T3, and Swiss 3T3 (mouse) | Embryonic fibroblasts | See Gilmore and Romer (1996)32, Emerson et al. (2014)33 and McNeil and Warder (1987)14 | *425–600 μm glass beads reported32 |
| *Used 200–300 μm glass beads33 | |||
| *75 μm glass beads gave better results than 400 μm14 | |||
| DM (Indian muntjac) | Skin fibroblasts | See Manders, Kimura, and Cook (1999)34 | |
| CHO (hamster) | Epithelial-like ovary cells | See Memedula and Belmont (2003)35 | *Used 425–600 μm glass beads35 |
| BAE (bovine) | Bovine aortic endothelial cells (BAEC-11) | See McNeil and Warder (1987)14 | *75 μm glass beads gave better results than 400 μm14 |
| PtK-2 (Potomus tridaclylis) | Epithelial kidney cells | See McNeil and Warder (1987)14 | *75 μm glass beads gave better results than 400 μm14 |
| HUVEC (human) | Umbilical vein endothelial cells | See Gilmore and Romer (1996)32 | *Used 425–600 μm glass beads32 |
| J774 and J774.2 (mouse) | monocyte macrophage cells | See Becker et al. (2005)36 and McNeil and Warder (1987)14 | *Gentle agitation (instead of tapping) and 425–600 μm glass beads36 |
| MS-5 (mouse) | bone marrow stromal cells | See Molenaar et al. (2003)37 | |
| WPE1-NB11 (human) | prostate epithelial cells | See Gilmore and Romer (1996)32 and | *Used 425–600 μm glass beads32 |
| Emerson et al. (2014)33 | *Used 200–300 μm glass beads33 |
Table 1: Bead loading in different cell lines. For the cell lines that have not yet been bead-loaded in this laboratory, references and notes on variations in the protocol are provided.
Supplemental Video 1: Example of a bead-loaded cell undergoing cell division. U2OS cells were loaded with 1.5 µg of the 14.4 kbp plasmid smFLAG-KDM5B-15xBoxB-24xMS2, 0.5 µg of Cy3-conjugated anti-FLAG Fab (green), 130 ng of HaloTag-MCP (magenta) in 8 µL of bead loading solution. Directly before imaging, the HaloTag was stained with JF646-HaloLigand. The MS2 stem-loops of the mRNA transcribed from the reporter plasmid are labeled by MCP (magenta spots), and FLAG-tagged translated reporter protein is labeled via anti-FLAG Fab (green colocalization to mRNA). Mature Fab-labeled protein localizes to the nucleus. This cell was imaged 4-15 h after bead loading. Scale bar = 10 µm. Abbreviation: MCP = MS2 coat protein. Please click here to download this Video.
Discussion
The bead loading technique described here is a cost-effective and time-efficient method for introducing macromolecules and other particles into adherent cells. This versatile process can load protein (Figure 2A)15,16,26,27, a combination of protein and plasmids (Figure 2B,C)22,25, RNA (Figure 4C), 100 and 250 nm polystyrene beads (personal correspondence), synthetic dyes39 or quantum dots34,40. Bead loading may have the capability to load other types of membrane-impermeable particles as well. Its most frequently used application is for loading antibodies or Fabs to target endogenous epitopes, such as post-translational modifications (PTMs), into live cells. Targets, such as PTMs, are often difficult to label in live cells without established PTM-specific, genetically encoded probes41,42. In contrast, bead loading can introduce multiple types of probes, reporters, or other molecular tools together into the same cell for monitoring multiple readouts simultaneously. We anticipate that bead loading will be a useful technique for loading a variety of macromolecules or particles.
A major advantage of bead loading is the low cost: each experiment costs less than 0.01 USD. A bead loader apparatus can be made easily using inexpensive materials costing in total ~$150, which is significantly less expensive than any other cell-loading method. The cost of a bead loader apparatus can be further reduced to under $10 by replacing the reusable metal chamber with a plastic one. For this, either drill a hole in a 35 mm chamber or remove the glass from a 35 mm glass-bottom chamber, then securely fasten the mesh in place with tape. In lieu of an apparatus, bead loading can even be performed using a wide-bore 1000 µL pipette tip to scoop and sprinkle beads onto cells, although this variation makes it difficult to sprinkle a monolayer of beads onto cells (step 4.6).
Another benefit of bead loading is that cells can retain normal overall morphology, recover rapidly, and continue to grow and divide, at least for the U2OS, RPE1, and HeLa cells studied here and for the other cell lines studied elsewhere (Figure 3; Figure 4A,B; Supplemental Video 1; and Table 1)31. During bead loading, cells undergo physical stress and sometimes dislodge and peel (~5% of cells peel under optimal conditions, but greater cell loss can happen if bead loading is performed too forcefully or too many glass beads are loaded atop the cells, as depicted in Figure 3B). However, bead-loaded cells that remain attached to the coverslip usually appear healthy and can be imaged as soon as 30 min after bead loading (Figure 3A). We generally allow cells a 30-min recovery period but anticipate that imaging sooner post-bead loading is feasible.
A major drawback of this technique is that the cells need to be capable of withstanding minor physical stress during loading and remain securely adherent to the coverslip. Poorly/non-adherent cell lines or cells grown on coated plates (e.g., HEK and stem cells) often detach upon gentle tapping during bead loading. Further, experience has shown that primary neurons are too sensitive for bead loading.
Bead loading is best suited for single-cell or single-molecule experiments. In our experience, bead loading has a roughly 20-40% protein loading efficiency, and ~20% of bead-loaded cells also expressed a co-loaded plasmid (Figure 2A,B). Thus, bead loading plasmids may be less efficient for protein expression than bead loading purified proteins because plasmids must not only enter cells but also be expressed (which involves, among other things, nuclear import, transcription, and translation, each of which can lower expression efficiency). The low efficiency of bead-loaded plasmid expression can be circumvented by using alternative transfection protocols, such as lipofection, before bead loading proteins or probes16,27. Additionally, incubating cells in optimal media for 30 min before bead loading may assist plasmid expression. Due to low plasmid expression, bead loading has not often been used as an alternative to lipofection-based transfection in this laboratory. The only exception is when a purified protein, such as Fab, is to be co-loaded, in which case it is quite convenient to bead-load the protein and plasmid at the same time. Moreover, for cells that are unresponsive or intolerant to lipofection, bead loading may provide an alternate, albeit low-efficiency, method for transient plasmid expression.
Disclosures
The authors have no conflicts of interest to disclose.
Acknowledgements
We are grateful to the members of the Stasevich lab for innumerable conversations that helped improve and develop this protocol. Specifically, Dr. Linda Forero and Dr. Phil Fox for advice on bead loading different cell types. We would like to sincerely thank Dr. Yoko Hayashi-Takanaka, Dr. Yuko Sato and Dr. Hiroshi Kimura for sharing their glass bead loading protocol. We are very grateful to Dr. Ashok Prasad and Dr. Diego Krapf for generously sharing their bead loading protocols for introducing inorganic particles into cells. We are grateful to Dr. Travis Sanders, Craig Marshall, and Dr. Thomas Santangelo for generously sharing their labeled RNA reagent. ALK, MNS, CAC, GG and TJS were supported by the National Institutes of Health (NIH) grant R35GM119728 and the National Science Foundation (NSF) CAREER grant MCB-1845761, both to TJS. CAC was also supported by the NSF NRT award DGE-1450032.
Materials
| Name | Company | Catalog Number | Comments |
| 10 cm cell culture dishes | VWR | 82050-916 | Use to culture cells |
| 35 mm cell culture dishes | Falcon | 353001 | Use to construct bead loader |
| Attofluor Cell Chamber | Thermo Fisher Scientific | A7816 | Use to construct the custom bead loader |
| DMEM, high glucose, no glutamine | Thermo Fisher Scientific | 11960069 | Use in general cell culture |
| Drierite Indicating Absorbents | Thermo Fisher Scientific | 07-578-3B | Store the bead loader in a desiccator with these absorbent pellets |
| Fetal Bovine Serum | Atlas Biologics | F-0050-A | Use in general cell culture and as a supplement before bead loading |
| Glass beads, acid washed, ≤106 µm* | Millipore Sigma | G4649 | Sprinkle on cells to bead load plasmid DNA and proteins |
| Glass bottom dishes, 35 mm, #1.5, 14 mm glass | MatTek Corporation | P35G-1.5-14-C | Seed cells onto these chambers for imaging |
| L-Glutamine-200 mM | Thermo Fisher Scientific | 25030081 | Use to make DMEM + media |
| Opti-MEM, Reduced Serum Medium | Thermo Fisher Scientific | 31985070 | Optimal media for incubating cells before bead loading (optional step) |
| Parafilm | VWR | 52858-032 | waxy film used to construct bead loader |
| Penicillin-Streptomycin | Thermo Fisher Scientific | 15140-122 | Use to make DMEM + media |
| Phenol-free DMEM | Thermo Fisher Scientific | 31053036 | Use on cells before imaging |
| Phosphate Buffered Saline (PBS) | Thermo Fisher Scientific | AM9625 | Working stock of sterile 1X PBS |
| Sodium Hydroxide (NaOH) | Thermo Fisher Scientific | S318-500 | Use 2 M solution to wash glass beads |
| Spectra Mesh Woven Filters, Polypropylene, 105 µm opening | Spectrum Labs | 148496 | Use to construct bead loader |
| Trypsin | Thermo Fisher Scientific | 25300062 | Use in general cell culture |
References
- Stewart, M. P., et al. In vitro and ex vivo strategies for intracellular delivery. Nature. 538 (7624), 183-192 (2016).
- Felgner, P. L., et al. Lipofection: a highly efficient, lipid-mediated DNA-transfection procedure. Proceedings of the National Academy of Sciences of the United States of America. 84 (21), 7413-7417 (1987).
- Schenborn, E. T., Goiffon, V. DEAE-dextran tansfection of mammalian cultured cells. Methods in Molecular Biology. 130, 147-153 (2000).
- Celis, J. E. Microinjection of somatic cells with micropipettes: comparison with other transfer techniques. Biochemical Journal. 223 (2), 281-291 (1984).
- Chakrabarti, R., Wylie, D. E., Schuster, S. M. Transfer of monoclonal antibodies into mammalian cells by electroporation. Journal of Biological Chemistry. 264 (26), 15494-15500 (1989).
- Wilson, A. K., Horwitz, J., De Lanerolle, P. Evaluation of the electroinjection method for introducing proteins into living cells. American Journal of Physiology. 260 (2), 355-363 (1991).
- Potter, H. Transfection by electroporation. Current Protocols in Molecular Biology. 62 (1), 1-6 (2003).
- Fawell, S., et al. Tat-mediated delivery of heterologous proteins into cells. Proceedings of the National Academy of Sciences of the United States of America. 91 (2), 664-668 (1994).
- Prior, T. I., FitzGerald, D. J., Pastan, I. Translocation mediated by domain II of Pseudomonas exotoxin A: transport of barnase into the cytosol. Biochemistry. 31 (14), 3555-3559 (1992).
- Walev, I., et al. Delivery of proteins into living cells by reversible membrane permeabilization with streptolysin-O. Proceedings of the National Academy of Sciences of the United States of America. 98 (6), 3185-3190 (2001).
- Pitchiaya, S., Androsavich, J. R., Walter, N. G. Intracellular single molecule microscopy reveals two kinetically distinct pathways for microRNA assembly. EMBO Reports. 13 (8), 709-715 (2012).
- McNeil, P. L., Murphy, R. F., Lanni, F., Taylor, D. L. A method for incorporating macromolecules into adherent cells. The Journal of Cell Biology. 98 (4), 1556-1564 (1984).
- Ortiz, D., Baldwin, M. M., Lucas, J. J. Transient correction of genetic defects in cultured animal cells by introduction of functional proteins. Molecular and Cellular Biology. 7 (8), 3012-3017 (1987).
- McNeil, P. L., Warder, E. Glass beads load macromolecules into living cells. Journal of Cell Science. 88 (5), 669-678 (1987).
- Hayashi-Takanaka, Y., et al. Tracking epigenetic histone modifications in single cells using Fab-based live endogenous modification labeling. Nucleic Acids Research. 39 (15), 6475-6488 (2011).
- Morisaki, T., et al. Real-time quantification of single RNA translation dynamics in living cells. Science. 352 (6292), 1425-1429 (2016).
- Tanenbaum, M. E., Gilbert, L. A., Qi, L. S., Weissman, J. S., Vale, R. D. A protein-tagging system for signal amplification in gene expression and fluorescence imaging. Cell. 159 (3), 635-646 (2014).
- Zhao, N., et al. A genetically encoded probe for imaging nascent and mature HA-tagged proteins in vivo. Nature Communications. 10 (1), 2947(2019).
- Jedlitzke, B., Mootz, H. D. Photocaged nanobodies delivered into cells for light activation of biological processes. ChemPhotoChem. 5 (1), 22-25 (2021).
- Coulon, A., et al. Kinetic competition during the transcription cycle results in stochastic RNA processing. eLife. 3, 03939(2014).
- Pichon, X., Robert, M. -C., Bertrand, E., Singer, R. H., Tutucci, E. New generations of MS2 variants and MCP fusions to detect single mRNAs in living eukaryotic cells. Methods in Molecular Biology. 2166, 121-144 (2020).
- Koch, A., et al. Quantifying the dynamics of IRES and cap translation with single-molecule resolution in live cells. Nature Structural & Molecular Biology. 27, 1095-1104 (2020).
- Moon, S. L., et al. Multicolor single-molecule tracking of mRNA interactions with RNP granules. Nature cell biology. 21 (2), 162-168 (2019).
- Moon, S. L. Coupling of translation quality control and mRNA targeting to stress granules. Journal of Cell Biology. 219 (8), 202004120(2020).
- Cialek, C. A., et al. Imaging translational control by Argonaute with single-molecule resolution in live cells. bioRxiv. , (2021).
- Forero-Quintero, L. S., et al. Live-cell imaging reveals the spatiotemporal organization of endogenous RNA polymerase II phosphorylation at a single gene. bioRxiv. , (2020).
- Lyon, K., Aguilera, L. U., Morisaki, T., Munsky, B., Stasevich, T. J. Live-cell single RNA imaging reveals bursts of translational frameshifting. Molecular Cell. 75 (1), 172-183 (2019).
- JoVE. Introduction to Fluorescence Microscopy. General Laboratory Techniques. JoVE Science Education Database. , JoVE. Cambridge, MA. (2021).
- Hayashi-Takanaka, Y., Yamagata, K., Nozaki, N., Kimura, H. Visualizing histone modifications in living cells: spatiotemporal dynamics of H3 phosphorylation during interphase. Journal of Cell Biology. 187 (6), 781-790 (2009).
- Kumar, P., Nagarajan, A., Uchil, P. D. Analysis of cell viability by the MTT assay. Cold Spring Harbor Protocols. 2018 (6), (2018).
- Stasevich, T. J., et al. Regulation of RNA polymerase II activation by histone acetylation in single living cells. Nature. 516 (7530), 272-275 (2014).
- Besteiro, S., Michelin, A., Poncet, J., Dubremetz, J. -F., Lebrun, M. Export of a Toxoplasma gondii rhoptry neck protein complex at the host cell membrane to form the moving junction during invasion. PLOS Pathogens. 5 (2), 1000309(2009).
- Gilmore, A. P., Romer, L. H. Inhibition of focal adhesion kinase (FAK) signaling in focal adhesions decreases cell motility and proliferation. Molecular Biology of the Cell. 7 (8), 1209-1224 (1996).
- Emerson, N. T., Hsia, C. -H., Rafalska-Metcalf, I. U., Yang, H. Mechanodelivery of nanoparticles to the cytoplasm of living cells. Nanoscale. 6 (9), 4538-4543 (2014).
- Manders, E. M. M., Kimura, H., Cook, P. R. Direct imaging of DNA in living cells reveals the dynamics of chromosome formation. Journal of Cell Biology. 144 (5), 813-822 (1999).
- Memedula, S., Belmont, A. S. Sequential recruitment of HAT and SWI/SNF components to condensed chromatin by VP16. Current Biology. 13 (3), 241-246 (2003).
- Becker, T., Volchuk, A., Rothman, J. E. Differential use of endoplasmic reticulum membrane for phagocytosis in J774 macrophages. Proceedings of the National Academy of Sciences of the United States of America. 102 (11), 4022-4026 (2005).
- Molenaar, C., et al. Visualizing telomere dynamics in living mammalian cells using PNA probes. The EMBO Journal. 22 (24), 6631-6641 (2003).
- Jones, S. A., Shim, S. -H., He, J., Zhuang, X. Fast, three-dimensional super-resolution imaging of live cells. Nature Methods. 8 (6), 499-505 (2011).
- Sabri, A., Xu, X., Krapf, W. M. Elucidating the origin of heterogeneous anomalous diffusion in the cytoplasm of mammalian cells. Physical Review Letters. 125 (5), 053901(2020).
- Sato, Y., et al. Genetically encoded system to track histone modification in vivo. Scientific Reports. 3, 2436(2013).
- Sato, Y., Stasevich, T. J., Kimura, H. Visualizing the dynamics of inactive X chromosomes in living cells using antibody-based fluorescent probes. X-Chromosome Inactivation. Methods in Molecular Biology. 1861, Humana. New York, NY, USA. 91-102 (2018).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved