
Un abonnement à JoVE est nécessaire pour voir ce contenu. Connectez-vous ou commencez votre essai gratuit.
Method Article
Analyse de la forme des lignes des spectres RMN dynamiques pour caractériser les réarrangements de sphère de coordination dans un complexe chiral de polyhydrure de rhénium
Dans cet article
Résumé
L’analyse de la forme des lignes des spectres RMN recueillis sur une plage de températures sert de guide pour le réarrangement des atomes de la sphère de coordination interne à un complexe chiral de polyhydrure de rhénium(V) à huit coordonnées, ReH5(PPh3)2 (sec-butylamine). L’analyse de la forme des lignes est également utilisée pour déterminer les paramètres d’activation ΔH‡, ΔS‡ et ΔG‡ pour ces réarrangements d’atomes.
Résumé
La spectroscopie par résonance magnétique nucléaire (RMN) en solution dynamique est la méthode typique de caractérisation des réarrangements dynamiques des atomes dans la sphère de coordination pour les complexes de polyhydrure de métaux de transition. L’ajustement de la forme des lignes des spectres RMN dynamiques peut conduire à des estimations des paramètres d’activation des processus de réarrangement dynamique. Une combinaison de la spectroscopie RMN dynamique 31 P-{1 H} d’atomes de phosphore liés aux métaux avec la spectroscopie RMN dynamique 1H-{31P} des ligands hydrures peut identifier les réarrangements de ligands hydrures qui se produisent en conjonction avec un réarrangement d’atomes de phosphore. Pour les molécules qui présentent une telle paire couplée de réarrangements, la spectroscopie RMN dynamique peut être utilisée pour tester des modèles théoriques pour les réarrangements de ligands. La spectroscopie RMN dynamique 1H-{31P} et l’ajustement de la forme de ligne peuvent également identifier la présence d’un processus d’échange qui déplace un ligand hydrure spécifique au-delà de la sphère de coordination interne du métal par un échange de protons avec une molécule de solvant telle que l’eau adventice. La préparation d’un nouveau composé, ReH5(PPh3)2(sec-butylamine), qui illustre de multiples processus de réarrangement dynamique, est présentée ainsi que l’ajustement de la forme des lignes des spectres RMN dynamiques du complexe. Les résultats de l’ajustement de la forme de la ligne peuvent être analysés par l’équation d’Eyring pour estimer les paramètres d’activation des processus dynamiques identifiés.
Introduction
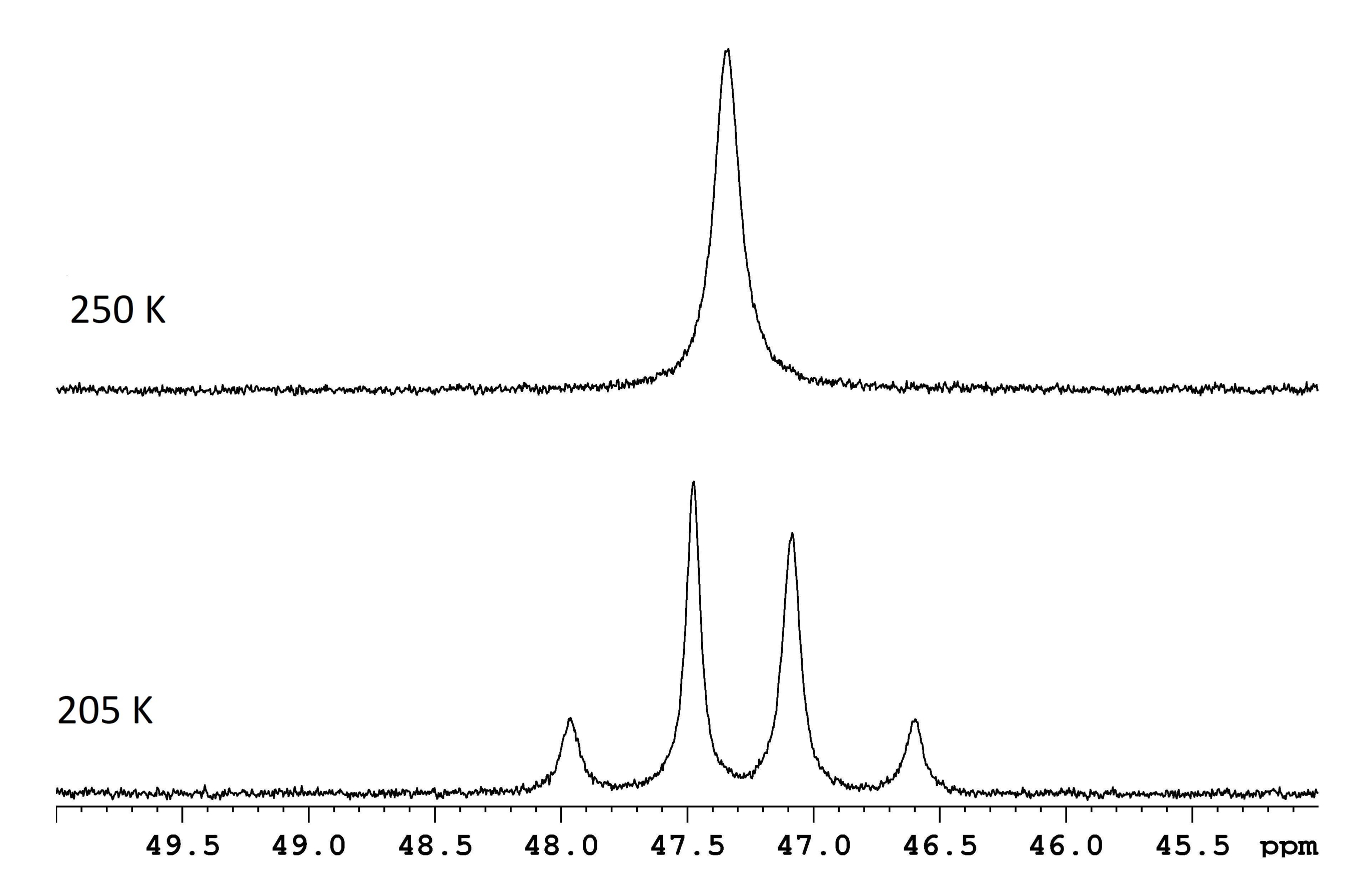
La spectroscopie RMN est couramment utilisée pour caractériser les processus dynamiques qui se produisent à l’intérieur ou entre les molécules. Pour de nombreux réarrangements intramoléculaires simples, l’estimation de ΔG‡ est aussi simple que la mesure de la différence de fréquence, Δν, entre deux résonances à la limite d’échange lent et la détermination de la température de coalescence pour ces mêmes résonances (Figure 1)1. La relation,
ΔG‡ = 4,575 x 10-3 kcal/mol x T c [9,972 + log (Tc/Δν)]
où Tc est la température de coalescence pour une paire de résonances qui représentent la forme d’échange lent d’un échantillon dynamique, peut être utilisé pour résoudre l’énergie libre d’activation pour un tel réarrangement dynamique. Les systèmes dynamiques plus complexes nécessitent l’ajustement de la forme des lignes des spectres RMN dynamiques ou une autre technique de RMN telle que la spectroscopie d’échange bidimensionnel (2D-EXSY) ou la spectroscopie d’effet Overhauser à cadre rotatif bidimensionnel (2D-ROESY) pour estimer les paramètres d’activation.

Figure 1 : Spectres RMN pour une solution d 8-toluène de ReH5(PPh3)2(sec-butylamine) à deux températures. La différence de fréquence entre les deux doublets à échange lent (trace inférieure, 117,8 Hz) et une température de coalescence de 250 K (trace supérieure) correspondent à une barrière d’énergie (ΔG‡) de 11,8 kcal/mol. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
L’ajustement de la forme des lignes des spectres RMN dynamiques est une technique courante utilisée depuis longtemps pour l’estimation des paramètres d’activation qui décrivent les réarrangements dynamiques pour les substances ayant une énergie d’activation d’environ 5 à 25 kcal/mol 2,3,4,5. La détermination des barrières énergétiques à l’échange de protons entre l’eau et les molécules d’amine6, la barrière énergétique à la rotation autour de la liaison C-N dans le diméthylformamide7, ou la taille générale des fractions organiques8 ne sont que quelques exemples des nombreuses propriétés qui ont été évaluées par ajustement de la forme des raies des spectres RMN dynamiques. Ce manuscrit démontre l’utilisation de l’ajustement de la forme des lignes pour caractériser les processus dynamiques intermoléculaires et intramoléculaires qui se produisent pour le complexe ReH5 (PPh3) 2 (sec-butyl amine). Les objectifs de cette expérience et d’expériences similaires d’ajustement de la RMN en forme de ligne sont les suivants: 1) caractériser tous les processus d’échange d’atomes dynamiques intramoléculaires observables RMN s’ils sont présents, 2) identifier et caractériser les processus d’échange d’atomes dynamiques intramoléculaires observables RMN s’ils sont présents, 3) identifier les échanges d’atomes intramoléculaires corrélés qui se produisent pour, dans cet exemple, les atomes d’hydrogène et de phosphore, et 4) pour l’exemple présenté ici, comparer deux modèles publiés pour les processus dynamiques qui se produisent dans le complexe ReH5(PPh3)2 (sec-butylamine).
Les systèmes de polyhydrure de rhénium(V) à huit coordonnées sont des systèmes dynamiques complexes dans lesquels les ligands participent à de multiples processus dynamiques et les atomes de phosphore peuvent participer à un processus dynamique unique qui est un deuxième aspect d’un processus d’échange de ligands hydrures 9,10,11,12,13,14,15,16,17,18 ,19,20,21,22,23,24,25,26,
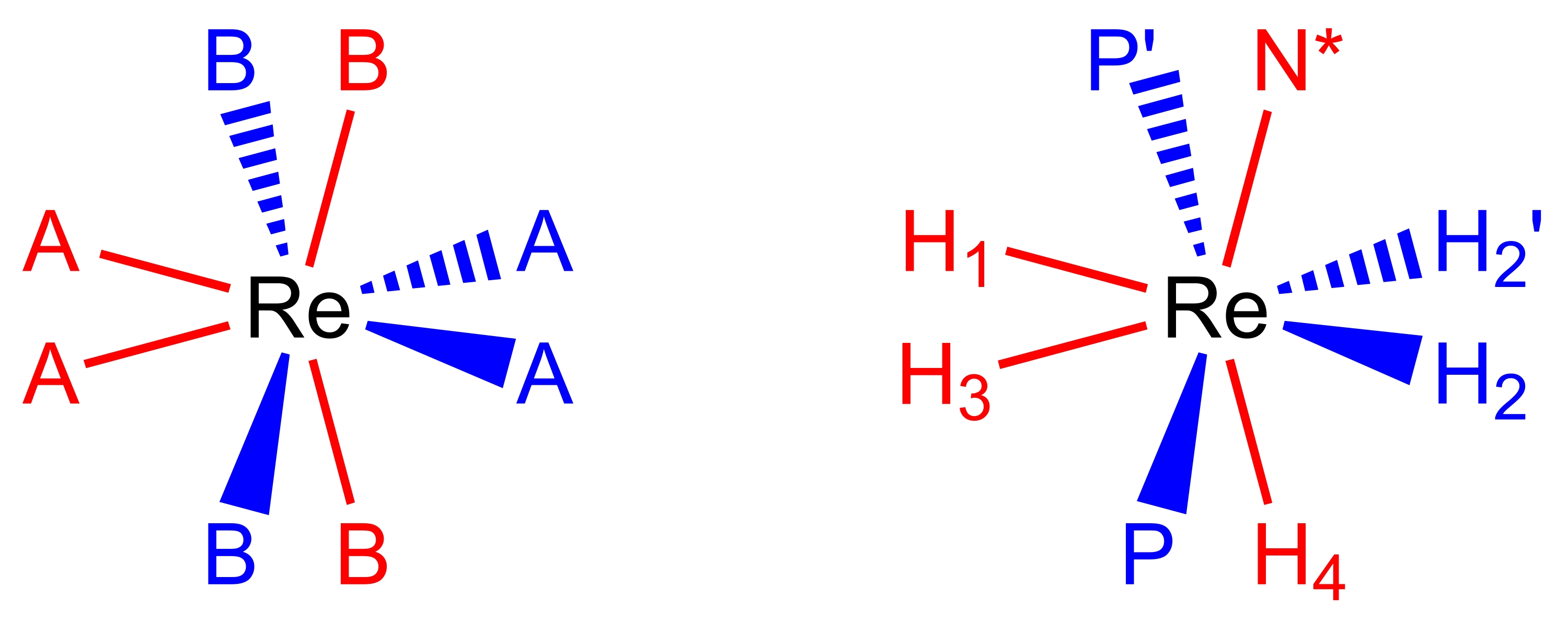
27,28,29. Les complexes de polyhydrure de rhénium(V) pseudododécaédriques à huit coordonnées adoptent une géométrie moléculaire (Figure 2), qui peut être décrite comme une paire de trapèzes orthogonaux de ligands17,26. Les sommets sur les longs bords des trapèzes sont communément étiquetés comme des sites B et, dans les complexes de polyhydrure de rhénium, sont généralement les sites occupés par des ligands donneurs neutres à deux électrons tels que les phosphines tertiaires ou les ligands amine. Les sommets sur les bords courts des trapèzes sont communément étiquetés comme des sites A et sont généralement occupés par des ligands anioniques à deux électrons, hydrures. Les spectres RMN à température ambiante des complexes polyhydrure de rhénium(V) sont, généralement, trompeusement simples en raison des nombreux processus dynamiques qui se produisent dans les solutions à température ambiante.

Figure 2 : Un ensemble de coordination dodécaédrique (à gauche) et le complexeReH5(PPh3)2(sec-butylamine) dans la même perspective (à droite). Les sites de couleur rouge représentent les sites de coordination qui forment un trapèze vertical, et les sites de couleur bleue représentent les sites de coordination qui forment un trapèze horizontal. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
Les complexes de la forme ReH5(PPh3)2(amine) sont la classe de complexes polyhydrure de rhénium la plus étudiée en ce qui concerne les processus dynamiques 9,10,12,13,16,30,31. Trois processus dynamiques (Figure 3) ont été identifiés pour les complexes ReH5(PPh3)2(amine) : 1) un échange de protons entre le seul ligand hydrure du site B et un proton d’une molécule d’eau (fortuit ou intentionnel)9,13, 2) un échange tourniquet d’une paire de ligands hydrure de site A avec unligand 9 hydrure de site B adjacent, 11,13,30,31, et 3) une inversion stérique (ou pseudorotation) qui se manifeste par un échange par paires des ligands hydrures du site A et un mouvement par paires des atomes du site B vers le côté opposé du centre du rhénium (comme illustré sur la figure 4)4,5,6,8,26,27 . Le mouvement des atomes du site B vers le côté opposé du rhénium est observable par spectroscopie RMN dynamique comme: 1) un processus qui rend les 3 et 5 protons inéquivalents de N = pyridine équivalents à température ambiante10,30,31, 2) un processus qui provoque un échange rapide des isomères E et Z de N = ligands amines aromatiques substitués de manière asymétrique à température ambiante9, 10,13,30,31, ou 3) un processus qui provoque un échange rapide des perspectives stériques d’une paire diastéréotopique d’atomes de phosphore par rapport à un centre chiral situé sur le ligand amine9,30,31. Le complexe chiral ReH5(PPh3)2(sec-butylamine) qui n’a pas encore été signalé auparavant offre l’occasion de décrire de façon générale les méthodes qui peuvent être utilisées pour identifier et caractériser les réarrangements dynamiques des complexes de polyhydrure de rhénium.

Figure 3 : Représentations des processus dynamiques observés par spectroscopie RMN pour les solutions deReH5(PPh3)2(sec-butylamine). La représentation A représente l’échange d’un seul proton d’eau adventive contre le ligand hydrure unique du site B. La représentation B représente l’échange de tourniquets de trois ligands hydrures adjacents, dont deux résident dans le site A tandis que le troisième est le ligand hydrure unique du site B. La représentation C représente à la fois l’échange par paires de ligands hydrures du site A ainsi que l’inversion stérique des atomes de phosphore par rapport au ligand amine chiral (N*). Il convient de noter que l’échange par paires du ligand de l’hydrure du site A ne nécessite pas de déplacement des ligands hydrure du site A vers le côté opposé du centre du rhénium. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
Pour les systèmes chimiques tels que les complexes de polyhydrure de rhénium, qui présentent un ensemble complexe de processus dynamiques, l’ajustement de la forme des lignes des spectres RMN dynamiques est la technique RMN la plus utilisée pour caractériser les processus 9,11,13,16,21,29. Les technologies bidimensionnelles EXSY 9,32 ou 2D-ROESY11 sont des techniques alternatives de RMN dynamique qui peuvent également être utilisées pour caractériser quantitativement les processus dynamiques. Les spectres EXSY bidimensionnels sont généralement mesurés dans le domaine de la température d’échange lent; les spectres ROESY bidimensionnels sont généralement mesurés dans le domaine de la température d’échange rapide. Les deux techniques bidimensionnelles peuvent nécessiter beaucoup de temps dans le spectromètre pour l’acquisition de données, en ce sens que chacune des techniques acquiert un ensemble de données beaucoup plus grand, à une température donnée, que les ensembles de données unidimensionnels nécessaires pour l’analyse de l’ajustement de la forme de la ligne. Des processus dynamiques simples qui sont bien compris, tels que l’échange dynamique des deux groupes méthyle du diméthylformamide, peuvent être facilement caractérisés par l’une des trois techniques de RMN. Des systèmes plus complexes, tels que ReH5 (PPh3) 2 (sec-butyl amine), dans lesquels des ligands d’hydrures individuels participent à de multiples processus dynamiques, ou des systèmes qui ne sont pas nécessairement bien compris, tels qu’un nouveau complexe polyhydrure de métal de transition qui peut ou non échanger des protons entre un ligand hydrure hydrure et de l’eau adventive, sont plus facilement caractérisés quantitativement par la méthode RMN ajustée en forme de ligne que par les méthodes de RMN bidimensionnelle. Contrairement aux méthodes de RMN bidimensionnelle, la méthode d’ajustement de la forme de ligne fournit une visualisation facilement interprétable de la correspondance entre un modèle testé et les données expérimentales ainsi que la preuve visuelle d’un échange qui déplace un ligand hydrure au-delà de la sphère de coordination interne du rhénium. Sur la base des hauteurs et des formes des pics dans les spectres d’échange lent, même un système dynamique complexe tel que ReH5 (PPh3) 2 (sec-butylamine) peut conduire à un ensemble initial de modèles d’échange facilement testés. De plus, lorsque plusieurs modèles théoriques ont été rapportés pour une transformation moléculaire, l’ajustement de la forme des lignes des spectres RMN dynamiques peut permettre une comparaison visuelle de chaque modèle par rapport aux spectres observés.
Au-delà des trois techniques de RMN mentionnées ci-dessus, des expériences de RMN de substitution isotopique impliquantD2Oou HD ont été utilisées pour démontrer qualitativement l’échange intermoléculaire d’atomes pour des systèmes complexes de polyhydrure de rhénium, mais n’ont pas été utilisées pour les caractérisations quantitatives 9,33,34,35. Les calculs théoriques présentent une méthode supplémentaire pour caractériser les processus dynamiques des systèmes dynamiques complexes30,31,36. Les calculs théoriques ont l’avantage sur l’ajustement de forme de ligne en ce sens qu’ils peuvent être utilisés pour différencier les possibilités qui ne peuvent pas être distinguées par l’analyse de l’ajustement de forme de ligne. Par exemple, des calculs théoriques ont été utilisés pour décrire un échange qui implique trois ligands hydrures adjacents sur certains complexes rhénium(V) comme un échange tourniquet des trois ligands hydrures, plutôt qu’une paire alternée d’échanges par paires avec chaque échange par paires, y compris un ligand hydrure unique et l’un des deux ligands hydrures chimiquement équivalents30, 31. Les résultats des calculs théoriques sont généralement comparés aux caractérisations quantitatives observées expérimentalement à partir de l’une des trois techniques de RMN mentionnées ci-dessus pour vérifier la validité des résultats calculés.
L’ajustement de la forme des lignes des spectres RMN dynamiques tire parti du changement d’apparence des spectres RMN qui se produit lorsque les noyaux actifs RMN se déplacent entre différents environnements chimiques lors d’une mesure RMN. Les spectres RMN à échange lent (spectres avec des résonances lorentziennes indépendantes pour chaque ensemble de noyaux échangeurs) se produisent à des températures où la différence de fréquence entre les résonances pour les noyaux qui s’échangent est grande par rapport à la vitesse d’échange des noyaux37. Les spectres RMN à échange rapide (spectres avec une seule résonance lorentzienne pour l’échange de noyaux) se produisent à des températures où la vitesse d’échange des noyaux est beaucoup plus grande que la différence de fréquence entre les résonances d’échange lent37. Les taux de change intermédiaires se produisent pour les températures comprises entre le domaine de température d’échange lent et le domaine de température d’échange rapide37. Si les paramètres fondamentaux de la fréquence de Larmor, du décalage chimique des noyaux échangeurs, des constantes de couplage (le cas échéant) pour les noyaux échangeurs et des populations relatives de chaque type de noyau sont connus, les constantes de vitesse pour les échanges putatifs entre noyaux peuvent être déterminées en comparant les spectres simulés aux spectres observés à plusieurs températures intermédiaires. De bons ajustements pour les simulations à plusieurs températures donnent des données de constante de température et de vitesse qui peuvent être utilisées avec l’équation d’Eyring pour estimer les paramètres d’activation pour le ou les échanges putatifs. Les résultats de la méthode se sont avérés à la fois exacts et reproductibles.
Protocole
1. Préparation de l’échantillon
- Préparation de ReH7(PPh3)235
- Mélanger 0,15 g de borohydrure de sodium et 0,41 g de ReOCl 3(PPh3)2 dans une fiole à fond rond de 100 ml à deux ou trois cols munie d’un septum en caoutchouc et d’un orifice à gaz, ou dans une fiole Kjeldahl de 100 mL (munie d’un orifice à gaz à bras latéral) munie d’un septum en caoutchouc (figure supplémentaire 1).
- Ajouter une barre de rotation à la cuve de réaction.
- Dans une hotte, utilisez un morceau de tube de pression en caoutchouc pour connecter l’orifice de gaz de la cuve de réaction à l’un des robinets d’arrêt d’un collecteur en verre double pour le vide et l’azote gazeux. Connectez le collecteur de vide en verre à une pompe à vide avec un tube de pression en caoutchouc et connectez le collecteur d’azote en verre à une bouteille d’azote gazeux régulée.
- Connectez le gaz de sortie du collecteur d’azote gazeux à un robinet d’arrêt qui peut être utilisé pour diriger le gaz évacué à travers une colonne d’huile minérale de 2 cm ou une colonne de mercure de 2 cm.
- Ouvrez le robinet de la bouteille d’azote et ajustez la pression sur le gaz en circulation à 34 livres par pouce carré. Évacuez le flux d’azote gazeux à travers le barboteur de mercure.
- Évacuer le gaz à l’intérieur de la cuve de réaction en ajustant le robinet d’arrêt sur le collecteur de verre pour connecter le récipient au collecteur à vide. Remplissez la cuve de réaction avec de l’azote gazeux en changeant le robinet d’arrêt du collecteur de verre de sorte qu’il relie le collecteur de gaz au récipient de réaction.
- Répétez les étapes 1.1.5 et 1.1.6 deux fois de plus pour remplacer complètement l’air dans la cuve de réaction par de l’azote gazeux. Refroidir la fiole et son contenu dans un bain de glace.
- Ajouter 8 mL d’eau désoxygénée et 8 mL de tétrahydrofurane désoxygéné aux solides dans la cuve de réaction à l’aide d’une seringue. Commutez le robinet d’arrêt de ventilation de gaz de sorte que le gaz passe à travers le barboteur d’huile minérale. Remuez légèrement la suspension dans le bain de glace pendant 15 min. Retirer la cuve de réaction du bain de glace après les 15 premières minutes d’agitation.
- Laisser le mélange continuer à remuer pendant encore 45 minutes. Notez la couleur du mélange réactionnel comme indicateur de la fin de la réaction. Une couleur de mélange réactionnel beige à orange (figure supplémentaire 1) indique que la réaction a atteint son point final.
- Après avoir obtenu une couleur orange à beige pour le mélange réactionnel, filtrer le mélange à travers un entonnoir en verre fritté moyen de 30 mL. Lavez le solide récupéré trois fois chacun avec des portions de 15 mL d’eau, de méthanol et d’éther éthylique. Sécher le solide sous vide pour éliminer tout solvant adsorbé.
NOTE: La réaction produit généralement entre 0,20 g et 0,25 g de produit.
- Préparation de ReH5(PPh3)2(sec-butylamine)
- Peser 0,070 g de ReH7(PPh3)2 et le transférer dans une fiole à fond rond à col unique de 50 ml contenant une barre d’essorage. Ajuster le ballon à un condenseur équipé d’un orifice de gaz. Désoxygéner le récipient de réaction à l’aide de la méthode de pompe et de remplissage des étapes 1.1.3 à 1.1.7.
- Ajouter un volume de 8 mL de tétrahydrofurane désoxygéné dans la cuve de réaction à l’aide d’une seringue en craquant le joint entre le ballon à fond rond et le condenseur. Ajouter un volume de 0,2 mL de sec-butylamine de la même manière. Réglez le robinet d’arrêt de ventilation de gaz de sorte que les évents de gaz vers le barboteur d’huile minérale.
- Chauffer le mélange réactionnel à reflux à 65 °C avec un manteau chauffant relié à un transformateur à courant alternatif variable réglé sur 40 sur une échelle de 0 à 140 pendant 40 min. Refroidir le mélange réactionnel à une température qui permet une manipulation pratique de la fiole.
- Verser le mélange réactionnel dans 25 mL de méthanol dans une fiole d’erlenmeyer de 125 mL. Remuer vigoureusement le mélange pendant 5 min. Ajouter 5 mL d’eau pour induire la formation d’un précipité jaune floculant.
- Recueillir le précipité jaune par filtration sous vide dans un entonnoir en verre fritté. Laver le solide avec 15 ml de méthanol. Sécher le solide sous vide. À la suite de ce processus, le rendement typique du produit est de 0,035 g.
2. Acquisition et analyse des spectres RMN
- Mesure des spectres RMN dynamiques
- Préparer un échantillon RMN avec environ 8 mg du complexe ReH5(PPh3)2(sec-butylamine) dans environ 0,8 mL de d8-toluène. Insérez l’échantillon dans l’instrument.
- Cliquez sur l’onglet Fichier et sélectionnez Nouveau parmi les choix qui s’affichent pour ouvrir une boîte de dialogue utilisée pour créer une expérience RMN.
- Créez une expérience de 1H en procédant comme suit.
- Attribuez un nom de dossier pour la nouvelle expérience en remplissant la zone de saisie Nom avec un nom de fichier unique. Attribuez un numéro d’expérience tel que 1 pour l’expérience 1H dans la zone EXPNO .
- Attribuez un numéro de processus de 1 à l’expérience dans la zone PROCNO . Affectez le dossier dans un répertoire à l’aide de la liste déroulante pour DIR. Identifiez le solvant sur lequel l’instrument verrouillera à partir de la liste déroulante Choix de solvants .
- Choisissez le répertoire qui contient les paramètres de l’expérience 1H dans la liste déroulante des répertoires dans les répertoires de l’expérience. Sélectionnez l’expérience Proton parmi les choix de la liste déroulante Expérience et (facultatif) ajoutez un titre pour les données dans la zone de remplissage du titre .
- Entrez une commande Eda dans la ligne de commande et ajustez les paramètres si nécessaire pour répondre aux descriptions de l’expérience fournies dans le deuxième paragraphe de la section Discussion ci-dessous.
- Cliquez sur l’onglet Fenêtre, sélectionnez Nouvelle fenêtre dans la liste et répétez les étapes 2.1.3.1-2.1.3.8 pour préparer une expérience 1 H-{31P} en utilisant une valeur EXPNO de 2 afin de différencier l’expérience de l’expérience 1H construite précédemment.
- Cliquez sur l’onglet Fenêtre, sélectionnez Nouvelle fenêtre dans la liste et répétez les étapes 2.1.3.1-2.1.3.8 pour préparer une expérience 31 P-{1 H} en utilisant une valeur EXPNO de 3 afin de différencier l’expérience des expériences 1 H et 1 H-{31P} construites précédemment (voir le tableau supplémentaire 1pour des informations détaillées sur les paramètres).
- Entrez une commande Verrouiller dans la ligne de commande et sélectionnez le choix d 8-toluène dans la liste. Cliquez sur OK pour accepter le choix du solvant. Entrez une commande Atma dans la ligne de commande, si nécessaire, en raison d’une sonde à noyau variable en bande X, pour minimiser l’énergie réfléchie aux fréquences de Larmor pendant 1H et 31P sur l’instrument.
- Entrez une commande Ro sur la ligne de commande, tapez une valeur de 20 dans la zone et cliquez sur le bouton Démarrer la rotation . Entrez une commande Shim sur la ligne de commande. Choisissez une routine autoshim appropriée telle que Topshim dans la liste des routines de shim et cliquez sur le bouton Démarrer .
- Entrez une commande Rga sur la ligne de commande. Choisissez la sélection Réglage automatique du récepteur et cliquez sur OK. À son tour, mesurez les trois spectres de l’échantillon à température ambiante en utilisant 64 balayages pour chaque spectre avec une commande Go sur la ligne de commande.
- Transformez les données d’une expérience en spectre avec une commande Efp entrée dans la ligne de commande.
- Ajustez le phasage du spectre à l’aide des commandes suivantes.
- Cliquez sur l’onglet Phase suivi d’un clic sur l’onglet Ajuster la phase. Placez le curseur sur le bouton 0 de la barre d’outils de phasage et maintenez le bouton gauche de la souris enfoncé pour que le bouton 0 devienne vert.
- Avec le bouton gauche de la souris enfoncé, faites rouler la souris vers l’avant ou vers l’arrière jusqu’à ce que la ligne de base soit plate sur tout le spectre et que toutes les résonances soient affichées sous forme d’absorbances (les pics s’élèvent au-dessus de la ligne de base).
- Si la ligne de base ne peut pas être rendue plate avec seulement le bouton 0, réglez le bouton 1 comme décrit aux étapes 2.1.10.1 et 2.1.10.2 ainsi que le bouton 0, jusqu’à ce que la ligne de base soit plate pour toute la fenêtre spectrale.
- Enregistrez le réglage de phase avec les données en cliquant sur le bouton Enregistrer et Retour dans la barre d’outils de phasage.
- Ajustez le nombre de balayages pour chaque mesure, au besoin, en fonction du rapport signal sur bruit dans le spectre, en gardant à l’esprit que le rapport signal/bruit diminue généralement à des températures plus basses en raison de la décoalescence des signaux en résonances individuelles (Figure 4).
- Préparez le spectromètre pour le contrôle de la température selon les instructions du fournisseur. Entrez un débit de 200 L/h pour le gaz de refroidissement et une température cible de 290 K pour la sonde. Laissez le spectromètre se stabiliser à la température cible pendant 2 min. Augmenter le débit de gaz de refroidissement, si nécessaire, à 210 ou 220 L/h pour stabiliser la température.
- Cale l’échantillon à 290 K comme à l’étape 2.1.7. Changez le nom de fichier pour chacun des spectres précédemment mesurés en ajoutant la température à la fin du nom de fichier (étapes 2.1.2 et 2.1.3.1) et obtenez un ensemble de trois spectres à 290 K.
- Augmenter le débit de gaz de refroidissement de ≥ 30 L/h, au besoin pour le stabiliser à la température suivante, et diminuer la température cible de 10 K. Laisser le spectromètre se stabiliser à la température suivante pendant 2 minutes, puis caler l’échantillon comme à l’étape 2.1.7. Mesurez l’ensemble des trois spectres.
- Répétez les étapes 2.1.13 et 2.1.14 au besoin pour acquérir des spectres jusqu’à la température la plus basse souhaitée.
REMARQUE: Une température de 200 K est généralement suffisante pour un ensemble complet de données qui convient pour déterminer les paramètres d’activation pour les processus dynamiques de l’échantillon. - Réchauffer l’échantillon à température ambiante par incréments de 10 K. Stabiliser la température pendant 2 minutes à chaque température avant de réchauffer à nouveau l’échantillon pour éviter d’endommager le revêtement en verre de la sonde.
- Analyse de la forme des lignes des spectres mesurés
- Dans le programme RMN, cliquez sur la barre de commandes en haut à gauche de la fenêtre et sélectionnez Ouvrir dans le menu déroulant. Sélectionnez Ouvrir les données RMN stockées au format standard. Cliquez sur OK pour ouvrir la fenêtre de l’explorateur de fichiers du programme.
- Accédez au dossier pour les données à analyser par ajustement de forme de ligne. Sélectionnez le numéro de fichier qui correspond au spectre à analyser et cliquez sur le bouton Afficher . Le spectre (s’il a déjà été traité) ou la courbe de désintégration par induction libre (FID) est affiché dans le logiciel RMN.
- Traitez le FID si nécessaire, en entrant une commande Efp (multiplication exponentielle, transformation de Fourier et correction de phase) dans la ligne de commande. Ajustez la phase du spectre (étape 2.1.10).
- Ajuster la ligne de base du spectre; S’il n’est pas plat sur tout le spectre, égalez la ligne d’intensité 0, comme suit.
- Cliquez sur l’onglet Processus , puis sur l’onglet Ligne de base . Placez le curseur sur le bouton A . Appuyez sur le bouton gauche de la souris et faites glisser la souris vers l’avant ou vers l’arrière pour niveler la ligne de réglage rouge avec l’extrémité gauche (champ inférieur) du spectre.
- Si la ligne de base n’est toujours pas au niveau de la ligne de réglage rouge, répétez le processus avec les boutons de lettre restants jusqu’à ce que la ligne de réglage rouge corresponde à la ligne de base du spectre. Utilisez le bouton Enregistrer et Retourner pour enregistrer l’ajustement lorsque la ligne de base ajustée en rouge correspond à la ligne de base réelle.
- Sélectionnez l’onglet Analyser dans le logiciel RMN. Dans les options d’analyse, sélectionnez le choix Formes de ligne suivi du choix Ajuster les modèles RMN dynamiques .
- Le spectre est maintenant affiché dans la fenêtre du module d’ajustement de forme de ligne. Utilisez les barres d’outils au-dessus du spectre pour ajuster la façon dont le spectre est affiché. La fenêtre à gauche du spectre gère l’ajustement de la forme de la ligne du spectre.
- Ajustez l’affichage du spectre à l’aide de l’outil Zoom lisse afin que la partie du spectre à installer soit affichée dans la fenêtre du spectre. Utilisez les boutons de barre d’outils Décaler le spectre gauche et droit pour centrer une partie du spectre dans la fenêtre d’affichage.
- Accédez à la fenêtre de changement chimique pour l’ajustement de la forme de ligne en sélectionnant l’onglet Spectre dans la fenêtre d’ajustement de forme de ligne.
- Cliquez sur le bouton Modifier la plage . Entrez les décalages chimiques supérieurs et inférieurs pour l’ajustement de la forme de ligne et cliquez sur le bouton OK pour accepter ces limites.
- Démarrez un modèle d’ajustement de forme de ligne en cliquant sur l’onglet Système de rotation dans la fenêtre d’ajustement de forme de ligne. Cliquez sur le bouton Ajouter pour permettre la construction d’un système de spin de modèle.
- Désélectionnez LB (pour l’élargissement de ligne) et entrez la valeur pour l’élargissement de ligne manuellement avec la souris et le bouton LB dans la barre d’outils d’ajustement de forme de ligne.
- Ajoutez le premier noyau dans le modèle en cliquant sur l’onglet Noyau puis en cliquant sur le bouton Ajouter . Un ensemble de valeurs par défaut apparaîtra pour Nucleus 1. Ajustez le décalage chimique pour Nucleus 1 en entrant une valeur pour le décalage chimique dans la zone Nu(iso) ou avec l’outil de décalage chimique dans la barre d’outils d’ajustement de forme de ligne.
REMARQUE: Si la boîte de sélection est laissée dans la forme cochée, le déplacement chimique de ce noyau sera modifié pour obtenir le meilleur ajustement. Les variables non vérifiées ne seront pas modifiées dans le processus d’ajustement de ligne. - Utilisez la case Pseudospin pour le noyau 1 pour entrer le nombre de noyaux équivalents pour le noyau 1 avec chaque noyau de spin 1/2 équivalent à 0,5 en comptage. Entrez la somme des spins dans la case Pseudospin afin de tenir compte de tous les noyaux équivalents.
- Utilisez la boîte In Molecule pour accueillir des modèles qui nécessitent plus d’une seule molécule pour participer à un processus dynamique. Attribuez des résonances provenant de différentes molécules à des molécules distinctes en utilisant des désignations telles que 1, 2, etc. pour différentes molécules. Pour les résonances qui proviennent d’une seule molécule, assignez 1 pour toutes les valeurs In Molecule .
- Ajoutez le deuxième noyau et tous les noyaux suivants au modèle en cliquant sur l’onglet Noyau , puis sur le bouton Ajouter . Incluez le couplage spin-spin entre les noyaux en entrant le couplage dans la case JN appropriée (où N est le noyau avec lequel le noyau ajouté est couplé, N = 1, 2, ...) ou en ajustant le bouton Couplage scalaire dans la barre d’outils d’ajustement de forme de ligne.
- Commencez le processus de description des échanges d’atomes en cliquant sur l’onglet Réaction . Cliquez sur la case à cocher si la constante de vitesse de l’échange doit être modifiée dans l’ajustement de la forme de la ligne. Entrez le nombre de noyaux à échanger (nombre par rapport à leurs onglets d’identification tels que Nucleus 1 et Nucleus 2) dans la zone Échanges pour le premier échange du modèle.
- Décrivez les échanges à tester dans les cases situées sous la case Échanges . Définissez les échanges entre les onglets Nucleus dans les zones ci-dessous. Un échange à deux noyaux serait entré en tant que noyau 1 à noyau 2 et noyau 2 à noyau 1. S’assurer que les échanges sont cycliques en ce sens que si un noyau est déplacé du noyau 1, un autre noyau doit être déplacé dans le noyau 1.
- Utilisez le bouton Vitesse d’échange de la barre d’outils d’ajustement de forme de ligne pour modifier la valeur initiale de k afin d’ajuster itérativement la valeur de k, même si la case à cocher est activée pour la constante de débit.
- Ajoutez d’autres échanges au modèle en cliquant sur l’onglet Réaction , puis sur le bouton Ajouter . Ajoutez des échanges au modèle si nécessaire. Utilisez les outils de la barre d’outils d’ajustement de forme de ligne pour ajuster les variables de départ, y compris l’intensité du spectre, afin qu’elles correspondent bien au spectre à ajuster.
- Commencez l’ajustement itératif de la forme de ligne en cliquant sur le bouton Démarrer l’ajustement du spectre dans la barre d’outils d’ajustement de forme de ligne. Continuez l’ajustement itératif jusqu’à ce qu’aucun changement ne soit trouvé dans le meilleur chevauchement entre le spectre et le modèle ou jusqu’à ce que 1000 itérations soient atteintes. Si le montage s’arrête à 1000 itérations, poursuivez les itérations suivantes avec le bouton Démarrer l’ajustement du spectre . Le spectre du modèle est affiché avec le spectre réel à des fins de comparaison.
- Enregistrez les valeurs de meilleur ajustement à partir des onglets appropriés. Enregistrez le spectre le mieux ajusté en cliquant sur l’onglet Spectre dans la fenêtre d’ajustement de la forme de ligne, puis en cliquant sur le bouton Enregistrer .
REMARQUE: Le spectre le mieux ajusté sera enregistré dans le même dossier que celui utilisé pour collecter les données. Le spectre le mieux ajusté sera distingué des données d’origine en étant enregistré avec un numéro de traitement différent qui est entré lors de la sauvegarde. - Enregistrez le modèle utilisé pour l’ajustement de la forme de ligne en cliquant sur l’onglet Principal , puis sur le bouton Enregistrer. Entrez un nom pour le modèle.

Figure 4 : Comparaison de 31intensités de signal P-{1H} pour un seul échantillon de ReH5(PPh3)2(sec-butylamine) dans le d8-toluène. Une démonstration représentative de la différence d’intensité du signal entre une résonance de phosphore unique à échange rapide et une paire de résonances de phosphore près de la température de coalescence pour ces résonances. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
3. Détermination des paramètres d’activation à partir d’un diagramme d’Eyring 1
- Entrez les données de l’ajustement de la forme de ligne pour un processus dynamique modélisé dans une feuille de calcul avec la variable indépendante entrée comme 1/T et la variable dépendante entrée comme ln(k/T).
- Insérez un nuage de points des données dans la feuille de calcul. Ajoutez une ligne de tendance à travers les données. Utilisez la pente et l’interception de la ligne de tendance pour résoudre ΔH‡ et ΔS‡. La pente de la ligne de tendance est -ΔH‡/R tandis que l’intersection de la ligne de tendance est ΔS‡/R + 23,76.
- Résoudre pour ΔG‡ à une température donnée en utilisant la relation
ΔG‡(T) = ΔH‡ - TΔS‡.
NOTE: Pour un simple échange de deux noyaux avec des résonances qui fusionnent, une vérification des valeurs de ΔH‡ et ΔS‡ peut être effectuée en comparant ΔG‡ calculée à la température de coalescence avec la valeur de ΔG‡ qui résulte de la différence de fréquence d’échange lente entre les résonances et la température de coalescence.
Résultats
Les caractérisations des deux produits de polyhydrure de rhénium décrites dans ce manuscrit sont mieux accomplies par spectroscopie RMN 1 H-{31 P} et 31P-{1H}. Dans une solution de 6-benzène d à température ambiante, la résonance du ligand hydrure de ReH7(PPh3)2 apparaît comme un triplet binomialà δ = -4,2 ppm avec 2 JPH = 18 Hz par spectroscopie RMN 1H (Figure supplémentaire 2).<...
Discussion
Il y a quatre éléments dans la préparation de ReH7(PPh3)2 qui peuvent avoir une incidence sur la quantité et la pureté du matériau produit. Tout d’abord, l’utilisation d’un bain de glace pendant les 15 premières minutes de la réaction est importante pour éliminer la chaleur de la réaction qui se produit entre le borohydrure de sodium et l’eau. Des températures initiales plus élevées entraînent une diminution du rendement du produit ReH7(PPh 3)2 en raison d...
Déclarations de divulgation
Les auteurs n’ont aucun conflit d’intérêts à divulguer.
Remerciements
Les auteurs remercient le Département de chimie et de physique et le Programme de subventions pour la créativité et la recherche (Naik, Moehring) de l’Université Monmouth pour leur soutien financier à ce travail.
matériels
| Name | Company | Catalog Number | Comments |
| Bruker Avance II 400 MHz NMR spectrometer | Bruker Biospin | The instrument includes a two channel probe (1H and X) with the X channel tunable from 162 MHz to 10 Mhz. The instrument is also VT capable with a dewar and heat exchanger for VT work. | |
| d8-toluene | MilliporeSigma | 434388 | |
| Powerstat variable transformer | Powerstat | ||
| sec-butyl amine | MilliporeSigma | B89000 | |
| Sodium borohydride | MilliporeSigma | 452882 | |
| Tetrahydrofuran | MilliporeSigma | 186562 | |
| Thermowell C3AM 100 mL | Thermowell | ||
| Topspin 3.0 or 4.1.4 with dNMR | Bruker Biospin | Data was acquired with Topspin version 3.0 and data handling was performed on a second computer that was running Topspin version 4.1.4.. | |
| Trichlorooxobis(triphenylphosphine) rhenium(V) | MilliporeSigma | 370193 | |
| Vacuubrand PC3000 vacuum pump with a CVC 3000 controller | Vacuubrand |
Références
- Zimmer, K. D., Shoemaker, R., Ruminski, R. R. Synthesis and characterization of a fluxional Re(I) carbonyl complex fac-[Re(CO)3(dpop')Cl] with the nominally tri-dentate ligand dipyrido(2,3-α:3',2'-j)phenazine (dpop). Inorganica Chimica Acta. 359 (5), 1478-1484 (2006).
- McGlinchey, M. J. Symmetry breaking in NMR spectroscopy: the elucidation of hidden molecular rearrangement processes. Symmetry. 6 (3), 622-654 (2014).
- Casarini, D., Luazzi, L., Mazzanti, A. Recent advances in stereodynamics and conformational analysis by dynamic NMR and theoretical calculations. European Journal of Organic Chemistry. 2010 (11), 2035 (2010).
- Palmer, A. G., Williams, J., McDermott, A. Nuclear magnetic resonance studies of biopolymer dynamics. Journal of Physical Chemistry. 100 (31), 13293-13310 (1996).
- Kern, D., Kern, G., Scherer, G., Fischer, G., Drakenberg, T. Kinetic analysis of cyclophilin-catalyzed prolyl cis/trans isomerization by dynamic NMR spectroscopy. Biochemistry. 34 (41), 13594-13602 (1995).
- Menger, F. M., Lynn, J. L. Fast proton transfer at a micelle surface. Journal of the American Chemical Society. 97 (4), 948-949 (1975).
- Pines, A., Rabinovitz, M. A nuclear magnetic resonance total line-shape treatment of internal rotation in dimethylformamide. Tetrahedron Letters. 9 (31), 3529-3532 (1968).
- Mancinelli, M., Bencivenni, G., Pecorari, D., Mazzanti, A. Stereochemistry and recent applications of axially chiral organic molecules. European Journal of Organic Chemistry. 2020 (27), 4070-4086 (2020).
- Streisel, D. J., et al. Fluxionality, substitution, and hydrogen exchange at eight-coordinate rhenium(V) polyhydride centers. Inorganica Chimica Acta. 496 (1), 119028 (2019).
- Jimenez, Y., Strepka, A. M., Borgohain, M. D., Hinojosa, P. A., Moehring, G. A. Ortho-metalation, rotational isomerization, and hydride-hydride coupling at rhenium(V) polyhydride complexes stabilized by aromatic amine ligands. Inorganica Chimica Acta. 362 (9), 3259-3266 (2009).
- Lee, J. C., Yao, W., Crabtree, R. H., Ruegger, H. Fluxionality in [ReH5(PPh3)2(pyridine)]. Inorganic Chemistry. 35 (3), 695-699 (1996).
- Patel, B. P., Kavallieratos, K., Crabtree, R. H. Effects of dihydrogen bonding on fluxionality in ReH5(PPh3)2L. Journal of Organometallic Chemistry. 528 (1), 205-207 (1997).
- Geetha, B., et al. Chiral amine ligands at rhenium(V) pentahydride complexes allow for characterization of an energetically accessible and reversible steric inversion of diastereotopic phosphorus atoms. Inorganica Chimica Acta. 531 (1), 120741 (2022).
- Paulo, A., Ascenso, J., Domingos, A., Galvao, A., Santos, I. Rhenium-(III) and -(V) hydride complexes with modified poly(pyrazolyl)borates. Journal of the Chemical Society, Dalton Transactions. 1999 (8), 1293-1300 (1999).
- Bianchini, C., et al. Synthesis and characterization of rhenium polyhydrides stabilized by the tripodal ligand MeC(CH2PPh2)3. Journal of Organometallic Chemistry. 451 (1), 97-106 (1993).
- Scorzelli, A. G., Macalush, B. E., Naik, D. V., Moehring, G. A. Comparative study of fluxional processes at two different classes of eight-coordinate rhenium(V) polyhydride complexes. Inorganica Chimica Acta. 516 (1), 120120 (2021).
- Luo, X. -. L., Crabtree, R. H. Synthesis and spectroscopic characterization of rhenium complexes ReH5(triphos)] and [ReH6(triphos)]+ [triphos = PPh(CH2CH2PPh2)2]. Journal of the Chemical Society. 1991 (5), 587-590 (1991).
- Kim, Y., Deng, H., Gallucci, J. C., Wojcicki, A. Rhenium polyhydride complexes containing PhP(CH2CH2CH2PCy2)2 (Cyttp): protonation, insertion, and ligand substitution reactions of ReH5(Cyttp) and structural characterization of ReH5(Cyttp) and [ReH4(η2-H2)(Cyttp)]SbF6. Inorganic Chemistry. 35 (24), 7166-7173 (1996).
- Bolano, S., et al. Synthesis, characterization, protonation studies and X-ray crystal structure of ReH5(PPh3)2(PTA) (PTA = 1,3,5-triaza-7-phosphaadamantane). Journal of Organometallic Chemistry. 691 (4), 629-637 (2006).
- Ginsberg, A. P., Abrahams, S. C., Jamieson, P. B. Nonrigid stereochemistry in eight-coordinate pentahydridorhenium complexes. Journal of the American Chemical Society. 95 (14), 4751-4752 (1973).
- Bolano, S., Bravo, J., Garcia-Fontan, S. Mono- and dinuclear rhenium polyhydride complexes bearing the chelating ligand 1,2-bis(dicyclohexylphosphinanyloxy)ethane. European Journal of Inorganic Chemistry. 2004 (24), 4812-4819 (2004).
- Leeaphon, M., Rohl, K., Thomas, R. J., Fanwick, P. E., Walton, R. A. Reactions of the polyhydride complex ReH7(PPh3)2 with quinoline, 2-hydroxyquinoline, and 2-mercaptoquinoline. The preparation and characterization of hydrido complexes of rhenium(V) and chloro complexes of rhenium(III). Inorganic Chemistry. 32 (24), 5562-5568 (1993).
- Mejia, E., Togni, A. Rhenium complexes containing the chiral tridentate ferrocenyl ligand pigiphos. Organometallics. 30 (17), 4765-4770 (2011).
- Moehring, G. A., Walton, R. A. Reactions of heptahydrobis(triphenylphosphine)rhenium with bidentate aromatic heterocycles. Inorganic Chemistry. 26 (17), 2910-2912 (1987).
- Kosanovich, A. J., Reibenspies, J. H., Ozerov, A. V. Complexes of high-valent rhenium supported by the PCP pincer. Organometallics. 35 (4), 513-519 (2016).
- Emge, T. J., Koetzle, T. F., Bruno, J. W., Caulton, K. G. Pentahydridorhenium: crystal and molecular structure of ReH5(PMePh2)3. Inorganic Chemistry. 23 (24), 4012-4017 (1984).
- Costello, M. T., Fanwick, P. E., Green, M. A., Walton, R. A. Reactions of Heptahydridobis(triphenylphosphine)rhenium with 1-(diphenylphosphino)-2-(diphenylarsino)ethane (arphos) and 1,2-bis(diphenylarsino)ethane (dpae). Structural characterization of ReH5(PPh3)2(arphos-As) and ReH5(PPh3)2(dpae-As). Inorganic Chemistry. 30 (4), 861-864 (1991).
- Alvarez, D., Lundquist, E. G., Ziller, J. W., Evans, W. J., Caulton, K. G. Synthesis, structure and applications of transition-metal polyhydride anions. Journal of the American Chemical Society. 111 (22), 8392-8398 (1989).
- Albinati, A., et al. Synthesis, characterization, and interconversion of the rhenium polyhydrides ReH3(η4-NP3)] and [ReH4(η4-NP3)]+ {NP3 = tris[2-(diphenylphosphanyl)ethyl]amine}. European Journal of Inorganic Chemistry. 2002 (6), 1530-1539 (2002).
- Bosque, R., et al. Site preference energetics, fluxionality, and intramolecular M−H···H−N hydrogen bonding in a dodecahedral transition metal polyhydride. Inorganic Chemistry. 36 (24), 5505-5511 (1997).
- Tao, Y., Sou, W., Luo, G. -. G., Kraka, E. Describing polytopal rearrangement processes of octacoordinate structures. I. renewed insights into fluxionality of the rhenium polyhydride complex ReH5(PPh3)2(Pyridine). Inorganic Chemistry. 60 (4), 2492-2502 (2021).
- Beringhelli, T., D'Alfonso, G., Minoja, A. P. Rhenium-platinum mixed metal clusters. Characterization in solution and dynamic behavior of the isomers of [Re3Pt(µ-H3)(CO)14]. An example of a labile metal fragment that undergoes intermolecular exchange. Organometallics. 13 (2), 663-668 (1994).
- Grieco, G., Blacque, O. Solution and solid-state structure of the first NHC-substituted rhenium heptahydrides. European Journal of Inorganic Chemistry. 2019 (34), 3810-3819 (2019).
- Wazio, J. A., Jimenez, V., Soparawalla, S., John, S., Moehring, G. A. Hydrogen exchange of rhenium(VII) heptahydridobis(triphenylphosphine) with water, aniline, methanol, and itself. Inorganica Chimica Acta. 362 (1), 159-165 (2009).
- Chatt, J., Coffey, R. S. Hydrido-complexes of rhenium-containing tertiary phosphines. Journal of the Chemical Society, A. 1969, 1963-1972 (1969).
- Tao, Y., Wang, X., Zou, W., Luo, G. -. G., Kraka, E. Unusual intramolecular motion of ReH92- in K2ReH9 crystal: circle dance and three-arm turnstile mechanisms revealed by computational study. Inorganic Chemistry. 61 (2), 1041-1050 (2022).
- Berger, X., Braun, S. . 200 and More NMR Experiments a Practical Course. , (2004).
- He, G., Chen, J., Sung, H. H. -. Y., Williams, I. D., Jia, G. Substituent effect on reactions of ReH5(PMe2Ph)3 with propargyl alcohols. Inorganica Chimica Acta. 518 (1), 120239 (2021).
- Donnelly, L. J., Parsons, S., Morrison, C. A., Thomas, S. P., Love, J. B. Synthesis and structures of anionic rhenium polyhydride complexes of boron-hydride ligands and their application in catalysis. Chemical Science. 11 (9), 9994-9999 (2020).
- Donnelly, L. J., et al. C-H borylation catalysis of heteroaromatics by a rhenium boryl polyhydride. ACS Catalysis. 11 (12), 7394-7400 (2021).
- Jin, H., et al. CO-enabled rhenium hydride catalyst for directed C(sp2)-H bond alkylation with olefins. Organic Chemistry Frontiers. 2 (4), 378-382 (2015).
- Takaya, H., Ito, M., Murahashi, S. -. I. Rhenium-catalyzed addition of carbonyl compounds to the carbon−nitrogen triple bonds of nitriles: α-C−H activation of carbonyl compounds. Journal of the American Chemical Society. 131 (31), 10824-10825 (2009).
- Carr, S. W., Fowles, E. H., Fontaine, X. L. R., Shaw, B. L. Multihydride complexes of rhenium, osmium or iridium containing monodentate ditertiary phosphine ligands: selective hydrogen-deuterium exchanges of the rhenium multihydrides. Journal of the Chemical Society, Dalton Transactions. 1990 (2), 573-579 (1990).
- Jin, H., et al. Rhenium-catalyzed acceptorless dehydrogenative coupling via dual activation of alcohols and carbonyl compounds. ACS Catalysis. 3 (10), 2195-2198 (2013).
- Loza, M. L., de Gala, S., Crabtree, R. H. Steric crowding in a rhenium polyhydride induced by a chelating disilyl ligand: synthesis, characterization, and reactivity of ReH5(disil)(PPh3)2 (disil = 1,2-Bis(dimethylsilyl)benzene and 1,2-Bis(dimethylsilyl)ethane). Inorganic Chemistry. 33 (22), 5073-5078 (1994).
- Lin, Y., Zhu, X., Xiang, M. Transition metal polyhydrides-catalyzed addition of activated nitriles to aldehydes and ketones via Knoevenagel condensation. Journal of Organometallic Chemistry. 448 (1-2), 215-218 (1993).
- Abdukader, A., Jin, H., Cheng, Y., Zhu, C. Rhenium-catalyzed amination of alcohols by hydrogen transfer process. Tetrahedron Letters. 55 (30), 4172-4174 (2014).
- Lin, Y., Zhou, Y. Selective transfer hydrogenation catalyzed by transition metal polyhydrides. Fenzi Cuihua. 5 (2), 119-124 (1991).
- Green, M. A., Huffman, J. C., Caulton, K. G., Rybak, W. K., Ziolkowski, J. J. Ligand scavenging and catalytic utilization of the phototransient ReH5(PMe2Ph)2. Journal of Organometallic Chemistry. 218 (2), 39-43 (1981).
- Komiya, S., Chigira, T., Suzuki, T., Hirano, M. Polymerization of alkyl methacrylate catalyzed by hydridorhenium complexes. Chemistry Letters. 4 (4), 347-348 (1999).
- Michos, D., Luo, X. L., Faller, J. W., Crabtree, R. H. Tungsten(VI) hexahydride complexes supported by chelating triphosphine ligands: protonation to give η2-dihydrogen complexes and catalytic dehydrogenation of cyclooctane to cyclooctene. Inorganic Chemistry. 32 (8), 1370-1375 (1993).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationExplorer plus d’articles
This article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.