
Un abonnement à JoVE est nécessaire pour voir ce contenu. Connectez-vous ou commencez votre essai gratuit.
Method Article
Isolement de petits follicules préantraux de l’ovaire bovin à l’aide d’une combinaison de fragmentation, d’homogénéisation et de filtration en série
Dans cet article
Résumé
Faire progresser l’étude de la folliculogenèse préantrale nécessite des méthodes efficaces d’isolement des follicules à partir d’ovaires simples. Un protocole mécanique simplifié pour l’isolement des follicules des ovaires bovins à l’aide d’un hachoir et d’un homogénéisateur de tissus est présenté ici. Cette méthode permet de prélever un grand nombre de follicules préantraux viables à partir d’un seul ovaire.
Résumé
Comprendre le processus complet de la folliculogenèse des mammifères est crucial pour améliorer les technologies de procréation assistée chez le bétail, les humains et les espèces en voie de disparition. La recherche s’est principalement limitée aux follicules antraux et aux grands follicules préantraux en raison de la difficulté d’isoler les follicules préantraux plus petits, en particulier chez les grands mammifères tels que les espèces bovines. Ce travail présente une approche efficace pour récupérer un grand nombre de petits follicules préantraux d’un seul ovaire bovin. Le cortex des ovaires bovins individuels a été tranché en cubes de 500 μm à l’aide d’un hachoir à tissus et homogénéisé pendant 6 minutes à 9 000-11 000 tr/min à l’aide d’une sonde de 10 mm. Les gros débris ont été séparés de l’homogénat à l’aide d’un tissu à fromage, suivi d’une filtration en série à travers des crépines cellulaires de 300 μm et 40 μm. Le contenu retenu dans la passoire de 40 μm a été rincé dans un plat de recherche, où les follicules ont été identifiés et recueillis dans une goutte de milieu. La viabilité des follicules collectés a été testée par coloration bleue de trypan. Cette méthode permet d’isoler un grand nombre de petits follicules préantraux viables d’un seul ovaire bovin en 90 minutes environ. Fait important, cette méthode est entièrement mécanique et évite l’utilisation d’enzymes pour dissocier le tissu, ce qui peut endommager les follicules. Les follicules obtenus à l’aide de ce protocole peuvent être utilisés pour des applications en aval telles que l’isolement de l’ARN pour la RT-qPCR, l’immunolocalisation de protéines spécifiques et la culture in vitro .
Introduction
Les follicules ovariens sont les unités fonctionnelles de l’ovaire, responsables de la production du gamète (ovocyte) ainsi que des hormones essentielles à la fonction de reproduction et à la santé globale. Les follicules primordiaux se forment dans l’ovaire au cours du développement fœtal ou dans la période néonatale selon les espèces1, et ils constituent la réserve ovarienne d’une femelle. La croissance folliculaire commence par l’activation des follicules primordiaux qui quittent la piscine de repos et entrent dans la phase de croissance. La folliculogenèse préantrale, englobant tous les stades folliculaires avant le développement de l’antre, est un processus hautement dynamique qui nécessite des changements morphologiques et métaboliques synchrones dans l’ovocyte et les cellules granulosa environnantes, entraînés par une communication étroite entre ces deux types de cellules 2,3. Les follicules préantraux constituent la majorité des unités folliculaires présentes dans l’ovaire à un moment donné4. On estime que le développement par les stades préantraux de la folliculogenèse est de plusieurs semaines plus long que le développement antral 5,6, et ce temps est nécessaire pour que les cellules ovocytaires et somatiques acquièrent une maturité suffisante pour entrer dans la phase finale de développement (c’est-à-dire le stade antral) et se préparer à l’ovulation, à la fécondation et au développement embryonnaire 7,8,9.
Une grande partie des connaissances actuelles sur la follicululogenèse préantrale ovarienne provient des modèles murins10,11,12,13, en partie en raison de la facilité de récupération d’un grand nombre de ces follicules à partir d’un ovaire plus petit et moins fibreux. Bien que les rapports d’isolement d’un grand nombre de follicules préantraux à partir d’ovaires bovins remontent à environ 30 ans14, une compréhension plus complète des processus régulant le développement de ces follicules à un stade précoce n’a pas été réalisée, en grande partie en raison du manque de méthodes optimisées, efficaces et reproductibles pour récupérer un nombre suffisant de follicules préantraux viables, en particulier aux premiers stades de développement. Avec l’intérêt croissant pour la préservation de la réserve ovarienne pour une utilisation future dans la procréation assistée chez l’homme, les vaches deviennent un modèle attrayant en raison de leur structure ovarienne plus similaire15. Cependant, l’ovaire bovin est nettement plus riche en collagène que l’ovaire de souris16, ce qui rend l’isolement mécanique à l’aide de méthodes décrites pour la souris très inefficace. Les efforts visant à étendre les techniques de préservation de la fertilité comprennent la croissance in vitro complète des follicules préantraux jusqu’au stade antral, suivie de la maturation in vitro (MIV) des ovocytes fermés, de la fécondation in vitro (FIV) et de la production et du transfert d’embryons17. Jusqu’à présent, tout ce processus n’a été réalisé que chez la souris18. Chez les bovins, les progrès vers la croissance des follicules in vitro se limitent à quelques rapports avec des stades folliculaires variables au début de la culture, ainsi qu’une durée variable de culture entre les protocoles17,19.
Les méthodes décrites dans la littérature pour la récolte des follicules préantraux de l’ovaire bovin ont principalement utilisé des techniques mécaniques et enzymatiques, isolées ou en combinaison 2,14,17,20. Le premier rapport d’un protocole d’isolement du follicule préantral bovin utilisait un homogénéisateur tissulaire et une filtration en série pour traiter les ovaires entiers20. Cette étude a été suivie de rapports combinant des procédures mécaniques et enzymatiques utilisant la collagénase14. Un thème récurrent lors de l’utilisation de la collagénase pour digérer le tissu ovarien est le risque potentiel de dommages à la membrane basale folliculaire, ce qui peut compromettre la viabilité du follicule 14,21,22,23. Par conséquent, différentes combinaisons de méthodes mécaniques ont été employées, telles que l’utilisation d’un hachoir à tissus et de pipetage répété ou d’un hachoir à tissus combiné à l’homogénéisation20,24,25,26. Une autre technique mécanique qui a été décrite utilise des aiguilles pour disséquer les follicules préantraux directement à partir du tissu ovarien, ce qui est particulièrement utile pour isoler des follicules secondaires plus grands (>200 μm). Cependant, ce processus prend du temps, est inefficace pour isoler les petits follicules préantraux et dépend des compétences lorsqu’il est tenté dans les ovaires bovins 19,27,28.
Tirant parti des différentes techniques décrites dans la littérature, ce protocole visait à optimiser l’isolement des follicules préantraux à partir d’ovaires monobovins d’une manière simple, cohérente et efficace qui évite l’incubation dans des solutions enzymatiques. L’amélioration des méthodes d’isolement des follicules préantraux permettra de mieux comprendre cette étape de la folliculogenèse et de permettre le développement de systèmes de culture efficaces pour développer les follicules préantraux jusqu’au stade antral. Les procédures détaillées décrites ici pour l’isolement des follicules préantraux d’un grand mammifère tel que l’espèce bovine seront vitales pour les chercheurs qui souhaitent étudier la folliculogenèse précoce chez une espèce non murine qui est transposable à l’homme.
Protocole
Les ovaires bovins (Bos taurus) ont été prélevés dans un abattoir local et transportés au laboratoire dans les 6 heures suivant la collecte. En raison du grand nombre d’animaux traités dans l’installation, l’âge, la race et le stade du cycle de l’oestrus des animaux sont inconnus. Étant donné qu’aucun animal vivant n’a été utilisé dans ces expériences, un protocole approuvé de soin et d’utilisation des animaux n’était pas requis.
1. Préparation de l’équipement et des réactifs
- Couvrez une section de 2 pieds de large d’une paillasse de laboratoire avec du papier de laboratoire.
- Obtenir un manche de scalpel, une lame de scalpel stérile, un hémostatique, une paire de pinces à dissection, une seringue luer-lock de 20 mL, une aiguille de 18 G, deux béchers de 200 mL, une fiole d’erlenmeyer de 500 mL, un entonnoir en plastique de 104 mm de diamètre, une planche à découper en plastique, une couche d’étamine de 22 cm2 (l’étamine peut être stérilisée par autoclavage avant utilisation) par ovaire en cours de traitement, une crépine cellulaire de 300 μm et une crépine cellulaire de 40 μm (voir le tableau des matériaux).
- Transférez tout l’équipement sur le papier paillassant.
- Utilisez l’hémostatique pour insérer la lame du scalpel sur la poignée du scalpel. Alignez la base inclinée de la lame sur l’indicateur incliné de la poignée, puis faites glisser la lame dans la rainure de la poignée.
- Placer l’entonnoir dans l’erlenmeyer et couvrir l’ouverture de l’entonnoir avec l’étamine.
- Placer un tube conique de 50 ml par ovaire à traiter dans un bain d’eau ou de billes réglé à 38,5 °C.
- Placer une boîte de Pétri carrée de 100 mm x 15 mm par ovaire en cours de traitement sur un chauffe-lames réglé à 38,5 °C.
- Ajouter 10 mL de pénicilline-streptomycine (PenStrep; 10 000 U/mL de pénicilline et 10 000 μg/mL de streptomycine) à 1 L de 1x solution saline tamponnée au phosphate (PBS). Réchauffer le PBS + PenStrep dans un bain d’eau ou de billes réglé à 38,5 °C au moins 2 h avant le traitement des ovaires.
REMARQUE: La solution PBS + PenStrep est impérative pour laver les ovaires lorsque des follicules isolés seront cultivés, et il est toujours recommandé pour toute expérience en aval afin d’atténuer la contamination microbienne. - Pour prélever le filtrat d’ovaire traité, utiliser un milieu de lavage folliculaire (FWM) composé de TCM199 avec des sels de Hank (voir le tableau des matières) contenant 3 mg/mL d’albumine sérique bovine (BSA), 25 mM de tampon HEPES, 100 UI de pénicilline/100 μg/mL de streptomycine, 1 mM de pyruvate de sodium (NaPyr) et 100 nM d’acides aminés non essentiels (NEAA).
- Transférer le TCM199 stérile, une bouteille de 250 ml et une bouteille graduée de 100 ml dans une enceinte de biosécurité (ESB). Transférer 194 ml de TCM199 dans le flacon.
- Retirer le bécher de TCM199 de l’ESB et porter à agitation. Ajouter 600 mg de BSA, 1,19 g de tampon HEPES et une barre d’agitation autoclavée dans le flacon et remuer jusqu’à dissolution.
- Une fois que le tampon BSA et HEPES est complètement dissous, ajouter 1 N d’hydroxyde de sodium (NaOH) au milieu jusqu’à ce qu’il atteigne un pH de 7,6-7,8, mesuré par un pH-mètre.
- Essuyez la bouteille de milieu, un dispositif de filtration sous vide, quatre tubes coniques de 50 ml et les bouteilles de PenStrep, NaPyr et NEAA contenant de l’éthanol à 70 % avant de les transférer à l’ESB.
- Ajouter 2 mL de PenStrep (10 000 U/mL de pénicilline et 10 000 μg/mL de streptomycine), 100 mM de NaPyr et 100x NEAA dans la bouteille de TCM199 + 3 mg/mL de BSA + 25 mM HEPES. Filtrer stérile le milieu final et l’aliquote dans les tubes coniques de 50 mL. Conserver le milieu à 4 °C jusqu’à 2 semaines.
- Réchauffer un tube conique de 50 ml de milieu par deux ovaires dans un bain de billes réglé à 38,5 °C au moins 1 h avant le traitement des ovaires.
2. Configuration du hachoir à tissus
- Assurez-vous que le hachoir en tissu (voir le tableau des matériaux) est branché et allumé.
- Réglez l’épaisseur de la tranche à 500 μm, le bouton de commande de la lame à 20° et le bouton de contrôle de vitesse à 90° selon les spécifications du fabricant.
- Insérez une boîte de Petri en plastique de 60 mm dans le porte-plaque et insérez le support de plaque sur sa scène.
- Soulevez le bras de hachage aussi haut qu’il le souhaite en tournant le bouton de commande manuelle dans le sens des aiguilles d’une montre.
- À l’aide d’une pince, placez une lame de rasoir à double tranchant (voir le tableau des matériaux) sur la vis insérée dans le bras de hachage. Placez le fermoir de la lame sur la lame et fixez avec la rondelle et l’écrou. Laissez l’écrou un quart se détacher.
- Faites pivoter le bouton de commande manuelle jusqu’à ce que le bras de hachage enclenche la lame à plat sur la boîte de Pétri. Serrez l’écrou le reste du chemin avec le tourne-écrou.
- Levez le bras de découpe aussi haut qu’il le souhaite à l’aide du bouton de commande manuelle. Déplacez le bouton de déverrouillage de la table tout vers la gauche jusqu’à ce qu’il se mette en place.
3. Préparation des ovaires
- Transférer les ovaires en laboratoire pour réchauffer (38,5 °C) PBS + PenStrep stérile.
REMARQUE: Il est recommandé de traiter les ovaires pour l’isolement du follicule dès que possible après le retrait de l’animal. Dans ce protocole, les ovaires ont été traités dans les 6 heures suivant la récolte. Les ovaires ont été transportés de l’abattoir au laboratoire dans des thermos contenant une solution saline stérile à 0,9 % à environ 38,5 °C. - Si possible, choisissez de petits ovaires (≤ 4 cm x 3 cm x 3 cm) contenant de petits follicules antraux (3-5 mm), pas de gros follicules antraux (≥8 mm) et pas de corps jaune proéminent (figure 1). Ces critères sont recommandés pour s’assurer qu’une quantité minimale de débris non folliculaires, tels que les cellules stromales et la matrice extracellulaire, est incluse dans la boîte carrée résultante contenant des follicules isolés.
REMARQUE: Les follicules antraux peuvent être identifiés comme des structures vésiculeuses sphériques remplies de liquide à la surface de l’ovaire. Les corps jaunes peuvent être identifiés comme des structures rigides rouges, oranges ou jaunes dépassant de la surface de l’ovaire. - Utilisez des ciseaux pour enlever tout excès de tissu conjonctif et de graisse des ovaires.
- Laver les ovaires pendant 30 s dans de l’éthanol à 70% dans un bécher.
- Lavez les ovaires 3x pendant 2 minutes chacun dans des béchers de PBS chaud (38,5 °C) + PenStrep, en utilisant PBS frais + PenStrep pour chaque lavage.
- Gardez les ovaires au chaud (38,5 °C) PBS + PenStrep jusqu’à ce qu’ils soient prêts pour le traitement.
REMARQUE : La distance entre le laboratoire et la source ovarienne peut être variable. Par conséquent, il est important de compléter le protocole en temps opportun pour assurer le maintien de la viabilité folliculaire.

Figure 1 : Anatomie de l’ovaire bovin. L’ovaire bovin est constitué de deux régions principales enfermées dans une couche épithéliale. Le cortex, composé du tissu à gauche de la ligne pointillée, contient des follicules ovariens du stade primordial au stade antral. Les follicules préantraux sont trop petits pour être vus à l’œil nu; Les follicules antraux sont marqués d’astérisques. La moelle, composée du tissu à droite de la ligne pointillée, contient des vaisseaux sanguins, des vaisseaux lymphatiques et des nerfs. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
4. Procédure de hachage
REMARQUE: Ne traitez qu’un seul ovaire à la fois. Traiter les ovaires rapidement pour éviter les baisses de température, ce qui peut affecter la viabilité du follicule.
- Transférer un ovaire sur la planche à découper sur le papier de paillasse (figure 2A) et préparer le hachoir à tissus (figure 2B).
- À l’aide d’une pince et d’un scalpel, couper l’ovaire en deux et retirer la moelle de chaque moitié, ne laissant que le cortex à une épaisseur d’environ 1 mm, comme le montre la figure 2C.
- Couper l’ovaire en deux longitudinalement d’un site de fixation ligamentaire au site de fixation opposé.
- Conserver la moitié de l’ovaire sur la planche à découper à traiter et remettre l’autre moitié de l’ovaire dans un PBS + PenStrep chaud (38,5 °C).
- Avec la moelle exposée tournée vers le haut, trancher le long de la courbure de l’ovaire à environ 2 mm de la surface de l’ovaire sans couper à travers le cortex.
- Utilisez la tranche le long de la courbure de l’ovaire comme guide pour approfondir la tranche, toujours en suivant la courbure de l’ovaire pour séparer le cortex de la moelle.
- Disséquer et éliminer tout corps jaune de l’ovaire en coupant le long du bord du corps jaune.
- Retournez l’ovaire à moitié de sorte que l’épithélium soit tourné vers le haut et utilisez le scalpel pour finir de couper la moelle loin du cortex. Coupez tout tissu conjonctif blanc restant autour du bord de la partie de l’ovaire qui était reliée aux ligaments.
- Une fois que la majorité de la moelle est enlevée, utilisez le scalpel pour couper le cortex à environ 1 mm d’épaisseur. Manipulez le scalpel avec de petits mouvements de va-et-vient pour raser le reste de la moelle.
REMARQUE: La moelle est la partie interne de l’ovaire contenant de gros vaisseaux sanguins. Le cortex est la partie externe de l’ovaire, située directement sous l’épithélium de surface le plus externe. Le cortex a une épaisseur d’environ 1 mm dans l’ovaire bovin, et ainsi couper l’ovaire à une épaisseur de 1 mm enlèvera la moelle.
- Couper le cortex en morceaux ne dépassant pas 2,5 cm x 2,5 cm. Gardez les morceaux de cortex dans un PBS + PenStrep chaud (38,5 °C) jusqu’à ce qu’ils soient prêts à être hachés.
- Remplissez un bécher avec au moins 50 ml de PBS chaud (38,5 °C) + PenStrep et procurez-vous une pipette de transfert en plastique.
- Transférer un seul morceau de cortex dans la boîte de Petri sur le hachoir à tissus et mouiller le tissu avec trois ou quatre gouttes de PBS chaud (38,5 °C) + PenStrep.
- Maintenez le morceau de tissu stable avec une paire de pinces et appuyez une fois sur le bouton de réinitialisation pour démarrer le hacheur de tissu. Stabiliser la boîte de Petri d’une main tout en continuant à stabiliser le tissu avec la pince. Déplacez les pinces vers la gauche le long du tissu au besoin pour éviter que la lame ne heurte la pince. Les bandes résultantes auront une longueur d’environ 500 μm.
- Une fois que tout le morceau de cortex a été coupé en bandes, utilisez le bouton du porte-lame pour soulever la lame de la boîte de Petri et les pinces pour enlever tout tissu de la lame.
- Faites pivoter le porte-plaque de 90°.
- Appuyez une fois sur le bouton de réinitialisation. Stabilisez la boîte de Petri d’une main tout en utilisant la pince pour pousser les bandes de tissu dans le chemin de la lame.
- Passez la lame entièrement à travers les bandes de tissu. Utilisez le bouton du porte-lame pour soulever la lame de la boîte de Petri et les pinces pour enlever tout tissu de la lame.
- Utilisez la pipette de transfert et le PBS + PenStrep chaud (38,5 °C) pour laver le tissu haché (taille finale du tissu : cubes de 500 μm x 500 μm x 1 mm) dans un tube conique préchauffé (38,5 °C) de 50 mL. Remettre le tube conique dans l’eau ou le bain de perles pour garder le tissu haché au chaud (38,5 °C).
- Utilisez le pilote d’écrou pour retirer l’écrou du bras de hachage et retirez la rondelle et le fermoir de la lame. À l’aide d’une pince, retirez la lame du bras de découpe, retournez-la de sorte que le bord inutilisé soit face à la boîte de Petri et replacez-la sur le bras de découpe. Replacez le fermoir de lame, la rondelle et l’écrou, puis réinitialisez le bouton de dégagement de la table comme décrit aux étapes 2.5 à 2.7.
- Répétez les étapes 4.5 à 4.12 pour tous les morceaux de cortex restants de l’ovaire, en remplaçant les lames par de nouvelles lames après l’utilisation de chaque tranchant.
- Jetez toutes les lames usagées dans un contenant en plastique à parois rigides.
5. Procédure d’homogénéisation
- Assurez-vous que l’homogénéisateur (voir le tableau des matériaux) est branché et que la vitesse est réglée sur la deuxième barre (9 000-11 000 tr/min). Insérez la sonde du générateur de 10 mm dans l’unité conformément aux spécifications du fabricant.
- Réglez une minuterie pendant 1 min et insérez la sonde dans le tube conique de 50 ml contenant le tissu cortex haché d’un ovaire (étape 4.11) et suffisamment de PBS + PenStrep pour remplir le tube jusqu’à la ligne de 25 ml. La profondeur à laquelle la sonde est insérée doit être égale à 1/3 de la hauteur du liquide mesurée à partir du fond de la chambre. Positionnez la sonde légèrement décentrée pour minimiser les tourbillons.
- Démarrez la minuterie et mettez l’homogénéisateur sous tension. Assurez-vous que le bas de la sonde ne touche pas le tube et maintenez le tube immobile pendant que l’homogénéisateur est allumé.
- Après 1 min d’homogénéisation, retirez la sonde du tube. À l’aide d’une pince, retirez tout tissu conjonctif obstruant les trous de ventilation et l’espace entre le couteau du rotor et le tube du rotor. Si des morceaux de cortex sont coincés dans la sonde, retirez-les avec une pince et replacez-les dans le tube.
- Répétez les étapes 5.2-5.4 5x supplémentaires pour un total de 6 minutes d’homogénéisation.
- Placer le tube contenant du tissu homogénéisé dans l’eau ou le bain de perles pour garder le tissu au chaud (38,5 °C). Après avoir traité le dernier ovaire, démontez, nettoyez et séchez immédiatement la sonde du générateur conformément aux spécifications du fabricant.
6. Procédure de filtration
- Verser le tissu dispersé dans l’entonnoir recouvert d’étamine inséré dans l’erlenmeyer. Rincer le contenu du tube dans l’entonnoir à l’aide de PBS + PenStrep chaud (38,5 °C) jusqu’à ce qu’il ne reste plus de fragments de tissu dans le tube.
- Forcez les fragments de tissu à passer à travers les trous du tissu en tordant l’étamine autour des fragments de tissu et en pressant jusqu’à ce que tout excès de liquide et de tissu soit retiré de l’étamine.
- Rouvrez l’étamine sur l’entonnoir, rincez l’étamine avec PBS + PenStrep à l’aide d’une pipette de transfert et pressez à nouveau les fragments de tissu résiduels à travers le chiffon.
- Utilisez un hémostatique pour maintenir la crépine cellulaire de 300 μm au-dessus d’un bécher de 200 mL. Verser le filtrat dans l’erlenmeyer à travers la crépine cellulaire. Rincer le contenu de la fiole dans la crépine cellulaire à l’aide de PBS chaud (38,5 °C) + PenStrep jusqu’à ce qu’il ne reste plus de fragments de tissu.
- Si la crépine cellulaire est obstruée par du tissu, tapotez doucement la passoire cellulaire contre le bécher pour vous assurer que tout le liquide a filtré dans le bécher, puis retournez la passoire cellulaire et tapotez les gros débris de tissu sur le papier de paillasse. Remettez la passoire cellulaire sur le bécher et continuez à verser le filtrat à travers elle. Répéter si nécessaire jusqu’à ce que tout le filtrat de l’erlenmeyer ait été filtré.
- Utilisez un hémostatique pour maintenir la passoire cellulaire de 40 μm au-dessus d’un deuxième bécher de 200 mL. Verser le filtrat dans le premier bécher de 200 ml à travers la crépine cellulaire. Rincer le contenu du bécher dans la passoire cellulaire à l’aide de PBS + PenStrep chaud (38,5 °C) jusqu’à ce qu’il ne reste plus de fragments de tissu. Ne jetez pas le contenu de la crépine à cellules de 40 μm.
- Placez l’aiguille de 18 G sur la seringue de 20 mL. Remplissez la seringue avec FWM. Retournez la passoire cellulaire de 40 μm sur une boîte de Petri carrée et utilisez la seringue pour laver le contenu de la passoire cellulaire dans la boîte. Remplissez la seringue et rincez la crépine cellulaire au besoin jusqu’à ce qu’il ne reste plus de fragments de tissu.
REMARQUE : En règle générale, 25 mL de FWM sont suffisants pour rincer complètement le contenu de la crépine à cellules de 40 μm.

Figure 2 : Configuration de l’espace de travail pour le traitement des ovaires et le flux de travail de protocole. (A) Installation de banc pour couper les ovaires avant la coupe et pour filtrer l’homogénat d’ovaire. (B) Hachoir à tissus et homogénéisateur mis en place, avec support en polystyrène expansé pour réduire les vibrations de l’étage d’homogénéisateur. (C) Schéma illustrant le déroulement du traitement d’un ovaire entier. Les ovaires sont coupés de l’excès de tissu conjonctif, puis coupés en deux, et la moelle est retirée jusqu’à ce qu’il reste une tranche de cortex de ~1 mm d’épaisseur. Le cortex est coupé en morceaux de 2,5 cm x 2,5 cm et haché dans un hachoir à tissu réglé à un intervalle de coupe de 500 μm. Les morceaux sont ensuite homogénéisés et l’homogénat est filtré à travers une étamine suivie d’une filtration à travers des crépines cellulaires de 300 μm et 40 μm. Le contenu de la crépine cellulaire de 40 μm est rincé dans une boîte de Petri carrée, qui est recherchée pour les follicules à l’aide d’un stéréomicroscope. Créé avec BioRender.com. Veuillez cliquer ici pour voir une version agrandie de cette figure.
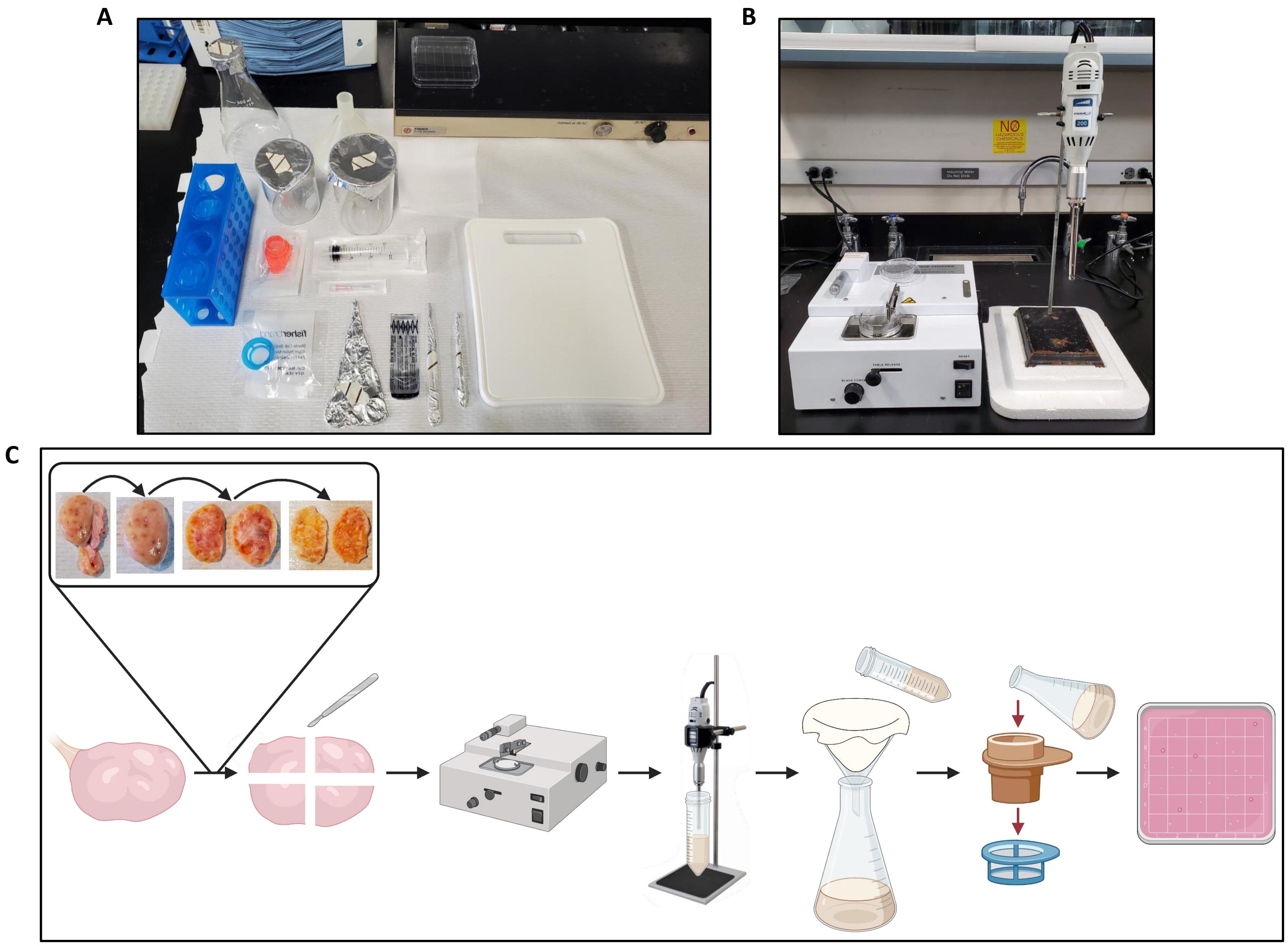{kind=link}
7. Recherche et collecte de follicules
- Transférer la boîte de Petri carrée (étape 6.6) dans un stéréoscope avec une scène chauffée réglée à 38,5 °C. Le grossissement du stéréoscope doit être réglé entre 1,25x et 3,2x selon les préférences du chercheur.
- Pipeter 10 μL de gouttes de FWM dans une boîte de Petri de 60 mm et couvrir les gouttes d’huile minérale pour éviter le dessèchement. Placer la boîte de Petri avec les gouttes de média sur une plaque chauffante réglée à 38,5 °C.
REMARQUE: Une plaque à 4 puits peut être utilisée pour recueillir les follicules. Ajouter 500 μL de produit de lavage à un ou deux puits. Placer sur la plaque chauffante réglée à 38,5 °C. - Procurez-vous un piston et une pointe de micropipette.
REMARQUE : Une micropipette en verre de 1 à 5 μL (voir le tableau des matériaux) est recommandée parce que les follicules sont moins susceptibles d’adhérer à la pipette en verre et d’être perdus lorsqu’ils sont transférés entre les solutions. C’est aussi un instrument assez petit pour permettre des micromanipulations plus faciles et plus précises des follicules. - Identifier les follicules de la boîte de Petri carrée et transférer dans le milieu (FWM) gouttes à l’aide de la micropipette. De nombreux follicules sont susceptibles d’être incorporés dans les débris tissulaires et peuvent être récupérés en utilisant l’une des deux méthodes décrites ci-dessous.
REMARQUE: Les follicules sont oblongs, plutôt que des sphères parfaites, et ont généralement un ovocyte, se présentant comme un cercle blanc solide dans des contrastes plus sombres, vers le centre du follicule (Figure 3A-C). Prenez soin d’éviter de confondre les follicules avec les ovocytes dénudés. Les ovocytes ont tendance à être des sphères parfaites et sont entourés d’une membrane épaisse et claire (la zone pellucide). Un microscope inversé avec un grossissement de 10x (ou plus) peut être utilisé pour examiner de plus près les follicules (Figure 3D).- Séparez soigneusement les follicules des débris à l’aide de l’extrémité de la micropipette ou de fines aiguilles (27 G).
- Alternativement, utilisez une pipette Pasteur en verre avec une ampoule en caoutchouc pour prendre et gicler les débris dans le plat plusieurs fois pour déloger les follicules des débris.
- Travaillez rapidement, en ne prenant pas plus de 30 minutes, pour rechercher la boîte de Petri afin d’aider à préserver la viabilité du follicule.
- Placez un maximum de seulement cinq follicules par goutte de 10 μL, car une densité plus élevée peut augmenter la probabilité que les follicules adhèrent ensemble.
8. Test de viabilité d’exclusion du bleu de trypan
REMARQUE: Utilisez le couvercle d’une boîte de Petri ou une assiette à 4 puits pour toutes les étapes suivantes, car les follicules adhèrent moins au plastique du couvercle qu’au plastique de la boîte réelle.
- Préparer PBS + 0,2% de polyvinylpyrrolidone (PVP) en dissolvant 100 mg de PVP dans 50 mL de PBS.
REMARQUE: PVP est utilisé ici pour réduire la probabilité que les follicules se fixent au plat. - Utilisez la micropipette pour transférer tous les follicules (moyenne de 40) des gouttes de média en une goutte de 50 μL de PBS + 0,2% PVP.
- Lavez les follicules 2x en les transférant séquentiellement sur des gouttes fraîches de 50 μL de PBS + 0,2% PVP.
- Transférer les follicules à une goutte de 285 μL de PBS + 0,2% PVP.
- Ajouter 15 μL de bleu de trypan à la goutte de 285 μL de PBS + 0,2 % de PVP (concentration finale de 0,05 % de bleu de trypan) et mélanger soigneusement la goutte à l’aide d’un embout de pipette de 200 μL réglé à 100 μL.
REMARQUE : Si vous utilisez la plaque à 4 puits pour l’essai de viabilité du trypan, ajoutez 475 μL de PBS + 0,2 % de PVP et 25 μL de bleu de trypan à un puits. - Incuber les follicules pendant 1 min dans la goutte bleue de trypan, puis transférer les follicules dans une goutte de 50 μL (ou puits de 500 μL) de PBS + 0,2% PVP.
- Laver les follicules 3x selon l’étape 8.3 avec des gouttes fraîches de 50 μL (ou 500 μL par puits) de PBS + 0,2% PVP.
- Jetez tous les follicules qui apparaissent encore bleus après trois lavages dans PBS + 0,2% PVP, car ils ne sont pas viables. Tous les follicules qui ne conservent pas la coloration bleue après trois lavages sont viables et peuvent être utilisés pour l’immunofluorescence, la culture ou d’autres procédures (Figure 3E). Congeler rapidement les follicules dans de l’azote liquide et conserver à -80 °C jusqu’à ce qu’ils soient utilisés si nécessaire.
- Effectuer une analyse RT-qPCR et une coloration par immunofluorescence des follicules comme décrit aux étapes 9 et 10.

Figure 3 : Follicules isolés et essai d’exclusion du bleu de trypan. (A-C) Les follicules isolés ont été imagés au stéréomicroscope à plusieurs grossissements. (A) Follicules isolés parmi les débris dans la boîte de recherche initiale. Les follicules individuels sont entourés en rouge. Barre d’échelle = 2 000 μm. (B) Follicules isolés et débris dans une gouttelette de produit de lavage de follicule recouverte d’huile minérale. Barre d’échelle = 1 000 μm. (C) Follicules isolés sans débris à un grossissement plus élevé. Barre d’échelle = 1 000 μm. (D) Follicules isolés imagés à l’aide d’un microscope à fond clair inversé. Barre d’échelle = 100 μm. (E) Images représentatives de follicules viables (non colorés) et non viables (coloration bleue) imagées à l’aide d’un microscope à fond clair inversé et d’un objectif 20x. Barre d’échelle = 100 μm. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
9. Analyse RT-qPCR
- Isoler l’ARN des follicules viables (à partir de l’étape 8.8) à l’aide d’un réactif d’isolement d’ARN (voir le tableau des matériaux). Purifier l’ARN et traiter avec de la DNase à l’aide d’une trousse de nettoyage disponible dans le commerce (voir le tableau des matériaux) conformément aux instructions du fabricant.
- Éluer l’ARN avec 14 μL d’eau exempte de RNase et le quantifier à l’aide d’un spectrophotomètre. L’ARN peut être stocké à -80 °C jusqu’à la synthèse de l’ADNc.
- Effectuer la synthèse de l’ADNc à partir de quantités égales d’ARN extrait des follicules primaires et secondaires précoces, en utilisant un kit de synthèse d’ADNc disponible dans le commerce (voir le tableau des matériaux) conformément aux instructions du fabricant. Incuber le mélange réactionnel pendant 5 min à 25 °C puis 60 min à 42 °C, puis terminer la réaction en chauffant à 70 °C pendant 5 min.
- Effectuer la RT-qPCR avec l’ADNc synthétisé (5 ng par réaction) et les amorces (tableau 1) en utilisant un mélange réactionnel disponible dans le commerce (voir le tableau des matériaux). Utiliser des conditions de cyclage thermique : 30 s à 95 °C pour l’activation de la polymérase, suivies de 40 cycles d’amplification, où chaque cycle comprenait 15 s à 95 °C pour la dénaturation et 30 s à 60 °C pour le recuit/extension. Analyser la RT-qPCR en quantifiant les valeurs de seuil de cycle (Ct) et/ou visualiser les produits de PCR à l’aide de l’électrophorèse sur gel d’agarose.
NOTE: L’expression transcrite du marqueur cellulaire de la granulosa FSHR et du marqueur des cellules germinales DAZL a été évaluée dans cette étude. Les gènes de référence étaient H2A et ACTB. - Effectuer une analyse de la courbe de fusion en augmentant la température de 65 °C par incréments de 0,5 °C toutes les 5 s jusqu’à ce qu’elle atteigne 95 °C.
10. Analyse d’immunofluorescence
- Fixer les follicules viables (à partir de l’étape 8.8) pendant 15 minutes dans une goutte de 100 μL de paraformaldéhyde (PFA) à 4 % (v/v) à température ambiante (RT), suivi d’un lavage 3x dans des gouttes de 100 μL de PBS + 0,1 % BSA + 0,1 % Tween 20.
- Bloquer les follicules pendant 1 h à TA dans un tampon de blocage composé de 1x PBS + 5% (v/v) de sérum normal d’âne (NDS). Après le blocage, incuber les follicules pendant une nuit à 4 °C dans une goutte de 100 μL d’anticorps CX37 anti-humain de lapin de 4 μg/mL ou d’isotype IgG (témoin négatif) de lapin de 4 μg/mL dilué dans un tampon de blocage.
- Laver les follicules 3x dans des gouttes de 100 μL de PBS + 0,1% BSA + 0,1% Tween 20, puis les incuber pendant 1 h à TA dans l’obscurité dans une goutte de 100 μL d’anticorps secondaire âne-anti-lapin AlexaFluor 488 2 μg/mL dilué dans un tampon bloquant.
- Incuber les follicules pendant 5 min à TA dans l’obscurité dans une goutte de 100 μL de 1 μg/mL Hoechst 33342 diluée dans un tampon bloquant pour marquer l’ADN.
- Transférer les follicules dans une goutte de 5 μL de support de montage (voir le tableau des matériaux) sur une lame de microscope en verre et couvrir avec un couvercle. Laissez les lames durcir à la RT pendant la nuit, puis scellez avec du vernis à ongles. Conservez-les à 4 °C jusqu’à l’imagerie.
- Imagez toutes les diapositives dans les 48 heures suivant le glissement du couvercle. Effectuer l’imagerie à l’aide d’un microscope à épifluorescence inversée (voir le tableau des matériaux) sous des filtres DAPI (excitation 380 nm et émission 450 nm) et FITC (excitation 470 nm et émission 525 nm).
- Fixez le temps d’exposition pour les deux canaux. Ajustez le temps d’exposition FITC (CX37) en fonction du témoin négatif isotype lapin. Utilisez un objectif 20x et le canal DAPI réglé sur un temps d’exposition de 50 ms pour identifier les follicules marqués par isotype de lapin.
- Imagez ces follicules sous le canal FITC et diminuez le temps d’exposition jusqu’à ce que tout signal vert de fond soit aboli. Notez ce temps d’exposition.
- Imagez tous les follicules marqués par des anticorps CX37 en utilisant le temps d’exposition défini pour le canal FITC isotype et le temps d’exposition de 50 ms pour le canal DAPI.
- Traiter l’intensité du signal, mesurée par la zone grise moyenne après seuillage, à l’aide d’un programme de traitement d’imageinformatique 29 (voir le tableau des matériaux).
- Ajustez le fichier tiff de l’image DAPI pour chaque follicule de sorte que le follicule entier soit souligné. Utilisez la fonction Analyser les particules du programme pour sélectionner l’ensemble du follicule comme région d’intérêt (ROI).
- Ouvrez le fichier tiff de l’image FITC pour le follicule correspondant et superposez le retour sur investissement généré à partir de l’image DAPI sur l’image FITC. Utilisez la fonction Mesure du programme pour quantifier la zone grise moyenne de l’image FITC, qui représente l’intensité du signal.
Résultats
Vue d’ensemble et étapes critiques
Grâce à ce protocole, les petits follicules préantraux bovins peuvent être isolés de manière fiable à partir d’ovaires simples en nombre pertinent expérimentalement. Sur un total de 30 répétitions, une moyenne de 41 follicules a été obtenue par répétition, avec une plage de 11 à 135 follicules (Figure 4A). Dans 14 réplications, les follicules ont été caractérisés pour le stade de développement décrit précédem...
Discussion
Le présent protocole détaille une méthode reproductible pour récupérer les follicules préantraux à un stade précoce, en particulier aux stades primaire et secondaire précoce, de l’ovaire bovin. Ce protocole s’appuie sur les rapports précédents 20,25,30,34,35,36 et fournit des optimisations qui permettent d’isoler un nombre significatif de follicules d’un ovaire individuel.
Déclarations de divulgation
Les auteurs n’ont rien à divulguer.
Remerciements
Ce projet a été partiellement financé par le projet multi-états W4112 de l’USDA et le prix UC Davis Jastro Shields à SM.
Les auteurs aimeraient remercier Central Valley Meat, Inc. d’avoir fourni les ovaires bovins utilisés dans toutes les expériences. Les auteurs remercient également Olivia Silvera pour son aide au traitement des ovaires et à l’isolement des follicules.
matériels
| Name | Company | Catalog Number | Comments |
| 5-3/4" Soda Lime Disposable Glass Pasteur Pipette | Duran Wheaton Kimble | 63A54 | Pasteur pipette that can be used to dislodge follicles from debris while searching within the petri dish |
| 16% Paraformaldehyde | Electron Microscopy Sciences | 15710 | Diluted to 4%; fixation of follicles for immunostaining |
| 20 mL Luer-lock Syringe | Fisher Scientific | Z116882-100EA | Syringe used with the 18 G needle to dislodge follicles from the 40 μm cell strainer |
| #21 Sterile Scalpel Blade | Fisher Scientific | 50-365-023 | Used to cut the ovaries and remove the medula |
| 40 μm Cell Strainer | Fisher Scientific | 22-363-547 | Used to filter the filtrate from the 300 μm cell strainer |
| 104 mm Plastic Funnel | Fisher Scientific | 10-348C | Size can vary, but ensure the cheese cloth is cut appropriately and that the ovarian homogenate will not spill over |
| 300 μm Cell Strainer | pluriSelect | 43-50300-03 | Used to filter the filtrate from the cheese cloth |
| 500 mL Erlenmeyer Flask | Fisher Scientific | FB500500 | Funnel and flask used to catch filtrate from the cheese cloth |
| Air-Tite Sterile Needles 18 G | Thermo Fisher Scientific | 14-817-151 | 18 G offers enough pressure to dislodge follicles from the 40 μm cell strainer |
| Air-Tite Sterile Needles 27 G 13 mm | Fisher Scientific | 14-817-171 | Needles that can be used to manipulate any debris in which follicles are stuck |
| BD Hoechst 33342 Solution | Fisher Scientific | BDB561908 | Fluorescent DNA stain |
| Bovine Serum Albumin (BSA) | Sigma-Aldrich | A7030-100G | Component of follicle wash media |
| Cheese Cloth | Electron Microscopy Sciences | 71748-00 | First filtering step of the ovarian homogenate meant to remove large tissue debris |
| Classic Double Edge Safety Razor Blades | Wilkinson Sword | N/A | Razor blades that fit the best in the McIlwain Tissue Chopper and do not dull quickly |
| Donkey-Anti-Rabbit Secondary Antibody, Alexa Fluor 488 | Fisher Scientific | A-21206 | Secondary antibody for immunostaining |
| Eisco Latex Pipette Bulbs | Fisher Scientific | S29388 | Rubber bulb to use with Pasteur pipettes |
| HEPES Buffer | Sigma-Aldrich | H3375 | Component of follicle wash media |
| Homogenizer | VWR | 10032-336 | Homogenize the ovarian tissue to release follicles |
| ImageJ/Fiji | NIH | v2.3.1 | Software used for analysis of fluorescence-immunolocalization |
| McIlwain Tissue Chopper | Ted Pella | 10184 | Used to cut ovarian tissue small enough for homogenization |
| Microscope - Stereoscope | Olympus | SZX2-ILLT | Dissection microscope used for searching and harvesting follicles from the filtrate |
| Microscope - Inverted | Nikon | Diaphot 300 | Inverted microscope used for high magnification brightfield visualization of isolated follicles |
| Microscope - Inverted | ECHO | Revolve R4 | Inverted microscope used for high magnification brightfield and epifluorescence visualization of isolated follicles |
| Mineral Oil | Sigma-Aldrich | M8410-1L | Oil to cover the drops of follicle wash medium to prevent evaporation during searching |
| Non-essential Amino Acids (NEAA) | Gibco | 11140-050 | Component of follicle wash medium |
| Normal Donkey Serum | Jackson ImmunoResearch | 017-000-001 | Reagent for immunostaining blocking buffer |
| Nunc 4-well Dishes for IVF | Thermo Fisher Scientific | 144444 | 4-well dishes for follicle isolation and washing |
| Penicillin-Streptomycin Solution 100x | Gibco | 15-140-122 | Component of follicle wash medium |
| Petri Dish 60 mm OD x 13.7 mm | Ted Pella | 10184-04 | Petri dish that fits the best in the McIlwain Tissue Chopper |
| Phosphate Buffered Saline (PBS) | Fisher Scientific | BP665-1 | Washing buffer for ovaries and follicles |
| Plastic Cutting Board | Fisher Scientific | 09-002-24A | Cutting board of sufficient size to safely cut ovaries |
| Polyvinylpyrrolidone (PVP) | Fisher Scientific | BP431-100 | Addition of PVP (0.1% w/v) to PBS prevents follicles from sticking to the plate or each other |
| ProLong Gold Antifade Mountant | Thermo Fisher Scientific | P36930 | Mounting medium for fluorescently labeled cells or tissue |
| Qiagen RNeasy Micro Kit | Qiagen | 74004 | RNA column clean-up kit |
| R | The R Foundation | v4.1.2 | Statistical analysis software |
| Rabbit-Anti-Human Cx37/GJA4 Polyclonal Antibody | Abcam | ab181701 | Cx37 primary antibody for immunostaining |
| RevertAid RT Reverse Transcription Kit | Thermo Fisher Scientific | K1691 | cDNA synthesis kit |
| Rstudio | RStudio, PBC | v2021.09.2 | Statistical analysis software |
| Sodium Hydroxide Solution (1N/Certified) | Fisher Scientific | SS266-1 | Used to increase media pH to 7.6-7.8 |
| Sodium Pyruvate (NaPyr) | Gibco | 11360-070 | Component of follicle wash medium |
| Square Petri Dish 100 mm x 15 mm | Thermo Fisher Scientific | 60872-310 | Gridded petri dishes allow for more efficient identification of follicles |
| SsoAdvanced Universal SYBR Green Supermix | BioRad | 1725271 | Mastermix for PCR reaction |
| Steritop Threaded Bottle Top Filter | Sigma-Aldrich | S2GPT02RE | Used to sterilize follicle wash medium |
| SYBR-safe DNA gel stain | Thermo Fisher Scientific | S33102 | Staining to visual PCR products on agarose gel |
| TCM199 with Hank’s Salts | Gibco | 12-350-039 | Component of follicle wash medium |
| Triton X-100 | Fisher Scientific | BP151-100 | Detergent for immunostaining permeabilization buffer |
| Trizol reagent | Thermo Fisher Scientific | 15596026 | RNA isolation reagent |
| Trypan Blue Solution, 0.4% | Gibco | 15-250-061 | Used for testing viability of isolated follicles |
| Tween 20 | Detergent for immunostaining wash buffer | ||
| Warmer Plate Universal | WTA | 20931 | Warm plate to keep follicles at 38.5 °C while searching under the microscope |
| Wiretrol II Calibrated Micropipets | Drummond | 50002-005 | Glass micropipettes to manipulate follicles |
Références
- Fortune, J. E., Yang, M. Y., Allen, J. J., Herrick, S. L. Triennial reproduction symposium: The ovarian follicular reserve in cattle: What regulates its formation and size. Journal of Animal Science. 91 (7), 3041-3050 (2013).
- Fair, T., Hulshof, S. C., Hyttel, P., Greve, T., Boland, M. Oocyte ultrastructure in bovine primordial to early tertiary follicles. Anatomy and Embryology. 195 (4), 327-336 (1997).
- Jaffe, L. A., Egbert, J. R. Regulation of mammalian oocyte meiosis by intercellular communication within the ovarian follicle. Annual Review of Physiology. 79, 237-260 (2017).
- Driancourt, M. A., Reynaud, K., Cortvrindt, R., Smitz, J. Roles of KIT and KIT LIGAND in ovarian function. Reviews of Reproduction. 5 (3), 143-152 (2000).
- Lussier, J. G., Matton, P., Dufour, J. J. Growth rates of follicles in the ovary of the cow. Journal of Reproductive Fertility. 81 (2), 301-307 (1987).
- Aerts, J. M. J., Bols, P. E. J. Ovarian follicular dynamics: a review with emphasis on the bovine species. Part I: Folliculogenesis and preantral follicle development. Reproduction in Domestic Animals. 45 (1), 171-179 (2010).
- Sugiura, K., Pendola, F. L., Eppig, J. J. Oocyte control of metabolic cooperativity between oocytes and companion granulosa cells: energy metabolism. Developmental Biology. 279 (1), 20-30 (2005).
- Eppig, J. J., Pendola, F. L., Wigglesworth, K., Pendola, J. K. Mouse oocytes regulate metabolic cooperativity between granulosa cells and oocytes: amino acid transport. Biology of Reproduction. 73 (2), 351-357 (2005).
- Sugimura, S., et al. Amphiregulin co-operates with bone morphogenetic protein 15 to increase bovine oocyte developmental competence: effects on gap junction-mediated metabolite supply. Molecular Human Reproduction. 20 (6), 499-513 (2014).
- Edson, M. A., Nagaraja, A. K., Matzuk, M. M. The mammalian ovary from genesis to revelation. Endocrine Reviews. 30 (6), 624-712 (2009).
- Matzuk, M. M., Burns, K. H. Genetics of mammalian reproduction: modeling the end of the germline. Annual Review of Physiology. 74, 503-528 (2012).
- McGee, E. A., Raj, R. S. Regulators of ovarian preantral follicle development. Seminars in Reproductive Medicine. 33 (3), 179-184 (2015).
- Chen, Y., et al. The factors and pathways regulating the activation of mammalian primordial follicles in vivo. Frontiers in Cell and Developmental Biology. 8, 575706 (2020).
- Figueiredo, J. R., et al. Development of a combined new mechanical and enzymatic method for the isolation of intact preantral follicles from fetal, calf and adult bovine ovaries. Theriogenology. 40 (4), 789-799 (1993).
- Sirard, M. A. The ovarian follicle of cows as a model for human. Animal Models and Human Reproduction. , 127-144 (2017).
- Parkes, W. S., et al. Hyaluronan and collagen are prominent extracellular matrix components in bovine and porcine ovaries. Genes. 12 (8), 1186 (2021).
- Araújo, V. R., Gastal, M. O., Figueiredo, J. R., Gastal, E. L. In vitro culture of bovine preantral follicles: a review. Reproductive Biology and Endocrinology. 12 (1), 1-14 (2014).
- Eppig, J. J., Schroeder, A. C. Capacity of mouse oocytes from preantral follicles to undergo embryogenesis and development to live young after growth, maturation, and fertilization in vitro. Biology of Reproduction. 41 (2), 268-276 (1989).
- McLaughlin, M., Telfer, E. E. Oocyte development in bovine primordial follicles is promoted by activin and FSH within a two-step serum-free culture system. Reproduction. 139 (6), 971-978 (2010).
- Nuttinck, F., Mermillod, P., Massip, A., Dessy, F. Characterization of in vitro growth of bovine preantral ovarian follicles: A preliminary study. Theriogenology. 39 (4), 811-821 (1993).
- Demeestere, I., et al. Effect of preantral follicle isolation technique on in-vitro follicular growth, oocyte maturation and embryo development in mice. Human Reproduction. 17 (8), 2152-2159 (2002).
- Fattahi, A., et al. Optimization of porcine ovarian follicle isolation methods for better developmental potential. Tissue Engineering Part A. 26 (13-14), 712-719 (2020).
- Nagashima, J. B., Hill, A. M., Songsasen, N. In vitro development of mechanically and enzymatically isolated cat ovarian follicles. Reproduction and Fertility. 2 (1), 35-46 (2021).
- Lucci, C. M., Rumpf, R., Figueiredo, J. R., Báo, S. N. Zebu (Bos indicus) ovarian preantral follicles: Morphological characterization and development of an efficient isolation method. Theriogenology. 57 (5), 1467-1483 (2002).
- Langbeen, A., et al. Characterization of freshly retrieved preantral follicles using a low-invasive, mechanical isolation method extended to different ruminant species. Zygote. 23 (5), 683-694 (2014).
- Candelaria, J. I., Denicol, A. C. Characterization of isolated bovine preantral follicles based on morphology, diameter and cell number. Zygote. 28 (2), 154-159 (2020).
- vanden Hurk, R., et al. Ultrastructure and viability of isolated bovine preantral follicles. Human Reproduction Update. 4 (6), 833-841 (1998).
- Paes, V. M., et al. Effect of heat stress on the survival and development of in vitro cultured bovine preantral follicles and on in vitro maturation of cumulus-oocyte complex. Theriogenology. 86 (4), 994-1003 (2016).
- Schindelin, J., et al. Fiji: An open-source platform for biological image analysis. Nature Methods. 9 (7), 676-682 (2012).
- de Aguiar, L. H., Hyde, K. A., Pedroza, G. H., Denicol, A. C. Heat stress impairs in vitro development of preantral follicles of cattle. Animal Reproduction Science. 213, 106277 (2020).
- Kristensen, S. G., Ebbesen, P., Andersen, C. Y. Transcriptional profiling of five isolated size-matched stages of human preantral follicles. Molecular and Cellular Endocrinology. 401, 189-201 (2015).
- Candelaria, J. I., Rabaglino, M. B., Denicol, A. C. Ovarian preantral follicles are responsive to FSH as early as the primary stage of development. Journal of Endocrinology. 247 (2), 153-168 (2020).
- Nuttinck, F., et al. Comparative immunohistochemical distribution of Connexin 37 and Connexin 43 throughout folliculogenesis in the bovine ovary. Molecular Reproduction and Development. 57 (1), 60-66 (2000).
- Itoh, T., Hoshi, H. Efficient isolation and long-term viability of bovine small preantral follicles in vitro. In Vitro Cellular and Developmental Biology-Animal. 36 (4), 235-240 (2000).
- Saha, S., Shimizu, M., Geshi, M., Izaike, Y. In vitro culture of bovine preantral follicles. Animal Reproduction Science. 63 (1-2), 27-39 (2000).
- Bus, A., et al. Preservation of connexin 43 and transzonal projections in isolated bovine pre-antral follicles before and following vitrification. Journal of Assisted Reproduction and Genetics. 38 (2), 479-492 (2021).
- Gougeon, A., Ecochard, R., Thalabard, J. C. Age-related changes of the population of human ovarian follicles: increase in the disappearance rate of non-growing and early-growing follicles in aging women. Biology of Reproduction. 50 (3), 653-663 (1994).
- Xu, D., et al. Raf-ERK1/2 signaling pathways mediate steroid hormone synthesis in bovine ovarian granulosa cells. Reproduction in Domestic Animals. 54 (5), 741-749 (2019).
- Santos, R. R., et al. Cryopreservation of ovarian tissue: an emerging technology for female germline preservation of endangered species and breeds. Animal Reproduction Science. 122 (3-4), 151-163 (2010).
- Leonel, E. C. R., Lucci, C. M., Amorim, C. A. Cryopreservation of human ovarian tissue: a review. Transfusion Medicine and Hemotherapy. 46 (3), 173-181 (2019).
- Bus, A., Langbeen, A., Martin, B., Leroy, J. I. M. R., Bols, P. E. J. Is the pre-antral ovarian follicle the 'holy grail' for female fertility preservation. Animal Reproduction Science. 207, 119-130 (2019).
- Chen, J., et al. Optimization of follicle isolation for bioengineering of human artificial ovary. Biopreservation and Biobanking. , (2021).
- Chiti, M. C., et al. A modified and tailored human follicle isolation procedure improves follicle recovery and survival. Journal of Ovarian Research. 10 (1), 1-9 (2017).
- Kristensen, S. G., Rasmussen, A., Byskov, A. G., Andersen, C. Y. Isolation of pre-antral follicles from human ovarian medulla tissue. Human Reproduction. 26 (1), 157-166 (2011).
- Oktay, K., et al. Isolation and characterization of primordial follicles from fresh and cryopreserved human ovarian tissue. Fertility and Sterility. 67 (3), 481-486 (1997).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationExplorer plus d’articles
This article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.