
Method Article
Genome-wide proteina-proteina interazione Screening per Protein-frammento Complementation Assay (PCA) in cellule viventi
* Questi autori hanno contribuito in egual misura
In questo articolo
Riepilogo
Proteins interact with each other and these interactions determine in a large part their functions. Protein interaction partners can be identified at high-throughput in vivo using a yeast fitness assay based on the dihydrofolate reductase protein-fragment complementation assay (DHFR-PCA).
Abstract
Proteins are the building blocks, effectors and signal mediators of cellular processes. A protein’s function, regulation and localization often depend on its interactions with other proteins. Here, we describe a protocol for the yeast protein-fragment complementation assay (PCA), a powerful method to detect direct and proximal associations between proteins in living cells. The interaction between two proteins, each fused to a dihydrofolate reductase (DHFR) protein fragment, translates into growth of yeast strains in presence of the drug methotrexate (MTX). Differential fitness, resulting from different amounts of reconstituted DHFR enzyme, can be quantified on high-density colony arrays, allowing to differentiate interacting from non-interacting bait-prey pairs. The high-throughput protocol presented here is performed using a robotic platform that parallelizes mating of bait and prey strains carrying complementary DHFR-fragment fusion proteins and the survival assay on MTX. This protocol allows to systematically test for thousands of protein-protein interactions (PPIs) involving bait proteins of interest and offers several advantages over other PPI detection assays, including the study of proteins expressed from their endogenous promoters without the need for modifying protein localization and for the assembly of complex reporter constructs.
Introduzione
Reti di interazione proteina (PIN) offrono una risoluzione bassa mappa di come le proteine sono funzionalmente organizzati nella cella 1. Ogni connessione fisica tra due proteine, o interazione proteina-proteina (PPI), possono rappresentare un'associazione che è stabile nel tempo, come quelli trovati all'interno complessi proteici e che contribuiscono alla organizzazione strutturale della cellula. Queste connessioni possono anche rappresentare associazioni transitori che regolano l'attività, la stabilità, la localizzazione e le interazioni dei due partner. Identificare i partner di interazione fisica di una certa proteina fornisce quindi ricca informazioni sulla funzione e la regolazione di quella proteina 2,3. Per queste ragioni, grandi sforzi sono stati messi verso la mappatura dei PIN in organismi modello, tra cui Escherichia coli 4-6, Arabidopsis thaliana 7, Saccharomyces cerevisiae 8-12, Drosophila melanogaster </ Em> 13, Caenorhabditis elegans 14 e Homo sapiens 15. Questi studi hanno fornito importanti informazioni come le proteine sono organizzati in cella e informazioni così chiave sulle proteine con funzioni precedentemente sconosciuti.
Diverse strategie sono state sviluppate nel corso degli anni per studiare PIN. Queste tecnologie possono essere sostanzialmente raggruppati in tre categorie a seconda del tipo di informazioni che forniscono sul PPI (rivisto nel 16-18). Il primo si basa su lieviti doppio ibrido e dei suoi derivati 19. Queste tecnologie forniscono informazioni sulla diretta associazione tra coppie di proteine, che permette di costruire reti binari. La seconda famiglia è basata sulla purificazione di affinità delle proteine esca e l'identificazione dei loro partner associati, come la purificazione di affinità seguita da spettrometria di massa 20. Questi approcci identificano gruppi di proteine che sono direttamenteo indirettamente associato, generalmente in modo stabile, e sono estremamente potenti per identificare complessi proteici. Il terzo approccio si basa su saggi di complementazione proteica-frammento (APC) 11,21. Questo approccio fornisce un livello intermedio di risoluzione tra i due approcci precedenti, in quanto permette di rilevare associazioni dirette e prossimale tra proteine. Ogni tecnica ha i suoi punti di forza e di debolezza, come recentemente rivisto 18.
Il PIN eucariotica meglio descritta è di gran lunga quello del lievito Saccharomyces cerevisiae erba, in parte perché la sua proteoma è relativamente meno complessi di quelli di altri eucarioti modello e per saggi high throughput per rilevare IPP sono stati analizzati prima e sono più efficiente implementato in questo organismo modello 9-12. Un metodo particolarmente potente per il sistema lievito è la diidrofolato reduttasi proteina-frammento complementazione assay (DHFR-PCA), un metodo che è statoutilizzato in contesti diversi per studiare il PIN lievito in condizioni standard e perturbate 11,22-26. Questo metodo si basa su un saggio di sopravvivenza che consente il rilevamento di IPP diretti e quasi diretta per una data proteina esca ad entrambi i livelli di espressione endogeni e localizzazioni subcellulari native dei partner di interazione 11,21 in maniera quantitativa 27. Il segnale ottenuto da questo test (cioè dimensioni delle colonie su array colonia alta densità) riflette quindi la quantità di complessi proteici formati tra l'esca e la preda in un ambiente cellulare quasi equivalente a quella delle cellule wild-type. Il saggio si basa sulla ricostituzione di un enzima coinvolto nel metabolismo giornalista folati, la diidrofolato reduttasi (DHFR), in cui due frammenti complementari della DHFR che sono fusi alle due proteine di interesse vengono portati in prossimità quando le due proteine interagiscono, che a sua volta porta alla ricostituzione reversibili dell'attività enzimatica 11 e la crescita del ceppo su un terreno contenente metotrexato (MTX; Figura 1). Questo composto inibisce l'enzima DHFR endogena, ma non quello mutato utilizzato nel test 28. Due raccolte di ceppi PCA, uno contenente ~ 4.300 Mata ceppi con ORF fusa alla DHFR F [1,2] frammento e una contenente ~ 4.800 MAT α ceppi con un ORF fusa alla DHFR [3] frammento, può essere acquistato a implementare DHFR-PCA a scala piccola o grande in qualsiasi laboratorio. Qui, descriviamo un protocollo generale, ma dettagliato per schermo per PPI tra una proteina esca e ~ 4.800 proteine preda da questo test.
Protocollo
1. Costruzione / verifica di Bait Ceppi
- Se il ceppo esca di interesse è disponibile in [1,2] collezione Mata DHFR F, recuperarlo dalla collezione come descritto al punto 1.1.1, altrimenti costruire il ceppo come descritto al punto 1.1.2.
NOTA: Il protocollo descritto qui utilizza un DHFR F [1,2] ceppo come esca e la DHFR F [3] raccolta come prede, come questa raccolta contiene più ceppi rispetto alla DHFR F [1,2] collezione. Tuttavia, è possibile eseguire la schermata viceversa se il ceppo esca è disponibile solo nella DHFR F [3] raccolta o in entrambi gli orientamenti, se si richiede una maggiore copertura dell'interattoma.- Scongelare la targa di glicerolo che contiene il ceppo esca sul ghiaccio per un'ora. Sterilizzare il foglio di alluminio che copre la piastra con etanolo al 95%. Pierce il foglio con una punta sterile, pipettare su e giù per sospendere nuovamente le cellule e striscia 2-3 ml di glicerolo magazzino dal estratto di lievito selettivo peptonedestrosio (YPD) + 100 ug / ml Nourseothricin (Nat) per isolare singole colonie. Incubare per due giorni a 30 ° C.
- Costruzione di un ceppo esca PCA (Mata DHFR F [1,2]).
- Utilizzando ad alta fedeltà polimerasi e un protocollo standard di PCR, amplificare il DHFR F [1,2] cassetta dal plasmide pAG25-linker-DHFR F [1,2] -ADHterm utilizzando oligonucleotidi con sovrastante finisce omologa per gli ultimi 40 bp della ORF del 3'-end escluso il codone di stop (primer forward) ed al primo 40 bp del gene 3'-UTR (primer reverse) (Figura 1A).
- Trasformare il prodotto di PCR in cellule di lievito competenti (di solito in un ceppo BY4741) utilizzando il protocollo di trasformazione di lievito LiOAc / PEG standard a 29 (Figura 1A).
- Piatto su YPD selettiva + Nat medio isolare trasformanti positivi.
- Eseguire un PCR colonia di diagnostica colonie isolate per confermare il corretto DHFR F [1,2] la fusione. Utilizzare prIMers ricottura 1) nella sequenza genica codificante (oligo Forward) circa 100 bp a monte della fusione DHFR e 2) nel terminatore ADH della cassetta (oligo inversa) (Figura 1B).
- Sequenza il prodotto di PCR da Sanger sequenziamento per confermare il corretto fusion gene.
- Archiviare il ceppo esca confermata nel 25% glicerolo a -80 ° C.
NOTA: Il protocollo può essere messo in pausa in questa fase.
2. Pin-strumento di sterilizzazione e di stampa Procedure
NOTA: La procedura di sterilizzazione descritta di seguito è stato ottimizzato per le pin-tools manipolate dal BM3-BC (S & P Robotics) piattaforma robotica, ma può essere adattato ad altre piattaforme. Questa sezione descrive le procedure di sterilizzazione e stampa Pin utensili che vengono utilizzati per trasferire le cellule da un mezzo ad un altro per il resto del protocollo. In-house script usati per eseguire queste procedure possono essere richiesti. Nota that tutte le misure possono essere eseguite senza la necessità di una piattaforma robotica con un pin-utensile manuale 30.
- Montare il perno-strumento appropriato sulla piattaforma robotica.
- Preparare pulizia e stazioni bagnate come segue:
- Aggiungere 500 ml di acqua sterile nella stazione bagnomaria.
- Aggiungere 320 ml di acqua sterile nella stazione pennello.
- Aggiungere 380 ml di etanolo al 70% nel sonicatore durante la replica dalla piastra di agar (86 x 128 millimetri omnitray, contenente 35 ml di terreno solidificato) alla piastra di agar, o 400 ml quando la replica da micropiastra contenente colture liquide ad una piastra di agar.
- Aggiungere 35 ml di acqua sterile nella stazione bagnato (costituito da una omnitray vuoto sterile).
- All'inizio di ogni giornata che richiede la piattaforma robotica, sterilizzare gli strumenti pin-cinque volte per un minuto nel bagno sonicatore. Nel frattempo, accendere la lampada UV per cinque minuti per sterilizzare il recinto robot.
NOTA: Questo non è necessaria se il robot è alloggiato sotto una cappa sterile. - Tra ogni turno di replica pin-strumento, sterilizzare il pin-strumento come segue:
- Mettere a bagno la pin-strumento cinque volte per 10 secondi nella stazione bagnomaria per rimuovere grumi di cellule.
- Mettere a bagno la pin-strumento due volte avanti e indietro nella stazione di pennello.
NOTA: La spazzola rotante rimuove le cellule residue. - Mettere a bagno la pin-strumento due volte per 20 secondi nella stazione sonicatore.
NOTA: Rimanendo cellule sui pin verrà rimosso mediante ultrasuoni o ucciso da etanolo. - Assicurarsi che la profondità di immersione dei perni aumenta ad ogni bagno successivo per garantire una corretta sterilizzazione.
- Asciugare la pin-strumento nella stazione di aria essiccatore per 25 sec.
- Prima di cellule sulle piastre source, bagnare i perni nella stazione bagnato, contenente 35 ml di acqua sterile in un omnitray.
- Bagnare i perni due volte nelle colonie dalla piastra sorgente.
- Stampa su un piatto di destinazione appena versato toccando l'agSuperficie ar due volte (di seguito indicato come l'azione di "stampa" di un array).
3. La condensazione della DHFR F [3] Collection in 1.536 matrici Utilizzo di un Robotic Platform automatizzata
- Scongelare su ghiaccio il DHFR F [3] raccolta (60 piastre a 96 pozzetti) e un ulteriore piastra a 96 pozzetti riempiti con il DHFR L-F [3] ceppo di controllo (Figura 1C), che contiene il DHFR F [3] frammento e il linker monte espresso solo come controllo negativo.
NOTA: In linea di principio, questo frammento non deve interagire con qualsiasi proteina di fusione DHFR-frammento (vedi la discussione per i dettagli). - Piastre centrifuga (giro veloce) prima di rimuovere il foglio di alluminio per evitare i rischi di contaminazione incrociata tra i pozzetti.
- Condensare la collezione su 16 matrici di 384 ceppi (Figura 2a). Per fare questo, per ogni array 384, stampa quattro piatti glicerolo su quattro quadranti di YPD selettiva + 250 mg / ml Hygromycin B (HygB) omnitray utilizzando the 96 pin-utensile (qui, una matrice 384 può essere suddiviso in quattro quadranti ugualmente distanziate, ciascuna composta da 96 posizioni in una disposizione 2 x 2 matrice). Inserire quattro piastre a 96 pozzetti contenenti il [3] controllo negativo L-DHFR F tra le altre piastre 60 in modo da avere una serie finale di 64 placche che riempiono esattamente quattro 1.536 matrici. Sterilizzare il-strumento pin tra ogni ciclo di replica come descritto al punto 2.
NOTA: Inserire la L-DHFR F [3] piastre in modo da avere una tale lastra su ciascuno dei quattro finali 1.536 matrici. - Incubare le piastre per due giorni a 30 ° C. NOTA: In questa fase, la raccolta DHFR può essere memorizzato in un formato 384 a 4 ° C fino ad un mese su piastre di agar.
- Condensare la collezione in quattro array di 1.536 ceppi (Figura 2a). A tale scopo, per ogni array 1.536, stampare quattro matrici di 384 ceppi sui quattro quadranti di una piastra dello stesso mezzo come al punto 3.2 utilizzando il perno-utensile 384 (qui, un array di 1.536 può essere suddiviso in quattro equally distanziate quadranti ognuno composto da 384 posizioni in un layout 2 x 2 matrice).
- Incubare le piastre per due giorni a 30 ° C. NOTA: In questa fase, la raccolta DHFR può essere memorizzato in un formato 1.536 a 4 ° C fino ad un mese su piastre di agar.
- Replicare le quattro matrici sullo stesso mezzo di standardizzare le dimensioni delle colonie con un pin-tool 1.536.
- Incubare le piastre per due giorni a 30 ° C.
4. High-throughput Procedura DHFR-PCA
- Seminare una coltura del ceppo esca (DHFR F [1,2] fusione) ottenuto nella fase 1.1.1 o 1.1.2 in 20 ml di liquido YPD + Nat in un tubo da 50 ml (Figura 2B).
- Incubare per due giorni a 30 ° C con agitazione a 250 rpm per consentire alla cultura di raggiungere la saturazione.
- Dopo due giorni di incubazione, piatto 5 ml di cultura su un YPD + Nat omnitray. Let cellule adsorbono sulla superficie per 5-10 min e rimuovere il liquido in eccesso (Figura 2B). Ripetere tWICE per fare tre repliche.
- Incubare per due giorni a 30 ° C.
- Stampa il ceppo esca dai passi 4.3 e 4.4 da 12 piastre YPD (abbastanza per accoppiamento quattro piatti della DHFR F [3] di raccolta × tre repliche) con una pin-strumento di 1.536 con ogni prato cella non più di quattro volte.
- Stampa la matrice appropriata della DHFR F [3] raccolta sopra le celle esca utilizzando il pin-utensile 1.536 (Figura 2C).
- Lasciate che i ceppi accoppiano incubando per due giorni a 30 ° C.
- Selezionare cellule diploidi stampando colonie omnitrays contenenti YPD + HygB + Nat (Figura 2C).
- Incubare per due giorni a 30 ° C.
- Selezione diploide Ripetere come descritto ai punti 4.8 e 4.9 (Figura 2B).
- Preparare piatti con supporti contenenti MTX (MTX media) un giorno prima dell'uso seguendo questi passaggi 21 (ATTENZIONE:. Fare attenzione quando si lavora con MTX in quanto è un composto tossico Indossare sempre guanti, proteocchiali ction e un camice da laboratorio nel maneggiarlo) (Figura 2C):
- Preparare i 10x -lys / incontrato / ade aminoacido drop-out in acqua deionizzata. Filtro-sterilizzare il drop-out in una bottiglia sterile utilizzando un filtro siringa sterile 0,2 micron o, se necessario, in grandi quantità, un imbuto di filtrazione superiore 0,2 micron bottiglia (Tabella 1).
- Preparare una / ml soluzione madre MTX 10 mg in dimetilsolfossido (DMSO). Utilizzare immediatamente dopo la preparazione della soluzione e congelare il resto a -20 ° C. Proteggere dalla luce che è fotosensibile. Non congelare MTX dopo scongelamento esso.
- Preparare il supporto come segue (ingredienti e le quantità medie sono uguali a quelli utilizzati in Tarassov et al. 11):
- Per un litro di media, mescolare in due palloni separati: 1) 6,69 g di base di azoto lievito senza aminoacidi e senza solfato di ammonio e 330 ml di acqua deionizzata; 2) 25 g di agar nobili e 500 ml di acqua deionizzata.
- Boccette autoclave a 121 ° C per 20 min.
- Equilibrare temperatura in un bagno di acqua a 55 ° C per almeno un'ora.
- Mescolare i due palloni insieme e aggiungere 50 ml di sterile il 40% di glucosio, 100 ml di 10x sterile drop-out, e 20 ml di MTX 10 mg / ml.
- Versare 35 ml (vedi nota sotto) di medie omnitrays. Lasciare solidificare per almeno un'ora e mezza. Piastre Shield dalla luce.
NOTA: Qui, versando 35 ml di media per omnitray è fondamentale per garantire la parità di spessore della piastra, che è importante per tutte le fasi a valle.
- Fosfori del secondo turno di selezione diploide con la piattaforma robotica o con una normale fotocamera digitale con l'uniforme illuminazione targa. Utilizzare queste immagini per identificare le posizioni vuote sulle matrici quando si esegue l'analisi a valle. Assicurarsi che i parametri della telecamera sono sempre le stesse e che la luce robot è accesa.
- Stampa cellule diploidi su MTX supporto USIng pin-tool 1.536.
- Incubare per quattro giorni a 30 ° C in sacchetti di plastica per evitare l'essiccazione.
- Preparare un secondo lotto di omnitrays contenenti MTX media come descritto al punto 4.11.
- Dopo quattro giorni di incubazione, le piastre di immagine utilizzando la piattaforma robotica o regolare la macchina fotografica digitale. Assicurarsi che i parametri della telecamera sono sempre le stesse e che la luce robot è accesa.
- Eseguire una seconda tornata di selezione MTX replicando le celle del secondo lotto di MTX media.
NOTA: Ciò farà diminuire la crescita di ceppi PCA sfondo e aumentare la risoluzione quantitativa. - Incubare per quattro giorni a 30 ° C in sacchetti di plastica per evitare l'essiccazione.
- Fosfori come descritto al punto 4.16.
Analisi 5. Immagine
- Analizzare le immagini di array colonia con ImageJ personalizzata 31 script o utilizzando software pubblicati quali Colonizer, Ht analizzatore griglia colonia, profiler cellule, Colony Immaginer, ScreenMill, YeastXtract e gitter (compilato in 32). L'analisi delle immagini deve uscita uno o più fogli contenenti formati colonie per ciascuna posizione di ciascuna matrice, utilizzare questi formati colonie per tutte le analisi a valle.
NOTA: In questo studio, abbiamo utilizzato uno script ImageJ personalizzato descritto in Leducq et al 33 (vedi la sezione di discussione per maggiori dettagli)..
Analisi 6. I dati
NOTA: I risultati di analisi di immagine possono essere elaborati in una tabulazione come Excel o usando un linguaggio di scripting come R 34. Di seguito viene descritto il procedimento con una personalizzata ImageJ 31 sceneggiatura.
- Utilizzando uno script personalizzato, concatenare i file di output di analisi di immagine e annotare ogni riga con la piastra e filtrare le informazioni, come nella Tabella supplementare 1.
- Log 2 trasformare i valori delle dimensioni delle colonie (densità integrata o zona colonia, qui, colonna "IntDenBackSub & #8221; dalla tabella integrativa 1 è stato utilizzato).
NOTA: Distribuzione Valore sarà come in Figura 3A. - Normalizzare i valori sottraendo il valore medio di ogni piatto.
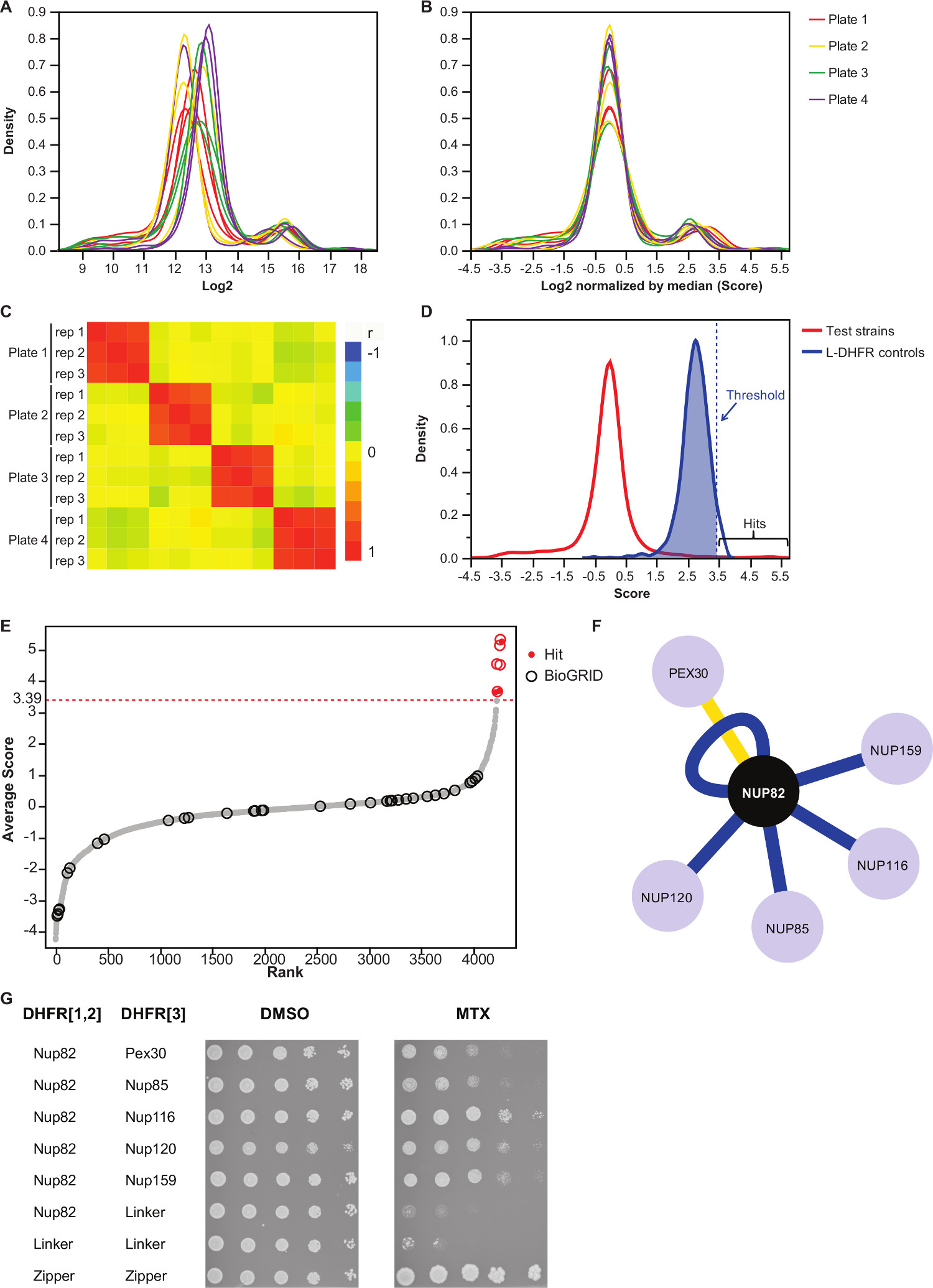
NOTA: Questo controllo passo per piastra di polarizzazione, che possono derivare da quantità supporti diseguale o variazione di acquisizione automatica delle immagini, e riduce la varianza inter-replicare (Figura 3B). - Verificare che correlano replicati tra loro (Figura 3C) per valutare la riproducibilità degli esperimenti.
- Per differenziare interagendo da coppie non interagenti esca-preda, impostare una soglia alta fiducia corrispondenti ai 95 ° percentile della distribuzione del L-DHFR F [3] controlli.
NOTA: In questo esperimento, ciò corrisponde a 3,39 (Figura 3D). In alternativa, può essere utilizzata una soglia basato sulla sovrapposizione con noti interattori fisici quali quelli riportati nella BIOGRID 35.Vedere la discussione per ulteriori dettagli. - Per qualsiasi esca, filtro prede identificato come coinvolti nelle interazioni falsi positivi in schermi DHFR-PCA (vedi la discussione per ulteriori dettagli) e riportate nella tabella integrativa 2 (identificato come "1" nella colonna "filtrato").
- La media dei log2 dimensioni colonia normalizzato delle tre repliche di ogni interazione (colonna "Ripartizione" in tabella integrativa 2).
7. Convalida dei Interactiani fisici utilizzando Esperimenti in piccola scala
NOTA: Qualsiasi PPI di particolare interesse avere un punteggio sopra o vicino alla soglia applicato può essere convalidato usando il saggio DHFR-PCA in un disegno sperimentale scala ridotta utilizzando un saggio di crescita su terreno solido o liquido MTX. I passi seguenti mostrano la procedura per costruire manualmente ceppi PCA diploidi ed eseguire test a campione su MTX media. Lo sperimentatore deve eseguire queste operazioni per tutti necessarControlli y (Bait-DHFR F [1,2] x L-DHFR F [3], Zipper-linker-DHFR ceppi diploidi e linker-DHFR ceppo diploide).
- Piatto 2-3 ml dello stock glicerolo del ceppo esca generata in 1.1.2.6), la L-DHFR F [3] ceppo di controllo, la Zipper-linker-DHFR e linker-DHFR ceppi diploidi su YPD + Nat, YPD + HygB e due volte YPD + Nat + HygB media, rispettivamente.
- Recuperare la preda di interesse per il DHFR F [3] la raccolta e seguire le istruzioni nel passaggio 1.1.1, ma striscia la tensione sul YPD + HygB medio invece di YPD + Nat media.
- Eseguire PCR diagnostico in 1.1.2.4 per confermare la DHFR F [3] fusa al locus prede e la sequenza del prodotto.
- Seminare 1 ml di terreno YPD liquido con ceppi aploidi di accoppiarsi (Bait x Prey, Bait x L-DHFR F [3] controllo) e crescere almeno due giorni a 30 ° C per permettere ai diploidi di formare.
- Selezionare diploidi strisciando 4-5 microlitri della cultura in 7.4 su terreno solido YPD + HygB + Nat. Crescere due giorni a 30 ° C.
- Select una colonia isolata e crescere durante la notte in coltura liquida (1 ml) per eseguire il test di crescita.
- Preparare piatti MTX e DMSO (stessi ingredienti MTX media, ma senza MTX) un giorno prima dell'uso. Vedere il punto 4.11 per ulteriori dettagli.
- Effettuare test crescita individuando diluizioni seriali delle diverse culture sul controllo (DMSO) e le piastre di selezione (MTX).
- Diluire precultures a OD = 1.
- Eseguire diluizioni cinque volte (fino a un fattore di diluizione di 625) in una piastra a 96 pozzetti sterili.
- Spot 4 ml di ogni diluizione sul supporto PCA (DMSO e MTX).
- Incubare a 30 ° C in sacchetti di plastica per evitare l'essiccazione.
- Fosfori da 1 a 7 giorni di incubazione che utilizzano la piattaforma robotica o una normale fotocamera digitale.
Risultati
Tabella integrativa 2 è un esempio di risultati rappresentativi ottenuti con la proteina di lievito Nup82 fusa alla DHFR F [1,2] frammento come esca. La soglia definita con la L-DHFR F [3] controlli può essere usato come una soglia empirica per determinare risultati confidenza elevati (figure 3D e 3E). In alternativa, la classifica punteggio può essere utilizzato per eseguire arricchimenti Gene Ontology o altro funzionale analizza 36 sulla base di standard di riferimento 37. Gli interattori fisiche conosciute della esca possono essere recuperati dal database come BIOGRID 35 e sovrapporre i dati (Figure 3E e 3F). In questo esempio, cinque degli otto colpi di alta fiducia sono stati precedentemente segnalato come interattori Nup82 e due fanno parte del subcomplesso Nup82, Nup116 e Nup159 (Figura 3F e 3G). L'altro membro del complesso, NSP1, non mostra alcun Interaczione nel nostro esperimento. Due prede, Ade17 e Tef2 (non mostrato in figura 3F), avevano punteggi al di sopra della soglia dura applicata, ma questi sono probabilmente falsi positivi mentre interagiscono quasi ogni proteina esca in schermi PCA abbiamo eseguito (risultati non pubblicati). D'altra parte, Pex30 può rappresentare un interattore fisica romanzo Nup82 e siamo stati in grado di confermare questa interazione con DHFR-PCA a bassa velocità (Figura 3G). Pex30 è una proteina di membrana perossisomica e poche interazioni dirette sono stati segnalati tra il complesso del poro nucleare (NPC) e questo organello. Uno schermo doppio ibrido identificato altre due proteine NPC, Nup53 e Asm4 (Nup59), come interattori fisiche di Pex30 38, e una interazione genetica tra Pex30 e Nup170 è stato segnalato 39. Due altri partner interazione rilevato, Nup120 e Nup85 (Figura 3F e 3G), non fanno parte del sub-complesso Nup82, illustrando la capacitàdella DHFR-PCA per rilevare le interazioni all'interno e tra Sottocomplessi in complessi più grandi 11.

Figura 1:. Ceppi di lievito di ingegneria per high-throughput DHFR-PCA (. Figura adattato da Leducq et al 2012 33) (A) Costruzione di aploidi Mata e MAT α ceppi di fondere Gene1 (G1) e Gene2 (G2) con la DHFR F [1,2] -NatMX e la DHFR F [3] -HPH cassette, rispettivamente. Le cassette sono amplificati da plasmidi pAG25-DHFR F [1,2] e pAG32-DHFR F [3] con primer avanti G1-5 'e G2-5', e reverse primer G1-3 'e G2-3', e poi inserito nel genoma all'estremità 3 'del gene bersaglio di ricombinazione omologa. Le proteine risultanti, P1 e P2, rispettivamente, sono fusi al DHFR F [1,2] frammento (Mata) e la DHFR F [3] frammento (MATα) attraverso un linker flessibile. (B) Verifica della costruzione di (A) viene eseguita mediante sequenziamento le giunzioni tra Gene1 e di ORF di Gene2 e le cassette DHFR. I ceppi PCA costruiti su opposti tipi di accoppiamento sono poi accoppiati per formare un diploide. Ceppi diploidi crescono su MTX medio se i due frammenti complementari DHFR vengono portati in prossimità da un'interazione tra P1 e P2, che ricostituisce la attività dell'enzima DHFR. (C) Costruzione del controllo diploidi PCA ceppi per schermi DHFR-PCA. I controlli negativi (L-DHFR) sono costruiti trasformando Mata e MATα ceppi separatamente aploidi con plasmidi P41-linker-DHFR F [1,2] e P41-linker-DHFR F [3] 11, rispettivamente. I due ceppi sono accoppiati con un conseguente controllo negativo ceppo diploide in cui i frammenti DHFR potutoper completarsi a vicenda (in alto). I controlli positivi sono costruiti utilizzando lo stesso approccio per i controlli negativi, ma i plasmidi trasformati in ceppi aploidi (P41-cerniera-linker-DHFR F [1,2] (P41-ZL-DHFR F [1,2]) e p41 -zipper-linker-DHFR F [3] (P41-ZL-DHFR F [3])) contengono due GCN4 leucina parallelo frammenti cerniera fusi ai frammenti complementari DHFR, che porta ad una interazione forte e costitutivo che ricostituisce l'attività DHFR (bottom ). Clicca qui per vedere una versione più grande di questa figura.
{kind=link}

Figura 2: High-throughput procedura DHFR-PCA (figura adattato da Leducq et al 33.) (A) Il MAT α DHFR F [3] collezione. è condensato in un formato 1536 attraverso due successivi cicli di condensazione. In primo luogo, le piastre glicerolo azionari sono combinati da gruppi di quattro su terreno selettivo YPD + HygB in un formato 384 tramite il pin-strumento 96. In secondo luogo, 384 matrici vengono combinati da gruppi di quattro su terreno selettivo YPD + HygB in formato 1536 con il pin-tool 384. (B) prati cellulari del ceppo esca MATA PCA sono preparati da crescere una cultura satura del ceppo esca in YPD selettiva + Nat medio e placcatura la cultura su un YPD + Nat omnitray. (C) Questi prati sono utilizzati per accoppiare il ceppo esca con la DHFR F [3] raccolta su YPD media. Le cellule vengono successivamente trasferiti due volte sul YPD + HygB + Nat medio di selezionare per diploidi e due volte su MTX medio per eseguire PCA. La crescita sarà osservata solo MTX medio se i frammenti DHFR si completano a vicenda a seguito di una interazione tra l'esca e le proteine preda.e.jpg "target =" _ blank "> Clicca qui per vedere una versione più grande di questa figura.

Figura 3:.. Analisi dei dati attraverso i passaggi di normalizzazione, la determinazione di una soglia di significatività e l'identificazione delle interazioni positive (A) la distribuzione di densità di dimensioni colonia su ciascuna piastra (log 2) (B) normalizzazione della mediana di ogni immagine corregge per bias associato con effetti piastra-piastra. (C) Heatmap che mostra il coefficiente di correlazione Spearman tra le piastre, confermando la riproducibilità della procedura. (D) Distribuzione dei punteggi per le IPP testati e [3] controlli L-DHFR F. Una soglia rigido può essere impostata sui 95 ° percentile della L-DHFR F [3] distribuzione per identificare PPI alta fiducia (rappresentato da una verticale tratteggiatadistribuzione di linea). (E) per Classifica del punteggio medio di ogni preda. Precedentemente riportati partner interazione fisiche del Nup82p nel BIOGRID 35 sono identificati da cerchi e quelli riportati in questo studio sono identificati da punti rossi. La soglia definita in (D) viene mostrata come una linea tratteggiata. (F) Rete mostrando le IPP ad elevata affidabilità identificati in questo studio. Bordi blu mostrano precedentemente segnalati interattori fisici (BIOGRID 35) e bordi gialli mostrano un'interazione precedentemente non dichiarata con Pex30. (G) Spot-diluizione di prova del PCA ceppi diploidi coinvolge Nup82-DHFR F [1,2] e preda-DHFR F [3 ] paia identificati come interattori fisiche del Nup82p nel presente studio. Test La crescita è stata eseguita in DMSO medio (MTX solvente, pannello di sinistra) e MTX medio (pannello di destra). Controlli negativi costituiti da Nup82-DHFR F [1,2] - Linker-DHFR F [3] e Linker-DHFR F [1,2] - Linker-DHFR F [3] e un controllo positivo costituito da tha una forte interazione tra due porzioni leucina cerniera (zip-DHFR F [1,2] - zip-DHFR F [3]) sono stati inclusi. La crescita delle cellule superiori ai controlli negativi in MTX terreno deve essere interpretata come una interazione fisica. Clicca qui per vedere una versione più grande di questa figura.
{kind=link}
| Aminoacidi | Quantità (g) |
| Adenina solfato * | 0.2 |
| L-triptofano | 0.4 |
| L-tirosina | 0.3 |
| L-fenilalanina | 0.5 |
| L'acido L-glutammico (sale monosodico) | 1.0 |
| L-Asparagine | 1.0 |
| L-Valina | 1.5 |
| L-treonina | 2.0 |
| L-Serina | 3.75 |
| Uracile | 0.2 |
| L-istidina HCl | 0.2 |
| L-Arginina HCl | 0.2 |
| L-metionina * | 0.2 |
| L-Lisina * | 0.2 |
| L-Leucina | 0.6 |
| * Ritirato durante l'esecuzione di serie PCA (come in questo protocollo), ma possono essere aggiunti per altri scopi. | |
Tabella 1:. Composizione del 10X -lys / incontrato / ade drop-out nel mezzo MTX quantità sono per un litro di 10x drop-out. Gli asterischi indicano i composti che devono essere esclusi per il mezzo MTX standard, ma che possono essere aggiunti per altri scopi.
Tabella supplementare 1:. I dati combinati dalle piastre 12 di prova La tabella 1 contiene il da concatenatota dall'analisi ImageJ eseguita utilizzando uno script ImageJ personalizzato con ogni riga corrisponde ad una sola posizione su ogni array 1.536. Inoltre, ogni riga è stato annotato con le informazioni relative al file immagine, piastra raccolta DHFR, replicare, ORF e il nome della proteina.
Tabella integrativa 2:. La crescita media normalizzata per ceppo tabella 2 contiene il registro 2 crescita normalizzata media per ogni ceppo della collezione con la deviazione standard. Prede filtrati, successi e noti interattori fisici sono identificati in colonne separate.
Discussione
Descriviamo un protocollo basato sul dosaggio DHFR-PCA che consentano l'identificazione sistematica delle interattori fisici per ogni proteina esca a high-throughput. Questo protocollo può essere adattato dallo screening altre esche, e questo a qualsiasi livello desiderato di replica. Dimostriamo l'affidabilità di questo protocollo per l'identificazione di partner di interazione fisica per una proteina esca coinvolta nel complesso nucleare Pore: Nup82. La nostra analisi ha permesso di trovare cinque interattori precedentemente segnalati e uno precedentemente non dichiarata Interactiano (Figure 3F e 3G), mettendo in evidenza la capacità del metodo per studiare il interattoma proteine del lievito.
Il protocollo qui descritto comprende diversi passaggi critici in cui lo sperimentatore deve prestare attenzione. Consigliamo a 1) Assicurarsi che il DHFR esca F [1,2] la fusione sia corretta (Figura 1B); ciò può essere ottenuto mediante sequenziamento costruzione e measuring espressione della proteina corretta usando un anti-DHFR F [1,2] o DHFR anti-F [3] anticorpo; 2) Prima di iniziare lo schermo, si consiglia di verificare se qualcuno esca di interesse presenta interazioni promiscue in schermi PCA. Questo può essere fatto eseguendo schermate di controllo con esche incrociate con il controllo L-DHFR appropriato o accoppiando manualmente l'esca con il controllo L-DHFR appropriata ed eseguendo un test crescita MTX medie. 3) Le piastre devono essere versato il giorno prima di essere utilizzate in modo che l'umidità è ottimale per l'adesione delle cellule sulla superficie agar durante il processo di stampa; 4) piastre di origine non dovrebbero essere usati più di quattro volte a trasferire abbastanza cellule sulla piastra di destinazione. Aumentando il numero di copie della piastra di destinazione può essere fatto da fasi successive di espansione (ad esempio 4 copie -> 16 copie -> 64 copie). In alternativa, le cellule possono essere ritirati in posizioni diverse sui prati o nella colonia tra i diversi cicli di replica; 5) Se più positions mancano dopo la selezione diploide (s), assicurarsi che le piastre di origine non sono stati utilizzati troppe volte nella fase di accoppiamento (passi 4,5-4,7); 6) Verificare che il medium MTX contiene tutti gli ingredienti essenziali alle concentrazioni di destra. Infatti, se nessuna crescita a tutti si osserva sul supporto MTX, può essere sia perché nessuna interazione è rilevabile con PCA per le proteine di interesse o perché il mezzo MTX non era preparato correttamente. Per garantire che il mezzo permette la crescita di ceppi che mostrano DHFR frammenti complementazione, un'interazione costitutiva può essere aggiunto in posizioni vuote della raccolta e usata come controllo positivo come DHFR-frammenti fusi a leucina frazioni cerniera 33 (Figura 1C). Test parallelo utilizzando i frammenti linker-DHFR o frammenti cerniera-linker-DHFR permetterà di discriminare tra le condizioni che consentono tutte le cellule di crescere (bassa concentrazione di MTX o proteine esca che tendono a rendere le interazioni falsi positivi, come described sotto) e le condizioni che impediscono la crescita di tutti i ceppi (Concentrazione MTX troppo alto o essenziale mancante nel mezzo); 7) Dato che PCA viene eseguita attraverso cicli successivi di repliche da un mezzo ad un altro, la contaminazione crociata tra ceppi tra piatti diversi si può verificare se, per esempio, la pin-utensile non è sterilizzato correttamente tra un round di replica e / o l'ultima acqua Bagno (station cioè umido) nella procedura di sterilizzazione è contaminato da colonie di precedenti round di replica. Diverse posizioni gli array sono vuoti e possono quindi essere utilizzati come posizioni di comando in cui deve essere osservato nessuna crescita per rilevare contaminazioni crociate.
L'analisi delle immagini può essere eseguita utilizzando diversi software pubblicati (vedi sezione 5 del protocollo) o qualsiasi script personalizzato. In questo studio, lo script personalizzato esegue i seguenti passi: 1) Lo script sottrae i valori dei pixel di un piatto vuoto a Pixel valori di ogni piatto in modo da corRect per illuminazione pregiudizi. 2) Lo script converte ogni immagine di sfondo con correzione per binario utilizzando una soglia di valore di pixel pari a 10. 3) Per ogni 1.536 posizioni di ogni piatto, determinati sovrapponendo un rettangolo sulle colonie bordo, lo script viene eseguito ImageJ "Analizza particelle ... Funzione "in una selezione circolare. La selezione circolare è impostata con un raggio pari all'intervallo tra due posizioni meno 10 pixel. 4) Lo script seleziona la particella più vicina dal centro della selezione e conferma come colonia se la sua posizione non è più di metà dell'intervallo tra due colonie di distanza dal centro della selezione. 5) Lo script misura valori dei pixel della particella selezionata sull'immagine sfondo corretta. 6) Per correggere ulteriormente i restanti pregiudizi retroilluminazione, lo script sottrae il valore medio di tutti i pixel dalla selezione circolare che non erano parte di una particella di valori dei pixel della colonia. La somma di questi valori di pixel correttos, memorizzati nella colonna "IntDenBackSub" della tabella complementare 1, sono usati come misura di dimensioni delle colonie.
Un punto critico all'interno della parte analisi è la scelta della soglia di significatività. Qui, abbiamo scelto una soglia basato sulla distribuzione del negativo L-DHFR F [3] controlli, ma a seconda dell'obiettivo dello schermo, tale soglia può essere troppo severe. Infatti, L-DHFR F [3] controlli vengono sovraespressi (promotore forte TEF) tale che i frammenti complementari possono spontaneamente complementari e questi non sono quindi rappresentativi della maggior parte delle proteine di espressione. Ciò è evidenziato dal fatto che la distribuzione della L-DHFR F [3] controlli è superiore alla media della crescita sfondo (Figura 3D). Così, alcune interazioni che hanno punteggi inferiori a questa soglia rigorosa ma che sono chiaramente al di fuori della distribuzione crescita di fondo può essere considerato come colpi presunti che possono rappresentare, per esempio, tra transitoria o deboleazioni. Questi possono essere ulteriormente studiate e cross-validati se, per esempio, le due proteine non sono espressi a livelli che possono consentire la complementazione spontanea dei frammenti DHFR come controlli L-DHFR. In alternativa, si può impostare una soglia di significatività in base alla percentuale di sovrapposizione con Interactiani fisici riportati in database come BIOGRID 35 al fine di massimizzare la percentuale di veri positivi su falsi positivi. Tuttavia, a differenza l'uso della distribuzione L-DHFR, questa alternativa può non essere sempre fattibile se, per esempio, il numero di noti interattori fisici non è sufficientemente elevato. Inoltre, la scelta della soglia di significatività ha un impatto sulla percentuale di falsi positivi e falsi negativi nel set di dati finale. Infatti, come qualsiasi altro test PPI rilevamento, falsi positivi possono derivare da interazione aspecifica di una proteina con la proteina di fusione DHFR-se, per esempio, la proteina è altamente abbondante come accennato in precedenza. Questoè esemplificato dal fatto che alcuni prede interagiscono sistematicamente con tutte le proteine esca in schermi PCA e, quindi, devono essere rimossi dall'analisi 11 (es Tef2 e Ade17 e Tabella integrativa 2). Per aggirare questo problema, una schermata di controllo PCA delle due collezioni contro il controllo L-DHFR appropriata (F [1,2] o F [3]) per identificare esche e prede espositrici spontanea DHFR frammenta complementazione può essere eseguita in condizioni specifiche di ogni schermata. Inoltre, l'esecuzione di un'analisi arricchimento Gene Ontology può aumentare la fiducia nei dati se la funzione di un determinato esca è noto. D'altra parte, DHFR-PCA può dar luogo a falsi negativi per diverse ragioni: 1) non tutte le proteine possono essere fuse ai frammenti DHFR quanto potrebbero destabilizzare proteine o modificare la loro localizzazione se, per esempio, la fusione della DHFR C-terminale interferisce con un segnale di localizzazione; 2) la ricostituzione DHFR in alcuni compartimenti cellulari may non producono folato se, per esempio, un elemento indispensabile per la sintesi folato non è disponibile; 3) C-termini necessità di essere all'interno di una distanza di 8 nm per DHFR complementazione si verifichi 11. Pertanto, un'interazione noto potrebbe non essere rilevato se la loro C-terminali non sono abbastanza vicino nello spazio. Questo è esemplificato qui dal fatto che una grande frazione di interazioni fisiche Nup82 riportati in banche dati, la maggior parte dei quali sono indiretto, non sono stati rilevati nel nostro test. Analogamente, le interazioni tra le proteine di membrana per cui il C-termini sono in trans rispetto alla membrana non comporteranno DHFR frammenta complementazione e non sarà rilevato 11. Limitazioni 1) e 3) può essere aggirato relativamente semplice fondendo il frammento DHFR alla N-terminali della proteina. Ciò potrebbe evitare di interferire con un segnale di localizzazione vicino al C-termini e può consentire di individuare una interazione tra le proteine di membrana la cui N e C-terminale sono in relativa cisalla membrana.
Diverse sfide rimangono nello studio di PIN (rivisto in 2,3). Le mappe di PIN prodotte finora sono stati ampiamente descritti in un'unica condizione sperimentale per ogni specie e quindi offrire una singola istantanea di come potrebbero essere organizzate reti di proteine. Vi è quindi una necessità per l'esplorazione di altre condizioni sperimentali per vedere come PIN possono essere riorganizzati in risposta ai cambiamenti ambientali, stimoli specifici, attraverso lo sviluppo o seguenti mutazioni. Queste sfide saranno superate dallo sviluppo di nuove tecnologie per interrogare IPP in tempo reale, in cellule viventi e adattando attuali tecniche in modo che possano essere utilizzati da un grande comunità di laboratori. Come una tecnica quantitativa in grado di rilevare i cambiamenti nella quantità di DHFR complementazione complessi 27, DHFR-PCA può essere adattato per superare queste sfide ed è stato utilizzato per studiare come i PPI sono interessate da un DNA agente 22 danni , agenti chimici 25, delezioni geniche 23,26 o in altre specie di lievito e loro ibridi 33. Exploring queste nuove dimensioni diventerà sempre più importante per rivelare la dinamica del PIN.
Divulgazioni
Parte delle tasse di pubblicazione ad accesso aperto per questo articolo sono stati pagati da S & P Robotics.
Riconoscimenti
Questo lavoro è stato sostenuto dal Canadian Institute of Health Research (CIHR) concede 191.597, 299.432 e 324.265, a scienze naturali e ingegneria Research Council del Canada Discovery concessione e un finanziamento Human Frontier Science Program al CRL. CRL è un CIHR Nuova Investigator. Guillaume Diss è sostenuto da una borsa di studio PROTEO. Samuel Rochette è supportato da NSERC e FRQNT borse.
Materiali
| Name | Company | Catalog Number | Comments |
| BioMatrix Robot, Bench-top Configuration | S&P Robotics Inc. | BM5-BC | |
| 96-format Pin-tool | S&P Robotics Inc. | PH-96-10 | Standard 96-format Pin-tool with 96 high-precision floating pins |
| 384-format Pin-tool | S&P Robotics Inc. | PH-384-10 | Standard 384-format Pin-tool with 384 high-precision floating pins |
| 1536-format Pin-tool | S&P Robotics Inc. | PH-1536-05 | Custom 1536-format Pin-tool with 0.5mm high-precision floating pins |
| Automated imaging module | S&P Robotics Inc. | IMG-02 | |
| Methotrexate | Bioshop Canada Inc. | MTX440 | CAUTION: toxic compound |
| Hygromycin B | Bioshop Canada Inc. | HYG003 | |
| Nourseothricin dihydrogen sulfate | Werner BioAgents | 5010000 | |
| Yeast-Interactome Collection | Thermo Scientific | YSC5849 | |
| Omni Tray w/lid sterile | Thermo Scientific | 242811 | |
| Anti-DHFR F[1,2] antibody | Sigma-Aldrich | D1067 | |
| Anti-DHFR F[3] antibody | Sigma-Aldrich | D0942 |
Riferimenti
- Alberts, B. The cell as a collection of protein machines: preparing the next generation of molecular biologists. Cell. 92, 291-294 (1998).
- Diss, G., et al. Integrative avenues for exploring the dynamics and evolution of protein interaction networks. Curr Opin Biotechnol. 24, 775-783 (2013).
- Vidal, M., Cusick, M. E., Barabasi, A. L. Interactome networks and human disease. Cell. 144, 986-998 (2011).
- Hu, P., et al. Global functional atlas of Escherichia coli encompassing previously uncharacterized proteins. PLoS biology. 7, e96 (2009).
- Arifuzzaman, M., et al. Large-scale identification of protein-protein interaction of Escherichia coli K-12. Genome Res. 16, 686-691 (2006).
- Rajagopala, S. V., et al. The binary protein-protein interaction landscape of Escherichia coli. Nature biotechnology. 32, 285-290 (2014).
- Arabidopsis-Interactome-Mapping-Consortium. Evidence for network evolution in an Arabidopsis interactome map. Science. 333, 601-607 (2011).
- Babu, M., et al. Interaction landscape of membrane-protein complexes in Saccharomyces cerevisiae. Nature. 489, 585-589 (2012).
- Krogan, N. J., et al. Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature. 440, 637-643 (2006).
- Gavin, A. C., et al. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature. 415, 141-147 (2002).
- Tarassov, K., et al. An in vivo map of the yeast protein interactome. Science. 320, 1465-1470 (2008).
- Uetz, P., et al. A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature. 403, 623-627 (2000).
- Guruharsha, K. G., et al. A protein complex network of Drosophila melanogaster. Cell. 147, 690-703 (2011).
- Li, S., et al. A map of the interactome network of the metazoan C. elegans. Science. 303, 540-543 (2004).
- Rual, J. F., et al. Towards a proteome-scale map of the human protein-protein interaction network. Nature. 437, 1173-1178 (2005).
- Landry, C. R., Levy, E. D., Abd Rabbo, D., Tarassov, K., Michnick, S. W. Extracting insight from noisy cellular networks. Cell. 155, 983-989 (2013).
- Berggard, T., Linse, S., James, P. Methods for the detection and analysis of protein-protein interactions. Proteomics. 7, 2833-2842 (2007).
- Wodak, S. J., Vlasblom, J., Turinsky, A. L., Pu, S. Protein-protein interaction networks: the puzzling riches. Current opinion in structural biology. 23, 941-953 (2013).
- Fields, S., Song, O. A novel genetic system to detect protein-protein interactions. Nature. 340, 245-246 (1989).
- Dunham, W. H., Mullin, M., Gingras, A. C. Affinity-purification coupled to mass spectrometry: basic principles and strategies. Proteomics. 12, 1576-1590 (2012).
- Michnick, S. W., Ear, P. H., Landry, C., Malleshaiah, M. K., Messier, V. A toolkit of protein-fragment complementation assays for studying and dissecting large-scale and dynamic protein-protein interactions in living cells. Methods Enzymol. 470, 335-368 (2010).
- Rochette, S., Gagnon-Arsenault, I., Diss, G., Landry, C. R. Modulation of the yeast protein interactome in response to DNA damage. Journal of proteomics. 100, 25-36 (2014).
- Diss, G., Dube, A. K., Boutin, J., Gagnon-Arsenault, I., Landry, C. R. A systematic approach for the genetic dissection of protein complexes in living cells. Cell Rep. 3, 2155-2167 (2013).
- Gagnon-Arsenault, I., et al. Transcriptional divergence plays a role in the rewiring of protein interaction networks after gene duplication. Journal of proteomics. 81, 112-125 (2013).
- Schlecht, U., Miranda, M., Suresh, S., Davis, R. W., St Onge, R. P. Multiplex assay for condition-dependent changes in protein-protein interactions. Proceedings of the National Academy of Sciences of the United States of America. 109, 9213-9218 (2012).
- Lev, I., et al. Reverse PCA, a systematic approach for identifying genes important for the physical interaction between protein pairs. PLoS Genet. 9, e1003838 (2013).
- Freschi, L., Torres-Quiroz, F., Dube, A. K., Landry, C. R. qPCA: a scalable assay to measure the perturbation of protein-protein interactions in living cells. Mol Biosyst. 9, 36-43 (2013).
- Pelletier, J. N., Campbell-Valois, F. X., Michnick, S. W. Oligomerization domain-directed reassembly of active dihydrofolate reductase from rationally designed fragments. Proceedings of the National Academy of Sciences of the United States of America. 95, 12141-12146 (1998).
- Gietz, R. D., Woods, R. A. Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzymol. 350, 87-96 (2002).
- Schuldiner, M., Collins, S. R., Weissman, J. S., Krogan, N. J. Quantitative genetic analysis in Saccharomyces cerevisiae using epistatic miniarray profiles (E-MAPs) and its application to chromatin functions. Methods. 40, 344-352 (2006).
- Schneider, C. A., Rasband, W. S., Eliceiri, K. W. NIH Image to ImageJ: 25 years of image analysis. Nature methods. 9, 671-675 (2012).
- Wagih, O., Parts, L. gitter: A Robust and Accurate Method for Quantification of Colony Sizes From Plate Images. G3 (Bethesda). 4 (3), 547-552 (2014).
- Leducq, J. B., et al. Evidence for the robustness of protein complexes to inter-species hybridization. PLoS Genet. 8, e1003161 (2012).
- . . Development Core Team: A language and environment for statistical computing. , (2008).
- Stark, C., et al. BioGRID: a general repository for interaction datasets. Nucleic Acids Res. 34, D535-D539 (2006).
- Vinayagam, A., et al. Protein complex-based analysis framework for high-throughput data sets. Science signaling. 6, rs5 (2013).
- Jansen, R., Gerstein, M. Analyzing protein function on a genomic scale: the importance of gold-standard positives and negatives for network prediction. Current opinion in microbiology. 7, 535-545 (2004).
- Ito, T., et al. A comprehensive two-hybrid analysis to explore the yeast protein interactome. Proceedings of the National Academy of Sciences of the United States of America. 98, 4569-4574 (2001).
- Costanzo, M., et al. The genetic landscape of a cell. Science. 327, 425-431 (2010).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati