È necessario avere un abbonamento a JoVE per visualizzare questo. Accedi o inizia la tua prova gratuita.
Method Article
Isolamento e imaging time-lapse di cellule mesenchimiche palatali embrionali di topo primarie per analizzare gli attributi del movimento collettivo
* Questi autori hanno contribuito in egual misura
In questo articolo
Riepilogo
Presentiamo un protocollo per l'isolamento e la coltura di cellule mesenchimali palatali embrionali embrionali primarie di topo per l'imaging time-lapse di test di crescita bidimensionale (2D) e di riparazione delle ferite. Forniamo anche la metodologia per l'analisi dei dati di imaging time-lapse per determinare la formazione del flusso cellulare e la motilità direzionale.
Abstract
Lo sviluppo del palato è un processo dinamico, che comporta la crescita verticale di ripiani palatali bilaterali accanto alla lingua seguita da elevazione e fusione sopra la lingua. I difetti in questo processo portano alla palatoschisi, un difetto di nascita comune. Recenti studi hanno dimostrato che l'elevazione della mensola palatale comporta un processo di rimodellamento che trasforma l'orientamento della mensola da uno verticale a uno orizzontale. Il ruolo delle cellule mesenchimali della mensola palatale in questo rimodellamento dinamico è stato difficile da studiare. L'analisi quantitativa basata sull'imaging time-lapse è stata recentemente utilizzata per dimostrare che le cellule mesenchimali palatali embrionali embrionali primarie di topo (MEPM) possono auto-organizzarsi in un movimento collettivo. Le analisi quantitative potrebbero identificare differenze nelle cellule MEPM mutanti da un modello murino con difetti di elevazione del palato. Questo documento descrive i metodi per isolare e colturare cellule MEPM da embrioni E13.5, in particolare per l'imaging time-lapse, e per determinare vari attributi cellulari del movimento collettivo, comprese le misure per la formazione del flusso, l'allineamento della forma e la persistenza della direzione. Ipotizza che le cellule MEPM possano servire come modello proxy per studiare il ruolo del mesenchima della mensola palatale durante il processo dinamico di elevazione. Questi metodi quantitativi consentiranno ai ricercatori nel campo craniofacciale di valutare e confrontare gli attributi di movimento collettivo nelle cellule di controllo e mutanti, il che aumenterà la comprensione del rimodellamento mesenchimale durante l'elevazione della mensola palatale. Inoltre, le cellule MEPM forniscono un raro modello cellulare mesenchimale per lo studio del movimento cellulare collettivo in generale.
Introduzione
Lo sviluppo del palato è stato ampiamente studiato in quanto i difetti della palatogenesi portano alla palatoschisi, un difetto di nascita comune che si verifica in casi isolati o come parte di centinaia di sindromi1,2. Lo sviluppo del palato embrionale è un processo dinamico che coinvolge il movimento e la fusione del tessuto embrionale. Questo processo può essere suddiviso in quattro fasi principali: 1) induzione di ripiani palatali, 2) crescita verticale dei ripiani palatali accanto alla lingua, 3) elevazione dei ripiani palatali sopra la lingua e 4) fusione dei ripiani palatali alla linea mediana1,3,4. Negli ultimi decenni, sono stati identificati molti mutanti di topo che manifestano palatoschisi5,6,7,8. La caratterizzazione di questi modelli ha indicato difetti nelle fasi di induzione, proliferazione e fusione della mensola palatale; tuttavia, i difetti di elevazione della mensola palatale sono stati rari. Pertanto, comprendere le dinamiche dell'elevazione della mensola palatale è un'area di ricerca intrigante.
Un'attenta analisi di alcuni mutanti di topo con difetti di elevazione della mensola palatale ha portato al modello attuale che mostra che la regione molto anteriore della mensola palatale sembra capovolgersi, mentre un movimento da verticale a orizzontale o "rimodellamento" dei ripiani palatali si verifica nelle regioni medio-posteriori del palato1,3,4, 9,10,11. L'epitelio del bordo mediale della mensola palatale probabilmente avvia la segnalazione necessaria per questo rimodellamento, che viene quindi guidato dal mesenchima della mensola palatale. Recentemente, molti ricercatori hanno identificato il ritardo di elevazione della mensola palatale in modelli murini che hanno mostrato aderenze orali transitorie che coinvolgono scaffali palatali12,13. Il rimodellamento mesenchimale comporta la riorganizzazione delle cellule per creare un rigonfiamento in direzione orizzontale, mentre contemporaneamente ritrae la mensola palatale nella direzione verticale9,10,14. Tra i diversi meccanismi proposti per influenzare l'elevazione della mensola palatale e il rimodellamento mesenchimale sottostante ci sono la proliferazione cellulare15,16,17,i gradienti chemiotattici18e i componenti della matrice extracellulare19,20. È sorta una domanda importante: il ritardo di elevazione della mensola palatale osservato nei topi carenti di Specc1lè anche in parte dovuto a un difetto nel rimodellamento della mensola palatale, e questo difetto di rimodellamento potrebbe manifestarsi in un difetto intrinseco nel comportamento delle cellule MEPM primarie21?
Le cellule MEPM primarie sono state utilizzate in campo craniofacciale per molti studi che coinvolgono l'espressione genica22, 23,24,25, 26,27,28,29e alcuni che coinvolgono la proliferazione30,31 e la migrazione25,31,32 , ma nessuno per l'analisi collettiva del comportamento cellulare. L'imaging time-lapse delle cellule MEPM è stato eseguito in coltura 2D e saggi di riparazione delle ferite per dimostrare che le cellule MEPM mostravano movimento direzionale e formavano flussi cellulari dipendenti dalla densità-attributi del movimento collettivo21. Inoltre, le cellule mutanti Specc1l hanno formato flussi cellulari più stretti e hanno mostrato traiettorie di migrazione cellulare altamente variabili. Si ritiene che questa mancanza di motilità coordinata contribuisca al ritardo dell'elevazione del palato negli embrioni mutanti Specc1l 13,21. Pertanto, questi saggi relativamente semplici che utilizzano cellule MEPM primarie possono servire come proxy per studiare il rimodellamento mesenchimale durante l'elevazione della piattaforma palatale. Questo documento descrive l'isolamento e la coltura delle cellule MEPM primarie, nonché l'imaging e l'analisi time-lapse, per i test 2D e di riparazione delle ferite.
Protocollo
Tutti gli esperimenti che coinvolgono animali sono stati condotti con un protocollo approvato dal KUMC Institutional Animal Care and Use Committee, in conformità con le loro linee guida e regolamenti (numero di protocollo: 2018-2447).
1. Raccogli embrioni E13.5
- Eutanasia di topi femmina gravide utilizzando una camera di inalazione di CO2 o con una procedura approvata dal Comitato istituzionale per la cura e l'uso degli animali. Procedere immediatamente alla dissezione.
- Esporre la metà inferiore della cavità addominale rimuovendo la pelle e il peritoneo. Asporta entrambe le corna dell'utero, che contengono gli embrioni E13.5.
- Posizionare brevemente l'utero in soluzione salina sterile tamponata con fosfato (PBS) prebellica a 37 °C per risciacquare il sangue, i capelli o altri detriti in eccesso. Metti l'utero in un piatto sterile di 10 cm pieno di PBS sterile.
- Usando piccole forbici, taglia la parete uterina lungo la lunghezza dell'utero per esporre ogni embrione, ancora nel suo sacco vitellino. Rimuovere il sacco vitellino che circonda l'embrione, ma salvarlo per la genotipizzazione, se necessario. Quando gli embrioni vengono rimossi, posizionare ogni embrione nel proprio pozzo di una piastra a 12 pozzetti piena di PBS.
2. Dissezione di scaffali palatali da embrioni (Figura 1)
NOTA: Sterilizzare gli strumenti di dissezione in acciaio inossidabile (vedi tabella dei materiali)dopo aver elaborato ogni embrione posizionando gli strumenti prima in un becher di alcol etilico al 100% (EtOH), poi in uno sterilizzatore per strumenti a 350 °C per 10 s, e quindi raffreddandoli in un secondo becher di 100% EtOH.
- Utilizzando un cucchiaio perforato sterilizzato, posizionare l'embrione in un nuovo piatto di 10 cm riempito con terreno di coltura MEPM costituito dal mezzo minimo essenziale di Dulbecco (DMEM) contenente il 10% di siero bovino fetale (FBS), L-glutammina (4 mM L-Glu) e gli antibiotici-penicillina e streptomicina (50 unità / ml).
- Decapitare l'embrione proprio sotto la linea della mascella usando forbici sterili (Figura 1A, linea tratteggiata rossa). Rimuovere la mascella inferiore inserendo un punto della pinzed fine #5 sterilizzata in bocca, mantenendolo appena all'interno della guancia. Spingere la punta della pinzaccia inserita attraverso fino a quando non esce dalla parte posteriore del cranio.
- Orientare la pinzpa, lungo la linea gialla in Figura 1B, in modo che l'altro lato della pinca (che è ancora al di fuori dell'embrione) sia in bilico appena sopra il condotto uditivo, quindi pizzicare la pinzcola chiusa per tagliare il tessuto. Se necessario, eseguire un'altra pinca fine lungo la cucitura della pinca ora chiusa per tagliare qualsiasi tessuto che non è stato completamente reciso dal pizzico.
- Ripeti il passaggio precedente per l'altro lato della testa dell'embrione. Continuare la procedura di taglio a pizzico per rimuovere completamente la mascella inferiore, la lingua e la parte inferiore del cranio ed esporre i ripiani palatali.
- Rimuovere il cranio del cranio tagliando appena sopra gli occhi, come mostrato nella Figura 1C (linea verde). Fallo posizionando la testa sul lato sinistro o destro e posizionando i punti delle piccole forbici in acciaio inossidabile davanti e dietro il cranio appena sopra il livello degli occhi dell'embrione. Taglia la parte superiore del cranio con un rapido taglio delle forbici, creando una superficie piana che sarà importante per la stabilità nei passaggi successivi e che dovrebbe assomigliare alla Figura 1D se vista di lato.
- Posizionare la parte rimanente della testa a testa in giù, con l'aspetto superiore della testa (cranio rimosso) appoggiato sul fondo del piatto, che fornirà una superficie stabile per la rimozione della mensola palatale. Prenditi un momento per identificare i ripiani palatali, che ora sono esposti e rivolti verso l'alto e appariranno come due creste rialzate su entrambi i lati di una scanalatura centrale nella metà anteriore della testa (Figura 1E).
- Fissare la parte rimanente della testa al piatto per immobilizzarlo mentre i ripiani vengono rimossi. Fallo inserendo un punto di una pinna fine attraverso il tessuto vicino alla regione nasale della testa, anteriore ai ripiani palatali, e inserisci l'altro punto della pinna attraverso la base del cranio, posteriore ai ripiani palatali. Tenerli in posizione durante l'esecuzione dell'escissione degli scaffali palatali.
- Immobilizzando la testa con una mano, scegliere uno qualsiasi dei due ripiani da rimuovere per primi e inserire entrambi i punti di una seconda coppia di pinna fine nel tessuto alla base della superficie laterale del ripiano e pizzicare per tagliare il tessuto (Figura 1F). Ripeti questo lungo la base della superficie mediale del ripiano e poi alle estremità anteriore e posteriore del ripiano per staccarti dalla testa.
- Sollevare delicatamente lo scaffale, facendo pizzichi aggiuntivi, se necessario, per liberare completamente il ripiano dal tessuto circostante.
- Ripetere i due passaggi precedenti per rimuovere il secondo ripiano palatale.
- Con i ripiani palatali ora liberati dal tessuto circostante e collocati in PBS (Figura G), utilizzare una pipetta di trasferimento a bulbo di plastica sterile per disegnare i ripiani nella pipetta e trasferirli in un tubo microcentrifuga da 1,5 ml insieme a circa 500 μL di PBS. Tenere i tubi contenenti ripiani palatali sul ghiaccio mentre il resto della lettiera viene elaborato nello stesso modo.
NOTA: In alternativa, i ripiani possono essere collocati in un tubo microcentrifuga da 1,5 ml contenente tripsina preriscalata (0,25%) immediatamente dopo la dissezione (invece di metterli sul ghiaccio). I campioni saranno più freschi, ma bisogna fare attenzione a cronomestrare tutte le fasi del procedimento per ogni singolo campione invece di trattare i campioni collettivamente.
3. Coltura di cellule MEPM
NOTA: Nelle condizioni qui descritte, le cellule epiteliali del palato non sopravvivono al primo passaggio, risultando in una coltura cellulare mesenchimale del palato puro. Utilizzare la tecnica sterile per eseguire tutte le fasi in un cappuccio di coltura tissutale.
- Aspirare e scartare il PBS dal tubo da 1,5 ml, facendo attenzione a non scartare i ripiani nel processo. Aggiungere immediatamente 200 μL di tripsina preriscalata (37 °C) (0,25%) a ciascun tubo che contiene ripiani palatali. Pipettare brevemente i ripiani su e giù nella tripsina usando una punta della pipetta da 1000 μL per accelerare la tripsinizzazione.
- Incubare le provette per 5 minuti a 37 °C, quindi pipettare ogni campione su e giù di nuovo per aiutare a rompere il tessuto. Incubare i tubi per altri 5 minuti a 37 °C e pipettare su e giù ancora una volta per completare la dissociazione del tessuto.
NOTA: I ripiani devono essere completamente o quasi completamente dissociati e sospesi nella tripsina senza che rimangano pezzi visibili di tessuto. - Aggiungere 800 μL di terreno di coltura MEPM (fase 2.1) a ciascun tubo da 1,5 ml. Centrifugare il tubo da 1,5 ml a 200 × g per 5 minuti per pellettizzare le celle. Rimuovere il surnatante e risuspenare il pellet cellulare in 1 mL di terreno di coltura MEPM.
- Placcare le cellule MEPM in una piastra trattata con coltura tissutale a 6 pozzetti contenente terreno di coltura MEPM. Lasciare che le celle aderiscano alla superficie plastica per 12 ore a 37 °C in un incubatore con il 5% di CO2.
NOTA: Dopo l'incubazione notturna, la stragrande maggioranza (~ 90%) delle cellule si attaccherà. A questo punto, le cellule aderenti appariranno abbastanza omogenee, con una forma triangolare o leggermente allungata. - Cambiare il mezzo ogni giorno aspirando delicatamente il vecchio mezzo e sostituendolo immediatamente con 1 mL di PBS sterile caldo senza calcio o magnesio per ~ 1 min. Aspirare il PBS e sostituirlo con 3 ml di terreno di coltura MEPM prebellico.
- Passare le cellule una volta che diventano confluenti al 100%.
NOTA: le cellule MEPM dovrebbero proliferare raddoppiando il numero quasi ogni giorno.- Per passare le cellule, aspirare delicatamente il vecchio mezzo e sostituirlo immediatamente con PBS caldo senza calcio o magnesio per ~ 1 minuto. Aspirare il PBS e sostituirlo con 0,5 ml di tripsina allo 0,25% preriscalata.
- Incubare a 37 °C per ~ 5 minuti, o fino a quando le cellule si staccano dalla superficie del piatto quando delicatamente oscillate avanti e indietro a mano. Una volta che le cellule si sono staccate, aggiungere immediatamente 5 ml di terreno di coltura MEPM preriscaldato alle cellule tripsinizzate.
- Utilizzando un pipetto sierologico da 10 mL, raccogliere delicatamente le cellule in un tubo conico da 15 mL e centrifugare il tubo a 200 × g per 5 minuti per pellettare le celle. Aspirare la tripsina e il mezzo e risospezionare le cellule in 3 ml di terreno di coltura MEPM prebellico. Pipettare delicatamente 1 mL di cellule in un singolo pozzetto di un piatto a 6 pozzetto e aggiungere 2 mL di terreno di coltura MEPM per portare il volume totale a 3 mL.
NOTA: questo costituisce una divisione 1:3 delle celle. I MEPM possono essere sostituiti fino a tre volte. La densità di semina dei MEPM è alquanto flessibile e il numero di cellule presenti varia a seconda del recipiente di coltivazione. Tuttavia, i MEPM non proliferano correttamente quando sono divisi troppo scarsamente e dovrebbero essere confluenti almeno al 20-25% nel loro nuovo piatto una volta aderiti.
4. Crioconservazione delle cellule MEPM
- Una volta che le cellule MEPM tripsinizzate sono state pellettate, risuscievole le cellule nel terreno di coltura MEPM per ottenere una concentrazione di ~ 1 × 106 cellule / ml. Pipettare le cellule in crioviali e aggiungere una concentrazione finale del 5% di dimetilsolfossido nel ceppo cellulare. Chiudere il crioviale, mescolare brevemente invertendo e posizionare immediatamente i flaconcini in un contenitore di congelamento che si raffredda ad una velocità di 1 °C/min.
- Posizionare il refrigeratore in un congelatore a -80 °C durante la notte. Il giorno successivo, spostare i crioviali in un serbatoio di azoto liquido per la conservazione a lungo termine.
- Scongelamento delle cellule MEPM crioconservate
- Rimuovere i crioviali dal serbatoio dell'azoto liquido e scongelare a temperatura ambiente fino a quando il contenuto inizia a diventare liquido. Svuotare il contenuto in un tubo conico da 15 ml contenente 9 mL di terreno di coltura MEPM preriscaldato.
- Centrifugare il tubo a 200 × g per pellettare le celle. Risuspenare le cellule in 1 mL di terreno di coltura MEPM e convogliare le cellule in un singolo pozzetto di una piastra a 6 pozzetti. Aggiungere 2 mL di terreno di coltura MEPM caldo per portare il volume totale a 3 ml.
- Colturare le cellule a 37 °C in un incubatore con il 5% di CO2. Cambia il mezzo ogni giorno.
5. Live-imaging di cellule MEPM - Saggio di migrazione collettiva 2D (Figura 2)
- Preparare una piastra da utilizzare per l'imaging dal vivo.
- Utilizzare piccole forbici chirurgiche o un bisturi affilato per accorciare un inserto sterile in silicone a 2 pozzetti ad un'altezza di ~ 1 mm. Preparare un inserto per ogni campione utilizzato.
- Usando la pinica, posizionare l'inserto in silicone a 2 pozzetti accorciato al centro di un pozzetto di una piastra a 6 pozzetti. Premere verso il basso lungo tutti i bordi per assicurarsi che sia completamente aderente.
- Scongelare le cellule crioconservate seguendo i passaggi descritti al paragrafo 4.3 di questo protocollo. Contare le cellule MEPM e seminare 300 celle/mm2 degli inserti in silicone accorciati in un volume totale di 40-50 μL di terreno di coltura MEPM per pozzetto. Coltura le cellule durante la notte a 37 °C in un incubatore con il 5% di CO2.
- Il giorno successivo, preparati per l'imaging time-lapse dal vivo.
- Utilizzare un microscopio a contrasto di fase con un incubatore sul palco e capacità di imaging automatico. Aggiungere acqua al serbatoio dell'incubatore sul palco per ridurre l'evaporazione del terreno di coltura; impostare la temperatura a 37 °C e CO2 al 5%. Lasciare ~ 30 minuti affinché l'umidità si accumuli prima di posizionare il piatto a 6 pozzedi nell'incubatrice sul palco.
- Utilizzare impostazioni equivalenti alle seguenti per l'imaging time-lapse.
- Selezionare l'obiettivo 4x per avere ampi campi di visualizzazione e filtro di contrasto di fase.
NOTA: la messa a fuoco automatica, il campione di ricerca automatica, lo z-stack e l'illuminazione automatica di solito non sono necessari. - Selezionare due campi microscopici per pozzo per catturare il lume degli inserti in silicone accorciati. Assicurarsi che tutte le posizioni di imaging abbiano la messa a fuoco corretta.
NOTA: i piani immagine possono essere regolati durante l'imaging, ma tale regolazione di solito non è necessaria. - Selezionare il tipo di file di output dell'immagine desiderato, quindi selezionare o deselezionare le opzioni di post-imaging, come la creazione automatica di video e filigrane, come desiderato. Se applicabile, selezionare il contrasto di fase come modalità di imaging.
NOTA: l'utilizzo di filigrane può impedire le successive fasi di elaborazione delle immagini. - Impostare la durata della registrazione su 72 h. Imposta il programma per catturare immagini ogni 10 minuti.
NOTA: di solito sono necessarie solo 48 ore di imaging, ma l'imaging può essere interrotto in qualsiasi momento prima del segno di 72 h senza perdere le immagini che sono già state scattate. - Assicurarsi che la camera ambientale sia operativa come richiesto al 5.3.1. Salvare queste impostazioni(la routine)e iniziare l'imaging.
- Selezionare l'obiettivo 4x per avere ampi campi di visualizzazione e filtro di contrasto di fase.
- Continuare l'imaging fino a 72 ore (o il tempo specificato).
6. Live-imaging di cellule MEPM in un test di riparazione della ferita (Figura 3)
- Preparare una piastra da utilizzare per l'imaging dal vivo. Usando una pinica, posizionare un inserto sterile in silicone a 2 pozzetti al centro di un pozzetto di una piastra a 6 pozzetti e premere verso il basso lungo tutti i bordi per assicurarsi che sia completamente aderente. Preparare un inserto a 2 fori per ogni campione utilizzato.
- Scongelare le cellule crioconservate seguendo i passaggi descritti al paragrafo 4.3 di questo protocollo. Contare le cellule MEPM e, se necessario, concentrare le cellule ad almeno 350 cellule/μL.
- Semina 1400 cellule/mm2 negli inserti in silicone in un volume di 100 μL di terreno di coltura MEPM per pozzetto. Colturare le cellule per 48 ore a 37 °C in un incubatore con il 5% di CO2e cambiare il mezzo ogni giorno.
- Dopo 48 ore preparare un microscopio per l'imaging time-lapse dal vivo come descritto nei sezioni 5.3 e 5.4. Immediatamente prima di posizionare le cellule nell'incubatore sul palco, aggiungere 3 ml di terreno di coltura MEPM preriscaldato al pozzo (ma all'esterno degli inserti), quindi rimuovere con cura gli inserti in silicone.
NOTA: La parete che separa le 2 camere lascia uno spazio che è la "ferita". - Avviare l'imaging time-lapse come descritto al punto 5.4, con le seguenti differenze:
- Utilizzare un obiettivo di ingrandimento più elevato (ad esempio, 10x). Per acquisire la chiusura della ferita, selezionare 5 campi di vista lungo ogni ferita, in modo che la ferita sia parallela all'asse verticale dell'immagine.
- Interrompere l'imaging dopo 72 ore o quando le ferite sono completamente chiuse.
7. Analisi computazionale di sequenze di immagini time-lapse
NOTA: eseguire le procedure seguenti su un computer dotato di strumenti di calcolo standard, ad esempio l'interprete python, il compilatore C e una shell (vedere la Tabella dei materiali).
- Analisi della confluenza
NOTA: Questa procedura può essere utilizzata per stimare la proliferazione cellulare all'interno di una coltura sparsa o per quantificare esperimenti di chiusura della ferita. Per rilevare le aree occupate dalle celle, viene applicata una soglia di segmentazione alla deviazione standard locale della luminosità dell'immagine. Il codice è stato descritto in precedenza da Wu et al.33 e Neufeld et al.34 ed è disponibile presso http://github.com/aczirok/cellconfluency.- Determinare la soglia di segmentazione per le immagini. Ad esempio, per visualizzare la segmentazione con una soglia 4, eseguire il comando
segment.py -i inout-image.jpg -S 4 -test output.jpg
e quindi controllare l'output (output.jpg).
NOTA: se la soglia è troppo bassa, le aree di sfondo nella micrografia vengono classificate come coperte da celle. Al contrario, se la soglia è troppo alta, le aree coperte da celle non sono classificate come tali. Il valore di soglia ottimale mantiene entrambi gli errori al minimo. - Utilizzare lo script di area.sh fornito per calcolare i valori di confluenza per una sequenza di immagini come
area.sh -S 4 img_001.jpg img_002.jpg .... > confluenza.dat
Nota : il valore della soglia di discriminazione 4 viene verificato nel passaggio 1 e i risultati vengono memorizzati nella confluenzadel file.dat . Ordinando le immagini in cartelle appropriate, l'elenco dei nomi dei file di immagine può essere sostituito dalla notazione con caratteri jolly:
area.sh -S4 *.jpg > confluenza.dat - Per gli esperimenti di chiusura della ferita, trasformare i dati di confluenza A(t)-la dimensione dell'area coperta da cellule espressa in percentuale in funzione del tempo-in velocità di propagazione del bordo della ferita V come

dove w denota la larghezza del campo e dA/dt è la derivata temporale di A(t), cioè il tasso di espansione dell'area coperta dalla cella.
- Determinare la soglia di segmentazione per le immagini. Ad esempio, per visualizzare la segmentazione con una soglia 4, eseguire il comando
- Mappa della motilità cellulare
- Eseguire un algoritmo di velocimetria dell'immagine delle particelle (PIV) per caratterizzare la motilità cellulare ed estrarre l'estensione del "flusso ottico" di movimento locale tra le coppie di immagini, ma non per identificare le singole cellule.
NOTA: Qui, viene utilizzata una dimensione iniziale della finestra di 50 μm, come descritto in dettaglio da Zamir et al.35 e Czirok et al.36, con una dimensione iniziale della finestra di 50 μm. L'analisi PIV produce un campo di velocità v(x,t) per ogni fotogramma dell'immagine t e posizione (all'interno dell'immagine) x. - Estrarre la velocità media della motilità cellulare da v(x,t) come media spaziale calcolata sull'area occupata dalla cellula, come determinato nel paragrafo 7.1.
- Eseguire un algoritmo di velocimetria dell'immagine delle particelle (PIV) per caratterizzare la motilità cellulare ed estrarre l'estensione del "flusso ottico" di movimento locale tra le coppie di immagini, ma non per identificare le singole cellule.
- Tracciamento manuale delle cellule
NOTA: mentre l'analisi PIV fornisce una valutazione automatica della motilità cellulare, concentrarsi sul comportamento delle singole cellule richiede spesso il tracciamento manuale. Sebbene diversi strumenti forniscano questa funzionalità, è molto utile se i marcatori posizionati manualmente possono essere modificati dopo il loro posizionamento iniziale e se il tracciamento può essere eseguito sia in avanti che indietro nel tempo.- Esegui il tracciamento delle celle con uno strumento python sviluppato su misura (http://github.com/donnagreta/cm_track), che fornisce anche funzioni di base dell'editor come l'eliminazione dei segmenti di traiettoria.
NOTA: questo strumento di rilevamento manuale produce le posizioni P(i,t) della cella i al tempo t in un file di testo e viene richiamato come
cm_track.py -i images/ -o track.dat
dove le immagini time-lapse si trovano nella cartella images/ e i dati di posizione vengono raccolti nella traccia del file.dat (Figura 4).
- Esegui il tracciamento delle celle con uno strumento python sviluppato su misura (http://github.com/donnagreta/cm_track), che fornisce anche funzioni di base dell'editor come l'eliminazione dei segmenti di traiettoria.

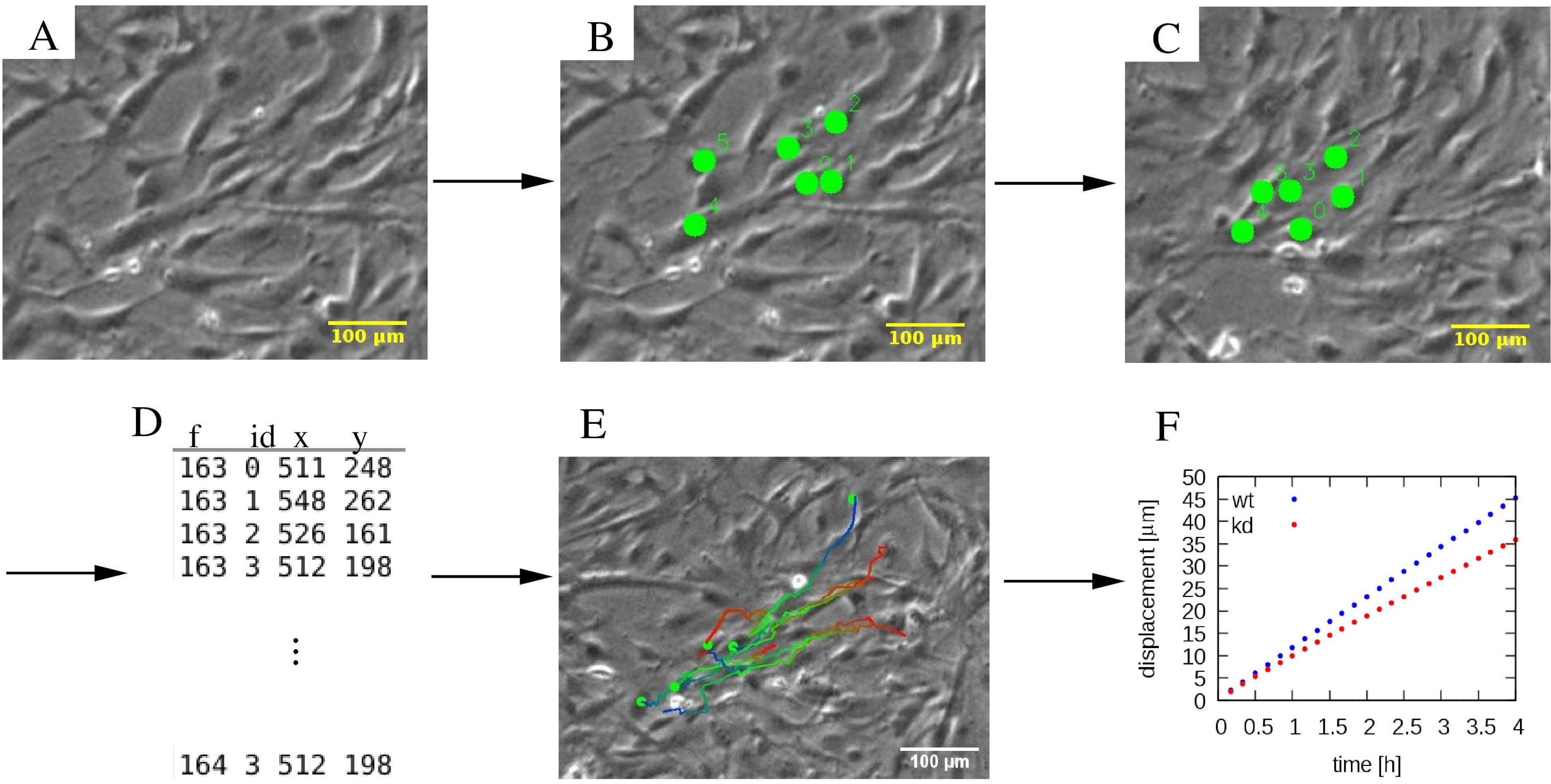
Figura 4: Analisi delle traiettorie delle singole cellule. (A) Le micrografie time-lapse a contrasto di fase sono sottoposte a (B, C) una procedura di tracciamento manuale, che contrassegna le celle (punti verdi). (D) Le posizioni delle celle (x,y) sono memorizzate per ogni cella distinta dal suo ID e per ogni fotogramma f. (E) Le traiettorie possono essere sovrapposte alle micrografie e codificate a colori per indicare informazioni temporali. Ad esempio, in ogni traiettoria, una tavolozza di colori da blu a rosso indica progressivamente i segmenti di traiettoria successivi, con il rosso e il blu che contrassegnano rispettivamente la posizione iniziale e quella finale della cella. (F) Varie proprietà statistiche delle traiettorie, come lo spostamento quadrato medio, possono essere estratte e utilizzate per caratterizzare la motilità di varie popolazioni cellulari, che in questo esempio includono cellule MEPM wildtype (wt, blu) e knockdown (kd, rosso). Le barre di scala rappresentano 100 μm. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
- Sovrapporre traiettorie sulle immagini utilizzando un secondo strumento, richiamato come
visdat.py traccia -d.dat -i immagini/ -o sovrapposizione/ -l999 -r3 -C2 - Raccogliete le immagini con le traiettorie sovrapposte nella sovrapposizione della cartella.
NOTA: in questo esempio, i dati sulla posizione della cella vengono memorizzati nella traccia del file.dat, mentre la sequenza di immagini time-lapse si trova all'interno delle immagini della cartella. Il resto dei parametri controlla la lunghezza massima delle traiettorie disegnate (-l), la dimensione dei simboli (-r) e la combinazione di colori (-C) utilizzata.
- Analisi delle traiettorie delle singole cellule
- Caratterizzare le traiettorie in modo dalla lunghezza totale del tracciato
 ,
,
e spostamento netto verso la ferita ,
,
dove X denota la proiezione di P nella direzione perpendicolare alla ferita: la coordinata x delle posizioni quando la ferita è parallela all'asse y. - Calcolare l'efficienza di guida
 come per ogni cella i e punto temporale t37. Caratterizzare le colture in base alla media di popolazione di queste misure a singola cellula, valutate in un punto temporale adatto t.
come per ogni cella i e punto temporale t37. Caratterizzare le colture in base alla media di popolazione di queste misure a singola cellula, valutate in un punto temporale adatto t.
- Caratterizzare le traiettorie in modo dalla lunghezza totale del tracciato
- Movimento di flusso collettivo delle cellule
- Caratterizzare le correlazioni spaziali locali dei movimenti cellulari dalla velocità media di altre cellule che si trovano nelle vicinanze di una cellula in movimento, come descritto in precedenza38,39.
NOTA: il codice computazionale è disponibile all'https://github.com/aczirok/flowfield.- Allineare un sistema di riferimento con le direzioni anteriore, posteriore, sinistra e destra a ciascun vettore v(x,t) (Figura 5A). Assegnare ciascuna delle celle circostanti o del vettore di velocità PIV alla cella spaziale appropriata del sistema di riferimento (Figura 5B). Ripetere la procedura in quanto ogni vettore funge da origine dei sistemi di riferimento (Figura 5C,D) in modo che un dato vettore velocità sia assegnato a più bin.
NOTA: un punto dati potrebbe trovarsi davanti a un vettore e a sinistra di un altro. - Ruotare i sistemi di riferimento in un orientamento comune (Figura 5C, F) e metterli in comune ( Figura5G).
NOTA: La media di ogni bin dei dati di velocità raggruppati è un vettore di velocità (U) che è indicativo di correlazione spaziale: la media è una misura di una componente di velocità condivisa (Figura 5H). - Adatta i campi di flusso U(x) con una funzione esponenziale
 , dove a, x0 e U0 sono parametri di adattamento. Dei tre parametri di adattamento, concentrati su x0, la lunghezza di correlazione (Figura 5I, J), che è la distanza caratteristica in cui scompaiono le correlazioni velocità-velocità locale.
, dove a, x0 e U0 sono parametri di adattamento. Dei tre parametri di adattamento, concentrati su x0, la lunghezza di correlazione (Figura 5I, J), che è la distanza caratteristica in cui scompaiono le correlazioni velocità-velocità locale.
- Allineare un sistema di riferimento con le direzioni anteriore, posteriore, sinistra e destra a ciascun vettore v(x,t) (Figura 5A). Assegnare ciascuna delle celle circostanti o del vettore di velocità PIV alla cella spaziale appropriata del sistema di riferimento (Figura 5B). Ripetere la procedura in quanto ogni vettore funge da origine dei sistemi di riferimento (Figura 5C,D) in modo che un dato vettore velocità sia assegnato a più bin.
- Caratterizzare le correlazioni spaziali locali dei movimenti cellulari dalla velocità media di altre cellule che si trovano nelle vicinanze di una cellula in movimento, come descritto in precedenza38,39.

Figura 5: Caratterizzazione della formazione di flussi di cellule in coltura. (A,D) Le immagini time-lapse a contrasto di fase della Figura 4A sono utilizzate per identificare i movimenti cellulari. Per ogni cella in movimento, un sistema di riferimento (blu) e contenitori spaziali (bianco) sono stati co-allineati per classificare le celle adiacenti come se fossero nella parte anteriore, posteriore, sinistra o destra. (B,E) La velocità delle celle adiacenti (vettori neri) era correlata allo stesso sistema di riferimento (C,F). Questa procedura è stata ripetuta per ogni cella e punto temporale. (G) Dopo aver raggruppato queste informazioni locali, ogni bin conterrà più vettori di velocità (grigio), che possono essere mediati per determinare la velocità media co-mobile (frecce magenta) in varie posizioni rispetto a una cellula mobile media. (H) La mappa della velocità media caratterizza quindi le velocità tipiche delle cellule in varie posizioni rispetto a una cella in movimento. (I,J) Infine, questo campo è stato campionato lungo l'asse anteriore-posteriore (parallelo) e anche lungo l'asse sinistra-destra (perpendicolare). Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
Risultati
La dissezione degli scaffali palatali è illustrata nella Figura 1. La sequenza di incisioni è progettata per ridurre al minimo lo slittamento del tessuto. Dopo la rimozione della testa (Figura 1A,B), la mascella inferiore viene rimossa (Figura 1B,C). L'incisione della parte superiore della testa (Figura 1C, D) viene eseguita per stabilizzare il tessut...
Discussione
L'elevazione della mensola palatale costituisce un evento di rimodellamento da verticale a orizzontale1,3,4,9,11. Si ipotizza che questo processo di rimodellamento richieda che le cellule mesenchimali della mensola palatale si comportino in modo coordinato. Le analisi con cellule MEPM wildtype mostrano che questo comportamento cellulare è intrinseco e può es...
Divulgazioni
Gli autori non hanno nulla da rivelare.
Riconoscimenti
Questo progetto è stato sostenuto in parte dalle sovvenzioni del National Institutes of Health DE026172 (I.S.) e GM102801 (A.C.). I.S. è stato anche sostenuto in parte dalla sovvenzione del Center of Biomedical Research Excellence (COBRE) (National Institute of General Medical Sciences P20 GM104936), dalla sovvenzione Kansas IDeA Network for Biomedical Research Excellence (National Institute of General Medical Sciences P20 GM103418) e dalla sovvenzione del Kansas Intellectual and Developmental Disabilities Research Center (KIDDRC) (U54 Eunice Kennedy Shriver National Institute of Child Health and Human Development, HD090216).
Materiali
| Name | Company | Catalog Number | Comments |
| Beaker, 250 mL (x2) | Fisher Scientific | FB-100-250 | |
| CO2 | Matheson Gas | UN1013 | |
| Conical tubes, 15 mL (x1) | Midwest Scientific | C15B | |
| Debian operating system | computational analysis of time-lapse images | ||
| Dulbecco's Modified Eagles Medium/High Glucose with 4 mM L-Glutamine and Sodium Pyruvate | Cytiva Life Sciences | SH30243.01 | |
| EtOH, 100% | Decon Laboratories | 2701 | |
| EVOS FL Auto | ThermoFisher Scientific | AMAFD1000 | |
| EVOS Onstage Incubator | ThermoFisher Scientific | AMC1000 | |
| EVOS Onstage Vessel Holder, Multi-Well Plates | ThermoFisher Scientific | AMEPVH028 | |
| Fetal Bovine Serum | Corning | 35-010-CV | |
| Fine point #5 Stainless Steel Forceps (x2) | Fine Science Tools | 11295-10 | Dissection |
| Instrument sterilizer bead bath | Fine Science Tools | 18000-45 | |
| Microcetrifuge tubes, 1.5mL | Avant | 2925 | |
| Micro-Dissecting Stainless Steel Scissors, Straight | Roboz | RS-5910 | Dissection |
| NucBlue (Hoechst) Live Ready Probes | ThermoFisher Scientific | R37605 | |
| Penicillin Streptomycin Solution, 100x | Corning | 30-002-CI | |
| Silicone Insert, 2-well | Ibidi | 80209 | |
| Small Perforated Stainless Steel Spoon | Fine Science Tools | MC17C | Dissection |
| Spring Scissors, 4 mm | Fine Science Tools | 15018-10 | |
| Sterile 10 cm dishe(s) | Corning | 430293 | |
| Sterile 12-well plate(s) | PR1MA | 667512 | |
| Sterile 6-well plate(s) | Thermo Fisher Scientific | 140675 | |
| Sterile PBS | Corning | 21-031-CV | |
| Sterile plastic bulb transfer pipette | ThermoFisher Scientific | 202-1S | |
| Trypsin, 0.25% | ThermoFisher Scientific | 25200056 |
Riferimenti
- Bush, J. O., Jiang, R. Palatogenesis: morphogenetic and molecular mechanisms of secondary palate development. Development. 139 (2), 231-243 (2012).
- Mossey, P. A., Little, J., Munger, R. G., Dixon, M. J., Shaw, W. C. Cleft lip and palate. Lancet. 374 (9703), 1773-1785 (2009).
- Lan, Y., Xu, J., Jiang, R. Cellular and molecular mechanisms of palatogenesis. Current Topics in Developmental Biology. 115, 59-84 (2015).
- Li, C., Lan, Y., Jiang, R. Molecular and cellular mechanisms of palate development. Journal of Dental Research. 96 (11), 1184-1191 (2017).
- Gritli-Linde, A. The etiopathogenesis of cleft lip and cleft palate: usefulness and caveats of mouse models. Current Topics in Developmental Biology. 84, 37 (2008).
- Meng, L., Bian, Z., Torensma, R., Vonden Hoff, J. W. Biological mechanisms in palatogenesis and cleft palate. Journal of Dental Research. 88 (1), 22-33 (2009).
- Dixon, M. J., Marazita, M. L., Beaty, T. H., Murray, J. C. Cleft lip and palate: understanding genetic and environmental influences. Nature Reviews Genetics. 12 (3), 167-178 (2011).
- Kousa, Y. A., Schutte, B. C. Toward an orofacial gene regulatory network. Developmental Dynamics. 245 (3), 220-232 (2016).
- Jin, J. Z., et al. Mesenchymal cell remodeling during mouse secondary palate reorientation. Developmental Dynamics. 239 (7), 2110-2117 (2010).
- Yu, K., Ornitz, D. M. Histomorphological study of palatal shelf elevation during murine secondary palate formation. Developmental Dynamics. 240 (7), 1737-1744 (2011).
- Chiquet, M., Blumer, S., Angelini, M., Mitsiadis, T. A., Katsaros, C. Mesenchymal remodeling during palatal shelf elevation revealed by extracellular matrix and F-actin expression patterns. Frontiers in Physiology. 7, 392 (2016).
- Paul, B. J., et al. ARHGAP29 mutation is associated with abnormal oral epithelial adhesions. Journal of Dental Research. 96 (11), 1298-1305 (2017).
- Hall, E. G., et al. SPECC1L regulates palate development downstream of IRF6. Human Molecular Genetics. 29 (5), 845-858 (2020).
- Walker, B. E., Fraser, F. C. Closure of the secondary palate in three strains of mice. Journal of Embryology and Experimental Morphology. 4 (2), 176-189 (1956).
- Jin, J. Z., Li, Q., Higashi, Y., Darling, D. S., Ding, J. Analysis of Zfhx1a mutant mice reveals palatal shelf contact-independent medial edge epithelial differentiation during palate fusion. Cell Tissue Research. 333 (1), 29-38 (2008).
- Kouskoura, T., et al. The etiology of cleft palate formation in BMP7-deficient mice. PLoS One. 8 (3), 59463 (2013).
- Lan, Y., Zhang, N., Liu, H., Xu, J., Jiang, R. Golgb1 regulates protein glycosylation and is crucial for mammalian palate development. Development. 143 (13), 2344-2355 (2016).
- He, F., et al. Wnt5a regulates directional cell migration and cell proliferation via Ror2-mediated noncanonical pathway in mammalian palate development. Development. 135 (23), 3871-3879 (2008).
- Lan, Y., Qin, C., Jiang, R. Requirement of hyaluronan synthase-2 in craniofacial and palate development. Journal of Dental Research. 98 (12), 1367-1375 (2019).
- Yonemitsu, M. A., Lin, T. Y., Yu, K. Hyaluronic acid is required for palatal shelf movement and its interaction with the tongue during palatal shelf elevation. Developmental Biology. 457 (1), 57-68 (2020).
- Goering, J. P., et al. SPECC1L-deficient palate mesenchyme cells show speed and directionality defect. Scientific Reports. 11 (1), 1452 (2021).
- Fantauzzo, K. A., Soriano, P. PI3K-mediated PDGFRalpha signaling regulates survival and proliferation in skeletal development through p53-dependent intracellular pathways. Genes and Development. 28 (9), 1005-1017 (2014).
- Vasudevan, H. N., Soriano, P. SRF regulates craniofacial development through selective recruitment of MRTF cofactors by PDGF signaling. Developmental Cell. 31 (3), 332-344 (2014).
- Vasudevan, H. N., Mazot, P., He, F., Soriano, P. Receptor tyrosine kinases modulate distinct transcriptional programs by differential usage of intracellular pathways. Elife. 4, 07186 (2015).
- Gao, L., et al. 2,3,7,8-Tetrachlorodibenzo-p-dioxin and TGFbeta3-mediated mouse embryonic palatal mesenchymal cells. Dose Response. 17 (1), 1559325818786822 (2019).
- Iyyanar, P. P. R., Nazarali, A. J. Hoxa2 inhibits bone morphogenetic protein signaling during osteogenic differentiation of the palatal mesenchyme. Frontiers in Physiology. 8, 929 (2017).
- Jiang, Z., Pan, L., Chen, X., Chen, Z., Xu, D. Wnt6 influences the viability of mouse embryonic palatal mesenchymal cells via the beta-catenin pathway. Experimental and Therapeutic Medicine. 14 (6), 5339-5344 (2017).
- Liu, X., et al. Negative interplay of retinoic acid and TGF-beta signaling mediated by TG-interacting factor to modulate mouse embryonic palate mesenchymal-cell proliferation. Birth Defects Research Part B: Developmental and Reproductive Toxicology. 101 (6), 403-409 (2014).
- Bush, J. O., Soriano, P. Ephrin-B1 forward signaling regulates craniofacial morphogenesis by controlling cell proliferation across Eph-ephrin boundaries. Genes & Development. 24 (18), 2068-2080 (2010).
- Mo, J., Long, R., Fantauzzo, K. A. Pdgfra and Pdgfrb genetically interact in the murine neural crest cell lineage to regulate migration and proliferation. Frontiers in Physiology. 11, 588901 (2020).
- He, F., Soriano, P. A critical role for PDGFRalpha signaling in medial nasal process development. PLoS Genetics. 9 (9), 1003851 (2013).
- Fantauzzo, K. A., Soriano, P. Generation of an immortalized mouse embryonic palatal mesenchyme cell line. PLoS One. 12 (6), 0179078 (2017).
- Wu, K., Gauthier, D., Levine, M. D. Live cell image segmentation. IEEE Transactions on Biomedical Engineering. 42 (1), 1-12 (1995).
- Neufeld, Z., et al. The role of Allee effect in modelling post resection recurrence of glioblastoma. PLoS Computational Biology. 13 (11), 1005818 (2017).
- Zamir, E. A., Czirok, A., Rongish, B. J., Little, C. D. A digital image-based method for computational tissue fate mapping during early avian morphogenesis. Annals of Biomedical Engineering. 33 (6), 854-865 (2005).
- Czirok, A., et al. Optical-flow based non-invasive analysis of cardiomyocyte contractility. Scientific Reports. 7 (1), 10404 (2017).
- Biggs, L. C., et al. Interferon regulatory factor 6 regulates keratinocyte migration. Journal of Cell Science. 127, 2840-2848 (2014).
- Czirok, A., Varga, K., Mehes, E., Szabo, A. Collective cell streams in epithelial monolayers depend on cell adhesion. New Journal of Physics. 15, 75006 (2013).
- Szabo, A., et al. Collective cell motion in endothelial monolayers. Physical Biology. 7 (4), 046007 (2010).
- Gulyas, M., Csiszer, M., Mehes, E., Czirok, A. Software tools for cell culture-related 3D printed structures. PLoS One. 13 (9), 0203203 (2018).
- Soderholm, J., Heald, R. Scratch n' screen for inhibitors of cell migration. Chemistry & Biology. 12 (3), 263-265 (2005).
- Riahi, R., Yang, Y., Zhang, D. D., Wong, P. K. Advances in wound-healing assays for probing collective cell migration. Journal of Laboratory Automation. 17 (1), 59-65 (2012).
- Svensson, C. M., Medyukhina, A., Belyaev, I., Al-Zaben, N., Figge, M. T. Untangling cell tracks: Quantifying cell migration by time lapse image data analysis. Cytometry Part A. 93 (3), 357-370 (2018).
- Fantauzzo, K. A., Soriano, P. PDGFRbeta regulates craniofacial development through homodimers and functional heterodimers with PDGFRalpha. Genes & Development. 30 (21), 2443-2458 (2016).
- Rafi, S. K., et al. Anti-epileptic drug topiramate upregulates TGFβ1 and SOX9 expression in primary embryonic palatal mesenchyme cells: Implications for teratogenicity. PLoS ONE. , (2021).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati