É necessária uma assinatura da JoVE para visualizar este conteúdo. Faça login ou comece sua avaliação gratuita.
Method Article
Isolamento e imagem de lapso de tempo das células mesenquima palatinas embrionárias do camundongo primário para analisar atributos de movimento coletivo
Neste Artigo
Resumo
Apresentamos um protocolo de isolamento e cultura de células mesenquimais palatinas embrionárias primárias para imagens de lapso de tempo de crescimento bidimensional (2D) e ensaios de reparação de feridas. Também fornecemos a metodologia para análise dos dados de imagem de lapso de tempo para determinar a formação de fluxo celular e a motilidade direcional.
Resumo
O desenvolvimento do paladar é um processo dinâmico, que envolve o crescimento vertical das prateleiras palatais bilaterais ao lado da língua seguidas de elevação e fusão acima da língua. Defeitos nesse processo levam à fissura palatina, um defeito comum de nascimento. Estudos recentes têm demonstrado que a elevação da prateleira palatal envolve um processo de remodelação que transforma a orientação da prateleira de uma vertical para uma horizontal. O papel das células mesenquimais da prateleira palatal nesta remodelagem dinâmica tem sido difícil de estudar. A análise quantitativa baseada em imagens de lapso de tempo foi recentemente usada para mostrar que as células mesenquimais palatinas embrionárias primárias do camundongo (MEPM) podem se auto-organizar em um movimento coletivo. Análises quantitativas poderiam identificar diferenças nas células mutantes mepm de um modelo de rato com defeitos de elevação do paladar. Este artigo descreve métodos para isolar e cultivar células MEPM de embriões E13.5 - especificamente para imagens de lapso de tempo - e determinar vários atributos celulares do movimento coletivo, incluindo medidas para formação de fluxo, alinhamento de forma e persistência de direção. Ele afirma que as células MEPM podem servir como um modelo proxy para estudar o papel do mesenquime de prateleira palatal durante o processo dinâmico de elevação. Esses métodos quantitativos permitirão aos pesquisadores do campo craniofacial avaliar e comparar atributos de movimento coletivo no controle e células mutantes, o que aumentará a compreensão da remodelagem mesenquimal durante a elevação da prateleira palatal. Além disso, as células MEPM fornecem um raro modelo de célula mesenquimal para investigação do movimento celular coletivo em geral.
Introdução
O desenvolvimento palaciano tem sido estudado extensivamente como defeitos na palatogênese levam à fissura palatina - um defeito de nascimento comum que ocorre em casos isolados ou como parte de centenas de síndromes1,2. O desenvolvimento do paladar embrionário é um processo dinâmico que envolve movimento e fusão de tecido embrionário. Este processo pode ser dividido em quatro etapas principais: 1) indução de prateleiras palatais, 2) crescimento vertical das prateleiras palatais ao lado da língua, 3) elevação das prateleiras palatais acima da língua, e 4) fusão das prateleiras palatais na linha média1,3,4. Ao longo das últimas décadas, muitos mutantes de camundongos foram identificados que manifestam fissura palatina5,6,7,8. A caracterização desses modelos indicou defeitos na indução da prateleira palatal, na proliferação e nas etapas de fusão; no entanto, defeitos de elevação da prateleira palatal têm sido raros. Assim, compreender a dinâmica da elevação da prateleira palatal é uma área intrigante de pesquisa.
A análise cuidadosa de alguns mutantes de camundongos com defeitos de elevação da prateleira palatal levou ao modelo atual mostrando que a região anterior da prateleira palatal parece virar para cima, enquanto um movimento vertical para horizontal ou "remodelagem" das prateleiras palatais ocorre nas regiões médias e posteriores do paladar1,3,4, 9,10,11. O epitélio de borda medial da prateleira palatal provavelmente inicia a sinalização necessária para esta remodelagem, que é então impulsionada pela mesenquime da prateleira palatal. Recentemente, muitos pesquisadores identificaram atraso na elevação da prateleira palatal em modelos de camundongos que mostraram aderências orais transitórias envolvendo prateleiras palatais12,13. A remodelagem mesenquimal envolve a reorganização das células para criar uma protuberância na direção horizontal, ao mesmo tempo em que retrai simultaneamente a prateleira palatal na direção vertical9,10,14. Entre os vários mecanismos propostos para afetar a elevação da prateleira palatal e a remodelagem mesenquimal subjacente estão a proliferação celular15,16,17, gradientes quimotacticos18e componentes da matriz extracelular19,20. Uma pergunta importante surgiu: o atraso de elevação da prateleira palatal é observado em camundongos deficientes specc1ltambém em parte devido a um defeito na remodelação da prateleira palatal, e esse defeito de remodelação poderia se manifestar em um defeito intrínseco no comportamento das células primárias do MEPM21?
Células mepm primárias têm sido utilizadas no campo craniofacial para muitos estudos envolvendo expressão genética22,23,24,25,26,27,28,29, e alguns envolvendo proliferação30,31 e migração25,31,32 , mas nenhum para análise de comportamento celular coletivo. A imagem de lapso de tempo das células MEPM foi realizada em estudos de cultura 2D e reparação de feridas para mostrar que as células MEPM apresentavam movimento direcional e formavam fluxos celulares dependentes da densidade do movimento coletivo21. Além disso, as células mutantes Specc1l formaram fluxos celulares mais estreitos e apresentaram trajetórias de migração celular altamente variáveis. Essa falta de motilidade coordenada é considerada para contribuir para o atraso de elevação do paladar nos embriões mutantes Specc1l 13,21. Assim, esses ensaios relativamente simples usando células MEPM primárias podem servir como um proxy para estudar a remodelagem mesenquimal durante a elevação da prateleira palatal. Este artigo descreve o isolamento e a cultura das células mepm primárias, bem como a imagem e análise de lapso de tempo, para os ensaios 2D e reparação de feridas.
Protocolo
Todos os experimentos envolvendo animais foram realizados com protocolo aprovado pelo Comitê Institucional de Cuidados e Uso de Animais da KUMC, de acordo com suas diretrizes e regulamentos (Número do Protocolo: 2018-2447).
1. Colher E13,5 embriões
- Eutanize camundongos gestantes usando uma câmara de inalação de CO2 ou por um procedimento aprovado pelo Comitê Institucional de Cuidados e Uso de Animais. Imediatamente proceder à dissecação.
- Exponha a metade inferior da cavidade abdominal removendo a pele e o peritônio. Extirpar ambos os chifres do útero, que contêm os embriões E13.5.
- Coloque brevemente o útero em solução salina pré-armada de 37 °C estéril (PBS) para enxaguar o excesso de sangue, cabelo ou outros detritos. Coloque o útero em um prato estéril de 10 cm recheado com PBS estéril.
- Usando uma tesoura pequena, corte a parede uterina ao longo do comprimento do útero para expor cada embrião, ainda em seu saco de gema. Remova o saco de gema ao redor do embrião, mas guarde-o para genotipagem, se necessário. À medida que os embriões são removidos, coloque cada embrião em seu próprio poço de uma placa de 12 poços cheia de PBS.
2. Dissecção de prateleiras palatais de embriões(Figura 1)
NOTA: Esterilize os instrumentos de dissecção de aço inoxidável (veja a Tabela de Materiais) após o processamento de cada embrião colocando os instrumentos primeiro em um béquer de 100% de álcool ettil (EtOH), depois em um esterilizador de instrumentos a 350 °C por 10 s, e depois resfriando-os em um segundo béquer de 100% EtOH.
- Utilizando uma colher perfurada esterilizada, coloque o embrião em um novo prato de 10 cm recheado com meio de cultura MEPM consistindo do meio essencial mínimo (DMEM) de Dulbecco contendo 10% de soro bovino fetal (FBS), L-glutamina (4 mM L-Glu) e antibióticos-penicilina e estreptomicina (50 unidades/mL).
- Decapitar o embrião logo abaixo da linha da mandíbula usando uma tesoura estéril(Figura 1A, linha pontilhada vermelha). Remova a mandíbula inferior inserindo um ponto das fórceps finas esterilizadas #5 na boca, mantendo-a apenas dentro da bochecha. Empurre o ponto das fórceps inseridas até que saia da parte de trás do crânio.
- Oriente os fórceps, ao longo da linha amarela na Figura 1B,de modo que o outro lado dos fórceps (que ainda está fora do embrião) esteja pairando sobre o canal auditivo, em seguida, belisque os fórceps fechados para cortar o tecido. Se necessário, execute outro fórceps fino ao longo da costura dos fórceps agora fechados para cortar qualquer tecido que não foi completamente cortado pela pitada.
- Repita a etapa anterior para o outro lado da cabeça do embrião. Continue o procedimento de corte de pinça para remover totalmente a mandíbula inferior, a língua e a parte inferior do crânio e expor as prateleiras palatais.
- Remova o crânio do crânio cortando logo acima dos olhos, como mostrado na Figura 1C (linha verde). Faça isso colocando a cabeça no lado esquerdo ou direito e posicionando os pontos da pequena tesoura de aço inoxidável na frente e atrás do crânio logo acima do nível do olho do embrião. Corte a parte superior do crânio com um corte rápido da tesoura, criando uma superfície plana que será importante para a estabilidade em etapas posteriores e que deve parecer a Figura 1D quando vista do lado.
- Coloque a parte restante da cabeça de cabeça de cabeça para baixo, com o aspecto superior da cabeça (crânio removido) descansando na parte inferior do prato, o que fornecerá uma superfície estável para a remoção da prateleira palatal. Tire um momento para identificar as prateleiras palatais, que agora estão expostas e viradas para cima e aparecerão como dois cumes elevados em ambos os lados de uma ranhura central na metade anterior da cabeça(Figura 1E).
- Fixar a porção restante da cabeça ao prato para imobilizá-la enquanto as prateleiras são removidas. Faça isso inserindo um ponto de um fórceps fino através do tecido próximo à região nasal da cabeça, anterior às prateleiras palatais, e insira o outro ponto das fórceps através da base do crânio, posteriormente às prateleiras palatais. Guarde-os no lugar enquanto realiza a excisão das prateleiras palatais.
- Imobilizando a cabeça com uma mão, escolha qualquer uma das duas prateleiras para remover primeiro, e insira os dois pontos de um segundo par de fórceps finos no tecido na base da superfície lateral da prateleira, e belisque para cortar o tecido(Figura 1F). Repita isso ao longo da base da superfície medial da prateleira e, em seguida, tanto nas extremidades anterior quanto posterior da prateleira para se soltar da cabeça.
- Levante suavemente a prateleira, fazendo pitadas adicionais, conforme necessário, para libertar completamente a prateleira do tecido circundante.
- Repita as duas etapas anteriores para remover a segunda prateleira palatal.
- Com as prateleiras palatais agora liberadas do tecido circundante e colocadas em PBS (Figura G),use uma pipeta de transferência de lâmpada plástica estéril para elaborar as prateleiras da pipeta, e transfira-as para um tubo de microcentrifuuge de 1,5mL, juntamente com aproximadamente 500 μL de PBS. Mantenha os tubos contendo prateleiras palatais no gelo enquanto o resto da ninhada é processada da mesma forma.
NOTA: Alternativamente, as prateleiras podem ser colocadas em um tubo de microcentrifuuge de 1,5mL contendo trippsina pré-armada (0,25%) imediatamente após a dissecção (em vez de colocá-las no gelo). As amostras serão mais frescas, mas deve-se ter cuidado ao tempo todos os passos de processo para cada amostra individual, em vez de tratar as amostras coletivamente.
3. Cultura das células MEPM
NOTA: Sob as condições aqui descritas, as células epiteliais do paladar não sobrevivem à primeira passagem, resultando em uma cultura celular mesenquimal do paladar puro. Use técnica estéril para realizar todas as etapas em uma capa de cultura de tecido.
- Aspire e descarte o PBS do tubo de 1,5 mL, tomando cuidado para não descartar as prateleiras no processo. Adicione imediatamente 200 μL de trippsina pré-armada (37 °C) a cada tubo que contenha prateleiras palatais. Em breve, encoste as prateleiras para cima e para baixo na trippsina usando uma ponta de pipeta de 1000 μL para acelerar a trippsinização.
- Incubar os tubos por 5 min a 37 °C, em seguida, tubo cada amostra para cima e para baixo novamente para ajudar a quebrar o tecido. Incubar os tubos por mais 5 min a 37 °C, e escoar para cima e para baixo mais uma vez para completar a dissociação do tecido.
NOTA: As prateleiras devem ser completamente ou quase completamente dissociadas e suspensas na trippsina sem pedaços visíveis de tecido restantes. - Adicione 800 μL de meio de cultura MEPM (passo 2.1) a cada tubo de 1,5 mL. Centrifugar o tubo de 1,5 mL a 200 × g por 5 min para pelotar as células. Remova o supernatante e resuspense a pelota celular em 1 mL de meio de cultura MEPM.
- Emplaque as células MEPM em uma placa tratada com cultura de tecido de 6 poços contendo meio de cultura MEPM. Permita que as células aderam à superfície plástica por 12 h a 37 °C em uma incubadora com 5% de CO2.
NOTA: Após a incubação durante a noite, a grande maioria (~90%) das células se anexará. Neste ponto, as células aderidas parecerão bastante homogêneas, com uma forma triangular ou ligeiramente alongada. - Troque o meio todos os dias aspirando suavemente o antigo meio e substituindo-o imediatamente por 1 mL de PBS quente estéril sem cálcio ou magnésio por ~1 min. Aspire o PBS, e substitua por 3 mL de meio de cultura MEPM pré-armado.
- Passagem as células uma vez que se tornam 100% confluentes.
NOTA: As células MEPM devem proliferar dobrando o número quase diariamente.- Para passar as células, aspire suavemente o meio antigo e substitua-o imediatamente por PBS quente sem cálcio ou magnésio por ~1 min. Aspire o PBS, e substitua-o por 0,5 mL de trippsina pré-armada de 0,25%.
- Incubar a 37 °C por ~5 min, ou até que as células se desprendem da superfície do prato quando suavemente balançadas para frente e para trás à mão. Uma vez que as células tenham se destacado, adicione imediatamente 5 mL de meio de cultura MEPM pré-armado às células experimentadas.
- Usando um tubo sorológico de 10 mL, colete suavemente as células em um tubo cônico de 15 mL e centrifugar o tubo a 200 × g por 5 min para pelotar as células. Aspire a trippsina e média, e resuspense as células em 3 mL de meio de cultura MEPM pré-armado. Tubo suavemente 1 mL de células em um único poço de um prato de 6 poços, e adicione 2 mL de meio de cultura MEPM para trazer o volume total para 3 mL.
NOTA: Isso constitui uma divisão de 1:3 de células. Os deputados podem ser aprovados em até três vezes. A densidade de semeadura dos MEPMs é um pouco flexível, e o número de células presentes varia dependendo do vaso de cultivo. No entanto, os MEPMs não proliferam adequadamente quando divididos muito escassamente e devem ser pelo menos 20-25% confluentes em seu novo prato uma vez que aderem.
4. Criopreservação de células MEPM
- Uma vez que as células MEPM experimentadas são pelotas, resuspendam as células no meio de cultura MEPM para obter uma concentração de ~ 1 × 106 células/mL. Encoba as células em criovials, e adicione uma concentração final de 5% de dimetilsulfoxida no estoque celular. Cubra o criovial, misture brevemente invertendo, e coloque imediatamente os frascos em um recipiente de congelamento que esfrie a uma taxa de 1 °C/min.
- Coloque o refrigerador em um congelador de -80 °C durante a noite. No dia seguinte, mova os criovials para um tanque de nitrogênio líquido para armazenamento a longo prazo.
- Descongelamento criopreservado células MEPM
- Remova os criovials do tanque de nitrogênio líquido e descongele à temperatura ambiente até que o conteúdo comece a ficar líquido. Esvazie o conteúdo em um tubo cônico de 15 mL contendo 9 mL de meio de cultura MEPM pré-armado.
- Centrifugar o tubo a 200 × g para pelotar as células. Resuspenque as células em 1 mL de meio de cultura MEPM, e encoste as células em um único poço de uma placa de 6 poços. Adicione 2 mL de meio de cultura MEPM quente para trazer o volume total para 3 mL.
- Cultura as células a 37 °C em uma incubadora com 5% de CO2. Mude o meio diariamente.
5. Imagem ao vivo das células MEPM - ensaio de migração coletiva 2D(Figura 2)
- Prepare uma placa para usar para imagens ao vivo.
- Use uma pequena tesoura cirúrgica ou um bisturi afiado para encurtar uma pastilha de silicone estéril de 2 poços a uma altura de ~1 mm. Prepare uma inserção para cada amostra que está sendo utilizada.
- Com fórceps, coloque a inserção encurtada de silicone de 2 poços no centro de um poço de uma placa de 6 poços. Pressione para baixo ao longo de todas as bordas para garantir que ele seja totalmente aderido.
- Descongele células criopreservadas seguindo as etapas da seção 4.3 deste protocolo. Conte células MEPM e sementes 300 células/mm2 das pastilhas de silicone encurtadas em um volume total de 40-50 μL MEPM cultura média por poço. Cultura as células durante a noite a 37 °C em uma incubadora com 5% de CO2.
- No dia seguinte, prepare-se para imagens de lapso de tempo ao vivo.
- Use um microscópio de contraste de fase com uma incubadora no palco e capacidade automática de imagem. Adicione água ao reservatório da incubadora no palco para reduzir a evaporação do meio de cultura; definir a temperatura para 37 °C e CO2 para 5%. Deixe ~30 min para que a umidade se acumule antes de colocar o prato de 6 poços na incubadora do palco.
- Use configurações equivalentes às seguintes para imagens de lapso de tempo.
- Selecione o objetivo 4x para ter grandes campos de visão e filtro de contraste de fase.
NOTA: Foco automático, amostra de auto-encontrar, pilha z e iluminação automática não são normalmente necessários. - Selecione dois campos microscópicos por poço para capturar o lúmen das pastilhas de silicone encurtadas. Certifique-se de que todas as posições de imagem tenham o foco correto.
NOTA: Os planos de imagem podem ser ajustados durante a imagem, mas esse ajuste geralmente não é necessário. - Selecione o tipo de arquivo de saída de imagem desejado e selecione ou dese selecione opções pós-imagem, como criação automática de vídeo e marcas d'água, conforme desejado. Se for aplicável, selecione o contraste de fase como modo de imagem.
NOTA: O uso de marcas d'água pode impedir etapas subsequentes de processamento de imagem. - Defina a duração da gravação para 72 h. Defina o programa para capturar imagens a cada 10 minutos.
NOTA: Geralmente apenas 48h de imagem são necessárias, mas a imagem pode ser interrompida a qualquer momento antes da marca de 72h sem perder imagens que já foram tiradas. - Certifique-se de que a câmara ambiental esteja operacional conforme necessário em 5.3.1. Salve essas configurações (a rotina) e comece a imagem.
- Selecione o objetivo 4x para ter grandes campos de visão e filtro de contraste de fase.
- Continue a imagem até 72 h (ou o tempo especificado).
6. Imagem ao vivo de células MEPM em um ensaio de reparo de feridas(Figura 3)
- Prepare uma placa para usar para imagens ao vivo. Usando fórceps, coloque uma pastilha de silicone estéril de 2 poços no centro de um poço de uma placa de 6 poços e pressione para baixo ao longo de todas as bordas para garantir que ela seja totalmente aderida. Prepare uma inserção de 2 poços para cada amostra que está sendo usada.
- Descongele células criopreservadas seguindo as etapas da seção 4.3 deste protocolo. Conte células MEPM e, se necessário, concentre as células em pelo menos 350 células/μL.
- Semente 1400 células/mm2 nas pastilhas de silicone em um volume de 100 μL de cultura MEPM meio por poço. Cultue as células por 48 h a 37 °C em uma incubadora com 5% de CO2, e mude o meio todos os dias.
- Após 48 h, prepare um microscópio para imagens de lapso de tempo ao vivo, conforme descrito nas seções 5.3 e 5.4. Imediatamente antes de colocar as células na incubadora no palco, adicione 3 mL de cultura MEPM pré-armada ao poço (mas fora das pastilhas) e, em seguida, remova cuidadosamente as pastilhas de silicone.
NOTA: A parede que separa as 2 câmaras deixa uma lacuna que é a "ferida". - Inicie imagens de lapso de tempo conforme descrito em 5.4, com as seguintes diferenças:
- Use um objetivo de ampliação mais elevado (por exemplo, 10x). Para capturar o fechamento da ferida, selecione 5 campos de visão ao longo de cada ferida, de modo que a ferida seja paralela ao eixo vertical da imagem.
- Pare de fotografar depois de 72 h ou quando as feridas estão totalmente fechadas.
7. Análise computacional de sequências de imagem de lapso de tempo
NOTA: Realize os seguintes procedimentos em um computador equipado com ferramentas computacionais padrão, como o intérprete python, o compilador C e uma concha (ver a Tabela de Materiais).
- Análise de confluência
NOTA: Este procedimento pode ser usado para estimar a proliferação celular dentro de uma cultura esparsa ou para quantificar experimentos de fechamento de feridas. Para detectar áreas ocupadas por células, um limiar de segmentação é aplicado ao desvio padrão local do brilho da imagem. O código foi descrito anteriormente por Wu et al.33 e Neufeld et al.34 e está disponível em http://github.com/aczirok/cellconfluency.- Determine o limiar de segmentação das imagens. Como exemplo, para ver a segmentação com um limiar 4, emitir o comando
segment.py -i inout-image.jpg-S 4 -saída de teste.jpg
e, em seguida, verifique a saída(saída.jpg).
NOTA: Se o limiar for muito baixo, as áreas de fundo no micrografo são classificadas como cobertas por células. Em contraste, se o limiar for muito alto, as áreas cobertas por células não são classificadas como tal. O valor de limiar ideal mantém ambos os erros no mínimo. - Use o script area.sh fornecido para calcular valores de confluência para uma sequência de imagens como
area.sh -S4 img_001.jpg img_002.jpg .... > confluência.dat
NOTA: O valor do limiar de discriminação 4 é verificado na etapa 1, e os resultados são armazenados na confluência de arquivos.dat. Ao classificar as imagens em pastas apropriadas, a lista de nomes de arquivos de imagem pode ser substituída por notação curinga:
area.sh -S4 *.jpg > confluência.dat - Para experimentos de fechamento de feridas, transforme os dados de confluência A(t)- o tamanho da área coberta por células expressa como uma porcentagem em função da velocidade de propagação de borda de ferida V como

onde w denota a largura do campo e dA/dt é o derivado de tempo de A(t), ou seja, a taxa de expansão da área coberta por células.
- Determine o limiar de segmentação das imagens. Como exemplo, para ver a segmentação com um limiar 4, emitir o comando
- Mapa de motilidade celular
- Execute um algoritmo de velocimetria de imagem de partículas (PIV) para caracterizar a motilidade celular e extrair a extensão do movimento local "fluxo óptico" entre pares de imagens, mas não para identificar células individuais.
NOTA: Aqui, é utilizado um tamanho inicial da janela de 50 μm, conforme descrito em detalhes por Zamir et al.35 e Czirok et al.36, com um tamanho inicial de janela de 50 μm. A análise piv produz um campo de velocidade v(x,t) para cada quadro de imagem t e localização (dentro da imagem) x. - Extrair a velocidade média da motilidade celular de v(x,t) como uma média espacial calculada sobre a área ocupada por células, conforme determinado na seção 7.1.
- Execute um algoritmo de velocimetria de imagem de partículas (PIV) para caracterizar a motilidade celular e extrair a extensão do movimento local "fluxo óptico" entre pares de imagens, mas não para identificar células individuais.
- Rastreamento manual de células
NOTA: Embora a análise do PIV forneça uma avaliação automática da motilidade celular, focar no comportamento de células individuais muitas vezes requer rastreamento manual. Embora várias ferramentas forneçam essa funcionalidade, é muito útil se os marcadores posicionados manualmente podem ser modificados após sua colocação inicial, e se o rastreamento pode ser realizado tanto para frente quanto para trás no tempo.- Execute o rastreamento de células com uma ferramenta python desenvolvida sob medida (http://github.com/donnagreta/cm_track), que também fornece funções básicas do editor, como a exclusão de segmentos de trajetória.
NOTA: Esta ferramenta de rastreamento manual produz as posições P(i,t) de célula i no momento em um arquivo de texto, e invocada como
cm_track.py -i imagens/ -o faixa.dat
onde as imagens de lapso de tempo estão nas imagens da pasta/ e os dados de posição são coletados na faixa de arquivo.dat (Figura 4).
- Execute o rastreamento de células com uma ferramenta python desenvolvida sob medida (http://github.com/donnagreta/cm_track), que também fornece funções básicas do editor, como a exclusão de segmentos de trajetória.

Figura 4: A análise das trajetórias celulares individuais. (A) Micrografógrafos de lapso de tempo de contraste de fase são submetidos a(B, C) um procedimento de rastreamento manual, que marca células (pontos verdes). (D) As posições celulares (x,y) são armazenadas para cada célula distinguida pelo seu ID e para cada quadro f. (E) Trajetórias podem ser sobrepostas nos micrografos e codificados por cores para indicar informações temporais. Como exemplo, em cada trajetória, uma paleta de cores azul a vermelha indica segmentos de trajetória progressivamente posteriores, com vermelho e azul marcando os locais iniciais e finais das células, respectivamente. (F) Várias propriedades estatísticas de trajetórias, como o deslocamento quadrado médio, podem ser extraídas e utilizadas para caracterizar a motilidade de várias populações celulares, que neste exemplo incluem células MEPM de tipo selvagem (wt, azul) e knockdown (kd, vermelho). As barras de escala representam 100 μm. Clique aqui para ver uma versão maior desta figura.
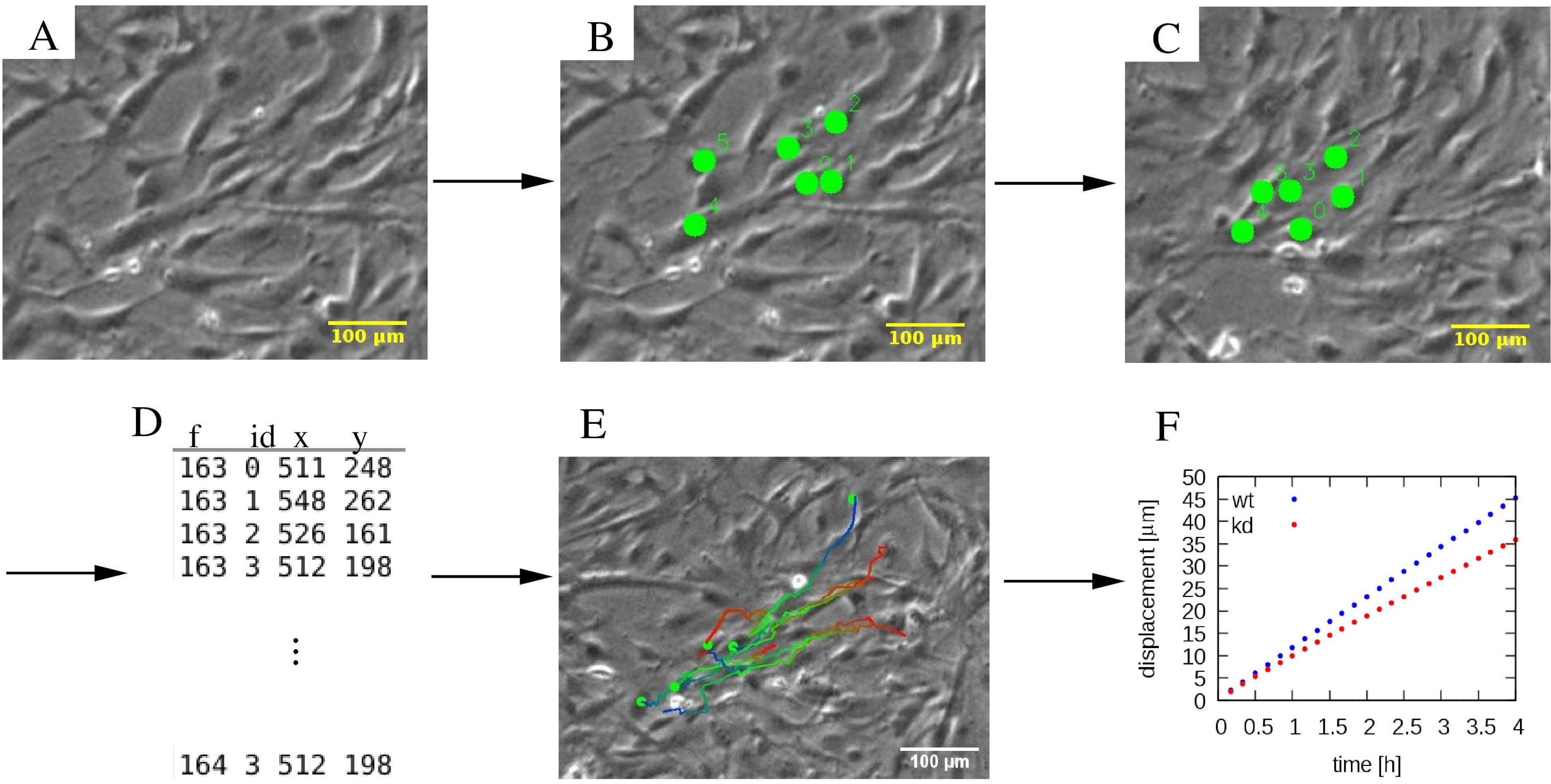{kind=link}
- Trajetórias de sobreposição em imagens usando uma segunda ferramenta, invocada como
visdat.py -d faixa.dat -i imagens/ -o sobreposição/ -l999 -r3 -C2 - Colete as imagens com as trajetórias sobrepostas na sobreposição da pasta.
NOTA: Neste exemplo, os dados de posição da célula são armazenados na faixa de arquivo.dat, enquanto a sequência de imagem de lapso de tempo está dentro das imagens da pasta. O restante dos parâmetros controla o comprimento máximo das trajetórias desenhadas (-l), o tamanho dos símbolos (-r) e o esquema de cores (-C) utilizado.
- Análise de trajetórias celulares individuais
- Caracterizar trajetórias pelo comprimento total do caminho
 ,
,
e deslocamento líquido em direção à ferida ,
,
onde X denota a projeção de P na direção perpendicular à ferida: a coordenada x das posições quando a ferida é paralela ao eixo y. - Calcule a eficiência de orientação quanto
 a cada célula i e ponto de tempo t37. Caracterizar culturas pela média populacional dessas medidas celulares únicas, avaliadas em um ponto de tempo adequado t.
a cada célula i e ponto de tempo t37. Caracterizar culturas pela média populacional dessas medidas celulares únicas, avaliadas em um ponto de tempo adequado t.
- Caracterizar trajetórias pelo comprimento total do caminho
- Movimento de streaming coletivo de células
- Caracterizar as correlações espaciais locais dos movimentos celulares pela velocidade média de outras células que estão nas proximidades de uma célula móvel, como descrito anteriormente38,39.
NOTA: O código computacional está disponível em https://github.com/aczirok/flowfield.- Alinhe um sistema de referência com direções dianteira, traseira, esquerda e direita a cada vetor v(x,t) (Figura 5A). Atribua cada uma das células circundantes ou vetor de velocidade PIV à célula espacial apropriada do sistema de referência (Figura 5B). Repita o procedimento, pois cada vetor serve como a origem dos sistemas de referência (Figura 5C,D) de modo que um determinado vetor de velocidade seja atribuído a várias caixas.
NOTA: Um ponto de dados pode estar na frente de um vetor, e à esquerda de outro. - Gire os sistemas de referência em uma orientação comum (Figura 5C,F) e acumule-os(Figura 5G).
NOTA: A média de cada bin dos dados de velocidade agrupado é um vetor de velocidade (U)que é indicativo de correlação espacial: a média é uma medida de um componente de velocidade compartilhada(Figura 5H). - Encaixe os campos de fluxo U(x) com uma função
 exponencial, onde a, x0 e U0 são parâmetros adequados. Dos três parâmetros de montagem, concentre-se no x0, o comprimento de correlação (Figura 5I,J), que é a distância característica onde as correlações de velocidade local desaparecem.
exponencial, onde a, x0 e U0 são parâmetros adequados. Dos três parâmetros de montagem, concentre-se no x0, o comprimento de correlação (Figura 5I,J), que é a distância característica onde as correlações de velocidade local desaparecem.
- Alinhe um sistema de referência com direções dianteira, traseira, esquerda e direita a cada vetor v(x,t) (Figura 5A). Atribua cada uma das células circundantes ou vetor de velocidade PIV à célula espacial apropriada do sistema de referência (Figura 5B). Repita o procedimento, pois cada vetor serve como a origem dos sistemas de referência (Figura 5C,D) de modo que um determinado vetor de velocidade seja atribuído a várias caixas.
- Caracterizar as correlações espaciais locais dos movimentos celulares pela velocidade média de outras células que estão nas proximidades de uma célula móvel, como descrito anteriormente38,39.

Figura 5: Caracterização da formação de fluxo de células cultivadas. (A,D) Imagens de lapso de tempo de contraste de fase da Figura 4A são usadas para identificar movimentos celulares. Para cada célula móvel, um quadro de referência (azul) e caixas espaciais (brancas) foram co-alinhados para categorizar as células adjacentes como sendo na frente, traseira, esquerda ou direita. (B,E) A velocidade das células adjacentes (vetores pretos) estava relacionada ao mesmo quadro de referência(C,F). Este procedimento foi repetido para cada célula e ponto de tempo. (G) Após a junção dessas informações locais, cada caixa conterá vários vetores de velocidade (cinza), que podem ser mediados para determinar a velocidade co-móvel média (setas magenta) em vários locais em relação a uma célula motil média. (H) O mapa de velocidade média caracteriza assim as velocidades típicas das células em vários locais em relação a uma célula móvel. (I,J) Finalmente, este campo foi amostrado ao longo do eixo frontal-traseiro (paralelo) e também ao longo do eixo esquerda-direita (perpendicular). Clique aqui para ver uma versão maior desta figura.
{kind=link}
Resultados
A dissecação das prateleiras palatais é ilustrada na Figura 1. A sequência de incisões é projetada para minimizar o deslizamento do tecido. Após a remoção da cabeça (Figura 1A,B), a mandíbula inferior é removida(Figura 1B,C). A incisão da parte superior da cabeça (Figura 1C,D) é feita para estabilizar o tecido quando colocado de cabeça p...
Discussão
A elevação da prateleira palatal constitui um evento de remodelação vertical para horizontal1,3,4,9,11. É postulado que este processo de remodelação requer que as células mesenquimais de prateleira palatal se comportem coordenadamente. As análises com células MEPM do tipo selvagem mostram que esse comportamento celular é intrínseco e pode ser quant...
Divulgações
Os autores não têm nada a revelar.
Agradecimentos
Este projeto foi apoiado em parte pelos Institutos Nacionais de Saúde de bolsas DE026172 (IS) e GM102801 (A.C.). O I.S. também foi apoiado em parte pelo Centro de Excelência em Pesquisa Biomédica (COBRE) (National Institute of General Medical Sciences P20 GM104936), Kansas IDeA Network for Biomedical Research Excellence grant (National Institute of General Medical Sciences P20 GM103418) e Kansas Intellectual and Developmental Disabilities Research Center (KIDDRC) (U54 Eunice Kennedy Shriver National Institute of Child Health and Human Development, HD090216).
Materiais
| Name | Company | Catalog Number | Comments |
| Beaker, 250 mL (x2) | Fisher Scientific | FB-100-250 | |
| CO2 | Matheson Gas | UN1013 | |
| Conical tubes, 15 mL (x1) | Midwest Scientific | C15B | |
| Debian operating system | computational analysis of time-lapse images | ||
| Dulbecco's Modified Eagles Medium/High Glucose with 4 mM L-Glutamine and Sodium Pyruvate | Cytiva Life Sciences | SH30243.01 | |
| EtOH, 100% | Decon Laboratories | 2701 | |
| EVOS FL Auto | ThermoFisher Scientific | AMAFD1000 | |
| EVOS Onstage Incubator | ThermoFisher Scientific | AMC1000 | |
| EVOS Onstage Vessel Holder, Multi-Well Plates | ThermoFisher Scientific | AMEPVH028 | |
| Fetal Bovine Serum | Corning | 35-010-CV | |
| Fine point #5 Stainless Steel Forceps (x2) | Fine Science Tools | 11295-10 | Dissection |
| Instrument sterilizer bead bath | Fine Science Tools | 18000-45 | |
| Microcetrifuge tubes, 1.5mL | Avant | 2925 | |
| Micro-Dissecting Stainless Steel Scissors, Straight | Roboz | RS-5910 | Dissection |
| NucBlue (Hoechst) Live Ready Probes | ThermoFisher Scientific | R37605 | |
| Penicillin Streptomycin Solution, 100x | Corning | 30-002-CI | |
| Silicone Insert, 2-well | Ibidi | 80209 | |
| Small Perforated Stainless Steel Spoon | Fine Science Tools | MC17C | Dissection |
| Spring Scissors, 4 mm | Fine Science Tools | 15018-10 | |
| Sterile 10 cm dishe(s) | Corning | 430293 | |
| Sterile 12-well plate(s) | PR1MA | 667512 | |
| Sterile 6-well plate(s) | Thermo Fisher Scientific | 140675 | |
| Sterile PBS | Corning | 21-031-CV | |
| Sterile plastic bulb transfer pipette | ThermoFisher Scientific | 202-1S | |
| Trypsin, 0.25% | ThermoFisher Scientific | 25200056 |
Referências
- Bush, J. O., Jiang, R. Palatogenesis: morphogenetic and molecular mechanisms of secondary palate development. Development. 139 (2), 231-243 (2012).
- Mossey, P. A., Little, J., Munger, R. G., Dixon, M. J., Shaw, W. C. Cleft lip and palate. Lancet. 374 (9703), 1773-1785 (2009).
- Lan, Y., Xu, J., Jiang, R. Cellular and molecular mechanisms of palatogenesis. Current Topics in Developmental Biology. 115, 59-84 (2015).
- Li, C., Lan, Y., Jiang, R. Molecular and cellular mechanisms of palate development. Journal of Dental Research. 96 (11), 1184-1191 (2017).
- Gritli-Linde, A. The etiopathogenesis of cleft lip and cleft palate: usefulness and caveats of mouse models. Current Topics in Developmental Biology. 84, 37 (2008).
- Meng, L., Bian, Z., Torensma, R., Vonden Hoff, J. W. Biological mechanisms in palatogenesis and cleft palate. Journal of Dental Research. 88 (1), 22-33 (2009).
- Dixon, M. J., Marazita, M. L., Beaty, T. H., Murray, J. C. Cleft lip and palate: understanding genetic and environmental influences. Nature Reviews Genetics. 12 (3), 167-178 (2011).
- Kousa, Y. A., Schutte, B. C. Toward an orofacial gene regulatory network. Developmental Dynamics. 245 (3), 220-232 (2016).
- Jin, J. Z., et al. Mesenchymal cell remodeling during mouse secondary palate reorientation. Developmental Dynamics. 239 (7), 2110-2117 (2010).
- Yu, K., Ornitz, D. M. Histomorphological study of palatal shelf elevation during murine secondary palate formation. Developmental Dynamics. 240 (7), 1737-1744 (2011).
- Chiquet, M., Blumer, S., Angelini, M., Mitsiadis, T. A., Katsaros, C. Mesenchymal remodeling during palatal shelf elevation revealed by extracellular matrix and F-actin expression patterns. Frontiers in Physiology. 7, 392 (2016).
- Paul, B. J., et al. ARHGAP29 mutation is associated with abnormal oral epithelial adhesions. Journal of Dental Research. 96 (11), 1298-1305 (2017).
- Hall, E. G., et al. SPECC1L regulates palate development downstream of IRF6. Human Molecular Genetics. 29 (5), 845-858 (2020).
- Walker, B. E., Fraser, F. C. Closure of the secondary palate in three strains of mice. Journal of Embryology and Experimental Morphology. 4 (2), 176-189 (1956).
- Jin, J. Z., Li, Q., Higashi, Y., Darling, D. S., Ding, J. Analysis of Zfhx1a mutant mice reveals palatal shelf contact-independent medial edge epithelial differentiation during palate fusion. Cell Tissue Research. 333 (1), 29-38 (2008).
- Kouskoura, T., et al. The etiology of cleft palate formation in BMP7-deficient mice. PLoS One. 8 (3), 59463 (2013).
- Lan, Y., Zhang, N., Liu, H., Xu, J., Jiang, R. Golgb1 regulates protein glycosylation and is crucial for mammalian palate development. Development. 143 (13), 2344-2355 (2016).
- He, F., et al. Wnt5a regulates directional cell migration and cell proliferation via Ror2-mediated noncanonical pathway in mammalian palate development. Development. 135 (23), 3871-3879 (2008).
- Lan, Y., Qin, C., Jiang, R. Requirement of hyaluronan synthase-2 in craniofacial and palate development. Journal of Dental Research. 98 (12), 1367-1375 (2019).
- Yonemitsu, M. A., Lin, T. Y., Yu, K. Hyaluronic acid is required for palatal shelf movement and its interaction with the tongue during palatal shelf elevation. Developmental Biology. 457 (1), 57-68 (2020).
- Goering, J. P., et al. SPECC1L-deficient palate mesenchyme cells show speed and directionality defect. Scientific Reports. 11 (1), 1452 (2021).
- Fantauzzo, K. A., Soriano, P. PI3K-mediated PDGFRalpha signaling regulates survival and proliferation in skeletal development through p53-dependent intracellular pathways. Genes and Development. 28 (9), 1005-1017 (2014).
- Vasudevan, H. N., Soriano, P. SRF regulates craniofacial development through selective recruitment of MRTF cofactors by PDGF signaling. Developmental Cell. 31 (3), 332-344 (2014).
- Vasudevan, H. N., Mazot, P., He, F., Soriano, P. Receptor tyrosine kinases modulate distinct transcriptional programs by differential usage of intracellular pathways. Elife. 4, 07186 (2015).
- Gao, L., et al. 2,3,7,8-Tetrachlorodibenzo-p-dioxin and TGFbeta3-mediated mouse embryonic palatal mesenchymal cells. Dose Response. 17 (1), 1559325818786822 (2019).
- Iyyanar, P. P. R., Nazarali, A. J. Hoxa2 inhibits bone morphogenetic protein signaling during osteogenic differentiation of the palatal mesenchyme. Frontiers in Physiology. 8, 929 (2017).
- Jiang, Z., Pan, L., Chen, X., Chen, Z., Xu, D. Wnt6 influences the viability of mouse embryonic palatal mesenchymal cells via the beta-catenin pathway. Experimental and Therapeutic Medicine. 14 (6), 5339-5344 (2017).
- Liu, X., et al. Negative interplay of retinoic acid and TGF-beta signaling mediated by TG-interacting factor to modulate mouse embryonic palate mesenchymal-cell proliferation. Birth Defects Research Part B: Developmental and Reproductive Toxicology. 101 (6), 403-409 (2014).
- Bush, J. O., Soriano, P. Ephrin-B1 forward signaling regulates craniofacial morphogenesis by controlling cell proliferation across Eph-ephrin boundaries. Genes & Development. 24 (18), 2068-2080 (2010).
- Mo, J., Long, R., Fantauzzo, K. A. Pdgfra and Pdgfrb genetically interact in the murine neural crest cell lineage to regulate migration and proliferation. Frontiers in Physiology. 11, 588901 (2020).
- He, F., Soriano, P. A critical role for PDGFRalpha signaling in medial nasal process development. PLoS Genetics. 9 (9), 1003851 (2013).
- Fantauzzo, K. A., Soriano, P. Generation of an immortalized mouse embryonic palatal mesenchyme cell line. PLoS One. 12 (6), 0179078 (2017).
- Wu, K., Gauthier, D., Levine, M. D. Live cell image segmentation. IEEE Transactions on Biomedical Engineering. 42 (1), 1-12 (1995).
- Neufeld, Z., et al. The role of Allee effect in modelling post resection recurrence of glioblastoma. PLoS Computational Biology. 13 (11), 1005818 (2017).
- Zamir, E. A., Czirok, A., Rongish, B. J., Little, C. D. A digital image-based method for computational tissue fate mapping during early avian morphogenesis. Annals of Biomedical Engineering. 33 (6), 854-865 (2005).
- Czirok, A., et al. Optical-flow based non-invasive analysis of cardiomyocyte contractility. Scientific Reports. 7 (1), 10404 (2017).
- Biggs, L. C., et al. Interferon regulatory factor 6 regulates keratinocyte migration. Journal of Cell Science. 127, 2840-2848 (2014).
- Czirok, A., Varga, K., Mehes, E., Szabo, A. Collective cell streams in epithelial monolayers depend on cell adhesion. New Journal of Physics. 15, 75006 (2013).
- Szabo, A., et al. Collective cell motion in endothelial monolayers. Physical Biology. 7 (4), 046007 (2010).
- Gulyas, M., Csiszer, M., Mehes, E., Czirok, A. Software tools for cell culture-related 3D printed structures. PLoS One. 13 (9), 0203203 (2018).
- Soderholm, J., Heald, R. Scratch n' screen for inhibitors of cell migration. Chemistry & Biology. 12 (3), 263-265 (2005).
- Riahi, R., Yang, Y., Zhang, D. D., Wong, P. K. Advances in wound-healing assays for probing collective cell migration. Journal of Laboratory Automation. 17 (1), 59-65 (2012).
- Svensson, C. M., Medyukhina, A., Belyaev, I., Al-Zaben, N., Figge, M. T. Untangling cell tracks: Quantifying cell migration by time lapse image data analysis. Cytometry Part A. 93 (3), 357-370 (2018).
- Fantauzzo, K. A., Soriano, P. PDGFRbeta regulates craniofacial development through homodimers and functional heterodimers with PDGFRalpha. Genes & Development. 30 (21), 2443-2458 (2016).
- Rafi, S. K., et al. Anti-epileptic drug topiramate upregulates TGFβ1 and SOX9 expression in primary embryonic palatal mesenchyme cells: Implications for teratogenicity. PLoS ONE. , (2021).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados