Для просмотра этого контента требуется подписка на Jove Войдите в систему или начните бесплатную пробную версию.
Method Article
Изоляция и покадровая визуализация первичных клеток эмбриональной небной мезенхимы мыши для анализа коллективных атрибутов движения
В этой статье
Резюме
Мы представляем протокол выделения и культивации первичных эмбриональных небных мезенхимальных клеток мыши для покадровой визуализации двумерного (2D) роста и ран-восстановительных анализов. Мы также предоставляем методологию анализа данных покадровой визуализации для определения образования клеточного потока и направленной подвижности.
Аннотация
Развитие неба – это динамичный процесс, который включает в себя вертикальный рост двусторонних небных полок рядом с языком с последующим возвышением и сращинием над языком. Дефекты в этом процессе приводят к расщелине неба, распространенному врожденному дефекту. Недавние исследования показали, что поднятие небной полки включает в себя процесс реконструкции, который преобразует ориентацию полки из вертикальной в горизонтальную. Роль мезенхимальных клеток небной полки в этом динамическом ремоделирование было трудно изучить. Количественный анализ на основе покадровой визуализации недавно был использован, чтобы показать, что первичные эмбриональные небные мезенхимальные клетки мыши (MEPM) могут самоорганизовываться в коллективное движение. Количественный анализ может выявить различия в мутантных клетках MEPM из мышиной модели с дефектами высоты неба. В этой статье описываются методы выделения и культивирования клеток MEPM из эмбрионов E13.5, специально для покадровой визуализации, и определения различных клеточных атрибутов коллективного движения, включая меры по формированию потока, выравниванию формы и сохранению направления. В нем утверждается, что клетки MEPM могут служить прокси-моделью для изучения роли мезенхимы небной полки во время динамического процесса подъема. Эти количественные методы позволят исследователям в черепно-лицевой области оценивать и сравнивать коллективные атрибуты движения в контрольных и мутантных клетках, что расширит понимание ремоделирования мезенхимальных мешков во время подъема небного шельфа. Кроме того, клетки MEPM обеспечивают редкую модель мезенхимальных клеток для исследования коллективного движения клеток в целом.
Введение
Развитие неба было широко изучено, поскольку дефекты небного генеза приводят к расщелине неба - распространенному врожденном дефекту, который возникает в единичных случаях или как часть сотен синдромов1,2. Развитие эмбрионального неба является динамическим процессом, который включает в себя движение и слияние эмбриональной ткани. Этот процесс можно разделить на четыре основных этапа: 1) индукция небных полок, 2) вертикальный рост небных полок рядом с языком, 3) возвышение небных полок над языком и 4) слияние небных полок на средней линии1,3,4. За последние несколько десятилетий у многих мышиных мутантов были выявлены проявления расщелины неба5,6,7,8. Характеристика этих моделей показала дефекты на этапах индукции, пролиферации и слияния небной полки; однако дефекты высоты небных шельфов были редкими. Таким образом, понимание динамики возвышения небного шельфа является интригующей областью исследований.
Тщательный анализ некоторых мышиных мутантов с дефектами высоты небной полки привел к текущей модели, показывающей, что сама передняя область небной полки, по-видимому, переворачивается вверх, в то время как вертикальное к горизонтальному движению или «ремоделирование» небных полок происходит в средней и задней областях неба1,3,4, 9,10,11. Эпителий медиальной кромки небной полки, вероятно, инициирует передачу сигналов, необходимую для этого ремоделирования, которое затем приводится в действие мезенхимой небной полки. В последнее время многие исследователи выявили задержку подъема небной полки в мышиных моделях, которые показали преходящие пероральные спайки с участием небных полок12,13. Мезенхимальное ремоделирование предполагает реорганизацию клеток для создания выпуклости в горизонтальном направлении, одновременно втягивая небную полку в вертикальном направлении9,10,14. Среди нескольких механизмов, предложенных для воздействия на высоту небной полки и лежащее в основе мезенхимальное ремоделирование, - пролиферация клеток15,16,17,хемотаксические градиенты18и компоненты внеклеточного матрикса19,20. Возник важный вопрос: наблюдается ли задержка подъема небной полки у мышей с дефицитом Specc1l такжечастично из-за дефекта ремоделирования небной полки, и может ли этот дефект ремоделирования проявляться во внутреннем дефекте поведения первичных клеток MEPM21?
Первичные клетки MEPM использовались в черепно-лицевой области для многих исследований, включающих экспрессию генов22,23,24,25,26,27,28,29,и несколько, включающих пролиферацию30,31 и миграцию25,31,32 , но не для коллективного анализа поведения клеток. Покадровую визуализацию клеток MEPM проводили в 2D-культуре и раневосточных анализах, чтобы показать, что клетки MEPM демонстрируют направленное движение и формируют зависящие от плотности клеточные потоки-атрибуты коллективного движения21. Кроме того, мутантные клетки Specc1l образовывали более узкие клеточные потоки и демонстрировали сильно изменчивые траектории миграции клеток. Считается, что это отсутствие скоординированной подвижности способствует задержке подъема неба у мутантных эмбрионов Specc1l 13,21. Таким образом, эти относительно простые анализы с использованием первичных клеток MEPM могут служить прокси для изучения ремоделирования мезенхимальных веществ во время подъема небного шельфа. В этой статье описывается изоляция и культура первичных клеток MEPM, а также покадровая визуализация и анализ для 2D и ран-восстановительных анализов.
протокол
Все эксперименты с участием животных проводились с протоколом, утвержденным Комитетом по уходу и использованию животных KUMC, в соответствии с их руководящими принципами и правилами (Номер протокола: 2018-2447).
1. Соберите эмбрионы E13.5
- Усыпляйте беременных самок мышей с помощью ингаляционной камерыCO2 или по процедуре, одобренной Институциональным комитетом по уходу за животными и их использованию. Немедленно приступайте к рассечению.
- Обнажить нижнюю половину брюшной полости путем удаления кожи и брюшины. Иссечение обоих рогов матки, которые содержат эмбрионы Е13.5.
- Кратковременно поместите матку в предварительно распавленный 37 °C стерильный фосфат-буферный физиологический раствор (PBS), чтобы смыть избыток крови, волос или другого мусора. Поместите матку в стерильную 10 см посуду, наполненную стерильным PBS.
- Используя небольшие ножницы, прорежьте стенку матки по всей длине матки, чтобы обнажить каждый эмбрион, все еще на свою желтковую мешок. Удалите желточный мешок, окружающий эмбрион, но сохраните его для генотипирования, если это необходимо. По мере удаления эмбрионов поместите каждый эмбрион в свой собственный колодец из 12-скважинной пластины, заполненной PBS.
2. Рассечение небных полок из эмбрионов(Рисунок 1)
ПРИМЕЧАНИЕ: Стерилизуйте инструменты для рассечения нержавеющей стали (см. Таблицу материалов)после обработки каждого эмбриона, помещая инструменты сначала в замок со 100% этиловым спиртом (EtOH), затем в инструментальный стерилизатор при 350 °C в течение 10 с, а затем охлаждая их во втором якере 100% EtOH.
- Используя стерилизованную перфорированную ложку, поместите эмбрион в новую 10-сантиметровую посуду, наполненную питательной средой MEPM, состоящей из минимальной необходимой среды Dulbecco (DMEM), содержащей 10% фетальной бычий сыворотки (FBS), L-глутамина (4 мМ L-Glu) и антибиотиков-пенициллина и стрептомицина (50 единиц / мл).
- Обезглавить эмбрион прямо под линией челюсти стерильными ножницами(рисунок 1А,красная пунктирная линия). Удалите нижнюю челюсть, вставив одну точку стерилизованных тонких щипцов No 5 в рот, удерживая ее только внутри щеки. Протолкните точку вставленных щипцов до тех пор, пока она не выйдет из задней части черепа.
- Наведите щипцы вдоль желтой линии на рисунке 1Bтак, чтобы другая сторона щипцов (которая все еще находится вне эмбриона) парила прямо над ушным каналом, затем зажмите щипцы, чтобы разрезать ткань. При необходимости провести еще один тонкий щипц по шву теперь уже закрытых щипцов, чтобы разрезать любую ткань, которая не была полностью разорвана щепоткой.
- Повторите предыдущий шаг для другой стороны головки эмбриона. Продолжайте процедуру щипки, чтобы полностью удалить нижнюю челюсть, язык и нижнюю часть черепа и обнажить небные полки.
- Удалите череп черепа, разрезав чуть выше глаз, как показано на рисунке 1C (зеленая линия). Сделайте это, поместив голову на левую или правую сторону и расположив точки небольших ножниц из нержавеющей стали перед и позади черепа чуть выше уровня глаз эмбриона. Отрежьте верхнюю часть черепа одним быстрым ножницами, создав плоскую поверхность, которая будет важна для стабильности на последующих этапах и которая должна выглядеть как рисунок 1D при взгляде сбоку.
- Поместите оставшуюся часть головы вверх ногами, при этом верхний аспект головы (череп удален) лежит ровно на дне тарелки, что обеспечит стабильную поверхность для удаления небной полки. Найдите минутку, чтобы определить небные полки, которые теперь выставлены и обращены вверх и будут выглядеть как два приподнятых гребня по обе стороны от центральной канавки в передней половине головы(рисунок 1E).
- Прикрепить оставшуюся часть головки к блюду, чтобы обездвижить ее, пока полки удалены. Сделайте это, вставив одну точку тонкого щипца через ткань около носовой области головы, с передней стороны к небным полкам, а другую точку щипцов вставьте через основание черепа, с задней части небных полок. Держите их на месте во время выполнения иссечения небных полок.
- Обездвижив головку одной рукой, выберите любую из двух полок для удаления первой, и вставьте обе точки второй пары тонких щипцов в ткань у основания боковой поверхности полки и защемление, чтобы разрезать ткань(рисунок 1F). Повторите это вдоль основания медиаль-поверхности полки, а затем на переднем и заднем концах полки, чтобы отсоедиться от головки.
- Аккуратно приподнимите полку, сделав дополнительные щипки, по мере необходимости, чтобы полностью освободить полку от окружающих тканей.
- Повторите предыдущие два шага, чтобы удалить вторую небную полку.
- Теперь, когда небные полки освобождены от окружающих тканей и помещены в PBS(рисунок G),используйте стерильную пластиковую лампу-переноску-пипетку, чтобы вытянуть полки в пипетке и перенести их в микроцентрифужную трубку объемом 1,5 мл вместе с примерно 500 мкл PBS. Держите трубки, содержащие небные полки, на льду, так как остальная часть подстилки обрабатывается таким же образом.
ПРИМЕЧАНИЕ: В качестве альтернативы, полки могут быть помещены в микроцентрифужную трубку 1,5 мл, содержащую предварительно расплавленный трипсин (0,25%) сразу после рассечения (вместо того, чтобы помещать их на лед). Образцы будут более свежими, но необходимо позаботиться о том, чтобы вовремя выполнить все дальнейшие шаги для каждого отдельного образца, а не обрабатывать образцы коллективно.
3. Культура клеток MEPM
ПРИМЕЧАНИЕ: В условиях, описанных здесь, эпителиальные клетки неба не выживают при первом прохождении, что приводит к чистой культуре мезенхимальных клеток неба. Используйте стерильную технику для выполнения всех шагов в вытяжке культуры тканей.
- Аспирировать и выбросить PBS из трубки 1,5 мл, заботясь о том, чтобы не выбросить полки в процессе. Немедленно добавьте 200 мкл предварительного (37 °C) трипсина (0,25%) в каждую трубку, содержащую небные полки. Кратковременно пипеткой полок вверх и вниз в трипсине, используя наконечник пипетки 1000 мкл, чтобы ускорить трипсинизацию.
- Инкубировать пробирки в течение 5 мин при 37 °C, затем пипетировать каждый образец вверх и вниз снова, чтобы помочь разрушить ткань. Инкубируют трубки еще 5 мин при 37 °C и еще раз пипетируют вверх и вниз, чтобы завершить диссоциацию ткани.
ПРИМЕЧАНИЕ: Полки должны быть полностью или почти полностью диссоциированы и взвешены в трипсине без видимых кусков ткани. - Добавляйте 800 мкл мкл питательной среды MEPM (этап 2.1) в каждую пробирку 1,5 мл. Центрифугируют пробирку 1,5 мл при 200 × г в течение 5 мин для гранулирования клеток. Удалите супернатант и повторно суспендируют гранулу клеток в 1 мл питательной среды MEPM.
- Налечите клетки MEPM в 6-скважинную тканевую культурную пластину, содержащую культурную среду MEPM. Дайте клеткам прилипать к пластиковой поверхности в течение 12 ч при 37 °C в инкубаторе с 5% CO2.
ПРИМЕЧАНИЕ: После ночной инкубации подавляющее большинство (~ 90%) клеток прикрепляются. В этот момент прилипшения клеток будут выглядеть достаточно однородно, с треугольной или слегка вытянутой формой. - Меняйте среду каждый день, осторожно аспирируя старую среду и немедленно заменяя ее 1 мл теплого стерильного PBS без кальция или магния в течение ~ 1 мин. Аспирировать PBS и заменить 3 мл досовой питательной среды MEPM.
- Проходит клетки, как только они становятся 100% сливаются.
ПРИМЕЧАНИЕ: Клетки MEPM должны размножаться, удваивая их количество почти ежедневно.- Чтобы пройти клетки, аккуратно аспирируйте старую среду и немедленно замените ее теплым PBS без кальция или магния в течение ~ 1 мин. Аспирировать PBS и заменить его 0,5 мл предварительного 0,25% трипсина.
- Инкубировать при 37 °C в течение ~5 мин или до тех пор, пока клетки не отсоедут от поверхности чашки при осторожном раскачиваниях вперед и назад вручную. Как только клетки отделятся, немедленно добавьте 5 мл предварительной культуральной среды MEPM к трипсинизированным клеткам.
- Используя серологический пипеток 10 мл, аккуратно соберите клетки в коническую трубку 15 мл и центрифугируйте трубку при 200 × г в течение 5 мин, чтобы гранулировать клетки. Аспирировать трипсин и среду и повторно суспендировать клетки в 3 мл предварительной питательной среды MEPM. Осторожно пипетируйте 1 мл клеток в одну скважину 6-скважинной чашки и добавьте 2 мл культуральной среды MEPM, чтобы довести общий объем до 3 мл.
ПРИМЕЧАНИЕ: Это представляет собой разделение клеток 1:3. MEPM могут быть проезжаемы до трех раз. Плотность посева MEPM несколько гибкая, а количество присутствующих клеток варьируется в зависимости от культурного сосуда. Тем не менее, MEPM не размножаются должным образом, когда они разделены слишком редко, и должны быть по крайней мере на 20-25% влиты в их новое блюдо, как только они прилипают.
4. Криоконсервация клеток MEPM
- После того, как трипсинизированные клетки MEPM гранулированы, повторно суспендируют клетки в культурально-питательной среде MEPM для получения концентрации ~ 1 ×10 6 клеток / мл. Пипетка клеток в криовиалы и добавление конечной концентрации 5% диметилсульфоксида в клеточный запас. Колпачок криовиала, ненадолго перемешайте путем инвертирования и немедленно поместите флаконы в морозильную емкость, которая охлаждается со скоростью 1 °C/мин.
- Поместите охладитель в морозильную камеру при -80 °C на ночь. На следующий день переместите криовиалы в резервуар для жидкого азота для длительного хранения.
- Оттаивание криоконсервных клеток MEPM
- Удалите криовиалы из резервуара с жидким азотом и разморозьте при комнатной температуре, пока содержимое не начнет становиться жидким. Опорожните содержимое в коническую трубку объемом 15 мл, содержащую 9 мл предварительной питательной среды MEPM.
- Центрифугирование трубки при 200 × г для гранулирования клеток. Повторно суспендировали клетки в 1 мл питательной среды MEPM и пипетировали клетки в одну скважину из 6-скважинной пластины. Добавьте 2 мл теплой питательной среды MEPM, чтобы довести общий объем до 3 мл.
- Культивьку клеток при 37 °C в инкубаторе с 5% CO2. Меняйте среду ежедневно.
5. Живая визуализация клеток MEPM - 2D коллективный миграционный анализ(рисунок 2)
- Подготовьте пластину для использования в реальном времени.
- Используйте небольшие хирургические ножницы или острый скальпель, чтобы укоротить стерильную 2-х скважинную силиконовую вставку до высоты ~1 мм. Подготовьте вставку для каждого используемого образца.
- Используя щипцы, поместите укороченную 2-скважинную силиконовую вставку в центр колодца 6-скважинной пластины. Прижмите по всем краям, чтобы убедиться, что он полностью приклеен.
- Оттаивайте криоконсервные клетки, следуя шагам, описанным в разделе 4.3 настоящего протокола. Подсчитайте клетки MEPM и посыпяйте300 клеток/мм2 укороченной силиконовой вставки в общем объеме 40-50 мкл культуральной среды MEPM на скважину. Культивь на ночь при 37 °C в инкубаторе с 5% CO2.
- На следующий день подготовьтесь к покадровой съемке в реальном времени.
- Используйте фазово-контрастный микроскоп с инкубатором на сцене и возможностью автоматической визуализации. Добавить воду в резервуар инкубатора на сцене для уменьшения испарения питательной среды; установить температуру на 37 °C и CO2 на 5%. Дайте ~30 мин, чтобы влажность нарасти, прежде чем поместить 6-скважинную посуду в инкубатор на сцене.
- Используйте параметры, эквивалентные приведенным ниже, для покадровой визуализации.
- Выберите цель 4x, чтобы иметь большие поля зрения и фильтр фазовой контрастности.
ПРИМЕЧАНИЕ: Автофокус, образец автоматического поиска, z-стек и автоматическое освещение обычно не нужны. - Выберите два микроскопических поля на лунку, чтобы захватить просвет укороченного силиконового вставок. Убедитесь, что все положения изображения имеют правильную фокусировку.
ПРИМЕЧАНИЕ: Плоскости изображения могут быть отрегулированы во время визуализации, но такая настройка обычно не требуется. - Выберите нужный тип выходного файла изображения, затем выберите или откажитесь от выбора параметров после создания изображения, таких как автоматическое создание видео и водяные знаки, по желанию. Если применимо, выберите фазовый контраст в качестве режима визуализации.
ПРИМЕЧАНИЕ: Использование водяных знаков может препятствовать последующим этапам обработки изображения. - Установите продолжительность записи на 72 ч. Настройте программу на захват изображений каждые 10 минут.
ПРИМЕЧАНИЕ: Обычно требуется только 48 ч визуализации, но визуализация может быть остановлена в любой момент до отметки 72 ч без потери изображений, которые уже были сделаны. - Убедитесь, что экологическая камера функционирует в соответствии с требованиями, предусмотренными в разделе 5.3.1. Сохраните этинастройки (рутину)и начните создание образа.
- Выберите цель 4x, чтобы иметь большие поля зрения и фильтр фазовой контрастности.
- Продолжайте съемку до 72 ч (или указанного времени).
6. Живая визуализация клеток MEPM в анализе восстановления ран(рисунок 3)
- Подготовьте пластину для использования в реальном времени. Используя щипцы, поместите стерильную силиконовую вставку из 2 скважин в центр колодца 6-скважинной пластины и прижмите по всем краям, чтобы она полностью приклеилась. Подготовьте одну 2-скважинную вставку для каждого используемого образца.
- Оттаивайте криоконсервные клетки, следуя шагам, описанным в разделе 4.3 настоящего протокола. Подсчитайте клетки MEPM и при необходимости сконцентрируйте клетки по меньшей мере до 350 клеток/мкл.
- Посейте 1400 клеток/мм2 в силиконовые вставки в объеме 100 мкл питательной среды MEPM на скважину. Культививляйте клетки в течение 48 ч при 37 °C в инкубаторе с 5%CO2и меняйте среду каждый день.
- Через 48 ч подготовьте микроскоп для покадровой визуализации в реальном времени, как описано в разделах 5.3 и 5.4. Непосредственно перед помещением клеток в инкубатор на сцене добавьте в колодец (но вне вкладыш) 3 мл предварительно распохлоенной культурального носителя MEPM, а затем аккуратно удалите силиконовые вставки.
ПРИМЕЧАНИЕ: Стенка, разделяющая 2 камеры, оставляет щель, которая является «раной». - Начните покадровую визуализацию, как описано в 5.4, со следующими отличиями:
- Используйте объектив с более высоким увеличением (например, 10x). Чтобы запечатлеть закрытие раны, выберите 5 полей зрения вдоль каждой раны, чтобы рана была параллельна вертикальной оси изображения.
- Прекратить визуализацию через 72 ч или когда раны полностью закрылись.
7. Вычислительный анализ покадровых последовательностей изображений
ПРИМЕЧАНИЕ: Выполните следующие процедуры на компьютере, оснащенном стандартными вычислительными инструментами, такими как интерпретатор python, компилятор C и оболочка (см. Таблицу материалов).
- Анализ слияний
ПРИМЕЧАНИЕ: Эта процедура может быть использована для оценки пролиферации клеток в разреженной культуре или для количественной оценки экспериментов по закрытию ран. Для обнаружения областей, занимаемых ячейками, к локальному стандартному отклонению яркости изображения применяется порог сегментации. Код был описан ранее Wu et al.33 и Neufeld et al.34 и доступен по адресу http://github.com/aczirok/cellconfluency.- Определите порог сегментации для изображений. Например, чтобы увидеть сегментацию с пороговым значением 4, выполните команду
segment.py -i inout-image.jpg -S 4 -тестовый выход.jpg
а затем проверьте вывод (output.jpg).
ПРИМЕЧАНИЕ: Если порог слишком низкий, фоновые области на микрофотографии классифицируются как покрытые ячейками. Напротив, если порог слишком высок, области, покрытые ячейками, не классифицируются как таковые. Оптимальное пороговое значение сводит обе ошибки к минимуму. - Используйте предоставленный сценарий area.sh для вычисления значений слияния последовательности изображений как
area.sh -S 4 img_001.jpg img_002.jpg .... > слияние.dat
ПРИМЕЧАНИЕ: Пороговое значение дискриминации 4 проверяется на шаге 1, и результаты сохраняются в файле слияния.dat. Сортируя изображения по соответствующим папкам, список имен файлов изображений можно заменить подстановочными знаками:
слияние area.sh -S4 *.jpg >.dat - Для экспериментов по закрытию раны преобразуйте данные о слиянии A(t)-размер площади, покрытой клетками, выраженный в процентах как функция времени- в скорость распространения раневой кромки V как

где w обозначает ширину поля, а dA/dt — производную по времени от A(t), т. е. скорость расширения площади, покрытой ячейкой.
- Определите порог сегментации для изображений. Например, чтобы увидеть сегментацию с пороговым значением 4, выполните команду
- Карта подвижности клеток
- Выполнение алгоритма велоциметрии изображения частиц (PIV) для характеристики подвижности клеток и извлечения степени локального движения «оптического потока» между парами изображений, но не для идентификации отдельных клеток.
ПРИМЕЧАНИЕ: Здесь используется начальный размер окна 50 мкм, как подробно описано Zamir et al.35 и Czirok et al.36,с начальным размером окна 50 мкм. Анализ PIV дает поле скорости v(x,t) для каждого кадра изображения t и местоположения (внутри изображения) x. - Извлеките среднюю скорость подвижности клеток из v(x,t) как пространственное среднее, рассчитанное по занимаемой клетке площади, как определено в разделе 7.1.
- Выполнение алгоритма велоциметрии изображения частиц (PIV) для характеристики подвижности клеток и извлечения степени локального движения «оптического потока» между парами изображений, но не для идентификации отдельных клеток.
- Ручное отслеживание ячеек
ПРИМЕЧАНИЕ: В то время как анализ PIV обеспечивает автоматическую оценку подвижности клеток, чтобы сосредоточиться на поведении отдельных клеток, часто требуется ручное отслеживание. Хотя несколько инструментов предоставляют эту функциональность, очень полезно, если маркеры, расположенные вручную, могут быть изменены после их первоначального размещения, и если отслеживание может быть выполнено как вперед, так и назад во времени.- Выполняйте отслеживание ячеек с помощью специально разработанного инструмента python (http://github.com/donnagreta/cm_track), который также предоставляет основные функции редактора, такие как удаление сегментов траектории.
ПРИМЕЧАНИЕ: Этот инструмент ручного отслеживания выдает позиции P(i,t) ячейки i в момент времени t в текстовом файле и вызывается как
cm_track.py -i изображения/ -o трек.dat
где покадровые изображения находятся в папке images/, а данные о положении собираются в дорожке файла.dat (рисунок 4).
- Выполняйте отслеживание ячеек с помощью специально разработанного инструмента python (http://github.com/donnagreta/cm_track), который также предоставляет основные функции редактора, такие как удаление сегментов траектории.

Рисунок 4:Анализ отдельных траекторий клеток. (A) Фазово-контрастные покадровые микроснимки подвергаются (B, C) ручной процедуре отслеживания, которая маркирует клетки (зеленые точки). (D) Положения ячеек (x,y) сохраняются для каждой ячейки, отличающейся ее идентификатором, и для каждого кадра f.(E)Траектории могут быть наложены на микроснимки и обозначены цветом для указания временной информации. Например, в каждой траектории сине-красная цветовая палитра указывает на постепенно более поздние сегменты траектории, причем красный и синий обозначают начальное и конечное расположение ячеек соответственно. (F)Различные статистические свойства траекторий, такие как среднее квадратное смещение, могут быть извлечены и использованы для характеристики подвижности различных клеточных популяций, которые в этом примере включают клетки дикого типа (wt, синий) и нокдаун (kd, красный) MEPM. Шкала представляет собой 100 мкм. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
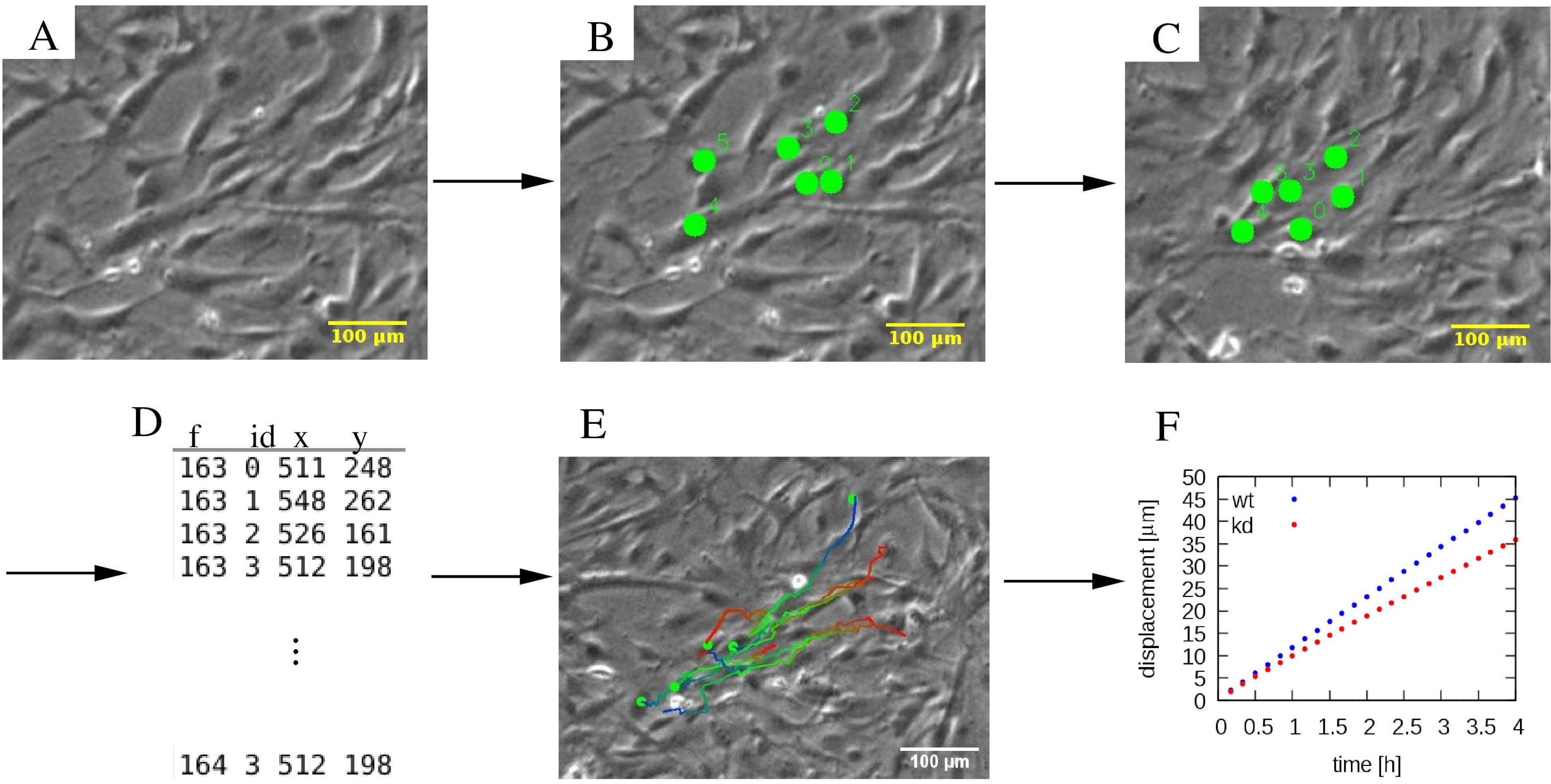{kind=link}
- Наложение траекторий на изображения с помощью второго инструмента, вызываемого как
visdat.py -d трек.dat -i изображения/ -o наложение/ -l999 -r3 -C2 - Соберите изображения с траекториями, наложенными в папку наложения.
ПРИМЕЧАНИЕ: В этом примере данные о положении ячейки хранятся в дорожке файла.dat, в то время как последовательность покадровых изображений находится в папке изображений. Остальные параметры управляют максимальной длиной нарисованных траекторий (-l), размером символов (-r) и используемой цветовой схемой (-C).
- Анализ индивидуальных траекторий клеток
- Характеристика траекторий по общей длине пути
 ,
,
и смещение сетки к ране ,
,
где X обозначает проекцию P в направлении, перпендикулярном ране: координата x положений, когда рана параллельна оси y. - Рассчитайте эффективность наведения как
 для каждой ячейки i, так и по времени t37. Охарактеризуйте культуры по популяции среднего значения этих одноклеточных мер, оцененных в подходящую точку времени t.
для каждой ячейки i, так и по времени t37. Охарактеризуйте культуры по популяции среднего значения этих одноклеточных мер, оцененных в подходящую точку времени t.
- Характеристика траекторий по общей длине пути
- Коллективное потоковое движение клеток
- Охарактеризуйте локальные пространственные корреляции движений клеток средней скоростью других клеток, которые находятся в непосредственной близости от движущейся клетки, как описаноранее 38,39.
ПРИМЕЧАНИЕ: Вычислительный код доступен по адресу https://github.com/aczirok/flowfield.- Выровняйте систему отсчета с направлениями спереди, сзади, влево и вправо к каждому вектору v(x,t) (рисунок 5A). Назначьте каждую из окружающих ячеек или вектор скорости PIV соответствующей пространственной ячейке системы отсчета(рисунок 5B). Повторите процедуру, так как каждый вектор служит источником систем отсчета(рисунок 5C, D), так что данный вектор скорости назначается нескольким бункерам.
ПРИМЕЧАНИЕ: Точка данных может находиться перед вектором и слева от другого. - Поверните системы отсчета в общую ориентацию(рисунок 5C,F)и объедините их(рисунок 5G).
ПРИМЕЧАНИЕ: Среднее значение каждого контейнера объединенных данных о скорости представляет собой вектор скорости(U),который указывает на пространственную корреляцию: среднее значение является мерой общей составляющей скорости(рисунок 5H). - Подогнать поля потока U(x) с экспоненциальной
 функцией , где a, x0 и U0 являются параметрами соответствия. Из трех параметров подгонки сосредоточьтесь на x0, длине корреляции(рисунок 5I,J),которая является характерным расстоянием, на котором исчезают локальные корреляции скорости и скорости.
функцией , где a, x0 и U0 являются параметрами соответствия. Из трех параметров подгонки сосредоточьтесь на x0, длине корреляции(рисунок 5I,J),которая является характерным расстоянием, на котором исчезают локальные корреляции скорости и скорости.
- Выровняйте систему отсчета с направлениями спереди, сзади, влево и вправо к каждому вектору v(x,t) (рисунок 5A). Назначьте каждую из окружающих ячеек или вектор скорости PIV соответствующей пространственной ячейке системы отсчета(рисунок 5B). Повторите процедуру, так как каждый вектор служит источником систем отсчета(рисунок 5C, D), так что данный вектор скорости назначается нескольким бункерам.
- Охарактеризуйте локальные пространственные корреляции движений клеток средней скоростью других клеток, которые находятся в непосредственной близости от движущейся клетки, как описаноранее 38,39.

Рисунок 5:Характеристика формирования потока культивированных клеток. (A,D) Фазоконтрастные покадровые изображения с рисунка 4А используются для идентификации движений клеток. Для каждой движущейся ячейки система отсчета (синий) и пространственные бункеры (белый) были выровнены, чтобы классифицировать соседние ячейки как на передние, задние, левые или правые. (Б,Э) Скорость соседних ячеек (черных векторов) была связана с одной и той же системой отсчета(C,F). Эта процедура повторялась для каждой клетки и точки времени. (G) После объединения этой локальной информации каждый бункер будет содержать несколько векторов скорости (серый), которые могут быть усреднены для определения средней скорости совместного перемещения (пурпурные стрелки) в различных местах относительно средней подвижной клетки. Такимобразом, карта средних скоростей характеризует типичные скорости ячейки в различных местах относительно движущейся ячейки. (I,J) Наконец, это поле было отобрано вдоль передне-задней (параллельной) оси, а также вдоль оси левый-правый (перпендикулярный). Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
{kind=link}
Результаты
Рассечение небных полок проиллюстрировано на рисунке 1. Последовательность разрезов предназначена для минимизации проскальзывания ткани. После удаления головы(Фиг.1А,В)нижняя челюсть удаляется(Фиг.1В,С). Разрез верхней...
Обсуждение
Возвышение небного шельфа представляет собой вертикальное и горизонтальное событиереконструкции 1,3,4,9,11. Постулируется, что этот процесс ремоделирования требует, чтобы клетки мезенхимального лож...
Раскрытие информации
Авторам нечего раскрывать.
Благодарности
Этот проект был частично поддержан грантами Национальных институтов здравоохранения DE026172 (I.S.) и GM102801 (A.C.). I.S. также частично поддерживался грантом Центра передовых биомедицинских исследований (COBRE) (Национальный институт общих медицинских наук P20 GM104936), грантом Kansas IDeA Network for Biomedical Research Excellence (Национальный институт общих медицинских наук P20 GM103418) и грантом Канзасского исследовательского центра интеллектуальных нарушений и нарушений развития (KIDDRC) (U54 Eunice Kennedy Shriver National Institute of Child Health and Human Development, HD090216).
Материалы
| Name | Company | Catalog Number | Comments |
| Beaker, 250 mL (x2) | Fisher Scientific | FB-100-250 | |
| CO2 | Matheson Gas | UN1013 | |
| Conical tubes, 15 mL (x1) | Midwest Scientific | C15B | |
| Debian operating system | computational analysis of time-lapse images | ||
| Dulbecco's Modified Eagles Medium/High Glucose with 4 mM L-Glutamine and Sodium Pyruvate | Cytiva Life Sciences | SH30243.01 | |
| EtOH, 100% | Decon Laboratories | 2701 | |
| EVOS FL Auto | ThermoFisher Scientific | AMAFD1000 | |
| EVOS Onstage Incubator | ThermoFisher Scientific | AMC1000 | |
| EVOS Onstage Vessel Holder, Multi-Well Plates | ThermoFisher Scientific | AMEPVH028 | |
| Fetal Bovine Serum | Corning | 35-010-CV | |
| Fine point #5 Stainless Steel Forceps (x2) | Fine Science Tools | 11295-10 | Dissection |
| Instrument sterilizer bead bath | Fine Science Tools | 18000-45 | |
| Microcetrifuge tubes, 1.5mL | Avant | 2925 | |
| Micro-Dissecting Stainless Steel Scissors, Straight | Roboz | RS-5910 | Dissection |
| NucBlue (Hoechst) Live Ready Probes | ThermoFisher Scientific | R37605 | |
| Penicillin Streptomycin Solution, 100x | Corning | 30-002-CI | |
| Silicone Insert, 2-well | Ibidi | 80209 | |
| Small Perforated Stainless Steel Spoon | Fine Science Tools | MC17C | Dissection |
| Spring Scissors, 4 mm | Fine Science Tools | 15018-10 | |
| Sterile 10 cm dishe(s) | Corning | 430293 | |
| Sterile 12-well plate(s) | PR1MA | 667512 | |
| Sterile 6-well plate(s) | Thermo Fisher Scientific | 140675 | |
| Sterile PBS | Corning | 21-031-CV | |
| Sterile plastic bulb transfer pipette | ThermoFisher Scientific | 202-1S | |
| Trypsin, 0.25% | ThermoFisher Scientific | 25200056 |
Ссылки
- Bush, J. O., Jiang, R. Palatogenesis: morphogenetic and molecular mechanisms of secondary palate development. Development. 139 (2), 231-243 (2012).
- Mossey, P. A., Little, J., Munger, R. G., Dixon, M. J., Shaw, W. C. Cleft lip and palate. Lancet. 374 (9703), 1773-1785 (2009).
- Lan, Y., Xu, J., Jiang, R. Cellular and molecular mechanisms of palatogenesis. Current Topics in Developmental Biology. 115, 59-84 (2015).
- Li, C., Lan, Y., Jiang, R. Molecular and cellular mechanisms of palate development. Journal of Dental Research. 96 (11), 1184-1191 (2017).
- Gritli-Linde, A. The etiopathogenesis of cleft lip and cleft palate: usefulness and caveats of mouse models. Current Topics in Developmental Biology. 84, 37 (2008).
- Meng, L., Bian, Z., Torensma, R., Vonden Hoff, J. W. Biological mechanisms in palatogenesis and cleft palate. Journal of Dental Research. 88 (1), 22-33 (2009).
- Dixon, M. J., Marazita, M. L., Beaty, T. H., Murray, J. C. Cleft lip and palate: understanding genetic and environmental influences. Nature Reviews Genetics. 12 (3), 167-178 (2011).
- Kousa, Y. A., Schutte, B. C. Toward an orofacial gene regulatory network. Developmental Dynamics. 245 (3), 220-232 (2016).
- Jin, J. Z., et al. Mesenchymal cell remodeling during mouse secondary palate reorientation. Developmental Dynamics. 239 (7), 2110-2117 (2010).
- Yu, K., Ornitz, D. M. Histomorphological study of palatal shelf elevation during murine secondary palate formation. Developmental Dynamics. 240 (7), 1737-1744 (2011).
- Chiquet, M., Blumer, S., Angelini, M., Mitsiadis, T. A., Katsaros, C. Mesenchymal remodeling during palatal shelf elevation revealed by extracellular matrix and F-actin expression patterns. Frontiers in Physiology. 7, 392 (2016).
- Paul, B. J., et al. ARHGAP29 mutation is associated with abnormal oral epithelial adhesions. Journal of Dental Research. 96 (11), 1298-1305 (2017).
- Hall, E. G., et al. SPECC1L regulates palate development downstream of IRF6. Human Molecular Genetics. 29 (5), 845-858 (2020).
- Walker, B. E., Fraser, F. C. Closure of the secondary palate in three strains of mice. Journal of Embryology and Experimental Morphology. 4 (2), 176-189 (1956).
- Jin, J. Z., Li, Q., Higashi, Y., Darling, D. S., Ding, J. Analysis of Zfhx1a mutant mice reveals palatal shelf contact-independent medial edge epithelial differentiation during palate fusion. Cell Tissue Research. 333 (1), 29-38 (2008).
- Kouskoura, T., et al. The etiology of cleft palate formation in BMP7-deficient mice. PLoS One. 8 (3), 59463 (2013).
- Lan, Y., Zhang, N., Liu, H., Xu, J., Jiang, R. Golgb1 regulates protein glycosylation and is crucial for mammalian palate development. Development. 143 (13), 2344-2355 (2016).
- He, F., et al. Wnt5a regulates directional cell migration and cell proliferation via Ror2-mediated noncanonical pathway in mammalian palate development. Development. 135 (23), 3871-3879 (2008).
- Lan, Y., Qin, C., Jiang, R. Requirement of hyaluronan synthase-2 in craniofacial and palate development. Journal of Dental Research. 98 (12), 1367-1375 (2019).
- Yonemitsu, M. A., Lin, T. Y., Yu, K. Hyaluronic acid is required for palatal shelf movement and its interaction with the tongue during palatal shelf elevation. Developmental Biology. 457 (1), 57-68 (2020).
- Goering, J. P., et al. SPECC1L-deficient palate mesenchyme cells show speed and directionality defect. Scientific Reports. 11 (1), 1452 (2021).
- Fantauzzo, K. A., Soriano, P. PI3K-mediated PDGFRalpha signaling regulates survival and proliferation in skeletal development through p53-dependent intracellular pathways. Genes and Development. 28 (9), 1005-1017 (2014).
- Vasudevan, H. N., Soriano, P. SRF regulates craniofacial development through selective recruitment of MRTF cofactors by PDGF signaling. Developmental Cell. 31 (3), 332-344 (2014).
- Vasudevan, H. N., Mazot, P., He, F., Soriano, P. Receptor tyrosine kinases modulate distinct transcriptional programs by differential usage of intracellular pathways. Elife. 4, 07186 (2015).
- Gao, L., et al. 2,3,7,8-Tetrachlorodibenzo-p-dioxin and TGFbeta3-mediated mouse embryonic palatal mesenchymal cells. Dose Response. 17 (1), 1559325818786822 (2019).
- Iyyanar, P. P. R., Nazarali, A. J. Hoxa2 inhibits bone morphogenetic protein signaling during osteogenic differentiation of the palatal mesenchyme. Frontiers in Physiology. 8, 929 (2017).
- Jiang, Z., Pan, L., Chen, X., Chen, Z., Xu, D. Wnt6 influences the viability of mouse embryonic palatal mesenchymal cells via the beta-catenin pathway. Experimental and Therapeutic Medicine. 14 (6), 5339-5344 (2017).
- Liu, X., et al. Negative interplay of retinoic acid and TGF-beta signaling mediated by TG-interacting factor to modulate mouse embryonic palate mesenchymal-cell proliferation. Birth Defects Research Part B: Developmental and Reproductive Toxicology. 101 (6), 403-409 (2014).
- Bush, J. O., Soriano, P. Ephrin-B1 forward signaling regulates craniofacial morphogenesis by controlling cell proliferation across Eph-ephrin boundaries. Genes & Development. 24 (18), 2068-2080 (2010).
- Mo, J., Long, R., Fantauzzo, K. A. Pdgfra and Pdgfrb genetically interact in the murine neural crest cell lineage to regulate migration and proliferation. Frontiers in Physiology. 11, 588901 (2020).
- He, F., Soriano, P. A critical role for PDGFRalpha signaling in medial nasal process development. PLoS Genetics. 9 (9), 1003851 (2013).
- Fantauzzo, K. A., Soriano, P. Generation of an immortalized mouse embryonic palatal mesenchyme cell line. PLoS One. 12 (6), 0179078 (2017).
- Wu, K., Gauthier, D., Levine, M. D. Live cell image segmentation. IEEE Transactions on Biomedical Engineering. 42 (1), 1-12 (1995).
- Neufeld, Z., et al. The role of Allee effect in modelling post resection recurrence of glioblastoma. PLoS Computational Biology. 13 (11), 1005818 (2017).
- Zamir, E. A., Czirok, A., Rongish, B. J., Little, C. D. A digital image-based method for computational tissue fate mapping during early avian morphogenesis. Annals of Biomedical Engineering. 33 (6), 854-865 (2005).
- Czirok, A., et al. Optical-flow based non-invasive analysis of cardiomyocyte contractility. Scientific Reports. 7 (1), 10404 (2017).
- Biggs, L. C., et al. Interferon regulatory factor 6 regulates keratinocyte migration. Journal of Cell Science. 127, 2840-2848 (2014).
- Czirok, A., Varga, K., Mehes, E., Szabo, A. Collective cell streams in epithelial monolayers depend on cell adhesion. New Journal of Physics. 15, 75006 (2013).
- Szabo, A., et al. Collective cell motion in endothelial monolayers. Physical Biology. 7 (4), 046007 (2010).
- Gulyas, M., Csiszer, M., Mehes, E., Czirok, A. Software tools for cell culture-related 3D printed structures. PLoS One. 13 (9), 0203203 (2018).
- Soderholm, J., Heald, R. Scratch n' screen for inhibitors of cell migration. Chemistry & Biology. 12 (3), 263-265 (2005).
- Riahi, R., Yang, Y., Zhang, D. D., Wong, P. K. Advances in wound-healing assays for probing collective cell migration. Journal of Laboratory Automation. 17 (1), 59-65 (2012).
- Svensson, C. M., Medyukhina, A., Belyaev, I., Al-Zaben, N., Figge, M. T. Untangling cell tracks: Quantifying cell migration by time lapse image data analysis. Cytometry Part A. 93 (3), 357-370 (2018).
- Fantauzzo, K. A., Soriano, P. PDGFRbeta regulates craniofacial development through homodimers and functional heterodimers with PDGFRalpha. Genes & Development. 30 (21), 2443-2458 (2016).
- Rafi, S. K., et al. Anti-epileptic drug topiramate upregulates TGFβ1 and SOX9 expression in primary embryonic palatal mesenchyme cells: Implications for teratogenicity. PLoS ONE. , (2021).
Перепечатки и разрешения
Запросить разрешение на использование текста или рисунков этого JoVE статьи
Запросить разрешениеСмотреть дополнительные статьи
This article has been published
Video Coming Soon
Авторские права © 2025 MyJoVE Corporation. Все права защищены