
Method Article
Visualizzazione del traffico citoscheletro-dipendente di organelli contenenti lipidi in embrioni di Drosophila
In questo articolo
Riepilogo
Nel primo embrione di Drosophila , molti organelli sono mobili. In linea di principio, possono essere ripresi dal vivo tramite specifiche sonde fluorescenti, ma il guscio d'uovo impedisce l'applicazione diretta all'embrione. Questo protocollo descrive come introdurre tali sonde tramite microiniezione e quindi analizzare il movimento degli organelli di massa tramite velocimetria dell'immagine delle particelle .
Abstract
Gli embrioni precoci di Drosophila sono grandi cellule contenenti una vasta gamma di organelli convenzionali ed embrionali specifici. Durante le prime tre ore di embriogenesi, questi organelli subiscono movimenti drammatici alimentati dallo streaming citoplasmatico a base di actina e dal traffico motorio lungo i microtubuli. Lo sviluppo di una moltitudine di piccole sonde fluorescenti (FP) specifiche per organelli consente di visualizzare una vasta gamma di diverse strutture contenenti lipidi in qualsiasi genotipo, consentendo l'imaging dal vivo senza richiedere un fluoroforo geneticamente codificato. Questo protocollo mostra come iniettare coloranti vitali e sonde molecolari negli embrioni di Drosophila per monitorare il traffico di organelli specifici mediante imaging dal vivo. Questo approccio è dimostrato etichettando le goccioline lipidiche (LD) e seguendo il loro movimento di massa mediante velocimetria dell'immagine delle particelle (PIV). Questo protocollo fornisce una strategia suscettibile allo studio di altri organelli, tra cui lisosomi, mitocondri, vescicole di tuorlo e ER, e per tracciare il movimento dei singoli LD lungo i microtubuli. L'uso di coloranti disponibili in commercio porta i benefici della separazione nelle regioni viola / blu e rosso lontano dello spettro. Mediante la co-etichettatura multiplex di organelli e/o elementi citoscheletrici tramite microiniezione, tutte le risorse genetiche in Drosophila sono disponibili per studi di traffico senza la necessità di introdurre proteine marcate in modo fluorescente. A differenza dei fluorofori geneticamente codificati, che hanno basse rese quantistiche e candeggina facilmente, molti dei coloranti disponibili consentono la cattura rapida e simultanea di diversi canali con elevate rese di fotoni.
Introduzione
I coloranti vitali e le sonde molecolari sono potenti strumenti per l'immagine di specifiche strutture cellulari e organelli vivi. Nell'embrione di Drosophila, molti organelli diversi mostrano la localizzazione guidata dal citoscheletro 1,2,3,4, ma l'applicazione di queste piccole molecole è impegnativa perché il guscio d'uovo è impermeabile a molti di loro. Questo protocollo descrive un metodo per utilizzare sonde fluorescenti (FP) in embrioni vivi tramite microiniezione al fine di rilevare il traffico su larga scala di organelli. La procedura comprende la preparazione della soluzione di iniezione, la raccolta degli ovuli e la preparazione degli embrioni, la microiniezione, l'imaging e l'analisi delle immagini.
Drammatici riarrangiamenti spaziali di organelli sono comuni in molti ovociti, uova ed embrioni animali, in parte a causa delle grandi dimensioni di queste cellule. Nell'embrione di Drosophila , ad esempio, le goccioline lipidiche (LD) e le vescicole del tuorlo si muovono verso il centro embrionale poco prima della cellularizzazione5. Questo movimento dipende dai microtubuli e lascia una regione di ~ 40 μm intorno alla periferia dell'embrione impoverito dei due organelli. Durante le prime fasi di scissione, molti organelli vengono trasportati da flussi citoplasmatici che sono guidati da contrazioni a base di actina-miosina sulla superficie dell'embrione6. Sebbene gli embrioni di molte specie mostrino riarrangiamenti simili, l'embrione di Drosophila è particolarmente adatto per seguire questi processi mediante imaging perché si sviluppa esternamente a "temperatura ambiente" di laboratorio standard, è relativamente trasparente, abbastanza piccolo da adattarsi alla maggior parte delle configurazioni del microscopio e può essere manipolato utilizzando potenti strumenti genetici.
Per alcuni organelli, sono disponibili proteine marcate fluorescentemente che etichettano specificamente queste strutture. Ad esempio, LSD-2 (noto anche come dPLIN2) è una proteina che negli embrioni si rivolge specificamente alla LD7. Sono disponibili linee volanti che trasportano transgeni inducibili che codificano per una fusione tra Green Fluorescent Protein (GFP) e LSD-28 o una trappola genica in cui la proteina fluorescente gialla (YFP) viene inserita nella regione codificante del gene endogeno LSD-2 9,10. Tuttavia, questo approccio ha dei limiti, incluso il fatto che queste proteine di fusione hanno basse rese quantistiche e tendono a sbiancare facilmente. Inoltre, etichettare più strutture diverse contemporaneamente può essere difficile: per molti organelli, è attualmente disponibile un solo tipo di tag fluorescente (spesso GFP o mCherry), quindi l'imaging di due organelli contemporaneamente potrebbe richiedere nuovi transgeni o inserzioni; inoltre, anche se sono disponibili tag compatibili, introdurli in un singolo ceppo può richiedere incroci che richiedono tempo. Rende anche meno conveniente l'uso delle molte potenti risorse genetiche, ad esempio, se due marcatori di organelli, un driver Gal4 e un costrutto RNAi inducibile devono essere presenti nella stessa madre.
In linea di principio, queste limitazioni possono essere superate con l'uso di FP, inclusi coloranti vitali (ad esempio, LysoTracker per contrassegnare i lisosomi), sonde molecolari (ad esempio, SiR-tubulina per etichettare i microtubuli) e molecole biologiche marcate fluorescentemente (ad esempio, C12 BODIPY per sondare il metabolismo degli acidi grassi11). Dall'uso in cellule coltivate, sono in genere ben convalidati come potenti strumenti per sondare la biologia cellulare. Gli FP sono versatili, hanno proprietà fotografiche superiori e sono compatibili con le proteine fluorescenti. Più coloranti possono essere miscelati e applicati contemporaneamente, spesso con i benefici della separazione nelle aree viola / blu e rosso lontano dello spettro e piccoli spostamenti di Stokes, impedendo il sanguinamento del canale. Piccoli spostamenti stokes consentono l'acquisizione simultanea di più canali di imaging, consentendo il tracciamento di più organelli contemporaneamente. Infine, possono anche essere applicati all'etichettatura degli organelli negli embrioni di altre specie di Drosophila o anche di altri insetti in cui non possono essere disponibili proteine marcate fluorescentemente.
Tuttavia, la maggior parte di questi FP non può attraversare l'elaborato guscio d'uovo dell'embrione di Drosophila . Si compone di cinque strati: tre strati coriali esterni (corion) che prevengono danni meccanici più uno strato ceroso che circonda la membrana vitellina che crea una barriera chimica12. Per semplicità, la combinazione dello strato ceroso e della membrana vitellina sarà indicata come la "membrana vitellina" di seguito. Per bypassare il guscio d'uovo, questo protocollo adatta un approccio di microiniezione embrionale stabilito per introdurre FP nell'embrione di Drosophila . Il protocollo descrive come monitorare il flusso citoplasmatico di LD negli embrioni in fase di scissione. Comprende la preparazione di aghi per iniezione e gabbie per la raccolta delle uova, il processo di raccolta delle uova e la rimozione meccanica del corion. Esamina come microiniettare e visualizzare gli embrioni e come analizzare il flusso di massa di LD usando la velocimetria dell'immagine delle particelle (PIV, adattata da6). Fornisce consigli sulla risoluzione dei problemi per garantire la sopravvivenza degli embrioni e creare il miglior sistema per l'imaging. Si discute anche di come il protocollo possa essere modificato per visualizzare contemporaneamente LD e microtubuli o per applicarlo allo studio di altri organelli, inclusi lisosomi, mitocondri, vescicole di tuorlo e reticolo endoplasmatico (ER).
Protocollo
1. Preparare i materiali necessari
NOTA: questi preparativi sono fatti meglio giorni o settimane prima del tempo.
- Preparare gli aghi per iniezione.
NOTA: gli aghi possono essere conservati a tempo indeterminato in un contenitore coperto. Gli aghi devono essere abbastanza sottili da erogare ~ 700 fL pur essendo abbastanza forti da perforare la membrana vitellina.- Posizionare un capillare nel supporto capillare sull'estrattore dell'ago. Fissare il capillare con i dadi alari. Allineare il centro del capillare con l'elemento riscaldante in modo da generare due aghi simmetrici.
- Scegliere le impostazioni appropriate sull'estrattore dell'ago in base alle istruzioni del produttore.
- Eseguire il controllo di qualità utilizzando un ambito di dissezione.
NOTA: le punte degli aghi devono essere il più sottili possibile. Le punte degli aghi incrinate, frastagliate o di grosso calibro vengono scartate. - Preparare lotti di 10 aghi (5 capillari) alla volta.
- Posizionare uno stucco deformabile, che è stato arrotolato in un cilindro, al centro del fondo di un contenitore con un coperchio. Premere con attenzione gli aghi nello stucco in modo che li tenga orizzontalmente con le punte dell'ago sospese in aria in modo sicuro per evitare danni. Coprire con il coperchio e conservare fino all'uso.
- Preparare i piatti di raccolta delle uova. Conservare a 4 °C.
NOTA: I piatti di raccolta delle uova sono piccoli piatti di agar integrati con succo di frutta per incoraggiare la deposizione delle uova. Come prepararli è descritto in molti protocolli, tra cui13. - Preparare la colla eptanica.
NOTA: Questa colla viene estratta dal nastro biadesivo e viene utilizzata per fissare saldamente gli embrioni a una copertina. Impedisce all'embrione di fluttuare fuori fuoco durante l'iniezione e l'imaging. La colla può essere preparata giorni o settimane prima del tempo.- Montare il nastro biadesivo a una dimensione maggiore di una pallina da golf e posizionarlo sul fondo di un contenitore di vetro da ~ 50 ml con un coperchio ermeticamente sigillato. Imballare il nastro strettamente in modo che sia presente abbastanza adesivo.
- In una cappa aspirante, riempire il contenitore di vetro con eptano per coprire tutto il nastro. Tenere il contenitore ben chiuso quando non in uso.
ATTENZIONE: L'eptano è infiammabile come liquido e vapore. Può causare irritazione agli occhi, alla pelle e alle vie respiratorie. I vapori respiratori possono causare sonnolenza, vertigini e danni ai polmoni. Tenere l'eptano lontano da fonti di accensione e conservarlo in un'area ben ventilata destinata agli infiammabili e lontano da sostanze incompatibili. - Lasciare riposare la colla eptanica durante la notte o più a lungo. Per ottenere i migliori risultati, posizionare il contenitore su uno shaker o un altro agitatore durante la notte per aiutare a sciogliere l'adesivo.
NOTA: il nastro stesso rimarrà, ma l'adesivo verrà sciolto nell'eptano. Nel passaggio 5.1.2, la colla viene applicata a una copertina; l'eptano evapora, lasciando la colla alle spalle. - Testare la preparazione della colla eptanica posizionandone una goccia su un vetrino e assicurandosi che rimanga un residuo appiccicoso una volta che l'eptano è evaporato. Se il residuo adesivo non è visibile in seguito, aggiungere altro nastro adesivo al contenitore di vetro e ripetere il passaggio precedente.
NOTA: Una volta preparata, la colla può essere utilizzata per mesi o addirittura anni.
- Preparare una soluzione madre BODIPY493/503 da 1 mg/mL (3,8 mM) diluendo la preparazione ottenuta commercialmente in DMSO anidro puro. Tienilo coperto per proteggerlo dalla luce e dall'acqua. Conservare a tempo indeterminato a -20 °C o a breve termine a 4 °C.
2. Preparare le gabbie per la raccolta delle uova
NOTA: Farlo almeno un giorno (preferibilmente 2 o 3 giorni) prima delle iniezioni pianificate.
- Preparare la pasta di lievito.
NOTA: La pasta di lievito integra proteine e altri nutrienti vitali non forniti nei piatti di succo di mela. Promuove la produzione di uova e la deposizione delle uova.- Aggiungere 2-10 g di lievito di birra secco e quindi 1 ml di acqua del rubinetto in un piccolo bicchiere. Mescolare usando una spatola.
- Continuare ad aggiungere acqua del rubinetto con incrementi di 1 ml fino a raggiungere la consistenza desiderata simile al dentifricio.
- Coprire il composto e conservarlo a 4 °C.
- Imposta gabbie per mosche per la raccolta delle uova.
NOTA: sono richieste mosche maschili e femminili del genotipo desiderato: 10-50 femmine di età inferiore a 2 settimane e un numero simile di maschi. Sono disponibili diverse opzioni per le gabbie per mosche, tra cui opzioni fatte in casa14,13. È importante che la dimensione dei piatti di succo di mela sia abbinata alle dimensioni delle gabbie di mosca.- Metti una piccola macchia di pasta di lievito su un piatto di succo di mela. Tienilo coperto e lascialo arrivare a temperatura ambiente perché le mosche non depongono le uova sul piatto se fa troppo freddo.
- Trasferisci le mosche in una gabbia di raccolta degli embrioni, sigilla con la piastra di succo di mela lievitata e fissa la piastra alla gabbia di raccolta degli embrioni.
- Lasciare che le mosche appena trasferite si acclimatino alla gabbia per 1-2 giorni, sostituendo la piastra con una fresca al giorno. Se tutto il lievito è stato mangiato entro il giorno successivo, aumentare la quantità di pasta di lievito per le raccolte future.
NOTA: Questo è un passo importante per aumentare la resa delle uova poiché le femmine ben nutrite depongono più uova.
3. Il giorno dell'iniezione, preparare la soluzione iniettabile e caricarla nell'ago
- Utilizzare la soluzione madre di BODIPY 493/503 (3,8 mM in DMSO) come soluzione iniettabile.
- Caricare un singolo ago con 1 μL della soluzione iniettabile utilizzando punte di carico dell'ago.
NOTA: Sforzati di portare il liquido fino alla punta senza intrappolare bolle d'aria. Essere pronti a sostituire rapidamente l'ago caricato in caso di rottura accidentale.- Attaccare una punta di carico a una micropipetta e aspirare 1 μL della soluzione iniettabile nella punta. Usando i guanti, tenere l'ago in una mano con la punta puntata verso il basso per ridurre al minimo il rischio di rottura.
- Inserire con attenzione la punta di carico nell'ago e spingerlo vicino alla punta dell'ago. Erogare il liquido vicino alla punta dell'ago. Dopo aver rimosso la pipetta, tenere l'ago verticalmente con la punta verso il basso fino a quando il liquido scorre verso la punta.
- Conservare gli aghi caricati in un contenitore separato con stucco come sopra (vedere punto 1.1.5).
NOTA: Gli aghi devono essere preparati in anticipo (fino a diverse ore) dall'iniezione, ma non devono essere utilizzati dopo più di un giorno. - Tenere gli aghi fuori dalla luce ambientale per evitare lo sbiancamento dei coloranti. Coprire il contenitore di stoccaggio con un foglio di alluminio senza danneggiare le punte dell'ago. Assicurati che tutta la luce sia oscurata.
4. Raccolta di embrioni per l'iniezione
NOTA: I tempi di raccolta dipendono dallo stadio degli embrioni in cui deve essere eseguita l'iniezione. Con lo schema temporale di seguito, gli embrioni al momento della preparazione per l'iniezione avranno 0-90 minuti, che corrisponde agli stadi di scissione15.
- Il giorno dell'iniezione, preparare piatti di succo di mela con lievito, come nel passaggio 2.2.1. Se sono previsti N cicli di iniezioni, preparare N + 2 piastre. Mantenere queste piastre a temperatura ambiente.
NOTA: Oltre alle piastre N per raccogliere gli embrioni per l'iniezione, una piastra è necessaria come piastra di pre-raccolta e un'altra per nutrire le mosche nella gabbia una volta completate le raccolte. - Sostituire la piastra sulla gabbia con una piastra lievitata fresca (piastra di pre-raccolta) e lasciarla sulla gabbia per 1-2 ore.
- Sostituire la piastra sulla gabbia con una piastra fresca (piastra di raccolta). Scartare la piastra di pre-raccolta, in quanto conterrà embrioni più vecchi del desiderato. Quindi, lasciare che le mosche depongano le uova per 1,5 ore.
NOTA: Poiché le mosche ben nutrite in genere depongono le uova poco dopo che le uova sono state fecondate, un tempo di raccolta di 1,5 ore assicura che alla fine della raccolta, la maggior parte degli embrioni sul piatto siano vecchi di 0-90 minuti, cioè siano in fase di scissione. Le mosche femmine che non sono state alimentate con lievito fresco dal giorno precedente tenderanno a trattenere le uova fecondate per un certo periodo di tempo indeterminato prima della deposizione. Quindi, la piastra di pre-raccolta può avere embrioni che sono stati fecondati prima dell'inizio della raccolta e quindi sono più vecchi di 60 minuti, a volte molto più vecchi. - Sostituire la piastra sulla gabbia con una piastra lievitata fresca. Coprire il piatto di raccolta in modo che le mosche vaganti non depongano le uova su di esse.
5. Preparare gli embrioni per la microiniezione
- Assemblare i materiali necessari.
- Attaccare un pezzo di nastro biadesivo a una diapositiva di vetro. Evitare di toccare il nastro con le dita in quanto ciò riduce la sua viscosità.
NOTA: questa diapositiva verrà utilizzata per rimuovere il corion e non per l'imaging. - Utilizzando una pipetta di trasferimento o una micropipetta (p200 o p1000), posizionare una piccola goccia (200 μL o meno) di colla eptanica all'incirca al centro di un coperchio rettangolare (60 x 25 mm). Lasciare evaporare l'eptano (questo richiede meno di un minuto).
NOTA: Questo coverslip verrà utilizzato per montare gli embrioni per l'iniezione e l'imaging. Le dimensioni sono scelte per adattarsi a un supporto metallico regolabile sul microscopio confocale. - Assemblare una camera di essiccazione, una camera sigillata (ad esempio, una scatola sandwich Tupperware) contenente perline di essiccazione. Utilizzare solo perline di essiccazione che non si sono idratate.
NOTA: Per garantire che gli embrioni assumano la soluzione iniettabile, l'embrione deve essere leggermente essiccato per ridurre la pressione interna. Questo viene fatto posizionando il coperchio con gli embrioni decorionati ed esposti all'aria nella camera di essiccazione.
- Attaccare un pezzo di nastro biadesivo a una diapositiva di vetro. Evitare di toccare il nastro con le dita in quanto ciò riduce la sua viscosità.
- Rimuovere meccanicamente il corion.
- Coprire la piastra embrionale opportunamente invecchiata con un sottile strato di Halocarbon Oil 27 per trasformare il guscio d'uovo trasparente.
NOTA: Gli embrioni diventano traslucidi entro decine di secondi15. Se questo passaggio non si verifica in modo efficiente, le piastre di succo di mela potrebbero essere troppo bagnate e devono essere asciugate prima dell'uso. - Visualizza la piastra sotto un telescopio di dissezione con transilluminazione (cioè la luce che attraversa la piastra negli oculari) per confermare lo stadio degli embrioni.
- Seleziona gli embrioni nelle fasi di scissione.
NOTA: gli embrioni dello stadio di scissione sono completamente opachi; i successivi stadi blastodermici possono essere riconosciuti da una banda di citoplasma trasparente tutto intorno al centro opaco15. Una buona introduzione su come riconoscere i vari stadi embrionali è disponibile sul sito web (dato nel riferimento16) gestito dalla Society of Developmental Biology. - Usando una pinzetta fine, afferrare un embrione dello stadio desiderato dalle sue appendici dorsali e trasferirlo sul vetrino preparato coperto da un pezzo di nastro biadesivo. Posizionare l'embrione sul nastro. Ridurre al minimo il trasferimento di olio.
- Il più delicatamente possibile, arrotolare l'embrione sulla superficie del nastro spingendo delicatamente l'embrione con il lato della punta della pinzetta. Non colpire l'embrione direttamente con le punte affilate della pinzetta.
NOTA: Se la forza applicata è troppo bassa, l'embrione non rotolerà. Se è troppo alto, scoppierà. Trovare la forza intermedia appropriata richiede esperienza e, quindi, questo passaggio dovrebbe essere praticato in anticipo. - Continuare a rotolare l'embrione fino a quando il corion aderisce transitoriamente al nastro e si incrina.
- Una volta che il corion si incrina, continua a rotolare per separare l'embrione (ancora all'interno della membrana vitellina) dal chorion mentre il chorion rimane attaccato al nastro.
- Confermare che il corion è completamente rimosso osservando la perdita delle appendici dorsali dall'embrione.
- Arrotolare l'embrione sul corion, che è meno adesivo del nastro. Strofinare delicatamente l'embrione con le pinzette fino a quando non si attacca alle pinzette per il trasferimento.
- Trasferire l'embrione al coverslip con la colla eptana e portarlo a contatto con la colla, che in genere lo stacca dalle pinzette.
- Regolare l'orientamento dell'embrione sulla coverslip.
- Incorporare la superficie laterale dell'embrione nella colla.
NOTA: La superficie dell'embrione incorporato nella colla è quella che verrà fotografata. Incorporare la superficie laterale è il più semplice, ma questo può essere regolato a seconda delle preferenze personali o della domanda biologica a cui rispondere. - Orientare l'asse lungo dell'embrione perpendicolare all'asse lungo del coverslip.
- Se l'embrione non giace nell'orientamento preferito, pulire le pinzette per rimuovere l'olio che staccherebbe l'embrione dalla colla. Quindi, tenta delicatamente di far rotolare l'embrione in posizione.
- Incorporare la superficie laterale dell'embrione nella colla.
- Preparare il numero desiderato di embrioni per l'iniezione nel modo sopra descritto.
NOTA: Con il passare del tempo dopo la rimozione del corion, l'aria ambiente inizierà a disidratare gli embrioni e quindi l'effetto del passaggio 5.3 sarà irregolare per gli embrioni sul coverslip, che sono stati deconizionati in momenti diversi. In genere, 1-3 embrioni sono i più gestibili.
- Coprire la piastra embrionale opportunamente invecchiata con un sottile strato di Halocarbon Oil 27 per trasformare il guscio d'uovo trasparente.
- Essiccare gli embrioni.
- Posizionare il coverslip con gli embrioni nella camera di essiccazione preparata e sigillarlo.
- Lasciare essiccare gli embrioni per 5-12 minuti.
NOTA: la tempistica di questa fase dipende dalla temperatura e dall'umidità ambiente e quindi deve essere determinata empiricamente. - Rimuovere il coperchio dalla camera e posizionare una goccia di Halocarbon Oil 700 su di esso, coprendo completamente gli embrioni, per evitare ulteriori essiccazioni.
- Ispezionare gli embrioni su un ambito di dissezione per giudicare il corretto disseccamento.
- Se gli embrioni sono solo leggermente avvizziti, ma non sgonfiati, procedere alla microiniezione (fase 6).
- Se gli embrioni non sono sufficientemente o eccessivamente essiccati, tornare alla fase 5.2 e preparare un nuovo lotto di embrioni poiché l'olio di alocarbonio 700 non può essere rimosso. Se gli embrioni sono stati eccessivamente essiccati, ridurre il tempo di essiccazione per il prossimo lotto di embrioni di 3 minuti; se erano sotto-essiccati, aumentare il tempo di essiccazione di 3 minuti.
6. Microiniettare embrioni
NOTA: assicurarsi che la configurazione della microiniezione includa un microscopio invertito, un micromanipolatore per tenere e posizionare l'ago per iniezione e un microiniettore commerciale per fornire volumi controllati.
- Accendere il microiniettore e inserire le impostazioni preferite.
NOTA: per questo protocollo, si consiglia di utilizzare l'uscita del movimento lineare e l'impostazione veloce, ma molti altri funzioneranno. L'operatore dovrebbe determinare le preferenze personali. - Caricare l'ago nel micromanipolatore. Per evitare che l'ago venga danneggiato durante il caricamento del coperchio contenente gli embrioni, spostarlo in una posizione sicura.
- Posiziona il coverslip sul palco, con gli embrioni in cima, verso l'ago. Quindi, spostare con attenzione l'ago in posizione per l'iniezione, con la punta ancora ben al di sopra del palco.
- Metti l'obiettivo 4x nel percorso della luce. Usando la manopola di messa a fuoco sul microscopio, metti a fuoco gli embrioni.
NOTA: Questo sarà il piano focale utilizzato per l'iniezione e sarà regolato solo in minima parte in futuro. - Usando i controlli dello stadio, sposta gli embrioni orizzontalmente al centro del campo visivo.
- Allontanarsi dagli embrioni, tenendoli ai margini del campo visivo, in preparazione per abbassare l'ago. Rimanere sullo stesso piano focale per assicurarsi che l'ago sia abbassato nella posizione corretta.
- Abbassare la punta dell'ago nel piano focale corretto e assicurarsi che sia visibile nel campo visivo insieme agli embrioni.
- Usando i controlli del micromanipolatore e guardando dall'alto (non ancora attraverso gli oculari), far cadere lentamente la punta dell'ago nell'olio, mirando all'area in cui punta l'obiettivo. Poiché l'olio è abbastanza viscoso, procedere lentamente per evitare di danneggiare l'ago.
- Una volta che l'ago è visibile attraverso gli oculari, continuare a utilizzare i controlli del micromanipolatore fino a quando la punta dell'ago è a fuoco e al centro del campo visivo.
- Spostando il palcoscenico, riporta gli embrioni al centro del campo visivo. Usa il controllo dello stadio, i controlli del micromanipolatore e la manopola di messa a fuoco per mettere a punto la posizione dell'embrione e della punta dell'ago l'uno rispetto all'altro mentre entrambi sono a fuoco. Mirare a eseguire iniezioni il più vicino possibile al coverslip.
- Eseguire il controllo della qualità dell'ago.
- Per garantire che l'ago sia funzionale, erogare parte della soluzione di iniezione nell'Halocarbon Oil 700 che circonda l'embrione, visibile come una bolla nell'olio.
- Se non fuoriesce nulla dall'ago, aumentare gradualmente la pressione sull'iniettore. Se questo non funziona, prendi un nuovo ago.
- Iniettare l'embrione.
NOTA: i volumi di iniezione accettabili vanno da ~0,06-1 pL. Il limite inferiore è impostato da ciò che può essere visualizzato entrando nell'embrione. Il limite superiore è fissato dal trauma che l'iniezione esercita sull'embrione.- Iniettare lungo i bordi laterali dell'embrione in quanto questo è il meno invasivo.
- Usando i controlli dello stadio, spostare l'embrione verso la punta dell'ago fino a quando quest'ultimo non perfora delicatamente l'embrione ed entra in esso.
- Allo stesso tempo, avviare il flusso della soluzione di iniezione e osservare la comparsa di un punto chiaro transitorio nel sito della punta dell'ago, che indica un trasferimento riuscito di liquido nell'embrione.
- Monitorare l'embrione. Se l'embrione resiste all'ago e si rompe (il citoplasma schizza fuori) all'ingresso, tornare al passaggio 5 e aumentare il tempo di essiccazione. Se l'embrione è piatto contro il coverslip e appare floscio durante l'iniezione, tornare al passaggio 5 e abbreviare il tempo di essiccazione.
- Se nessun colorante è entrato nell'embrione, confermare che il colorante può ancora fluire liberamente dall'ago come nel passaggio 6.7. Se il colorante non scorre, sostituire l'ago. Se il colorante scorre, inietta un nuovo embrione e inizia a rilasciare il colorante poco prima che l'ago perfori l'embrione.
NOTA: Le iniezioni ripetute dello stesso embrione non sono raccomandate in quanto rompe la ferita/sito di iniezione precedente.
- Ripetere fino a quando tutti gli embrioni vengono iniettati.
7. Embrioni di immagine
NOTA: Questo protocollo utilizza un microscopio confocale a scansione laser.
- Posizionare il coverslip nel supporto metallico sul microscopio confocale in modo che gli embrioni vengano ripresi direttamente dal basso attraverso il coverslip; sopra il coperchio, c'è solo olio e nessun'altra barriera.
- Come fase di controllo qualità, l'immagine prima utilizza la funzione di epifluorescenza sul microscopio confocale, con un obiettivo 40x. Assicurarsi che il fluoroforo sia visibile nell'embrione prima di procedere.
- Se il piano di imaging desiderato è diverso dal sito di iniezione, lasciare abbastanza tempo affinché il colorante si diffonda nell'area target.
NOTA: per BOPIPY 493/503, questa volta varia in genere da 30-60 minuti. Poiché il tempo di diffusione dipende molto dal colorante, altri coloranti possono richiedere l'ottimizzazione del sito di iniezione e del tempo di attesa. - Utilizzare obiettivi diversi per l'imaging su scale diverse. Usa un obiettivo 40x per visualizzare tutto l'embrione e un obiettivo 63x per visualizzare sottosezioni per scale più piccole.
- Per l'imaging dal vivo durante le fasi sinciziali prima della cellularizzazione, utilizzare le seguenti condizioni per iniziare: dimensioni dell'immagine di 512 x 512 pixel, media della linea di 3, frame rate di 0,1 fotogrammi al secondo (cioè 1 fotogramma acquisito ogni 10 s). Regolare le condizioni in base alle capacità del microscopio impiegato.
NOTA: queste condizioni consentono di acquisire oltre ~ 500 immagini da BOPIPY 493/503 e di catturare la maggior parte del movimento.
8. Analizza il flusso LD usando prima FIJI per preparare una serie temporale di immagini e poi python per l'analisi PIV
- Ottimizza l'acquisizione delle immagini.
- Eseguire iniezioni del colorante desiderato nella fase appropriata. Ripeti questo passaggio fino a quando la variazione giornaliera è trascurabile.
- Ottimizza le impostazioni di acquisizione delle immagini, tra cui ingrandimento, zoom, risoluzione e frequenza fotogrammi.
NOTA: esiste un compromesso tra immagini di alta qualità (risoluzione + media linea) e velocità di acquisizione (frame rate).
- Preparare i dati in FIJI.
- Acquisire una serie temporale di alta qualità da analizzare.
- Apri un fotogramma della serie temporale per generare una maschera con FIJI. Duplicare l'immagine. Convertire il tipo dell'immagine in un bit 8.
- Definite il limite dell'embrione utilizzando lo strumento di selezione poligonale o a mano libera. Utilizzate Cancella esterno per impostare i valori dei pixel al di fuori della selezione su 0. Deselezionare e quindi invertire all'interno della selezione per impostare il valore dei pixel all'interno della selezione sul valore massimo (255).
- Deseleziona, quindi utilizza il comando Istogramma per assicurarti che siano presenti solo valori di pixel pari a 0 e 255.
- Salvare questa nuova maschera immagine per la serie temporale specifica.
- Apri la serie temporale di interesse. Scegli quali dieci fotogrammi catturano meglio il tempo di interesse, ad esempio il ciclo nucleare 9 all'inizio della contrazione corticale. Duplica i dieci fotogrammi di interesse, formando un sottostack.
- Utilizzare la funzione Stack to Images per generare 10 immagini da analizzare con PIV.
- Salva i singoli file in modo da preservarne l'ordine (SeriesX_1, SeriesX_2, SeriesX_3 ...).
- Utilizzare la maschera preparata e i singoli frame per l'analisi PIV, utilizzando lo script python di esempio fornito (dati supplementari) o uno script personalizzato generato dall'utente.
NOTA: lo script fornito è basato sull'applicazione python di OpenPIV17. Emette la distanza in pixel che possono essere convertiti utilizzando la larghezza dei pixel e la frequenza dei fotogrammi per generare velocità o velocità. - Generare repliche completando i passaggi precedenti per embrioni aggiuntivi e compilando i valori di output.
Risultati
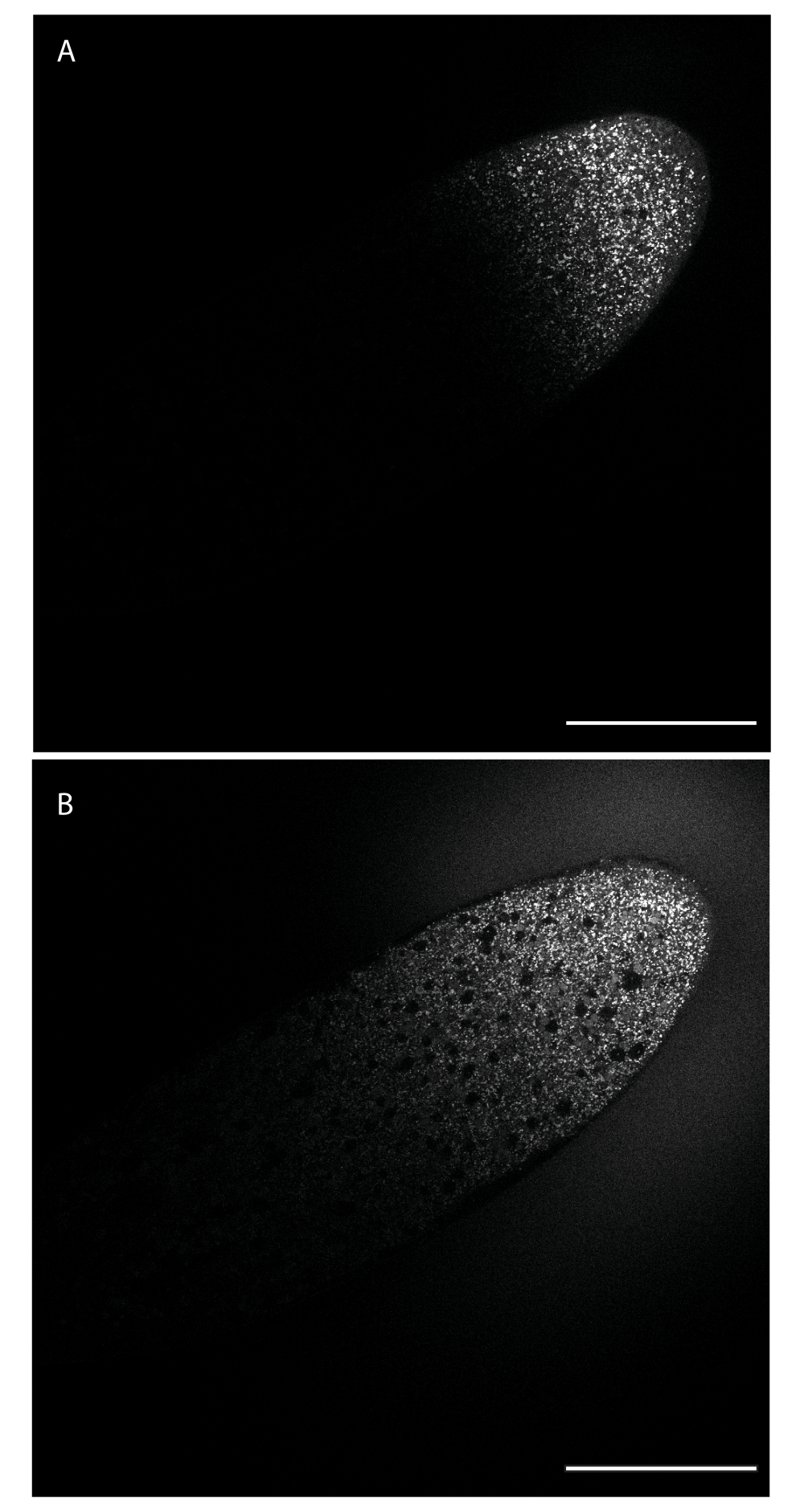
Dopo l'iniezione, il colorante sarà localizzato solo nel sito in cui è stata inserita la punta dell'ago. Il colorante si diffonderà quindi lontano dal sito di iniezione a seconda delle sue caratteristiche diffusive. La Figura 1 mostra l'iniezione di BODIPY 493/503, subito dopo l'iniezione (pannello A) e 24 minuti dopo (pannello B). Dopo 24 minuti, il colorante è arrivato all'incirca al punto medio dell'asse lungo dell'embrione.
L'analisi della motilità degli organelli può essere ottenuta attraverso iniezioni di coloranti e imaging time-lapse. Nella Figura 2, un embrione è stato co-iniettato con BODIPY 493/503 (Figura 2A) e LysoTracker Red (Figura 2B) e ripreso utilizzando l'eccitazione laser a 488 e 596 nm, rispettivamente. Questo embrione è stato poi ripreso in time-lapse (un fotogramma ogni 30 s per 30 min, 5 min analizzati). La serie temporale è stata quindi eseguita attraverso l'analisi PIV, il cui output è mostrato tramite streamline nella Figura 2A, B. Si noti che le linee di flusso non rappresentano la traiettoria delle singole particelle, ma il flusso citoplasmatico viene dedotto dall'analisi di tutte le particelle in quella regione del citoplasma. Attraverso l'etichettatura di due strutture cellulari indipendenti (LD e organelli acidi), l'analisi PIV trova flussi simili, con entrambe le etichette che convergono sulla regione centrale dell'embrione dove il citoplasma scorre nell'interno dell'embrione6.
Gli FP attualmente disponibili consentono l'etichettatura di molti altri organelli e strutture cellulari. La Figura 3 mostra l'etichettatura dell'ER tramite il tracker ER Green. Il tracker ER fornisce una buona risoluzione dell'involucro nucleare, consentendo la visualizzazione delle principali fasi del ciclo cellulare. Il tracker ER Green viene ripreso utilizzando l'eccitazione a 488 nm.
L'etichettatura dei mitocondri è complicata, poiché la maggior parte dei coloranti testati sembra essere intrappolata nel primo mitocondrio in cui entrano. D'altra parte, non è stato rilevato alcun segno di tossicità del colorante, rendendo possibile seguire i mitocondri marcati attraverso la cellularizzazione e il ciclo nucleare ectodermico 15. La Figura 4 mostra una cellula ectodermica diverse ore dopo l'iniezione con Mitoview 633 (lunghezza d'onda di eccitazione 633 nm).
BODIPY, Lysotracker e LipidSpot sono robusti e possono essere utilizzati per acquisire ~ 500 + immagini a 512 x 512, media di linea 3, frame rate da 1 / s a 0,1 / s. ER tracker Green, SiR-tubulina e i coloranti mitocondriali menzionati sono meno robusti e producono ~ 50-200 immagini nelle stesse condizioni.
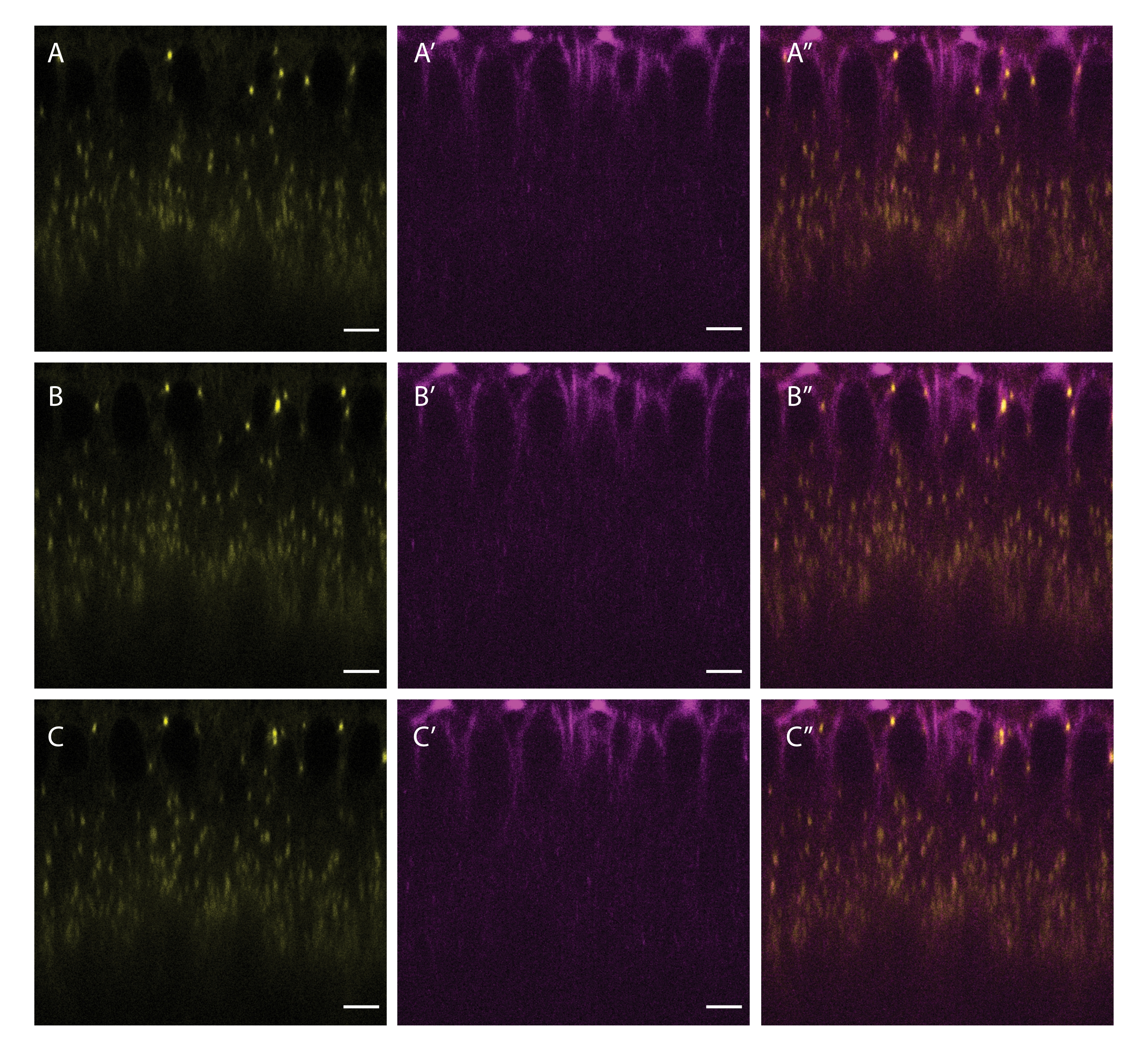
A partire dagli stadi di blastoderma, gli LCD si muovono bidirezionalmente lungo i microtubuli, alimentati dai motori a polarità opposta kinesin-1 e dineina citoplasmatica5. Questo movimento può essere visualizzato co-etichettando LD e microtubuli iniettando sia BODIPY 493/503 che SiR-Tubulin (Figura 5). Poiché gli LCD spesso invertono la loro direzione di movimento (mentre passano tra kinesin-1 e dineina citoplasmatica), frame rate più elevati durante l'acquisizione catturano meglio i dettagli critici della motilità LD.
L'imaging dal vivo delle vescicole autofluorescenti del tuorlo è possibile senza alcuna forma di iniezione di colorante (Figura 6). Tuttavia, l'autofluorescenza è debole e il laser di eccitazione è fototossico. Pertanto, l'imaging dal vivo del tuorlo autofluorescente ha uno scarso rapporto segnale-rumore rispetto all'iniezione di colorante.

Figura 1: Diffusione di BODIPY 493/503 attraverso l'embrione. Il colorante è stato iniettato lungo il bordo laterale verso l'estremità anteriore (in alto a destra) e si diffonde da questo sito di iniezione nell'embrione, etichettando LDs. (A) Il colorante si è diffuso attraverso porzioni dell'embrione adiacenti al sito di iniezione. (B) Circa 24 min/2 cicli nucleari dopo, il colorante si è diffuso oltre il punto medio dell'embrione. Barra di scala: 100 μm. Un telaio 1024 x 1024 (linea media 4) è stato acquistato ogni 30 s. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 2: Velocimetria a immagini di particelle (PIV) per LD e organelli acidi. Un embrione è stato iniettato sia con BODIPY 493/503 che con LysoTracker Red. (A) Canale BODIPY. (B) Canale rosso LysoTracker. (A',B') Semplifica i diagrammi generati dall'analisi PIV dei due canali generati da 10 fotogrammi sequenziali, inclusi quelli mostrati in A e B. A' corrisponde al flusso di LD, e B' corrisponde al flusso di organelli acidi. Si noti che sia A' che B' mostrano una confluenza a sinistra del centro in cui il contenuto embrionale fluisce fuori dal piano visivo, nel centro dell'embrione. Inoltre, si noti che BODIPY ha diffuso più di LysoTracker poiché diversi coloranti hanno proprietà diffusive diverse. Barra di scala: 100 μm. Un telaio 1024 x 1024 (linea media 4) è stato acquistato ogni 30 s. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 3: Il tracker ER etichetta le divisioni nucleari sinciziali. Un embrione di blastoderma sinciziale è stato microiniettato con ER tracker e una parte della sua superficie è stata fotografata nel tempo. (A) Assemblaggio del mandrino durante una divisione nucleare. (B) Abscissione dell'involucro nucleare durante la stessa divisione. (C) Successive interfasi. (D) L'inizio della divisione successiva è indicato dall'aspetto centrosomico (presenza di regioni circolari prive di ER, contrassegnate da punte di freccia). Si noti il graduale sbiancamento della tintura. Lunghezza d'onda di eccitazione: 488 nm. Barra della scala: 5 μm. A: fotogramma iniziale, B: 3 minuti trascorsi, C: 10 minuti trascorsi, D: 13 minuti trascorsi. Un telaio 1024 x 1024 (linea media 4) è stato acquistato ogni 30 s. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 4: Mitoview 633 etichettatura dei mitocondri. Un embrione è stato iniettato con Mitoview 633 durante lo stadio di blastoderma sinciziale e fotografato 4 ore dopo, dopo la cellularizzazione. L'immagine mostra una cellula neuroectodermica di un embrione in estensione della banda germinale. Barra scala: 5 μm. Un telaio 1024 x 1024 (linea media 4) è stato acquistato ogni 30 s. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 5: Co-etichettatura di LD e microtubuli. Un embrione cellularizzante è stato iniettato con una miscela di BODIPY 493/503 (giallo A,B,C) e SiR Tubulina (magenta A',B',C'). A'', B'', C'' mostrano i canali uniti. I pannelli A, A'' e A'' mostrano la cornice iniziale, i pannelli B, B' e B'' mostrano la cornice dopo 5 s, e i pannelli C, C' e C'' mostrano la cornice dopo 10 s. Barra scala: 5 μm. Un telaio 512 x 512 (linea media 3) è stato acquistato ogni 2,5 s. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 6: Imaging dell'autofluorescenza della vescicola del tuorlo durante le fasi di scissione sinciziale. (A) All'inizio dell'acquisizione. (B) Dopo 8 min. (C) Dopo 16 min. Lunghezza d'onda di eccitazione: 405 nm. Una bassa intensità di eccitazione è stata utilizzata per mantenere in vita l'embrione. Barra di scala: 100 μm. Un telaio 1024 x 1024 (linea media 4) è stato acquistato ogni 30 s. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
Dati supplementari. Fare clic qui per scaricare questo file.
Discussione
L'embrione di Drosophila è un modello potente e conveniente per studiare questioni fondamentali nella biologia cellulare e organismica. La sua relativa semplicità, la potente genetica e le piccole dimensioni lo rendono un sistema eccellente per l'imaging sia dei processi cellulari che dello sviluppo. Qui, un protocollo di microiniezione standard viene adattato per consentire l'utilizzo di FP negli embrioni. Questo approccio consente l'imaging fluorescente di specifiche strutture cellulari senza la necessità di fluorofori geneticamente codificati, aprendo molti background genetici all'imaging. La combinazione di più coloranti e proteine marcate fluorescenti scelte strategicamente può aprire l'imaging live multicanale che copre l'intero spettro della luce visibile.
Passaggi critici nel protocollo:
Questo protocollo utilizza BODIPY 493/503 per etichettare i LD. Questo approccio può essere facilmente adattato per contrassegnare altre strutture cellulari. Per la successiva analisi dell'immagine, uno dei fattori più importanti è il rapporto segnale-rumore, cioè la luminosità del colorante rispetto al segnale di fondo. I lisosomi sono stati ripresi con successo (LysoTracker Red, 1 mM), così come i mitocondri (Mitoview 633, 200 μM), l'ER (ER tracker Green, 10 μM) e i microtubuli (tubulina SiR, 200 nM in DMSO), come mostrato in Figura 2, Figura 3, Figura 4, Figura 5. Inoltre, le vescicole di tuorlo sono autofluorescenti ed emettono luce blu dopo l'eccitazione UV (immagine che utilizza 405 nm come lunghezza d'onda di eccitazione (Figura 6)). Per altri coloranti, mirare a che la concentrazione del colorante sia 100-1.000x di ciò che sarebbe necessario per colorare le cellule coltivate vive; questo è simile alla concentrazione di una soluzione madre che verrebbe diluita in terreni di coltura cellulare. Poiché questo protocollo richiede un'iniezione di 100 fL e l'embrione di Drosophila è di circa 9 nL nel volume18, queste concentrazioni di colorante saranno in media a una concentrazione embrionale interna inferiore a 1/100 di ciò che è presente nei terreni di coltura cellulare. Temporaneamente, la concentrazione locale sarà più alta nel sito di iniezione, che è più rilevante per i FP che non si diffondono bene (cioè coloranti mitocondriali e SiR-tubulina). Per questi FP, iniziare dalle alte concentrazioni raccomandate; se si osserva una morte inaspettata, diluire successivamente due volte fino a raggiungere un compromesso accettabile tra sopravvivenza e potenza del segnale.
Quando si co-iniettano più coloranti, entrambi i coloranti devono essere nello stesso solvente o entrambi i solventi e i coloranti devono essere compatibili con la miscela (non sono raccomandate concentrazioni di alcol superiori a ~ 10%).
La qualità degli aghi è essenziale per il successo di questa procedura, poiché la punta deve essere il più fine possibile. Altrimenti, il danno causato dalla ferita da iniezione può compromettere il successivo sviluppo dell'embrione. Poiché gli estrattori di aghi commerciali differiscono, è importante seguire i suggerimenti del produttore e provare più parametri di trazione fino a raggiungere la forma desiderata. È fondamentale eseguire la fase di controllo qualità 1.1.3 poiché lavorare con una punta dell'ago incrinata, frastagliata o di grandi dimensioni renderà l'iniezione di successo più difficile o addirittura impossibile.
L'embrione deve essere parzialmente essiccato in modo che ulteriori volumi di liquido possano essere aggiunti durante l'iniezione. Se l'embrione è sotto-essiccato, l'ago non entrerà facilmente e il citoplasma schizzerà fuori mentre l'ago penetra o mentre la soluzione viene iniettata. Se l'embrione è troppo essiccato, sembrerà sgonfiato e non si svilupperà correttamente. Il tempo esatto di asciugatura dipende dalle condizioni locali, ad esempio l'umidità dell'aria, e può cambiare di giorno in giorno. Deve essere determinato empiricamente per ogni sessione.
La capacità dell'embrione di sopravvivere dopo la microiniezione dipende in modo critico dalla qualità dell'ago, dal corretto essiccamento e dalla limitazione del volume di iniezione a meno di 1 pL (idealmente 100 fL). Finché questi parametri sono ottimizzati, non è evidente alcuna tossicità significativa quando i coloranti descritti vengono iniettati alle concentrazioni raccomandate. Se gli embrioni sopravvivono alle fasi di essiccazione e iniezione, in genere si sviluppano con successo nell'estensione della banda germinale, con un'eccezione delle sonde microtubule e ER che hanno causato difetti di cellularizzazione ad alti livelli (iniezione di 100 fL della concentrazione di riserva di ciascuno). I test non hanno riscontrato evidenti difetti dello sviluppo quando DMSO, acqua e miscele dei due sono stati iniettati ai volumi raccomandati, con un tasso di sopravvivenza embrionale attraverso l'estensione della banda germinale di ~ 75% o più. Volumi di iniezione superiori a 1 pL hanno causato difetti e gli embrioni iniettati con volumi di ~ 4 pL sviluppati per meno di 1 ora. Pertanto, i volumi di iniezione devono essere mantenuti bassi, il che significa che le concentrazioni di colorante devono essere elevate.
Generalmente, le iniezioni lungo il bordo laterale dell'embrione sono raccomandate in quanto provocano il minor danno. Tuttavia, potrebbe essere necessario regolare il sito di iniezione in base alle proprietà diffusive dei FP impiegati. BODIPY 493/503 e LysoTracker si diffondono più velocemente sull'intero embrione rispetto al Lipid Spot 610 (un altro colorante per contrassegnare gli LD), mentre SiR-Tubulin e Mitoview 633 non si diffondono mai completamente attraverso l'embrione (imaging fino a 7 ore dopo l'iniezione). Pertanto, può essere necessaria l'iniezione all'interno o vicino al sito di interesse. Quando si inietta nelle regioni anteriore o posteriore, si raccomanda un ago particolarmente sottile.
L'acquisizione di immagini si basa sulla microscopia confocale per sezionare e risolvere otticamente piccoli organelli e tutti i componenti citoscheletrici. Le tecniche che richiedono l'analisi di molte immagini (ad esempio, STORM o PALM) non funzioneranno perché il contenuto embrionale è in movimento e i fluorofori non sono ottimizzati per il fotocceleramento. La microscopia a epifluorescenza manca della risoluzione laterale e assiale per distinguere la maggior parte degli organelli e delle strutture cellulari più piccole. Per questi motivi, si consiglia vivamente di utilizzare un microscopio confocale o di utilizzare la tecnologia dei fogli di luce.
La riproducibilità dell'analisi delle immagini si basa in gran parte su dati di imaging coerenti. Per le maggiori possibilità di successo, è necessaria l'ottimizzazione della tecnica di iniezione e l'acquisizione delle immagini. Stabilire e praticare una tecnica in cui i coloranti di interesse, il sito di iniezione, l'età dell'embrione, il volume di iniezione e la configurazione dell'acquisizione sono tutti coerenti genererà i dati più robusti per l'analisi delle immagini.
Modifiche e risoluzione dei problemi del metodo
Questo protocollo dimostra un metodo per analizzare il flusso di massa di LD in embrioni in stadio di scissione utilizzando la velocimetria dell'immagine delle particelle. Lo stesso approccio può essere utilizzato per altri organelli, altri stadi di sviluppo e altri metodi di analisi. Ad esempio, la Figura 2 mostra l'analisi di LD e organelli acidi che fluiscono nelle fasi sinciziali dell'embriogenesi, visualizzate co-iniettando BODIPY 493/503 e LysoTracker Red. È stata ottenuta un'ulteriore imaging di successo del movimento LD negli embrioni fino a 7 ore dopo la fecondazione; questi embrioni mantengono la ferita da iniezione ma sono in grado di svilupparsi per diverse ore.
I dati raccolti utilizzando questo protocollo sono stati utilizzati per la velocimetria delle immagini di particelle, ma sono disponibili molte altre tecniche di analisi. Ad esempio, i programmi di tracciamento delle particelle come quelli che si trovano in ImageJ, Imaris o il tracciamento manuale possono essere utilizzati per ottenere velocità e direzionalità delle strutture in movimento. Si noti che la maggior parte di questi software di tracciamento sono costruiti per funzionare con i dati provenienti da sistemi di coltura cellulare planare e non sempre si adattano bene a strutture 3D come l'embrione di Drosophila . Inoltre, per la generazione dei dati di tracciamento delle particelle della migliore qualità, dovrebbero essere ripresi più piani Z; questo dovrebbe essere fattibile se i tempi di acquisizione dello stack di immagini sono inferiori a ~ 2 s. Questo benchmark dovrebbe essere raggiungibile su dischi confocali rotanti, fogli luminosi reticolari e recenti sistemi confocali a scansione laser. Tuttavia, la fattibilità del tracciamento delle particelle per organelli abbondanti come LD, mitocondri e lisosomi è bassa in quanto la quantità di segnale positivo in un campo visivo è troppo alta per gli attuali metodi di tracciamento. Può essere possibile il monitoraggio di strutture meno abbondanti come nuclei o vescicole di tuorlo. PIV per l'analisi del flusso funziona bene per LD e organelli acidi nelle fasi di scissione perché entrambi gli organelli si muovono liberamente. Organelli come nuclei, ER e mitocondri sono legati ad altre strutture cellulari e quindi non si muovono liberamente e non sono adatti all'analisi software che presuppone il movimento libero. Lo sperimentatore dovrebbe scegliere le tecniche più adatte all'organello di interesse.
Durante gli stadi di blastoderma sinciziale e cellulare, gli LCD (così come alcuni altri organelli) si muovono lungo i microtubuli orientati radialmente5. È quindi possibile trovare viste trasversali (come nella Figura 5) in cui singoli microtubuli sono a fuoco per lunghe distanze, consentendo il tracciamento delle particelle in 2D. Poiché questi piani ottici sono profondi all'interno dell'embrione, la potenza complessiva del segnale è diminuita e il rapporto segnale-rumore è ridotto.
Per l'analisi del tracciamento, l'imaging il più velocemente possibile può rivelare dettagli cruciali del movimento e quindi del macchinario mobile. Ad esempio, il movimento lipidico-goccioline è una miscela di due stati mobili, movimento lento-corto (~ 200 nm / s; distanza media di viaggio ~ 100 nm) e movimento veloce-lungo (~ 450 nm / s; distanza media di viaggio ~ 1.000 nm) 19; quindi, se le immagini vengono scattate ogni secondo o anche meno frequentemente, lo stato lento-corto diventa non rilevabile. Tuttavia, l'imaging frequente induce anche lo sbiancamento del fluoroforo e la fototossicità. Le condizioni di imaging, quindi, devono essere regolate in base alla domanda esatta da affrontare.
Limitazioni del metodo
A seconda del colorante desiderato, il metodo può essere limitato dalla compatibilità tra la solubilità del colorante e la tossicità della soluzione iniettabile. Alcoli come l'isopropanolo e l'etanolo sono difficili da maneggiare all'interno di un ago a causa della loro viscosità inferiore e sembrano danneggiare i componenti cellulari e uccidere l'embrione.
Il metodo non è adatto anche per visualizzare i primi passi nell'embriogenesi perché ci vogliono più di 30 minuti per preparare gli embrioni per l'iniezione. A temperatura ambiente, i cicli cellulari iniziali dell'embrione durano solo ~ 10 minuti ciascuno; quindi, anche se si dovesse scegliere un uovo appena fecondato nel passaggio 5.2.3, i primi cicli cellulari sarebbero già completati nel momento in cui l'embrione è pronto per l'imaging.
L'illuminazione con luce nella gamma UV/blu è considerevolmente più fototossica rispetto alle lunghezze d'onda più lunghe. In tali condizioni (ad esempio, seguire le vescicole autofluorescenti del tuorlo; Figura 6), si deve limitare il tempo di imaging (portando a serie temporali più brevi) o utilizzare una potenza laser inferiore (con conseguente riduzione del rapporto segnale-rumore).
Dopo la cellularizzazione, i coloranti iniettati in una posizione specifica tendono a diffondersi male, poiché devono attraversare molte membrane cellulari. Ciò limita la regione di osservazione nelle fasi successive dello sviluppo.
Il significato del metodo rispetto ai metodi esistenti/alternativi
Il movimento degli LD e di altri organelli contenenti lipidi nei primi embrioni può essere visualizzato con fluorofori geneticamente codificati, tecniche prive di etichette e mediante l'introduzione di FP. Quest'ultimo può essere ottenuto mediante permeabilizzazione della membrana vitellina12 o l'approccio di microiniezione discusso qui.
I fluorofori geneticamente codificati sono marcatori versatili i cui livelli sono tipicamente altamente riproducibili da embrione a embrione. Tuttavia, hanno rendimenti quantistici inferiori e candeggina più facilmente rispetto agli FP. In genere, sono disponibili solo in una o due versioni con tag (ad esempio, GFP o mCherry), limitando la scelta di quali strutture possono essere visualizzate contemporaneamente. I FP, d'altra parte, spesso esistono in una grande varietà; ad esempio, sono disponibili vari coloranti specifici per goccioline lipidiche con spettri di emissione da Autodot nello spettro UV / blu a Lipidtox e LipidSpot 610 nello spettro rosso lontano. Gli FP possono anche essere applicati direttamente a qualsiasi ceppo di interesse e quindi non richiedono la costruzione di ceppi per, ad esempio, introdurre il marcatore organello desiderato in un ceppo mutante di interesse. Questo vantaggio è particolarmente pronunciato quando più strutture devono essere etichettate contemporaneamente; invece di incroci che richiedono tempo che coprono più generazioni, questo può essere ottenuto in un solo giorno mescolando i coloranti pertinenti e introducendoli allo stesso tempo. Infine, se i processi cellulari devono essere sondati con inibizione farmacologica, farmaci e coloranti possono essere introdotti insieme.
I metodi senza etichetta sono approcci molto potenti per rilevare specifiche strutture cellulari. Ad esempio, gli LCD possono essere rilevati specificamente negli embrioni precoci mediante microscopia di terza generazione armonica20 o microscopia a perdita raman stimolata a femtosecondi21. Come gli FP, questi approcci possono essere applicati in qualsiasi background genetico e, poiché non causano lo sbiancamento, consentono potenzialmente un'acquisizione più rapida delle immagini. Tuttavia, sono in genere limitati a specifici organelli e quindi non supportano da soli l'imaging multiplex; richiedono anche microscopi specializzati.
Esistono due strategie generali per introdurre piccole molecole negli embrioni. Uno è l'approccio di microiniezione impiegato qui; l'altro è il trattamento chimico (terpenico) per permeabilizzare la membrana vitellina. Quest'ultimo approccio12 è meno coinvolto della microiniezione, ma anche più variabile da embrione a embrione. Inoltre, dopo la permeabilizzazione, la protezione fornita dalla membrana vitellina è compromessa e l'embrione vero e proprio è accessibile al mezzo esterno, rendendo più difficile mantenerlo in vita. La microiniezione ha molte meno probabilità di far deragliare lo sviluppo embrionale rispetto alla permeabilizzazione. Tuttavia, la permeabilizzazione è raccomandata se molti embrioni devono essere monitorati contemporaneamente, ad esempio per scopi di screening farmacologico. Per seguire il movimento delle strutture cellulari e ottenere serie di immagini riproducibili adatte all'analisi delle immagini, la microiniezione è il metodo di scelta.
Importanza e potenziali applicazioni del metodo in specifiche aree di ricerca
L'embrione di Drosophila è un importante sistema modello per lo studio di molti processi biologici e di sviluppo cellulare 1,5,6. L'etichettatura degli organelli con proteine fluorescenti ha dato importanti contributi alla comprensione di come si sviluppa l'embrione precoce, di come trafficano i vari organelli e di come tale traffico sia modulato dallo sviluppo e geneticamente. Tuttavia, la loro propensione allo sbiancamento e le sfide di generare ceppi in cui più organelli sono etichettati con colori diversi limitano l'applicazione di questo approccio. L'uso di FP introdotti dalla microiniezione risolve molte di queste sfide e può anche essere combinato con proteine marcate fluorescentemente. Questa tecnica consente l'imaging di più organelli, strutture cellulari e componenti citoscheletrici in qualsiasi background genetico. Di conseguenza, diversi genotipi possono essere confrontati tramite imaging dal vivo, rendendo possibile determinare l'effetto delle mutazioni sul traffico di più organelli.
Questo protocollo dimostra l'approccio di iniezione FP per gli embrioni di Drosophila melanogaster, ma in linea di principio, questo approccio si applica a tutte le uova di insetto per le quali sono state stabilite tecniche di microiniezione, comprese altre specie di Drosophila, grilli22 e afidi23.
Divulgazioni
Gli autori non hanno conflitti di interesse da divulgare.
Riconoscimenti
Ringraziamo Pakinee Phromsiri, Brian Jencik, Jinghong (James) Tang e Roger White per i loro commenti sul manoscritto. Ringraziamo Patrick Oakes, Stefano Di Talia e Victoria Deneke per aver condiviso le loro competenze su come eseguire l'analisi PIV. Questo lavoro è stato sostenuto dalle sovvenzioni del National Institutes of Health F31 HD100127 (a M. D. K.) e R01 GM102155 (a M. A. W).
Materiali
| Name | Company | Catalog Number | Comments |
| AUTODO | abcepta | SM1000a | Stains lipid droplets in violet/blue |
| BODIPY 493/503 | ThermoFisher | D3922 | Stains lipid droplets in green |
| Desiccant Beads –Desiccant-Anhydrous Indicating Drierite | W.A. Hammond Drierite Company | 21001 | |
| Dissecting microscope SteREO DiscoveryV20 with transillumination base | Zeiss | 4350030000000000 | |
| Double sided tape-Permanent Double-Sided Tape | Scotch (3M) | - | Sold by many vendors |
| ER-Tracker Green (BODIPY FL Glibenclamide) | ThermoFisher | E34251 | Stains the ER/nuclear envelope |
| Femptotip II | Eppendorf | H129354N | Ready to use |
| Glass capillaries -Glass Thinw w/fil 1.0mm 4in | World Precision Instruments | TW100F-4 | Must be pulled |
| Glass coverslip | - | - | Buy the appropriate refractive index for your objective lens |
| Glass slides -Double Frosted Microscope Slides precleaned | FisherBrand | 12-550-343 | |
| Halocarbon oil 27 | Sigma-Aldrich | H8773-100ML | |
| Halocarbon oil 700 | Sigma-Aldrich | H8898-50ML | |
| Heptane greener alternative anhydrous, 99% | Sigma-Aldrich | 246654-1L | Heptane is considered toxic by the USA's OSHA |
| Confocal microscope | Leica | Sp5 | Many different types of confocal microscopes will work. |
| LipidSpot 610 | Biotium | #70069 | Stains lipid droplets in far red |
| LysoTracker Red | ThermoFisher | L7528 | Stains lysosomes in red |
| MitoView 633 | Biotium | #70055 | Stains mitochondria |
| Needle loading pipette tips - 20uL microloader tips | Eppendorf | # 5242956.003 | |
| P-1000, Next Generation Micropipette Puller | Sutter Instruments | P-1000 | The settings used are heat-470, pull-70, velocity-60, delay-90, pressure-200, ramp-479 at 1x1. They refer to the intensity and length of the heating, the timing of pulling force and whether the force is applied linearly. Using these conditions, one capillary generates two usable needles with fine openings at the tip. |
| SiR-Tubulin | Cytosketon Inc | CY-SC014 | Made by Spirochrome; Cytoskeleton Inc. is the North American distributor. Stains microtubules in far red |
| TransferMan | Eppendorf | 5178 NK | |
| ViaFluor 405 | Biotium | #70064 | Stains microtubules in violet/blue |
Riferimenti
- Chowdhary, S., Tomer, D., Dubal, D., Sambre, D., Rikhy, R. Analysis of mitochondrial organization and function in the Drosophila blastoderm embryo. Scientific Reports. 7 (1), 5502 (2017).
- Mavor, L. M., et al. Rab8 directs furrow ingression and membrane addition during epithelial formation in Drosophila melanogaster. Development. 143 (5), 892-903 (2016).
- Riggs, B., et al. Actin cytoskeleton remodeling during early Drosophila furrow formation requires recycling endosomal components Nuclear-fallout and Rab11. Journal of Cell Biology. 163 (1), 143-154 (2003).
- Welte, M. A., Gross, S. P., Postner, M., Block, S. M., Wieschaus, E. F. Developmental regulation of vesicle transport in Drosophila embryos: forces and kinetics. Cell. 92 (4), 547-557 (1998).
- Welte, M. A. As the fat flies: The dynamic lipid droplets of Drosophila embryos. Biochimica et Biophysica Acta. 1851 (9), 1156-1185 (2015).
- Deneke, V. E., et al. Self-organized nuclear positioning synchronizes the cell cycle in Drosophila embryos. Cell. 177 (4), 925-941 (2019).
- Welte, M. A., et al. Regulation of lipid-droplet transport by the perilipin homolog LSD2. Current Biology. 15 (14), 1266-1275 (2005).
- Bailey, A. P., et al. Antioxidant role for lipid droplets in a stem cell niche of Drosophila. Cell. 163 (2), 340-353 (2015).
- Johnson, M. R., et al. H2Av buffering via dynamic sequestration to lipid droplets in Drosophila embryos. Elife. 7, 36021 (2018).
- Lowe, N., et al. Analysis of the expression patterns, subcellular localisations and interaction partners of Drosophila proteins using a pigP protein trap library. Development. 141 (20), 3994-4005 (2014).
- Rambold, A. S., Cohen, S., Lippincott-Schwartz, J. Fatty acid trafficking in starved cells: regulation by lipid droplet lipolysis, autophagy, and mitochondrial fusion dynamics. Developmental Cell. 32 (6), 678-692 (2015).
- Rand, M. D., Kearney, A. L., Dao, J., Clason, T. Permeabilization of Drosophila embryos for introduction of small molecules. Insect Biochemistry and Molecular Biology. 40 (11), 792-804 (2010).
- Tran, S. L., Welte, M. A. In-vivo centrifugation of Drosophila embryos. Journal of Visualized Experiments. (40), (2010).
- Rothwell, W. F., Sullivan, W. Drosophila embryo collection. CSH Protocols. 2007, (2007).
- Wieschaus, E., Nüsslein-Volhard, C., Roberts, D. B. . Drosophila: A Practical Approach. , 179-214 (1998).
- . The Interactive Fly: Stages of Development and Mitotic Domains Available from: https://www.sdbonline.org/sites/fly/aimain/2stages.htm (1999)
- Liberzon, A., et al. . OpenPIV/openpiv-python: OpenPIV - Python (v0.22.2) with a new extended search PIV grid option. , (2020).
- Markow, T. A., Beall, S., Matzkin, L. M. Egg size, embryonic development time and ovoviviparity in Drosophila species. Journal of Evolutionary Biology. 22 (2), 430-434 (2009).
- Gross, S. P., Welte, M. A., Block, S. M., Wieschaus, E. F. Dynein-mediated cargo transport in vivo. A switch controls travel distance. Journal of Cell Biology. 148 (5), 945-956 (2000).
- Debarre, D., et al. Imaging lipid bodies in cells and tissues using third-harmonic generation microscopy. Nature Methods. 3 (1), 47-53 (2006).
- Dou, W., Zhang, D., Jung, Y., Cheng, J. X., Umulis, D. M. Label-free imaging of lipid-droplet intracellular motion in early Drosophila embryos using femtosecond-stimulated Raman loss microscopy. Biophysical Journal. 102 (7), 1666-1675 (2012).
- Barry, S. K., et al. Injecting Gryllus bimaculatus Eggs. Journal of Visualized Experiments. (150), (2019).
- Le Trionnaire, G., et al. An integrated protocol for targeted mutagenesis with CRISPR-Cas9 system in the pea aphid. Insect Biochemistry and Molecular Biology. 110, 34-44 (2019).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneEsplora altri articoli
This article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati