
このコンテンツを視聴するには、JoVE 購読が必要です。 サインイン又は無料トライアルを申し込む。
Method Article
透過処理検出機能付き連続フローマイクロエレクトロポレーションシステムの構築と運用
要約
このプロトコルでは、ラボオンチップのマイクロ流体エレクトロポレーションデバイスを構築するために必要な微細加工技術について説明します。実験セットアップは、制御された単一細胞レベルのトランスフェクションを連続フローで実行し、集団ベースの制御により、より高いスループットに拡張することができます。細胞膜透過の程度をリアルタイムで電気的に監視する機能を示す分析が提供されます。
要約
CAR-T細胞療法などの現在の治療革新は、ウイルスを介した遺伝子送達に大きく依存しています。この技術は効率的ですが、製造コストが高いため、遺伝子導入に代替方法を使用することに関心が寄せられています。エレクトロポレーションは、遺伝子やその他の外因性物質の細胞内送達のための電気物理学的非ウイルス的アプローチです。電界を印加すると、細胞膜は一時的に細胞内への分子送達を可能にする。典型的には、エレクトロポレーションは、多数の細胞を処理するためにマクロスケールで実行される。ただし、このアプローチでは広範な経験的プロトコル開発が必要であり、初代細胞タイプやトランスフェクションが困難な細胞タイプを扱う場合はコストがかかります。長いプロトコル開発は、細胞を透過するのに十分な電界強度を達成するための大きな電圧の要件と相まって、マイクロスケールのエレクトロポレーションデバイスの開発につながりました。これらのマイクロエレクトロポレーションデバイスは、一般的な微細加工技術を使用して製造されており、高いスループット能力を維持する可能性のある、より優れた実験制御を可能にします。この研究は、連続フロー下で単一細胞レベルで細胞膜透過処理のレベルを検出できるマイクロ流体エレクトロポレーション技術を構築しています。しかし、この技術は毎秒4セルの処理に制限されていたため、システムのスループットを向上させるための新しいアプローチが提案され、ここで提示されます。この新しい手法は、細胞集団ベースのフィードバック制御と呼ばれ、さまざまなエレクトロポレーションパルス条件に対する細胞透過応答を考慮し、テスト対象の細胞タイプに最適なエレクトロポレーションパルス条件を決定します。次に、この「最適な」パルスが輸送中の細胞懸濁液に適用されるハイスループットモードが使用されます。デバイスの作製、マイクロ流体実験のセットアップと実行、および結果の分析の手順が詳細に示されています。最後に、このマイクロエレクトロポレーション技術は、緑色蛍光タンパク質(GFP)をコードするDNAプラスミドをHEK293細胞に送達することによって実証されます。
概要
CAR-T(キメラ抗原受容体改変T細胞)細胞療法やCRISPR(クラスター化された規則的に間隔を空けた短い回文反復DNA配列)/Cas9を使用した遺伝子編集など、生物医学研究における現在の治療革新は、外因性物質を細胞内空間にうまく効率的に送達する能力に大きく依存しています1。CAR-T療法において、細胞治療製造における遺伝子送達工程を行うゴールドスタンダードは、ウイルスベクター2を用いることである。ウイルス媒介遺伝子送達は効率的な送達モダリティであるが、いくつかの欠点もある。これらには、製造コスト、細胞毒性、免疫原性、突然変異誘発/腫瘍形成の可能性、および送達される遺伝子のサイズ制限が含まれます3。これらの制限は、代替の非ウイルス送達技術の研究開発につながっています。
ウイルスを介した遺伝子送達に代わるエレクトロポレーションは、細胞のDNA、RNA、およびタンパク質トランスフェクションを実行するために、最適な電気パルス波形の適用に依存しています。外部電界の適用後、細胞膜は一時的に損なわれ、細胞は、そうでなければ不透過性の外因性物質の細胞内送達の影響を受けやすくなる4。ウイルス媒介送達と比較して、エレクトロポレーションは、一般的に安全で、操作が容易で、運用コストが低いため、有利です。エレクトロポレーションは、低分子と高分子の両方の貨物を送達でき、系統に関係なく細胞のトランスフェクションに効率的です5。エレクトロポレーション後に望ましい結果、すなわち良好な生存率と良好なエレクトロトランスフェクション効率を達成するには、さまざまな実験パラメータを共最適化する必要があります。これらには、細胞タイプ6、細胞密度、分子濃度7、エレクトロポレーションバッファー特性(分子組成、導電率、浸透圧など)8、電極サイズ/形状9、および電気パルス波形(形状、極性、パルス数)10 が含まれます(図 1 を参照)。これらの各パラメータはエレクトロポレーション実験の結果に大きな影響を与える可能性がありますが、印加されたパルスの電気エネルギーが、結果として生じる細胞生存率とエレクトロトランスフェクション効率との間の固有のトレードオフの根源であるため、パルス波形は特に詳細に研究されています8。
通常、エレクトロポレーション実験はマクロスケールで行われ、細胞はエレクトロポレーションキュベット内の大きな平行平板電極のセットの間に数百マイクロリットルのバッファーに懸濁されます。電極は通常、電極距離が1〜4mmのアルミニウムで製造されます。細胞がピペットで手動でロードされると、キュベットはかさばる電気パルス発生器に電気的に接続され、ユーザーはパルス波形パラメータを設定して適用し、細胞懸濁液をエレクトロポレーションすることができます。マクロスケールまたはバルクエレクトロポレーションは細胞密度>106 cells/mLを処理できますが、この機能は電気パルス波形設定を最適化するときに無駄になる可能性があります。これは、細胞集団数が制限される可能性のある初代細胞タイプをエレクトロポレーションする場合に特に懸念される。さらに、電極間の距離が大きいため、パルス発生器は電界強度>1kV / cm11を達成するために大きな電圧を供給できなければなりません。これらの高電圧は、電解質バッファーを介した抵抗性電力散逸を引き起こし、ジュール加熱をもたらし、結果として生じるセル生存率に有害であり得る12。最後に、細胞の高密度懸濁液に対してエレクトロポレーションを行うことは、結果として生じるエレクトロトランスフェクション効率と細胞生存率の生来の変動性を一貫して負担します。懸濁液中の各細胞は、周囲の細胞のために異なる電界強度を経験する可能性があります。経験した電界強度が増加するか減少するかに応じて、結果として生じる細胞の生存率または電気トランスフェクション効率はそれぞれ悪影響を受ける可能性があります11。マクロスケールのエレクトロポレーションのこれらの欠点は、マイクロスケールで動作し、単一細胞レベルでのより良い制御を可能にする代替技術の追求と開発につながっています。
BioMEMS、または生物医学マイクロ電気機械システムの分野は、マイクロエレクトロニクス業界で行われた技術の進歩に由来しています。具体的には、微細加工プロセスを利用して、バイオメディカル研究の進歩のためのマイクロデバイスを開発します。これらの進歩には、in vivo電気モニタリング用の微小電極アレイ13、in situエレクトロポレーション用の容量性微小電極14、小型化された臓器オンチップデバイス15、マイクロ流体ポイントオブケア診断16、バイオセンサー17、およびナノおよびマイクロエレクトロポレーションデバイス19,20,21を含む薬物送達システム18の開発が含まれます。.生物学的細胞と同じサイズスケールでデバイスを設計および製造する能力のために、ナノおよびマイクロエレクトロポレーション技術は、それらのマクロスケールの対応物と比較して有利である22,23。これらのエレクトロポレーションデバイスは、通常、数十から数百マイクロメートルの間隔の電極セットが統合されているため、高電圧パルスアプリケーションの要件を排除します。この機能により、電解液を流れる電流が大幅に減少し、有毒な電解生成物の蓄積とこれらのシステムでのジュール加熱の影響が減少します。マイクロスケールチャネルはまた、パルス印加中にはるかに均一な電界が細胞に確実に印加されることを保証し、より一貫した結果をもたらす24。さらに、マイクロエレクトロポレーションデバイスがマイクロ流体プラットフォームに統合されることも一般的であり、これは完全に自動化された技術への将来の統合に役立ち、細胞治療製造において非常に望ましい機能である25。最後に、マイクロスケールのエレクトロポレーションは、エレクトロポレーションイベントの電気的尋問を可能にする。例えば、細胞膜透過処理の程度は、単一細胞レベルでリアルタイムでモニターすることができる26、27。この方法の目的は、エレクトロポレーションプロトコルを最適化するために細胞膜透過の程度を測定できるマイクロ流体シングルセルマイクロエレクトロポレーションデバイスの微細加工、システム操作、および分析を説明することですが、以前の最先端よりもスループットを向上させます。
シングルセルレベルのエレクトロポレーションを実行することは、静的セルエレクトロポレーション技術の開発により2001年にRubinskyらによって最初に実証されたため、もはや新しい技術ではありません28。彼らのマイクロデバイスは、エレクトロポレーションのイベントを電気的に監視する能力を最初に実証したため、革新的でした。これにより、細胞膜透過の程度を並列に電気的に検出してデバイスのスループットを向上させることができる静的な単一細胞エレクトロポレーション技術の開発にさらにつながりました。ただし、並列化とバッチ処理を行っても、これらのデバイスは、単位時間29,30あたりに処理できるセルの総数を大幅に欠いています。この制限は、はるかに高いスループットで単一セルレベルのマイクロエレクトロポレーションを行うことができるフロースルーデバイスの開発につながった31。静的環境からフロースルー環境へのこのデバイス移行は、エレクトロポレーションパルスの適用後の細胞膜透過の程度を電気的に監視する能力を制限する。この研究で説明した方法は、これら2つの技術、つまり個々の細胞の細胞膜透過の程度を連続的に連続的に検出、パルス、および監視できるマイクロエレクトロポレーション技術の間のギャップを埋めます。
この技術は最近、Zhengらで説明されました。その作業では、この技術の機能は、エレクトロポレーションパルスの振幅と持続時間の両方を変化させ、細胞膜透過処理を示すその後の電気信号が調査されたパラメトリック研究の完了とともに導入されました32。結果は、エレクトロポレーションパルスの強度の増加(すなわち、印加電界の増加またはパルス持続時間の増加)が、測定された細胞膜透過処理の増加を引き起こすことを示した。系をさらに検証するために、エレクトロポレーションの成功を示す一般的な蛍光指標であるヨウ化プロピジウム33を細胞懸濁液に添加し、電気パルスを印加した直後に蛍光画像を撮影しました。光信号、すなわち細胞内のヨウ化プロピジウムの蛍光強度は、細胞膜透過度の電気的測定と強く相関し、この電気的測定の信頼性を検証した。しかし、この研究では、翻訳可能な重要性がほとんどまたはまったくないヨウ化プロピジウムの送達のみを考慮しました。
この研究では、生物学的に活性なプラスミドDNA(pDNA)ベクターを送達し、エレクトロポレーション後に再播種および培養された細胞の電気トランスフェクション効率を評価しながら、システムのスループットを改善するために、この技術の新しいアプリケーションを紹介します。以前の研究は、エレクトロポレーションのイベントを電気的に測定できる既存のマイクロエレクトロポレーション技術よりも優れていますが、デバイスの現在の状態では、細胞検出、パルス印加、および細胞膜透過測定を実行するために、電極セット間の長い細胞通過時間(~250ミリ秒)が必要です。単一チャネルでは、スループットが 4 セル/秒に制限されます。この制限に対処するために、細胞集団ベースのフィードバック制御エレクトロポレーションの新しい概念が導入され、pDNAエレクトロトランスフェクションが実行されます。低生理伝導率エレクトロポレーションバッファーを使用することにより、このシステムは、多数のエレクトロポレーションパルスアプリケーションにわたって単一細胞の電気的問い合わせを可能にします。次に、電気的応答に基づいて、「最適な」エレクトロポレーションパルスが決定されます。次に、「ハイスループット」モードが実装され、細胞膜透過処理の決定が無効になり、流量が増加し、エレクトロポレーションパルスデューティサイクルが細胞の通過時間に一致して、電極間を通過するセルあたり1パルスが保証されます。この作業では、マイクロデバイスの製造のための微細加工手順、実験を実行するために必要な材料/機器とそのセットアップ、およびデバイスの操作/分析とその電気トランスフェクション効率(eTE)について詳しく説明します。

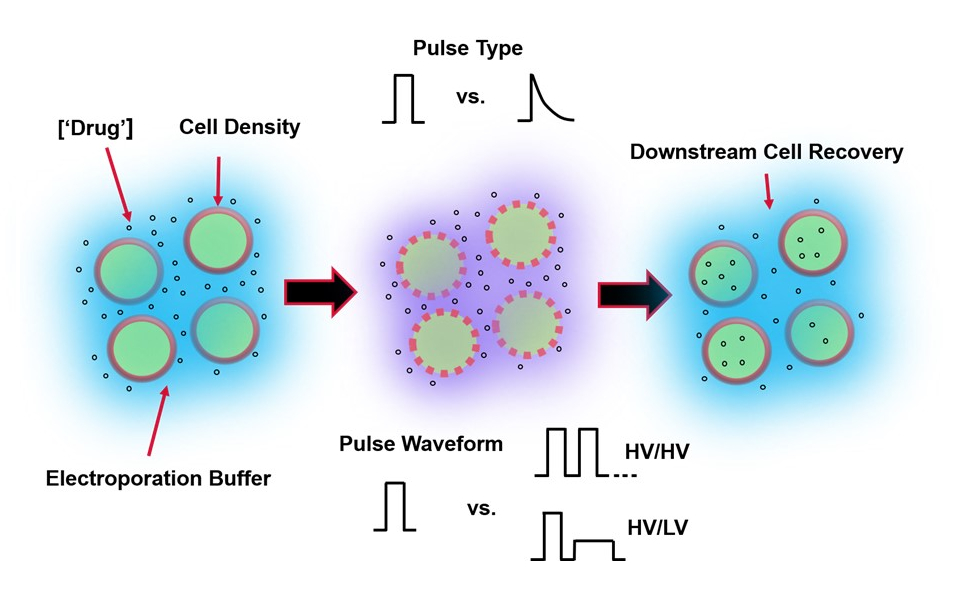
図1:エレクトロポレーションの結果に影響を与える実験的要因 。 (左)細胞懸濁液-エレクトロポレーションの開始前に考慮すべき重要な要素には、ペイロード(この場合はpDNA)、濃度、細胞密度、およびエレクトロポレーションバッファーの特性が含まれます。考慮すべきエレクトロポレーションバッファーの特性は、導電率、浸透圧、およびこれらの値に寄与する正確な分子組成です。(中)パルスアプリケーション-正確なパルスタイプ(方形波対指数関数的減衰)およびパルス波形(単一パルス対パルス列)を最適化して、結果として得られる細胞生存率と電気トランスフェクション効率の両方を最大化する必要があります。エレクトロポレーションプロセスで実装される一般的なパルス列は、通常、一連の高電圧(HV)パルス、またはHVパルスと低電圧(LV)パルス振幅の間で回転する一連のパルスで構成されます。(右)細胞回収-下流処理ステップ、特に細胞が転送される回収細胞培養培地を最適化する必要があります。特徴のない(左端)、エレクトロポレーションプロセス全体の最適化のために、追加のアップストリームセル処理ステップを実装できます。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}
Access restricted. Please log in or start a trial to view this content.
プロトコル
注意: ユーザーは、このプロトコルで使用されている材料と消耗品について、すべてのMSDSを確認する必要があります。各ステップで適切なPPEを着用し、実験中に滅菌技術を使用する必要があります。セクション1〜7では、デバイスの製造について説明します。
1.デバイス製造-マスク設計
注:微細加工プロセスの図については、 図2 を参照してください。微細加工ステップは、クリーンルーム環境で実施する必要があります。追加のPPEが必要です(ヘアネット、フェイシャルヘアネット、マスク、クリーンルームスーツ、靴カバー)。
- 選択したCADソフトウェアをインストールし、マイクロ流体チャネルと電極の両方の2次元「マスク」を設計し、設計を目的のファイル形式(.dxf、.dwgなど)で保存します。
注:2次元マスク回路図の例については、 補足図1 を参照してください。 - 印刷するサプライヤーに送付します。設計の寸法がサプライヤーの解像度能力の範囲内であることを確認してください。
2. デバイス作製:フォトリソグラフィ
注:提供されている微細加工レシピは、フォトレジストのメーカーの推奨事項から採用されており、出発点としてのみ使用する必要があります34。ベーキング時間、露光時間などの正確な値は、製造プロトコルごとに最適化する必要があります。シリコンウェーハとスライドガラスの両方を取り扱うには、ウェーハピンセットを使用することをお勧めします。
- マイクロ流体チャネル作製
- シリコンウェーハとソーダライムガラススライドクリーニング:手順2.1.2〜2.1.3に従って、シリコンウェーハと1インチ×3インチのソーダライムガラススライドクリーニングを実行します(どちらも「基板」と呼びます)。
- 基板をアセトン浴、イソプロパノール(IPA)浴、脱イオン水浴にそれぞれ10分間沈めます。この3ステップの洗浄を室温で連続して実行します。
- 加圧窒素またはろ過された空気ガス源を使用して表面を取り除き、乾燥させます。基板を150°Cのオーブンに最低30分間入れて、残りの水分を蒸発させます。
- シリコンウェーハ上のSU-8フォトリソグラフィ:手順2.1.5〜2.1.14に従って、シリコンウェーハ上でフォトリソグラフィを実行します。
注:20μmのマイクロ流体チャネル高さを達成するために、SU-8 2000シリーズネガフォトレジストを使用しました。正確なスピン速度は、SU-8の配合(すなわち、2010、2015など)によって異なります。但し、以下の条件はSU−8 2010製剤35に関するものである。 - シリコンウェーハを150°Cのオーブンから取り出し、室温(RT)まで冷却します。
- スピンコーターの真空システムを使用して、ウェーハをウェーハスピンコーターのチャックに固定します。スピナーをプログラムします。ステップ1-100rpm/sの加速度で10秒間500rpm、ステップ2-300rpm/sの加速度で30秒間1000rpm。
- 4 mLのSU-8 2010フォトレジストをシリコンウェーハの中央に分注します。プログラムを実行します。システムが停止したら、掃除機をオフにします。
- ピンセットを使用して、SU-8コーティングされたシリコンウェーハを95°Cのホットプレートに4〜5分間移し、ソフトベークします。次に、ウェーハをホットプレートから取り外し、RTまで冷却します。
注意: ラボ固有のフォトリソグラフィマスクアライナーの適切な起動手順に従ってください。 - 2Dマイクロ流体チャネル設計のフォトマスクをマスクホルダーに固定します。SU-8コーティングを上に向けてシリコンウェーハをウェーハチャックに挿入します。
- 露出設定を150mJ/cm2 に設定し、機械を稼働させます。
注意: 目の損傷を防ぐため、UV光源を直接見ないでください。 - SU-8コーティングされたシリコンウェーハを95°Cのホットプレートに4〜5分間置き、露光後ベークを行います。
- シリコンウェーハをSU-8現像液( 材料の表を参照)に3〜4分間浸します。穏やかな攪拌を適用します。溶液からウェーハを取り出し、IPAで表面をすすぎます。
- 加圧窒素またはろ過された空気ガス源を使用して表面を乾燥させます。UVフィルターを使用して顕微鏡下で特徴を検査し、マイクロ流体チャネルに明らかな欠陥がないことを確認します。
- シリコンウェーハを150°Cのオーブンに最低30分間入れて、ハードベイクします。
- RTまで冷却し、スタイラス形状測定を使用して、チャネル側壁の正確な高さと傾斜を測定します。
- スライドガラスのフォトリソグラフィ
注:ヘキサメチルジシラザン(HMDS)は、S1818ポジ型フォトレジスト36の接着促進剤として使用される。- スライドガラスを150°Cのオーブンから取り出し、RTまで冷却します。
- 真空を使用してスライドガラスをスピナーのチャックに固定し、スピナーをプログラムします。ステップ1 - 100 rpm/sの加速度で10秒間500 rpm。ステップ2-3000rpmで300rpm、300rpm/sの加速度で300rpm/sの加速度を記録しました。
- スライドガラスの表面に3〜4滴のHMDSを塗布します。プログラムを実行します。
注:~3μmの表面コーティングを実現するには、S1800ポジ型フォトレジストシリーズを使用する必要があります。正確なスピン速度は製剤によって異なります。以下の推奨事項は、S1818製剤34に関するものです。 - スライドガラスの表面に1 mLのフォトレジストを分注します。表面積をカバーするのに十分なことを確認してください。
- プログラムを実行します。システムが停止したら、掃除機をオフにし、スライドガラスを取り外します。
- S1818コーティングされたスライドガラスを120°Cのホットプレートに4分間置き、ソフトベイクします。取り外してRTに来るのを許可します。
- 2D電極設計のフォトマスクをマスクホルダーに固定します。
- スライドガラスをS1818コーティングを上に向けてウェーハチャックに挿入して位置合わせします。露光設定を250mJ/cm2 に設定し、機械を稼働させます。
注:異なるコンタクトアライナーモデルは、多かれ少なかれ非円形のさまざまな厚さの基材に対応する場合があります。 - スライドガラスをMF-319現像液に2分間浸します。穏やかな攪拌を適用します。スライドガラスの表面を脱イオン水ですすいでください。
- 加圧窒素またはろ過された空気ガス源を使用して表面を乾燥させ、UVフィルターを使用して顕微鏡下で特徴を観察します。リソグラフィパターンに明らかな欠陥がないことを確認してください。
- スライドガラスを150°Cのオーブンに入れ、目的の基板表面が上を向いていることを確認します。オーブンから取り出し、光から保護してください。
3.デバイス製造:フッ化水素酸(HF)エッチング
注意: この手順には、深く痛みを伴う化学火傷を引き起こす可能性のあるフッ化水素酸(HF)の取り扱いと廃棄が含まれます。ハンドラーを保護するために、追加のPPEを使用する必要があります(フェイスシールド、肘の長さの耐薬品性手袋、袖付きの耐薬品性エプロン)。グルコン酸カルシウム中和剤とスキンジェルは、ラボベンチの近くに保管する必要があります。この手順は単独で実行しないでください。HFは、容器が酸によってエッチングされるため、ガラス容器に保管したり、分注したりしないでください。
注意: HFは、露出したガラス(つまり、電極設計)を均一にエッチングしてガラスにくぼみを形成し、金属蒸着後の電極パターンのエッジ解像度を向上させます(セクション4)。
- スライドガラスを10:1緩衝HF溶液に1分間、ポリテトラフルオロエチレン容器に浸します。スライドガラスを脱イオン水に移して洗浄します。洗浄手順を3回繰り返します。
- 加圧窒素またはろ過された空気ガス源を使用して表面を乾燥させます。ガラス基板を65°Cのオーブンに一晩入れて、残っている水分を取り除きます。基板を光から覆います。
4.デバイス製造:物理蒸着
注:このステップでは、スライドガラス基板への金属蒸着を行い、電極パターンを定義します。一般的に使用される金属電極は、クロム/金とチタン/白金です。金および白金はガラス基板に付着しないため、接着を促進するためにそれぞれクロムまたはチタンのシード付着層が必要である37。
- クリーンルーム固有のプロトコルに従って、社内PVDシステムを操作してください。この作業では、DCスパッタリングシステムを使用し、100 SCCMのアルゴンガスを~8 mTorrの圧力と200 Wの電力でスパッタします。
- チタンを~100 Å/分の速度で8分間スパッタします。白金を~200 Å/分の速度で10分間スパッタします。基板をPVDチャンバーから取り出します。
5.デバイス製造:フォトレジストリフトオフ
注:このステップでは、フォトレジスト層をアセトン浴に溶解し、接着した白金電極をスライドガラスにパターン化します。
- 金属コーティングされたスライドガラスをアセトン浴に~10分間沈めます。
- 浴を超音波処理して攪拌を導入し、付着していない金属膜を破壊する。アセトンに浸したワイプを使用して、必要に応じて残留物を取り除きます。
- フォトレジスト/金属をすべて除去したら、電極パターンをイオン交換水で洗浄し、65°Cのオーブンに一晩入れて、残っている表面の水分を取り除きます。
- スタイラスプロファイラー(表面形状測定機)を使用して、パターン化された電極のプロファイルを測定します。
6.デバイス製造:ソフトリソグラフィ
注:このステップでは、エラストマーであるポリジメチルシロキサン(PDMS)を使用して、マイクロ流体チャネルをSU-8マスターレリーフ構造にレプリカ成形します。
- シリコンウェーハシラン化
注: これはオプションの手順です。ただし、サブセクション8で製造されたSU-2.1レリーフ構造の寿命が延びます。このステップは、化学ヒュームフードで実行する必要があります。- ウェーハをペトリ皿の底に固定し、ペトリ皿をデシケーターに入れます。
- シリコンウェーハの周囲を約50μLのトリクロロ(1H,1H,2H,2H-パーフルオロオクチル)シランで囲みます。真空(真空ポンプまたはハウス真空ライン)を接続し、20分間実行します。
- PDMSレプリカ成形
- 使い捨て容器(計量ボート、プラスチックカップなど)で、電子天秤の上にPDMSエラストマーベースと硬化剤を10:1の重量比で混合します。PDMS溶液をシリコンウェーハ上に注ぎ、混合物を真空下に置いてすべての気泡を除去します。
- 65°Cで最低4時間硬化させ、PDMSを固化させます。カミソリの刃の先端を使って、成形したPDMSを切り出し、シリコンウェーハから剥がします。
- 鋭利な生検パンチを使用して、デバイスの入口/出口からPDMSを取り外します。この装置では、入口と出口にそれぞれ0.75mmと3mmの生検パンチを使用しました。
注意: 使用する生検パンチは、リザーバー内のチューブをしっかりと密閉するために、相互接続チューブの外径よりもわずかに小さい直径にする必要があります。
- PDMSの超音波処理洗浄
- PDMSデバイスをIPAに沈め、超音波処理器に30〜45分間入れて、入口/出口からPDMSの破片を取り除きます。PDMSはIPAソリューションで膨らむ可能性があります。
- 脱イオン水ですすぎ、65°Cのオーブンに一晩入れて、PDMSを通常のサイズに戻します。
注意: 破片が残っていると、実験中にデバイスが詰まる可能性があります。破片の大きな部分は、超音波処理の前にスコッチテープを使用してPDMS表面から除去することができます。
7.デバイス製造:PDMSボンディング/ワイヤアタッチメント
注:このステップでは、PDMSとガラス基板の表面を酸素プラズマで処理して、PDMSとガラス38の間に不可逆的な結合を形成します。提供されるレシピは、実験室で使用される正確なシステムに適合させる必要があるかもしれません。
- デバイスをサイズに合わせてカットし、PDMSデバイスの表面がきれいであることを確認します。再洗浄しない場合は、サブセクション6.3の手順に従います。
- プラズマ発生器をプログラムします。 電力 を70 W、 時間を 35秒、 圧力 を325 mTorr、酸素ガスの 流量 を60 SCCMに設定します。PDMSと電極ガラススライドを機能上に向けてシステムに配置し、プログラムを実行します。
- デバイスを取り外し、ステレオスコープを使用してチャンネル機能を電極にすばやく位置合わせします。PDMSの中心から側面に向かってしっかりと圧力をかけ、ボンディング界面の不要な気泡を取り除きます。
- 95°Cの高温の場所に少なくとも2分間置き、ボンディング手順を完了し、デバイスをRTで冷却します。
- 22Gの単線を~6インチの長さで2本切断し、両端から絶縁体を剥がします。
- 銀導電性エポキシを使用してワイヤを電極パッドに接着します。完成したデバイスを65°Cのオーブンに一晩入れます。

図2:マイクロデバイスの製造。 (A)マイクロ流体チャネルの製造-重要なステップ:シリコンウェーハの洗浄(ステップ2.1.1-2.1.3)、フォトレジストコーティングとソフトベーク(ステップ2.1.7-2.1.8)、UV露光(ステップ2.1.10)、現像(ステップ2.1.12)、およびPDMS注入(サブセクション6.2)。(B)電極製造-重要なステップ:ガラススライドクリーニング(ステップ2.1.1-2.1.3)、HMDSコーティングおよびフォトレジストコーティング(ステップ2.2.3-2.2.4)、UV露光(ステップ2.2.8)、現像(ステップ2.2.9)、HFエッチング(セクション3)、物理蒸着(セクション4)、およびフォトレジストリフトオフ(セクション5)。(C)デバイスのファイナライゼーション-重要なステップ:入口/出口アクセスおよび超音波処理(ステップ6.2.3およびセクション6.3)、PDMSボンディング、およびワイヤアタッチメント(セクション7)。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}
8.細胞培養と収穫
注:標準的な細胞培養および滅菌処理手順を利用する必要があります。.細胞培養のための細胞タイプ固有のプロトコルに従ってください。
- 細胞培養
- 細胞継代:ステップ8.1.2〜8.1.5に従って細胞を培養および継代する。
- HEK293細胞を完全DMEM溶液(88%DMEM、10%熱不活化ウシ胎児血清、1%L-グルタミン、1%ペニシリン-ストレプトマイシン)で、37°C、95%O2、5%CO2のインキュベーター内のT25フラスコに培養します。 ~80%のコンフルエントに達したときにスケジュール通りに細胞を継代します。
- ピペットまたは真空システムを使用して培地を吸引し、細胞を0.25%トリプシン-EDTA(2 mL-T25フラスコ)中で37°Cで2分間インキュベートします。 2倍の量の培地でトリプシンを中和します。
- 細胞懸濁液を15 mLコニカルチューブに移し、HEK293細胞を770 x g で2分間遠心分離します。ピペットまたは真空システムを使用して上清を吸引します
- HEK293細胞を1 mLの予温DMEMに再懸濁します。
- セルプレーティング:ステップ8.1.7-8.1.8に従ってセルをプレートします
- T25フラスコ(5 mL のDMEM)で10:1から20:1の希釈で細胞をプレートし、培養を続行します。
- エレクトロポレーション実験のために回収する6ウェルプレート(ウェルあたり2 mLのDMEM)で細胞を5:1から20:1の希釈でプレートします。
注:HEK293細胞は、エレクトロポレーション実験の24時間前に播種し、細胞回収時に~70%のコンフルエンシーを達成しました(サブセクション8.3)。収穫スケジュールに一貫性がないと、エレクトロポレーションの結果にばらつきが生じる可能性があります。
- エレクトロポレーションバッファー
- エレクトロポレーションバッファーの調製
注:エレクトロポレーションバッファー調製の詳細については、Sherbaらを参照してください8。緩衝液組成は、285 mM スクロース、0.7 mM MgCl2、1 mM KCl、10 mM HEPES、3 mM NaOH(pH:7.4、浸透圧:310 mOsm、導電率:500 μS/cm)であった。エレクトロポレーションバッファーは、滅菌方法で処方し、4°Cで保存して~1ヶ月の保存期間とする必要があります。エレクトロポレーションバッファーの製剤は、細胞タイプごとに最適化する必要があります。
- エレクトロポレーションバッファーの調製
- 細胞採取とpDNA添加
- 細胞継代(8.1.2-8.1.4)と同じ手順に従います。
- 滅菌した1x PBSで細胞を洗浄し、細胞懸濁液を15 mLコニカルチューブに移し、細胞を770 x g で2分間遠心分離します。
- HEK293細胞ペレットをエレクトロポレーションバッファーで洗浄し、770 x g で2分間遠心分離します。細胞をエレクトロポレーションバッファーに~500万細胞/mLで再懸濁します。
注:細胞密度は、細胞の種類ごとに最適化する必要があります。 - 緑色蛍光タンパク質(GFP)をコードするpDNAを最終濃度20 μg/mLまで添加します。pDNA/細胞懸濁液を穏やかに混合し、実験のために懸濁液を1 ccシリンジに移します。
9. ハードウェア/実験セットアップ
注:実験のために細胞を回収する前に、細胞がエレクトロポレーションバッファーに懸濁される時間を最小限に抑えるために、実験のセットアップが完了していることを確認してください。実験の20〜30分前に電子機器の電源を入れてウォームアップします。シングルセル検出モジュールの動作に関する実験セットアップの概略図については、 図3 を参照してください。
注:カスタムビルドのPA90オペアンプ回路は、ロックインアンプを使用したシングルセルレベル検出に必要な感度と、十分に強力なエレクトロポレーションパルスを印加するために必要な高電圧の両方に対応するために開発されました。推奨回路39の仕様については、PA90データシートを参照してください。
- ロックインアンプを現在のプリアンプ設定で初期化し、アルゴリズムを介して設定します。ロックイン設定の詳細については、Zhengらを参照してください32。
- 電源、関数発生器、アンプ
- 電源1:回路の負の端に電力を供給するには、-15Vに設定します。
- 電源2(ファンクションジェネレータ):DC信号を出力するように設定し、振幅を2Vに設定します。 50倍のアンプ入力に接続します。
- 矩形波用のエレクトロポレーションパルス発生器をプログラム:目的のパルス幅(デューティサイクル)と目的のパルス振幅(ボルト)を設定します。
- 出力をトリガーモード(1パルス)に設定します。出力を50倍アンプの入力に接続します。
注意: パルス振幅をプログラムするときは、50倍のゲインを覚えておいてください。つまり、1kV/cmの電界強度を実現するには、合計30V、30V/300μm(電極間距離)が必要であるため、関数発生器の出力は30/50、つまり600mVに設定する必要があります。 - オシロスコープを使用して50倍アンプの出力を確認します。電源2(9.2.2)から1-100Vを出力します。エレクトロポレーションパルスの出力2可変振幅(9.2.4)。
- 10倍プローブをオシロスコープのチャンネルと、エレクトロポレーションパルスを印加するステップ7.6で完成したマイクロデバイス(被試験デバイス、DUT)に接続します。実験中にシステムを監視して、パルスが適用されていることを確認します。
- ロックインUSBが接続され、登録されていることを確認します。アルゴリズムコード内のすべてのロックイン設定(最も重要なのはロックイン出力周波数)を再確認してください。
- 顕微鏡/CCDカメラ
- スライドホルダーを介してマイクロデバイスを顕微鏡のステージに置きます。CCDカメラの電源を入れ、マイクロ流体チャネルに焦点を合わせます。4倍または10倍の対物レンズを使用します。

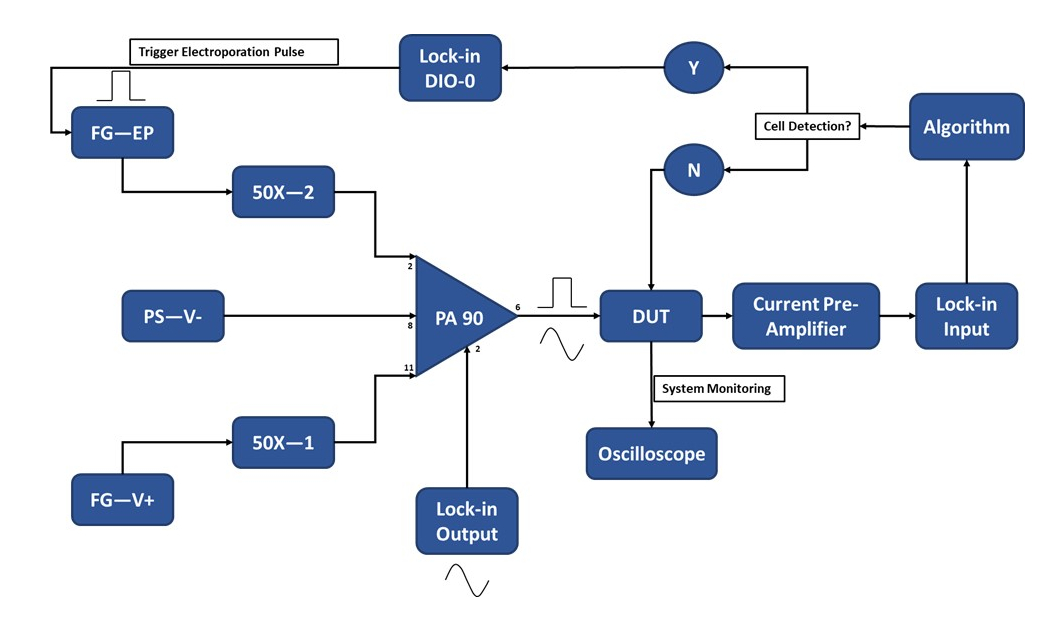
図3:実験セットアップ回路図-単一細胞検出。 高出力オペアンプ(PA-90)により、シングルセル検出に必要なロックイン出力AC信号に高電圧エレクトロポレーションパルスを重ね合わせることができます。この励起信号はマイクロエレクトロポレーションデバイス(被試験デバイス、DUT)を通過し、電流は電流プリアンプによって増幅され、アルゴリズムに供給されます。システムは、セル検出イベントを継続的に監視します。セルが侵入すると、ロックインアンプによってデジタル信号が生成され、輸送中のセルへのエレクトロポレーションパルスの適用がトリガーされます。凡例:PA-90(ハイパワーオペアンプ)、DUT(被試験デバイス)、DIO(デジタル入力/出力)、FG-EP(ファンクションジェネレータ/エレクトロポレーションパルス)、50X(50Xアンプ)、PS-V-(電源/PA 90の負電圧)、FG-V+(ファンクションジェネレータ、PA 90の正電圧)。この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}
10.実験操作
- マイクロ流体チャネルプライミング
- セルロードシリンジからすべての気泡を取り除きます。30 Gの針をセルロードシリンジに取り付けます。
- ピンセットを使用して、タイゴンチューブを針の長さにスライドさせます。出口リザーバーに回収培地(抗生物質なしのステップ8.1.2と同じ)、~40-50 μLを事前に充填します。
- 親指を使用して、流体がチューブラインの端にゆっくりと到達するようにプランジャーにゆっくりと圧力をかけます。
- シリンジをシリンジポンプに固定します。シリンジポンプをオンにして、前方灌流に設定されていることを確認します。
- 流量が正確であることを確認するために、シリンジの適切な直径にポンプをプログラムします。シリンジの直径の詳細については、ポンプのマニュアルを参照してください。
注意: 細胞がシリンジに沈降するのを防ぐために、シリンジポンプをクランプスタンドに固定して、シリンジの端を下に向けて垂直位置で動作できるようにします。 - シリンジポンプの流量を~10-20 μL/minに設定し、流体がチューブラインの端に達するまでポンプを稼働させます。チューブをマイクロ流体デバイスに固定します。
- シリンジポンプの流量を~5-10 μL/minに下げ、マイクロ流体デバイスからすべての空気が排出され、細胞がデバイスの出口まで通過するまでポンプを作動させます。
- ピペット吸引で出口から細胞を取り除きます。出口リザーバーに回収培地(抗生物質なしのステップ8.1.2と同じ)、~40-50μLを再充填します。
- 単一細胞エレクトロポレーション-細胞膜透過マッピング
注:細胞膜透過処理と細胞膜透過処理マッピングを示す電気データをそれぞれ理解するには、図 4 と 図5 を参照してください。- シリンジポンプの流量を~0.1-0.3 μL/minに設定して、電極セットを通る単一セルの流れを確保します。電極間のセル通過時間は~250ミリ秒である必要があります。
- [ファイル名を指定して実行]をクリックしてコンピュータプログラムを起動します。システムが電気データを保存していることを確認します。
- システムがセルを確実に検出して、コンピューター制御のパルスアプリケーションをトリガーしていることを確認します。それに応じて 検出しきい値 を調整します。
- 初期の最低電気エネルギーエレクトロポレーションパルスのパルスパラメータを設定します。この研究におけるエレクトロポレーションパルスパラメータについては 、表1 を参照してください。
- エレクトロポレーションパルス発生器の出力チャンネルをオンにします(ステップ9.2.3)。
- 事前に決定された数の細胞検出/パルスアプリケーションに従います(n = 100)。テストされた各条件の最後に、マイクロデバイスのコンセントから細胞を吸引し、コンセントに回収培地を補充します。
- 次のエレクトロポレーションパルス条件まで反復します。すべてのエレクトロポレーションパルス条件がテストされるまで繰り返します。
- 試験した各パルスアプリケーションについて細胞膜透過処理の程度を決定する。(後処理の検証については、サブセクション11.1で説明します)。細胞膜透過処理マップを生成します(図5)。
- エレクトロポレーションパルスパラメータを決定し、ハイスループットのポピュレーションベースのフィードバックを実現します。
- シリンジポンプの電源を切り、出口リザーバーから細胞を取り除き、出口に回収培地を補充します。
- ポピュレーションベースのフィードバック制御エレクトロポレーション-ハイスループット
注:母集団ベースのフィードバックプロセスを示す概略図については、 図6 を参照してください。- シリンジポンプの流量を~1-3 μL/minに設定して、電極セットを通る単一セルの流れを確保します。電極間のセル通過時間は~25ミリ秒である必要があります。
- パルス振幅を「最適化」条件(10.2.9)に設定し、トリガーモードをオフにして、セルの通過時間に一致するようにパルス幅を設定します。
- パルスオン時間が「最適化された」条件と一致するようにデューティサイクルを設定します。 表 1 を参照してください。
- 出力チャンネル関数発生器を ONに設定し、シリンジポンプをオンにして、必要な数のセルがエレクトロポレーションされるまでシステムを稼働させます。
- 完了したら、シリンジポンプとファンクションジェネレーターの両方をオフにします。
- 細胞を出口リザーバーから、事前に温めた回収培地で満たされた適切なサイズの細胞培養フラスコ/プレートに移し、培養フラスコ/プレートをインキュベーターに移します。
第11章 分析
- 単一細胞レベルの膜透過処理検出
注意: ハイスループットモジュール中に「最適な」パルスが使用されたことを確認するには、実験後の分析を実行して、サブセクション10.2からエクスポートされた電気データを検証する必要があります。エレクトロポレーションによる膜透過処理を代表する電気信号のグラフ表現については 、図4 を参照してください。- データを解析ソフトウェア(MATLAB、Pythonなど)にロードします。各パルス条件の電流対時間のプロットを生成します。
- 細胞膜透過処理の程度(ΔIP / ΔIC)を手動で決定します。図4を参照してください。テストしたすべてのパルス条件で細胞膜透過マップ(ΔIP / ΔI C 対電気エネルギー、図5)を生成します。「最適な」パルス状態を確認します。
- エレクトロトランスフェクション効率(eTE)
- 24時間のインキュベーション期間の後、エレクトロポレーションされた細胞をインキュベーターから取り出します。
- 生細胞染色を行います。DRAQ5を細胞培養容器内で終濃度5 μMに1:1000に希釈します。細胞/染色液を穏やかに混合し、37°Cで5〜30分間インキュベートします。
注:このステップでは、別の染色を実装できます。蛍光特性が、エレクトロトランスフェクションの成功を示す蛍光マーカーと重ならないようにします(GFPは緑色の波長で、DRAQ5は遠赤色です)。 - エピ蛍光顕微鏡、ランプ、およびカメラの電源を入れます(材料の表を参照)。
- インキュベーターから細胞を取り出し、顕微鏡に焦点を合わせます。
- 選択したフィールドの位相差画像(明視野)をキャプチャします。
- FITC(GFP)および遠赤色(DRAQ5)フィルタを使用して、同じフィールドのエピ蛍光画像をキャプチャします。手動またはアルゴリズムを使用して画像セットを分析します。
メモ: 代表的な画像については、 図7 を参照してください。 - すべての画像中のGFP陽性細胞の総数をカウントします。すべての画像でDRAQ5染色細胞の総数をカウントします。eTE(DRAQ5染色細胞に対するGFP陽性細胞の比率)を計算します。
Access restricted. Please log in or start a trial to view this content.
結果
図4は、単一パルス振幅に対する単一細胞レベルの膜透過処理検出の背後にある動作原理を示しています。エレクトロポレーション実験の開始後、細胞検出アルゴリズムは、ポイントごとのスロープベースの検出方法を介して、細胞検出に最適な閾値を決定します。次に、システムは、セルの進入を示す測定された電流の有意な負の変化について(1)を継続的に監視します...
Access restricted. Please log in or start a trial to view this content.
ディスカッション
このプロトコル内で提示される方法論は、主にマイクロ流体デバイスの微細加工に焦点を当てており、その後、特殊なエレクトロポレーション実験セットアップに統合されます。微細加工プロセスの詳細を説明するときによく使用される「レシピ」という用語は、機能するデバイスを正常に製造するために各ステップに従う/最適化することの重要性を示唆しています。ただし、UV露光時間/エ...
Access restricted. Please log in or start a trial to view this content.
開示事項
著者は開示するものは何もありません。
謝辞
著者らは、全米科学財団(NSF CBET 0967598、DBI IDBR 1353918)および米国教育省の精密および個別化医療の新興分野における大学院トレーニング(P200A150131)による、大学院生J.J.S.のフェローシップへの資金提供に対する財政的支援に感謝したいと思います。
Access restricted. Please log in or start a trial to view this content.
資料
| Name | Company | Catalog Number | Comments |
| 150-mm diameter petri dishes | VWR | 25384-326 | step 6.1.1 to secure wafer |
| 24-well tissue culture plates | VWR | 10062-896 | step 10.3.6 to plate electroporated cells |
| 33220A Waveform/Function generator | Agilent | step 9.2.3 electroporation pulse generator | |
| 4'' Si-wafers | University Wafer | subsection 2.1 for microfluidic channel fabrication | |
| 6-well tissue culture plates | VWR | 10062-892 | step 8.1.8 to plate cells |
| Acetone | Fisher Scientific | A18-4 | step 2.1.2 for cleaning and step 5.1 photoresist lift-off |
| Allegra X-22R Centrifuge | Beckman Coulter | steps 8.1.4 , 8.3.2. and 8.3.3. to spin down cells | |
| AutoCAD 2018 | Autodesk | subsection 1.1. to design transparency masks | |
| Buffered oxide etchant 10:1 | VWR | 901621-1L | subsection 3.1 for HF etching |
| CCD Monochrome microscope camera | Hamamatsu | Orca 285 C4742-96-12G04 | step 11.2.3. for imaging |
| CMOS camera- Sensicam QE 1.4MP | PCO | subsection 9.3 part of the experimental setup | |
| Conductive Epoxy | CircuitWorks | CW2400 | subsection 7.6. for wire attachement |
| Conical Centrifuge Tubes, 15 mL | Fisher Scientific | 14-959-70C | step 8.1.4. for cell centrifuging |
| Dektak 3ST Surface Profilometer | Veeco (Sloan/Dektak) | step 2.1.15 and 5.4 for surface profilometry | |
| Disposable biopsy punch, 0.75 mm | Robbins Instruments | RBP075 | step 6.2.3 for inlet access |
| Disposable biopsy punch, 3 mm | Robbins Instruments | RBP30P | step 6.2.3 for outlet access |
| DRAQ5 | abcam | ab108410 | step 11.2.2. for live cell staining |
| Dulbecco’s Modified Eagle’s Medium | ThermoFisher Scientific | 11885084 | step 8.1.2. part of media composition |
| E3631A Bipolar Triple DC power supply | Agilent | step 9.2.1.-9.2.2.part of the experimental setup | |
| Eclipse TE2000-U Inverted Microscope | Nikon | subsection 9.3. part of the experimental setup | |
| EVG620 UV Lithography System | EVG | step 2.1.9. and 2.2.7. for UV Exposure | |
| Fetal Bovine Serum | Neuromics | FBS001 | step 8.1.2. part of media composition |
| FS20 Ultrasonic Cleaner | Fisher Scientific | subsection 5.1. for photoresist lift-off | |
| Glass Media Bottle with Cap, 100mL | Fisher Scientific | FB800100 | step 8.2.1. for buffer storage |
| Glass Media Bottle with Cap, 500mL | Fisher Scientific | FB800500 | step 8.1.2.for media storage |
| HEK-293 cell line | ATCC | CRL-1573 | subsection 8.1 for cell culturing |
| HEPES buffer solution | Sigma Aldrich | 83264-100ML-F | step 8.2.1 part of electroporation buffer composition |
| Hexamethyldisilazane | Sigma Aldrich | 379212-25ML | step 2.2.3 adhesion promoter |
| HF2LI Lock-in Amplifier | Zurich Instruments | subsection 9.2 part of the experimental setup | |
| HF2TA Current amplifier | Zurich Instruments | subsection 9.2 part of the experimental setup | |
| Isopropyl Alcohol | Fisher Scientific | A459-1 | step 2.1.2 for cleaning, step 2.1.14 for rinsing wafer following SU-8 development, and step 6.3.1 for cleaning PDMS |
| IX81 fluorescence microscope | Olympus | step 11.2.3 for imaging | |
| L-Glutamine Solution | Sigma Aldrich | G7513-20ML | step 8.1.2. part of media composition |
| M16878/1BFA 22 gauge wire | AWC | B22-1 | subsection 7.5 for device fabrication |
| Magnesium chloride | Sigma Aldrich | 208337-100G | step 8.1.2 part of electroporation buffer composition |
| MF 319 Developer | Kayaku Advanced Materials | 10018042 | step 2.2.9. photoresist developer |
| Microposit S1818 photoresist | Kayaku Advanced Materials | 1136925 | step 2.2.4 positive photoresist for electrode patterning |
| Microscope slides, 75 x 25 mm | VWR | 16004-422 | step 2.2.1 electrode soda lime glass substrate |
| Model 2350 High voltage amplifier | TEGAM | 2350 | step 9.2.5. part of the experimental setup |
| National Instruments LabVIEW | National Instruments | data acquisition | |
| Needle, 30G x 1 in | BD Scientific | 305128 | step 10.1.1. part of the system priming |
| PA90 IC OPAMP Power circuit | Digi-key | 598-1330-ND | Part of the custom circuit |
| Penicillin-Streptomycin | Sigma Aldrich | P4458-20ML | step 8.1.2. part of media composition |
| Plasmid pMAX-GFP | Lonza | VCA-1003 | step 8.3.4. for intracellular delivery |
| Plastic tubing, 0.010'' x 0.030" | VWR | 89404-300 | step 10.1.2. for system priming |
| Platinum targets | Kurt J. Lesker | subsection 4.2. for physical vapor deposition | |
| Potassium chloride | Sigma Aldrich | P9333-500G | step 8.2.1. part of electroporation buffer composition |
| Pump 11 PicoPlus microfluidic syringe pump | Harvard Apparatus | MA1 70-2213 | step 10.1.4. for system priming |
| PVD75 Physical vapor deposition system | Kurt J. Lesker | subsection 4.1. for physical vapor deposition | |
| PWM32 Spinner System | Headway Research | steps 2.1.6 and 2.2.2. for substrate coating with photoresist | |
| PX-250 Plasma treatment system | March Instruments | subsection 7.2 for PDMS and glass substrate bonding | |
| SDG1025 Function/Waveform generator | Siglent | step 9.2.2. part of the experimental setup | |
| Sodium hydroxide | Sigma Aldrich | S8045-500G | step 8.2.1. part of electroporation buffer composition |
| SU-8 2010 negative photoresist | Kayaku Advanced Materials | Y111053 | step 2.1.7. for microfluidic channel patterning |
| SU-8 developer | Microchem | Y010200 | step 2.1.12. for photoresist developing |
| Sucrose | Sigma Aldrich | S7903-1KG | step 8.2.1. part of electroporation buffer composition |
| Sylgard 184 elastomer kit | Dow Corning | 3097358-1004 | step 6.2.1 10 : 1 mixture of PDMS polymer and hardening agent |
| Syringe, 1 ml | BD Scientific | 309628 | step 8.3.4. part of system priming |
| SZ61 Stereomicroscope System | Olympus | subsection 7.3. for channel and electrode alignment | |
| Tissue Culture Treated T25 Flasks | Falcon | 353108 | step 8.1.2 for cell culturing |
| Titanium targets | Kurt J. Lesker | subsection 4.2. for physical vapor deposition | |
| Transparency masks | CAD/ART Services | steps 2.1.9. and 2.2.7. for photolithography | |
| Trichloro(1H,1H,2H,2H-perfluorooctyl)silane | Sigma Aldrich | 448931-10G | step 6.1.2. for wafer silanization |
| Trypsin-EDTA solution | Sigma Aldrich | T4049-100ML | steps 8.1.3. and 8.3.1. for cell harvesting |
参考文献
- Gao, Q. Q., et al. Therapeutic potential of CRISPR/Cas9 gene editing in engineered T-cell therapy. Cancer Medicine. 8 (9), 4254-4264 (2019).
- Aijaz, A., et al. Biomanufacturing for clinically advanced cell therapies. Nature Biomedical Engineering. 2 (6), 362-376 (2018).
- Milone, M. C., O'Doherty, U. Clinical use of lentiviral vectors. Leukemia. 32 (7), 1529-1541 (2018).
- Weaver, J. C., Chizmadzhev, Y. A. Theory of electroporation: A review. Bioelectrochemistry and Bioenergetics. 41 (2), 135-160 (1996).
- Kotnik, T., Rems, L., Tarek, M., Miklavcic, D. Membrane electroporation and electropermeabilization: mechanisms and models. Annual Review of Biophysics. 48, 63-91 (2019).
- Rosazza, C., Meglic, S. H., Zumbusch, A., Rols, M. P., Miklavcic, D. Gene electrotransfer: A mechanistic perspective. Current Gene Therapy. 16 (2), 98-129 (2016).
- Clauss, J., et al. Efficient non-viral T-cell engineering by sleeping beauty minicircles diminishing DNA toxicity and miRNAs silencing the endogenous T-cell receptors. Human Gene Therapy. 29 (5), 569-584 (2018).
- Sherba, J. J., et al. The effects of electroporation buffer composition on cell viability and electro-transfection efficiency. Scientific Reports. 10 (1), 3053(2020).
- Lu, H., Schmidt, M. A., Jensen, K. F. A microfluidic electroporation device for cell lysis. Lab on a Chip. 5 (1), 23-29 (2005).
- Kar, S., et al. Single-cell electroporation: current trends, applications and future prospects. Journal of Micromechanics and Microengineering. 28 (12), (2018).
- Shi, J. F., et al. A review on electroporation-based intracellular delivery. Molecules. 23 (11), (2018).
- Wang, S. N., Zhang, X. L., Wang, W. X., Lee, L. J. Semicontinuous flow electroporation chip for high-throughput transfection on mammalian cells. Analytical Chemistry. 81 (11), 4414-4421 (2009).
- Wei, W. J., et al. An implantable microelectrode array for simultaneous L-glutamate and electrophysiological recordings in vivo. Microsystems & Nanoengineering. 1, (2015).
- Maschietto, M., Dal Maschio, M., Girardi, S., Vassanelli, S. In situ electroporation of mammalian cells through SiO2 thin film capacitive microelectrodes. Scientific Reports. 11 (1), (2021).
- Wu, Q. R., et al. Organ-on-a-chip: recent breakthroughs and future prospects. Biomedical Engineering Online. 19 (1), (2020).
- Pandey, C. M., et al. Microfluidics Based Point-of-Care Diagnostics. Biotechnology Journal. 13 (1), (2018).
- Vigneshvar, S., Sudhakumari, C. C., Senthilkumaran, B., Prakash, H. Recent advances in biosensor technology for potential applications - An overview. Frontiers in Bioengineering and Biotechnology. 4, (2016).
- Nuxoll, E. BioMEMS in drug delivery. Advanced Drug Delivery Reviews. 65 (11-12), 1611-1625 (2013).
- Kang, S., Kim, K. H., Kim, Y. C. A novel electroporation system for efficient molecular delivery into Chlamydomonas reinhardtii with a 3-dimensional microelectrode. Scientific Reports. 5, (2015).
- Zheng, M. D., Shan, J. W., Lin, H., Shreiber, D. I., Zahn, J. D. Hydrodynamically controlled cell rotation in an electroporation microchip to circumferentially deliver molecules into single cells. Microfluidics and Nanofluidics. 20 (1), (2016).
- Santra, T. S., Kar, S., Chang, H. Y., Tseng, F. G. Nano-localized single-cell nano-electroporation. Lab on a Chip. 20 (22), 4194-4204 (2020).
- Lee, W. G., Demirci, U., Khademhosseini, A. Microscale electroporation: challenges and perspectives for clinical applications. Integrative Biology. 1 (3), 242-251 (2009).
- Santra, T. S., Chang, H. Y., Wang, P. C., Tseng, F. G. Impact of pulse duration on localized single-cell nano-electroporation. Analyst. 139 (23), 6249-6258 (2014).
- Geng, T., Lu, C. Microfluidic electroporation for cellular analysis and delivery. Lab on a Chip. 13 (19), 3803-3821 (2013).
- Hsi, P., et al. Acoustophoretic rapid media exchange and continuous-flow electrotransfection of primary human T cells for applications in automated cellular therapy manufacturing. Lab on a Chip. 19 (18), 2978-2992 (2019).
- Khine, M., Ionescu-Zanetti, C., Blatz, A., Wang, L. P., Lee, L. P. Single-cell electroporation arrays with real-time monitoring and feedback control. Lab on a Chip. 7 (4), 457-462 (2007).
- Ye, Y. F., et al. Single-cell electroporation and real-time electrical monitoring on a microfluidic chip. 2020 33rd Ieee International Conference on Micro Electro Mechanical Systems (Mems 2020). , 1040-1043 (2020).
- Huang, Y., Rubinsky, B. Microfabricated electroporation chip for single cell membrane permeabilization. Sensors and Actuators a-Physical. 89 (3), 242-249 (2001).
- Guo, X. L., Zhu, R. Controllable in-situ cell electroporation with cell positioning and impedance monitoring using micro electrode array. Scientific Reports. 6, (2016).
- Punjiya, M., Nejad, H. R., Mathews, J., Levin, M., Sonkusale, S. A flow through device for simultaneous dielectrophoretic cell trapping and AC electroporation. Scientific Reports. 9, (2019).
- Wang, H. Y., Lu, C. Microfluidic electroporation for delivery of small molecules and genes into cells using a common DC power supply. Biotechnology and Bioengineering. 100 (3), 579-586 (2008).
- Zheng, M. D., et al. Continuous-flow, electrically-triggered, single cell-level electroporation. Technology. 5 (1), 31-41 (2017).
- Batista Napotnik, T., Miklavcic, D. In vitro electroporation detection methods - An overview. Bioelectrochemistry. 120, 166-182 (2018).
- MICROPOSIT™ S1800® G2 Series Photoresists. KAYAKU. , Available from: https://kayakuam.com/wp-content/uploads/2019/09/S1800-G2.pdf (2021).
- SU-8 2000 Permanent Negative Epoxy Photoresist. KAYAKU. , Available from: https://kayakuam.com/wp-content/uploads/2020/08/KAM-SU-8-2000-2000.5-2015-Datasheet-8.13.20-final.pdf (2001).
- Substrate Preparation. MicroChemicals. , Available from: https://www.microchemicals.com/technical_information/subtrate_cleaning_adhesion_photoresist.pdf (2021).
- Lisinenkova, M., Hahn, L., Schulz, J. 4M 2006 - Second International Conference on Multi-Material Micro Manufacture. , Elsevier. 91-94 (2006).
- Beh, C. W., Zhou, W. Z., Wang, T. H. PDMS-glass bonding using grafted polymeric adhesive - alternative process flow for compatibility with patterned biological molecules. Lab on a Chip. 12 (20), 4120-4127 (2012).
- PA90 High Voltage Power Operational Amplifiers. APEX. , Available from: https://www.apexanalog.com/resources/products/pa90u.pdf (2021).
- Lissandrello, C. A., et al. High-throughput continuous-flow microfluidic electroporation of mRNA into primary human T cells for applications in cellular therapy manufacturing. Scientific Reports. 10 (1), 18045(2020).
Access restricted. Please log in or start a trial to view this content.
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請さらに記事を探す
This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved