
Для просмотра этого контента требуется подписка на Jove Войдите в систему или начните бесплатную пробную версию.
Method Article
Изготовление и эксплуатация системы микроэлектропорации непрерывного потока с обнаружением пермеабилизации
В этой статье
Резюме
Этот протокол описывает методы микропроизводства, необходимые для создания микрофлюидного электропорационного устройства «лаборатория на чипе». Экспериментальная установка выполняет контролируемые одноклеточные трансфекции в непрерывном потоке и может быть расширена до более высокой пропускной способности с популяционным контролем. Представлен анализ, демонстрирующий возможность электрического мониторинга степени пермеабилизации клеточной мембраны в режиме реального времени.
Аннотация
Современные терапевтические инновации, такие как CAR-T-клеточная терапия, в значительной степени зависят от вирусно-опосредованной доставки генов. Несмотря на эффективность, этот метод сопровождается высокими производственными затратами, что вызвало интерес к использованию альтернативных методов доставки генов. Электропорация — это электрофизический, невирусный подход к внутриклеточной доставке генов и других экзогенных материалов. При приложении электрического поля клеточная мембрана временно допускает молекулярную доставку в клетку. Как правило, электропорация выполняется на макромасштабе для обработки большого количества клеток. Однако такой подход требует обширной разработки эмпирического протокола, что является дорогостоящим при работе с первичными и труднотрансфектируемыми типами клеток. Длительная разработка протокола в сочетании с требованием больших напряжений для достижения достаточной напряженности электрического поля для проницаемости ячеек привела к разработке микромасштабных электропорационных устройств. Эти микроэлектропорационные устройства изготавливаются с использованием распространенных методов микрофабрикации и позволяют осуществлять более широкий экспериментальный контроль с потенциалом для поддержания высокой пропускной способности. Эта работа основана на микрофлюидно-электропорационной технологии, способной обнаруживать уровень пермеабилизации клеточной мембраны на уровне одной клетки при непрерывном потоке. Однако эта технология была ограничена 4 ячейками, обработанными в секунду, и поэтому здесь предложен и представлен новый подход к увеличению пропускной способности системы. Этот новый метод, обозначаемый как контроль обратной связи на основе клеточной популяции, рассматривает реакцию пермеабилизации клеток на различные условия пульсации электропорации и определяет наиболее подходящие условия импульса электропорации для тестируемого типа клеток. Затем используется режим с более высокой пропускной способностью, где этот «оптимальный» импульс подается на суспензию ячейки в пути. Подробно представлены этапы изготовления устройства, настройки и проведения микрофлюидных экспериментов, а также анализа результатов. Наконец, эта технология микроэлектропорации демонстрируется путем доставки плазмиды ДНК, кодирующей зеленый флуоресцентный белок (GFP), в клетки HEK293.
Введение
Современные терапевтические инновации в биомедицинских исследованиях, такие как CAR-T (Chimeric Antigen Receptor Engineered T cell) клеточная терапия и генетическое редактирование с использованием CRISPR (кластеризованные регулярно чередующиеся короткие палиндромные повторяющиеся последовательности ДНК) / Cas9, в значительной степени зависят от способности успешно и эффективно доставлять экзогенный материал во внутриклеточное пространство1. В терапии CAR-T золотым стандартом для выполнения этапа доставки генов в производстве клеточной терапии является использование вирусных векторов2. Хотя вирусно-опосредованная доставка генов является эффективным методом доставки, она также имеет несколько недостатков. К ним относятся производственные затраты, цитотоксичность, иммуногенность, потенциал мутагенеза/опухолегенеза и ограничения по размеру гена (генов), который будет доставлен3. Эти ограничения привели к исследованиям и разработкам альтернативных, невирусных технологий доставки.
Электропорация, альтернатива вирусно-опосредованной доставке генов, опирается на применение оптимальной формы сигнала электрического импульса для выполнения трансфекции ДНК, РНК и белка клеток. После применения внешнего электрического поля клеточная мембрана ненадолго компрометируется, что делает клетку восприимчивой к внутриклеточной доставке в противном случае непроницаемых экзогенных материалов4. По сравнению с вирусно-опосредованной доставкой, электропорация выгодна, поскольку она, как правило, безопасна, проста в эксплуатации и имеет низкие эксплуатационные расходы. Электропорация может доставлять как мелкие, так и большие молекулярные грузы и может быть эффективной в трансфекции клеток независимо от линии5. Для достижения желаемых результатов после электропорации, т.е. хорошей жизнеспособности и хорошей эффективности электротрансфекции, необходимо совместно оптимизировать различные экспериментальные параметры. К ним относятся тип ячейки6, плотность клеток, концентрациямолекулы 7, свойства буфера электропорации (например, молекулярный состав, проводимость и осмолярность)8, размер /геометрия электрода9 и форма сигнала электрического импульса (форма, полярность, количество импульсов)10 (см. Рисунок 1 для иллюстрации). Хотя каждый из этих параметров может оказать значительное влияние на результаты экспериментов с электропорацией, форма импульсного сигнала была особенно изучена очень подробно, поскольку электрическая энергия приложенного импульса (импульсов) является корнем внутреннего компромисса между результирующей жизнеспособностью ячейки и эффективностью электротрансфекции8.
Как правило, эксперименты с электропорацией проводятся в макромасштабе, где ячейки суспендируются в 100 микролитров буфера между набором больших параллельно пластинчатых электродов внутри электропорационной кюветы. Электроды обычно изготавливаются из алюминия с расстоянием до электрода 1-4 мм. После того, как ячейки загружены вручную с помощью пипетки, кювета электрически подключается к громоздкому генератору электрических импульсов, где пользователь может установить и применить параметры импульсного сигнала для электропорации суспензии ячейки. Хотя макромасштабная или объемная электропорация может обрабатывать плотность ячеек >106 ячеек /мл, эта функция может быть расточительной при оптимизации настроек формы сигнала электрического импульса. Это особенно беспокоит при электропорации первичных типов клеток, где количество клеточных популяций может быть ограничено. Кроме того, из-за большого расстояния между электродами генератор импульсов должен быть в состоянии подавать большие напряжения для достижения напряженности электрического поля >1 кВ / см11. Эти высокие напряжения вызывают рассеивание резистивной мощности через буфер электролита, что приводит к джоулевому нагреву, что может нанести ущерб резистирующей жизнеспособности ячейки12. Наконец, выполнение электропорации на плотной суспензии ячеек будет последовательно обременено врожденной изменчивостью в результирующей эффективности электротрансфекции и жизнеспособности клеток. Каждая ячейка в суспензии может испытывать различную напряженность электрического поля из-за окружающих клеток. В зависимости от того, увеличивается или уменьшается напряженность испытываемого электрического поля, результирующая жизнеспособность ячейки или эффективность электротрансфекции могут отрицательно влиять накаждую из них 11. Эти недостатки макромасштабной электропорации привели к поиску и развитию альтернативных технологий, которые работают в микромасштабе и позволяют лучше контролировать на уровне одной ячейки.
Область BioMEMS, или биомедицинских микроэлектромеханических систем, проистекает из технологических достижений, сделанных в микроэлектронной промышленности. В частности, использование процессов микропроизводства для разработки микроустройств для продвижения биомедицинских исследований. Эти достижения включают разработку микроэлектродных матриц для электрического мониторинга in vivo 13, емкостных микроэлектродов для электропорации in situ14, миниатюрных устройств15 «орган на кристалле», микрофлюидной диагностики в местах оказания медицинской помощи16, биосенсоров17 и систем доставки лекарств18, включая нано- и микроэлектропорационные устройства 19,20,21 . Благодаря способности проектировать и производить устройства в том же масштабе, что и биологические клетки, нано- и микроэлектропорационные технологии являются выгодными по сравнению с их макромасштабным аналогом22,23. Эти электропорационные устройства устраняют необходимость высоковольтных импульсных применений, поскольку электродные наборы с интервалами от 10 до 100 секунд микрометров обычно интегрируются. Эта особенность резко снижает ток через электролит, что, в свою очередь, уменьшает накопление токсичных продуктов электролиза и эффекты джоульного нагрева в этих системах. Микромасштабные каналы также гарантируют, что гораздо более однородное электрическое поле надежно прикладывается к ячейкам во время применения импульса, что приводит к более последовательным результатам24. Кроме того, микроэлектропорационные устройства также являются обычным явлением для интеграции в микрофлюидную платформу, которая позволяет в будущем интегрироваться в полностью автоматизированную технологию, что является весьма желательной возможностью в производстве клеточной терапии25. Наконец, микромасштабная электропорация позволяет проводить электрический опрос событий электропорации. Например, степень пермеабилизации клеточной мембраны может контролироваться в режиме реального времени на уровне одной клетки26,27. Целью этого метода является описание микропроизводства, работы системы и анализа микрофлюидного одноэлектического микроэлектропорационного устройства, способного измерять степень пермеабилизации клеточной мембраны для оптимизации протоколов электропорации, но увеличивая пропускную способность по сравнению с предыдущим уровнем техники.
Выполнение одноэлементной электропорации больше не является новой техникой, как это было впервые продемонстрировано Рубинским и др. в 2001 году с разработкой технологии электропорации статических ячеек28. Их микроустройство было инновационным, поскольку они были первыми, кто продемонстрировал способность электрически контролировать событие электропорации. Это также привело к разработке статических одноэлементных электропорационных технологий, способных электрически определять степень пермеабилизации клеточной мембраны параллельным образом для увеличения пропускной способности устройств. Однако даже при распараллеливании и периодической обработке этим устройствам сильно не хватает общего количества ячеек, которые они могут обрабатывать в единицу времени29,30. Это ограничение привело к разработке проточных устройств, способных выполнять микроэлектропорацию на уровне одной ячейки при гораздо большей пропускной способности31. Этот переход устройства от статической к проточной среде ограничивает возможность электрического мониторинга степени пермеабилизации клеточной мембраны после применения электропорационного импульса. Метод, описанный в этой работе, устраняет разрыв между этими двумя технологиями, микроэлектропорационной технологией, способной электрически обнаруживать, пульсировать и контролировать степень пермеабилизации клеточной мембраны отдельных клеток непрерывным потоком, последовательным способом.
Эта технология была недавно описана в Zheng et al. В этой работе возможности этой технологии были представлены с завершением параметрического исследования, где как амплитуда, так и продолжительность электропорационного импульса варьировались, а последующий электрический сигнал, указывающий на пермеабилизацию клеточной мембраны, был исследован32. Результаты показали, что увеличение интенсивности электропорационного импульса (т.е. увеличение приложенного электрического поля или увеличение длительности импульса) вызывало увеличение измеряемой пермеабилизации клеточной мембраны. Для дальнейшей проверки системы к суспензии ячейки добавляли общий флуоресцентный индикатор успешной электропорации, йодид пропидия33, и сразу после применения электрического импульса было получено флуоресцентное изображение. Оптический сигнал, т.е. интенсивность флуоресценции йодида пропидия внутри ячейки, был сильно коррелирован с электрическим измерением степени пермеабилизации клеточной мембраны, что подтверждало надежность этого электрического измерения. Однако в этой работе рассматривалась только доставка небольшой молекулы йодида пропидия, которая практически не имеет переводимого значения.
В этой работе введено новое применение этой технологии для улучшения пропускной способности системы при доставке биологически активного вектора плазмидной ДНК (пДНК) и оценке эффективности электротрансфекции клеток, покрытых и культивируемых после электропорации. Хотя предыдущая работа превосходит существующие микроэлектропорационные технологии, которые способны электрически измерять событие электропорации, текущее состояние устройства по-прежнему требует длительного времени прохождения ячейки между набором электродов (~ 250 мс) для выполнения обнаружения ячейки, применения импульсов и измерения пермеабилизации клеточной мембраны. При использовании одного канала это ограничивает пропускную способность до 4 ячеек/с. Для борьбы с этим ограничением вводится новая концепция электропорации с обратной связью на основе клеточной популяции для выполнения электротрансфекции пДНК. Используя гипофизиологический буфер электропорации, эта система позволяет проводить электрический опрос отдельных клеток во множестве применений электропорационных импульсов. На основе электрического отклика затем определяется «оптимальный» импульс электропорации. Затем реализуется режим «высокой пропускной способности», в котором определение пермеабилизации клеточной мембраны сводится к нулю, скорость потока увеличивается, а рабочий цикл электропорационного импульса сопоставляется со временем прохождения ячейки, чтобы обеспечить один импульс на ячейку при прохождении между электродами. Эта работа предоставит подробную информацию о этапах микрофабрикации для изготовления микроустройства, материале / оборудовании и их настройке, необходимой для выполнения экспериментов, а также о работе / анализе устройства и его эффективности электротрансфекции (eTE).

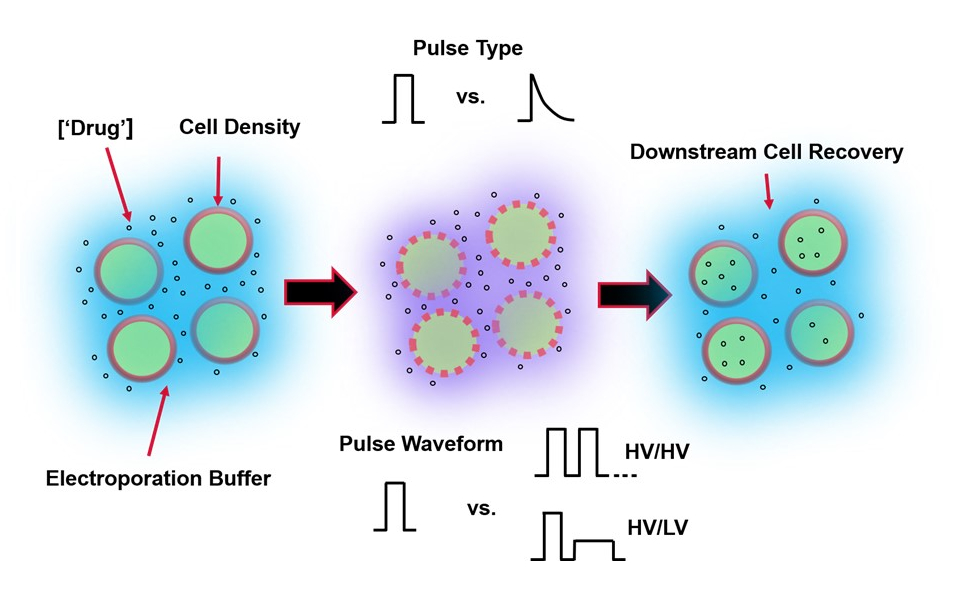
Рисунок 1: Экспериментальные факторы, влияющие на результаты электропорации. (Слева) Суспензия клетки - Важные факторы, которые следует учитывать до начала электропорации, включают: полезную нагрузку (в данном случае пДНК), концентрацию, плотность клеток и свойства буфера электропорации. Следует учитывать свойства электропорационного буфера: проводимость, осмолярность и точный молекулярный состав, способствующий этим значениям. (Средний) Применение импульса - Точный импульсный тип (квадратная волна против экспоненциального распада) и форма импульсного сигнала (одиночный импульс против импульсного поезда) должны быть оптимизированы для максимизации как результирующей жизнеспособности ячейки, так и эффективности электротрансфекции. Общие импульсные цепи, реализуемые в электропорационных процессах, обычно состоят из серии импульсов высокого напряжения (HV) или серии импульсов, вращающихся между величинами импульсов HV и низкого напряжения (LV). (Справа) Этапы обработки восстановления клеток вниз по потоку, в частности, среда культивирования клеток восстановления, в которую переносятся клетки, должны быть оптимизированы. Не показанные (крайний левый), дополнительные этапы обработки вышестоящих ячеек могут быть реализованы для общей оптимизации процесса электропорации. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
{kind=link}
Access restricted. Please log in or start a trial to view this content.
протокол
ПРИМЕЧАНИЕ: Пользователи должны проверить все MSDS на наличие материалов и расходных материалов, используемых в этом протоколе. Соответствующие СИЗ следует носить на каждом этапе, а стерильную технику использовать во время экспериментов. В разделах 1-7 обсуждается изготовление устройства.
1. Изготовление устройства - Дизайн маски
ПРИМЕЧАНИЕ: Обратитесь к рисунку 2 для иллюстрации процесса микропроизводства. Этапы микрофабрикации должны проводиться в условиях чистой комнаты. Необходимы дополнительные СИЗ (сетка для волос, сетка для волос на лице, маска, костюм для чистой комнаты, бахилы).
- Установите программное обеспечение CAD по выбору, спроектируйте 2-мерную «маску» как микрофлюидного канала, так и электродов и сохраните дизайн в желаемом формате файла (т. Е. .dxf, .dwg).
ПРИМЕЧАНИЕ: Пример схемы 2-мерной маски см. на дополнительном рисунке 1 . - Отправить выбранному поставщику для печати. Убедитесь, что размеры конструкций находятся в пределах разрешающих возможностей поставщика.
2. Изготовление приборов - Фотолитография
ПРИМЕЧАНИЕ: Предоставленные рецепты микрофабрикации взяты из рекомендаций производителя фоторезистов и должны использоваться только в качестве отправной точки34. Точные значения времени выпечки, времени экспозиции и т. Д. Должны быть оптимизированы для каждого протокола изготовления. Рекомендуется использовать вафельный пинцет для обработки как кремниевых пластин, так и стеклянных слайдов.
- Изготовление микрофлюидных каналов
- Очистка стекла из кремниевой пластины и натриево-известкового стекла: Выполните шаги 2.1.2-2.1.3 для выполнения очистки силиконовой пластины и 1" × 3" содово-известкового стекла (оба называются «подложкой»).
- Погрузите субстраты в ацетоновую ванну, изопропанольную (IPA) ванну и деионизированную водяную баню на 10 минут каждая. Выполняйте эту 3-ступенчатую стирку последовательно при комнатной температуре.
- Удалите и высушите поверхность с помощью источника азота под давлением или газированного фильтрованного воздуха. Поместите подложки в духовку с температурой 150 °C не менее чем на 30 минут, чтобы обеспечить испарение оставшейся влаги.
- Фотолитография СУ-8 на кремниевой пластине: Выполните фотолитографию на кремниевой пластине, выполнив шаги 2.1.5-2.1.14.
ПРИМЕЧАНИЕ: Для достижения высоты микрофлюидного канала 20 мкм использовался отрицательный фоторезист серии SU-8 2000. Точные скорости отжима будут варьироваться в зависимости от рецептуры СУ-8 (т.е. 2010, 2015 и т.д.); однако для формулы 35 СУ-8 2010 годасуществуют следующие условия. - Извлеките кремниевую пластину из духовки с температурой 150 °C и дайте ей остыть до комнатной температуры (RT).
- Закрепите пластину на патроне вафельного прядильного котла с помощью вакуумной системы отжима. Запрограммируйте спиннер. Шаг 1 - 500 об/мин в течение 10 с при ускорении 100 об/с, шаг 2 - 1000 об/мин в течение 30 с при разгоне 300 об/с.
- Нанесите 4 мл фоторезиста SU-8 2010 на центр кремниевой пластины. Запустите программу. Как только система остановится, выключите вакуум.
- С помощью пинцета переложите кремниевую пластину с покрытием SU-8 на конфорку при 95 °C в течение 4-5 мин для мягкой выпечки. Затем снимите пластину с конфорки и дайте ей остыть до RT.
ПРИМЕЧАНИЕ: Следуйте надлежащей процедуре запуска для лабораторного фотолитографического выравнивателя масок. - Закрепите фотомаску с помощью 2D-микрофлюидных каналов на держателе маски. Вставьте кремниевую пластину с покрытием SU-8, обращенным вверх, на вафельный патрон.
- Установите настройки экспозиции на 150 мДж/см2 и запустите машину.
ВНИМАНИЕ: Не смотрите непосредственно на источник ультрафиолетового света, чтобы избежать потенциального повреждения глаз. - Поместите силиконовую пластину с покрытием SU-8 на конфорку при температуре 95 °C в течение 4-5 мин для выпекания после воздействия.
- Погрузите кремниевую пластину в решение разработчика СУ-8 (см. Таблицу материалов) на 3-4 мин. Нанесите мягкое перемешивание. Извлеките пластину из раствора и промойте поверхность IPA.
- Высушите поверхность с помощью источника азота под давлением или газообразного фильтрованного воздуха. Осмотрите особенности под микроскопом с помощью УФ-фильтра и убедитесь в отсутствии явных дефектов в микрофлюидных каналах.
- Поместите силиконовую в духовку с температурой 150 °C не менее чем на 30 минут для твердой выпечки.
- Дайте остыть до RT и используйте профилометрию стилуса для измерения точной высоты и наклона боковых стенок канала.
- Фотолитография на стеклянных слайдах
ПРИМЕЧАНИЕ: Гексаметилдисилазан (HMDS) используется в качестве промотора адгезии для положительного фоторезиста S181836.- Извлеките стеклянную горку из духовки с температурой 150 °C и дайте ей остыть до RT.
- Закрепите затвор стекла на патроне спиннера с помощью вакуума и запрограммируйте спиннер. Шаг 1 - 500 об/мин в течение 10 с при разгоне 100 об/с. Шаг 2 - 3000 об/мин для 30 сидящих с ускорением 300 об/с.
- Распределите 3-4 капли HMDS по поверхности стеклянной горки. Запустите программу.
ПРИМЕЧАНИЕ: Для достижения поверхностного покрытия ~3 мкм следует использовать серию положительных фоторезистов S1800. Точные скорости отжима будут варьироваться в зависимости от состава; приводимые ниже рекомендации относятся к формуле34 S1818. - Нанесите 1 мл фоторезиста на поверхность стеклянного предметного стекла. Убедитесь, что достаточно, чтобы покрыть площадь поверхности.
- Запустите программу. Как только система остановится, выключите вакуум и удалите стекло.
- Поместите стеклянную горку S1818 с покрытием на конфорку при температуре 120 °C в течение 4 минут для мягкой выпечки. Удалить и позволить прийти на RT.
- Закрепите фотомаску с помощью 2D-электродов на держателе маски.
- Вставьте и выровняйте стеклянный затвор с покрытием S1818, обращенным вверх, на вафельный патрон. Установите настройки экспозиции на 250 мДж/см2 и запустите машину.
ПРИМЕЧАНИЕ: Различные модели контактных элайнеров могут быть более или менее приспособлены к некруглым, изменяющимся по толщине подложкам. - Погрузите стеклянную горку в решение разработчика MF-319 на 2 минуты. Нанесите мягкое перемешивание. Промойте поверхность стеклянной горки деионизированной водой.
- Высушите поверхность с помощью источника азота под давлением или отфильтрованного воздушного газа и наблюдайте за особенностями под микроскопом с помощью УФ-фильтра. Убедитесь, что в литографических рисунках нет явных дефектов.
- Поместите стеклянный слайд в духовку с температурой 150 °C, гарантируя, что интересующая поверхность подложки обращена вверх, не менее чем на 30 минут для твердой выпечки. Достаньте из духовки и защитите от света.
3. Изготовление устройства: травление фтористоводородной кислоты (HF)
ВНИМАНИЕ: Этот шаг включает в себя обработку и утилизацию фтористоводородной кислоты (HF), которая может вызвать глубокие, болезненные химические ожоги. Для защиты дрессировщика следует использовать дополнительные СИЗ (лицевой щиток, химически стойкие перчатки длиной до локтя, химически стойкий фартук с рукавами). Нейтрализатор глюконата кальция и гель для кожи следует хранить в непосредственной близости от лабораторного стенда. Этот шаг не следует выполнять в одиночку. HF никогда не следует хранить или выдавать в стеклянные контейнеры, так как контейнер будет травиться кислотой.
ПРИМЕЧАНИЕ: ВЧ равномерно вытравливает открытое стекло (т.е. конструкцию электрода) с образованием углубления в стекле, что позволяет улучшить разрешение краев электродного рисунка после осаждения металла (раздел 4).
- Погрузите стеклянный слайд в буферизованный HF-раствор 10:1 на 1 мин в контейнер для политетрафторэтилена. Переложите и вымойте стеклянные горки в деионизированной воде. Повторите шаг стирки 3 раза.
- Высушите поверхность с помощью источника азота под давлением или газообразного фильтрованного воздуха. Поместите стеклянные подложки в духовку с температурой 65 °C на ночь, чтобы удалить оставшуюся влагу. Накройте подложки от света.
4. Изготовление устройства: физическое осаждение из пара
ПРИМЕЧАНИЕ: Этот этап включает в себя осаждение металла на стеклянные скользящие подложки для определения рисунков электродов. Обычно используются металлические электроды хрома / золота и титана / платины. Золото и платина не прилипают к стеклянной подложке, поэтому для содействия адгезии37 требуется слой адгезии семян хрома или титана соответственно.
- Следуйте протоколу, специфичному для чистых помещений, чтобы управлять собственной PVD-системой. В этой работе используется система напыления постоянного тока и напыления с газом аргона 100 SCCM при давлении ~8 мТорр и мощности 200 Вт.
- Напыляйте титан в течение 8 мин со скоростью ~100 Å/мин. Распыляйте платину в течение 10 мин со скоростью ~200 Å/мин. Извлеките подложки из камеры PVD.
5. Изготовление устройства: фоторезистический взлет
ПРИМЕЧАНИЕ: Этот этап включает в себя растворение слоя фоторезиста в ацетоновой ванне, оставляя приклеенные платиновые электроды с рисунком на стеклянных слайдах.
- Погрузите стеклянные горки с металлическим покрытием в ацетоновую ванну на ~10 мин.
- Обработайте ванну звуком, чтобы ввести перемешивание, чтобы разбить неприклеенную металлическую пленку. Используйте пропитанную ацетоном салфетку, чтобы удалить любые остатки, если это необходимо.
- После того, как все фоторезисты / металл удалены, промойте электродные узоры деионизированной водой и поместите их в духовку с температурой 65 ° C на ночь, чтобы удалить оставшуюся поверхностную влагу.
- Используйте профилометрию стилуса для измерения профиля узорчатых электродов.
6. Изготовление устройства: Мягкая литография
ПРИМЕЧАНИЕ: Этот этап включает в себя реплику формования микрофлюидного канала на главную рельефную структуру SU-8 с использованием эластомера, полидиметилсилоксана (PDMS).
- Силанизация кремниевых пластин
ПРИМЕЧАНИЕ: Это необязательный шаг; однако это увеличит срок службы рельефной конструкции СУ-8, которая была изготовлена в подразделе 2.1. Этот этап должен быть выполнен в химическом вытяжном шкафу.- Закрепите на дне чашки Петри и поместите чашку Петри в осушитель.
- Окружите периметр кремниевой пластины приблизительно 50 мкл трихлор(1H,1H,2H,2H-перфтороктил) силана. Подключите вакуум (вакуумный насос или домашнюю вакуумную линию) и работайте в течение 20 минут.
- Формование реплик PDMS
- В одноразовом контейнере (например, весовой лодке, пластиковом стаканчике) смешайте эластомерное основание PDMS с отвердителем в весовом соотношении 10:1 поверх электронных весов. Налейте раствор PDMS на кремниевую пластину и поместите смесь под вакуум, чтобы удалить все пузырьки воздуха.
- Отверждение при 65 °C в течение как минимум 4 ч позволяет PDMS затвердевать. Используя кончик лезвия бритвы, вырежьте формованный PDMS и отклейте кремниевую пластину.
- Используя заостренный биопсийный перфоратор, удалите PDMS из входных /выходных отверстий устройства. Для этого устройства использовались 0,75 мм и 3 мм биопсийные перфораторы для входных и выходных отверстий соответственно.
ПРИМЕЧАНИЕ: Используемый биопсийный перфоратор должен иметь несколько меньший диаметр, чем наружный диаметр соединительной трубки, чтобы обеспечить плотное уплотнение трубки в резервуарах.
- Обработка ультразвуком PDMS
- Погрузите устройства PDMS в IPA и поместите их в ультразвуковой аппарат на 30-45 минут, чтобы удалить любой мусор PDMS из входов/выходов. PDMS может набухать в растворе IPA.
- Промыть деионизированной водой и поместить в духовку с температурой 65 °C на ночь, чтобы PDMS разбухла до нормального размера.
ПРИМЕЧАНИЕ: Любой оставшийся мусор может засорить устройство во время экспериментов. Большие куски мусора могут быть удалены с поверхности PDMS с помощью куска скотча перед обработкой ультразвуком.
7. Изготовление устройства: склеивание PDMS / крепление провода
ПРИМЕЧАНИЕ: Этот этап включает обработку поверхности PDMS и стеклянной подложки кислородной плазмой с образованием необратимой связи между PDMS и стеклом38. Предоставленный рецепт, возможно, потребуется адаптировать к точной системе, используемой в лаборатории.
- Отрежьте устройства по размеру и убедитесь, что поверхность устройства PDMS является чистой. Если это не так, выполните действия, описанные в подразделе 6.3.
- Запрограммируйте генератор плазмы. Установите мощность 70 Вт, время до 35 с, давление до 325 мТорр, расход газообразного кислорода до 60 SCCM. Поместите PDMS и электродное стекло в систему с функциями, обращенными вверх, и запустите программу.
- Извлеките устройства и быстро выровняйте элементы канала с электродами с помощью стереоскопа. Плотно приложите давление от центра PDMS к бокам, чтобы удалить нежелательные пузырьки воздуха на интерфейсе склеивания.
- Поместите в горячее место при 95 °C в течение не менее 2 мин, чтобы завершить процедуру склеивания и дать устройству остыть при RT.
- Отрежьте 2 куска сплошного провода 22-G длиной ~ 6 дюймов и снимите изолятор с обоих концов.
- Прикрепите провода к электродным прокладкам с помощью серебристой проводящей эпоксидной смолы. Поместите готовые устройства в печь с температурой 65 °C на ночь.

Рисунок 2: Изготовление микроустройств. (A) Изготовление микрофлюидных каналов - Ключевые этапы: очистка кремниевых пластин (этапы 2.1.1-2.1.3), фоторезистическое покрытие и мягкая выпечка (этапы 2.1.7-2.1.8), УФ-экспозиция (этап 2.1.10), разработка (этап 2.1.12) и заливка PDMS (подраздел 6.2). (B) Основные этапы изготовления электродов: очистка стекла (этапы 2.1.1-2.1.3), покрытие HMDS и фоторезистическое покрытие (этапы 2.2.3-2.2.4), воздействие ультрафиолета (этап 2.2.8), разработка (этап 2.2.9), ВЧ травление (раздел 3), физическое осаждение из паровой фазы (раздел 4) и фоторезистическое извлечение (раздел 5). (C) Этапы окончательной доработки устройства: доступ на вход/выход и обработка ультразвуком (этап 6.2.3 и раздел 6.3), склеивание PDMS и крепление провода (раздел 7). Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
{kind=link}
8. Культивирование и сбор клеток
ПРИМЕЧАНИЕ: Следует использовать стандартные клеточные культуры и процедуры стерильной обработки. Следуйте протоколу, специфичному для типа клетки, для клеточной культуры.
- Клеточная культура
- Клеточный пассаж: Культивирование и прохождение клеток в соответствии с этапами 8.1.2-8.1.5.
- Культивирование клеток HEK293 в полном растворе DMEM (88% DMEM, 10% термоинактивированная фетальная бычья сыворотка, 1% L-глутамин, 1% пенициллин-стрептомицин) в колбе T25 в инкубаторе при 37 °C, 95% O2, 5% CO2. Прохождение клеток по графику при достижении ~80% конфюлюзии.
- Аспирировать среду с помощью пипетки или вакуумной системы и инкубировать клетки в 0,25% трипсина-ЭДТА (колба 2 мл-Т25) в течение 2 мин при 37 °C. Нейтрализуют трипсин с удвоенным объемом питательных сред.
- Переносят клеточную суспензию в коническую трубку объемом 15 мл и центрифугу HEK293 при 770 х г в течение 2 мин. Аспирация супернатанта с помощью пипетки или вакуумной системы
- Повторное суспендирование клеток HEK293 в 1 мл предварительно подогретого ДМЭМ.
- Покрытие ячеек: Пластинчатые ячейки в соответствии с шагами 8.1.7-8.1.8
- Обложите клетки в разведении от 10:1 до 20:1 в колбе T25 (5 мл DMEM), чтобы продолжить культуру.
- Разложите ячейки в разведении от 5:1 до 20:1 в 6-луночной пластине (2 мл DMEM на скважину), которая будет собрана для экспериментов по электропорации.
ПРИМЕЧАНИЕ: Ячейки HEK293 покрываются за 24 ч до электропорационных экспериментов для достижения ~70% слияния при сборе клеток (подраздел 8.3). Непоследовательный график сбора урожая может привести к изменчивости результатов электропорации.
- Электропорационный буфер
- Подготовка электропорационного буфера
ПРИМЕЧАНИЕ: Обратитесь к Sherba et al. для получения информации о электропорационном буферном препарате8. Буферный состав составлял 285 мМ сахарозы, 0,7 мМMgCl2, 1 мМ KCl, 10 мМ HEPES, 3 мМ NaOH (рН: 7,4; осмоляльность: 310 мОсм, проводимость: 500 мкСм/см). Электропорационный буфер должен быть составлен стерильным образом и храниться при 4 °C в течение срока годности ~ 1 месяц. Состав электропорационного буфера должен быть оптимизирован на основе типа ячейки.
- Подготовка электропорационного буфера
- Сбор клеток и добавление пДНК
- Выполните те же действия, что и прохождение клеток (8.1.2-8.1.4).
- Промыть клетки в стерильных 1x PBS, перенести клеточную суспензию в коническую трубку 15 мл и центрифужные ячейки при 770 х г в течение 2 мин.
- Промывайте гранулу ячейки HEK293 в электропорационном буфере и центрифуге при 770 х г в течение 2 мин. Повторное суспендирование клеток в электропорационном буфере при ~5 млн клеток/мл.
ПРИМЕЧАНИЕ: Плотность ячеек должна быть оптимизирована для каждого типа клеток. - Добавьте кодирование пДНК для зеленого флуоресцентного белка (GFP) до конечной концентрации 20 мкг/мл. Осторожно смешайте пДНК/клеточную суспензию и перенесите суспензию в шприц объемом 1 куб. см для экспериментов.
9. Аппаратная/экспериментальная настройка
ПРИМЕЧАНИЕ: Перед сбором ячеек для экспериментов убедитесь, что экспериментальная установка завершена, чтобы свести к минимуму количество времени, в течение которого клетки подвешиваются в буфере электропорации. Включите электронику за 20-30 минут до экспериментов, чтобы разогреться. Схему экспериментальной установки для работы модуля обнаружения одной ячейки см. на рисунке 3 .
ПРИМЕЧАНИЕ: Специально разработана схема PA90 Op-Amp для обеспечения как чувствительности, необходимой для определения уровня в одной ячейке с помощью запирающего усилителя, так и высоких напряжений, необходимых для подачи достаточно сильных электропорационных импульсов. Обратитесь к техническому описанию PA90 для получения спецификаций на рекомендуемую схему39.
- Инициализируйте блокируемый усилитель с настройками тока предварительного усилителя и настройте его с помощью алгоритма. Обратитесь к Zheng et al. для получения подробной информации о настройках блокировки32.
- Блоки питания, генератор функций и усилитель
- Источник питания 1: Установите -15 В для питания отрицательного конца цепи.
- Источник питания 2 (генератор функций): Установите выходной сигнал постоянного тока и установите амплитуду на 2 В. Подключите к входу усилителя 50x.
- Программа Генератор электропорационных импульсов для квадратной волны: Установите желаемую ширину импульса (рабочий цикл) и желаемую амплитуду импульса (вольты).
- Установите выход в режим срабатывания (1 импульс). Подключите выход к входу 50-кратного усилителя.
ПРИМЕЧАНИЕ: Помните о 50-кратном усилении при программировании амплитуды импульса. Т.е. для достижения напряженности электрического поля 1 кВ/см требуется в общей сложности 30 В, 30 В/300 мкм (расстояние между электродами), поэтому выход генератора функций должен быть установлен на 30/50, или 600 мВ. - Проверьте выходы 50-кратного усилителя с помощью осциллографа. Выход 1-100 В от блока питания 2 (9.2.2). Выходная 2-переменная амплитуда для импульса электропорации (9.2.4).
- Подключите 10-кратный зонд к каналу осциллографа и к готовому микроустройству (тестируемому устройству, DUT) на этапе 7.6, где будет применяться электропорационный импульс. Контролируйте систему во время экспериментов, чтобы убедиться, что импульсы применяются.
- Убедитесь, что USB-накопитель подключен и зарегистрирован. Дважды проверьте все настройки блокировки в коде алгоритма (самое главное, частоту блокировки вывода).
- Микроскоп/ПЗС-камера
- Поместите микроустройство на сцену микроскопа с помощью держателя слайда. Включите ПЗС-камеру и поместите микрофлюидный канал в фокус. Используйте цель 4x или 10x.

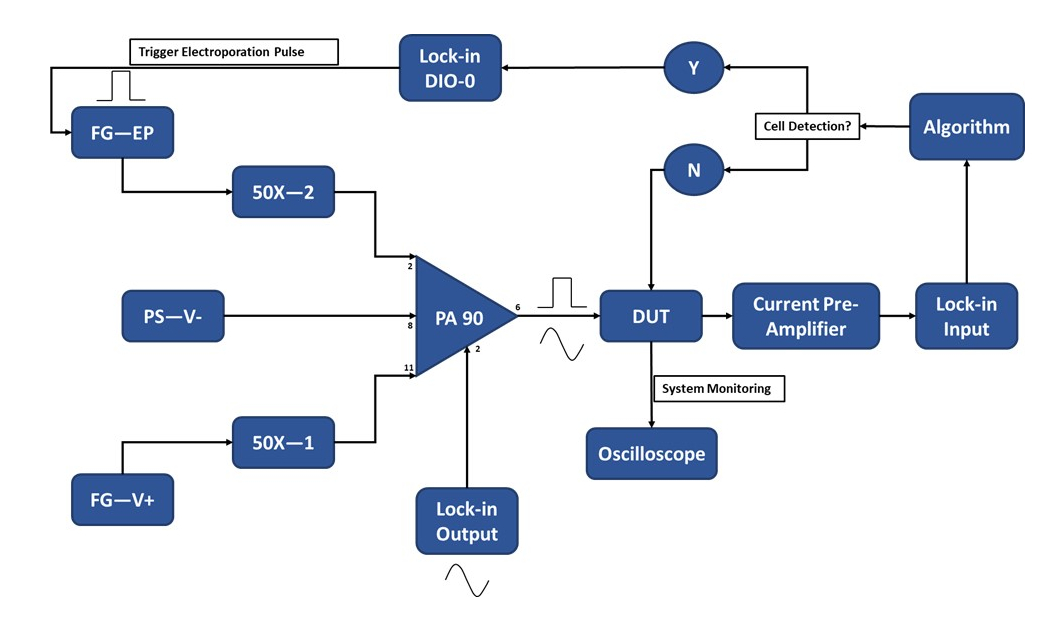
Рисунок 3: Схема экспериментальной установки - Обнаружение одиночных ячеек. Мощный операционный усилитель (PA-90) позволяет накладывать импульс электропорации высокого напряжения на фиксированный выходной сигнал переменного тока, необходимый для обнаружения одной ячейки. Этот сигнал возбуждения проходит через микроэлектропорационное устройство (Device Under Test, DUT), где ток затем усиливается предварительным усилителем тока и подается в алгоритм. Система непрерывно отслеживает событие обнаружения ячеек. При входе в ячейку цифровой сигнал генерируется блокирующим усилителем, чтобы инициировать подачу электропорационного импульса к ячейке (ячейкам) в пути. Условные обозначения: PA-90 (мощный операционный усилитель), DUT (тестируемое устройство), DIO (цифровой вход/выход), FG-EP (генератор функций / электропорационный импульс), 50X (усилитель 50X), PS-V- (питание / отрицательное напряжение для PA 90), FG-V+ (генератор функций, положительное напряжение для PA 90). Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
{kind=link}
10. Экспериментальная эксплуатация
- Микрофлюидная грунтовка каналов
- Удалите все пузырьки воздуха из шприца с ячеистой нагрузкой. Прикрепите иглу весом 30 г к шприцу с клеточной нагрузкой.
- Используя пинцет, сдвиньте трубку tygon вниз по длине иглы. Предварительно заполните выходной резервуар восстановительной средой (аналогично этапу 8.1.2 без антибиотиков), ~40-50 мкл.
- Используя большой палец, осторожно надавите на плунжер так, чтобы жидкость медленно достигала конца линии трубки.
- Прикрепите шприц к шприцевому насосу. Включите шприцевой насос и убедитесь, что он настроен на перфузию вперед.
- Запрограммируйте насос для правильного диаметра шприца, чтобы обеспечить точность расхода. Обратитесь к руководству по насосу для уточнения диаметров шприцев.
ПРИМЕЧАНИЕ: Чтобы ячейки не оседали в шприце, закрепите шприцевой насос на зажимной подставке таким образом, чтобы он мог работать в вертикальном положении с концом шприца, обращенным вниз. - Установите скорость потока шприцевого насоса, ~10-20 мкл/мин, и дайте насосу работать до тех пор, пока жидкость не достигнет конца линии трубок. Закрепите трубку на микрофлюидном устройстве.
- Уменьшите расход шприцевого насоса, ~5-10 мкл/мин, и позвольте насосу работать до тех пор, пока весь воздух не будет вытеснен из микрофлюидного устройства и ячейки не пройдут к выходу устройства.
- Извлеките ячейки из розетки с помощью аспирации пипетки. Повторно заполните выходной резервуар восстановительной средой (аналогично этапу 8.1.2 без антибиотиков), ~40-50 мкл.
- Картирование пермеабилизации одноэлементной электропорации клеточной мембраны
ПРИМЕЧАНИЕ: Обратитесь к Фиг.4 и Фиг.5 для лучшего понимания электрических данных, указывающих на пермеабилизацию клеточной мембраны и картирование пермеабилизации клеточной мембраны, соответственно.- Установите скорость потока шприцевого насоса на ~0,1-0,3 мкл/мин, чтобы обеспечить поток отдельных ячеек через электродный набор. Время прохождения ячейки между электродами должно составлять ~250 мс.
- Запустите компьютерную программу, нажав кнопку Выполнить. Убедитесь, что система сохраняет электрические данные.
- Убедитесь, что система надежно обнаруживает ячейки для запуска управляемых компьютером импульсных приложений. Соответствующим образом отрегулируйте порог обнаружения .
- Установите параметры импульса для начального электропорационного импульса с наименьшей электрической энергией. Параметры пульсации электропорации приведены в таблице 1 в данном исследовании.
- Включите выходной канал для генератора электропорационных импульсов (шаг 9.2.3.).
- Следуйте заранее определенному количеству приложений обнаружения клеток/импульсов (n = 100). В конце каждого испытуемого состояния аспирируют клетки из выхода микроустройства и пополняют выход восстановительными средами.
- Итерация к следующему состоянию электропорационного импульса. Повторяйте до тех пор, пока не будут проверены все условия электропорационного импульса.
- Определите степень пермеабилизации клеточной мембраны для каждого тестируемого импульсного применения. (Постпроцессная валидация описана в подразделе 11.1). Сгенерируйте карту пермеабилизации клеточной мембраны (рисунок 5).
- Определите параметры электропорационного импульса для высокопроизводительной обратной связи на основе популяции.
- Выключите шприцевой насос, извлеките ячейки из выпускного резервуара и пополните выходное отверстие восстановительными средами.
- Электропорация с управляемой обратной связью на основе населения и высокая пропускная способность
ПРИМЕЧАНИЕ: Схему, иллюстрирующую процесс обратной связи на основе населения, см. на рисунке 6 .- Установите скорость потока шприцевого насоса на ~1-3 мкл/мин, чтобы обеспечить поток отдельных ячеек через электродный набор. Время прохождения ячейки между электродами должно составлять ~25 мс.
- Установите амплитуду импульса в «оптимизированное» состояние (10.2.9), выключите режим триггера и установите ширину импульса в соответствии со временем прохождения ячейки.
- Установите рабочий цикл таким образом, чтобы время включения импульса соответствовало «оптимизированному» условию. См. таблицу 1.
- Установите для генератора функции выходного канала значение ВКЛ, включите шприцевой насос и позвольте системе работать до тех пор, пока нужное количество ячеек не будет электропорировано.
- По завершении выключите шприцевой насос и генератор функций.
- Переместите клетки из выходного резервуара в колбу/пластину для посева клеток соответствующего размера, заполненную предварительно нагретой восстановительной средой, и перенесите колбу/пластину культуры в инкубатор.
11. Анализ
- Обнаружение пермеабилизации мембраны на уровне одной клетки
ПРИМЕЧАНИЕ: Для обеспечения того, чтобы во время модуля высокой пропускной способности использовался «оптимальный» импульс, следует провести послеэкспериментальный анализ для проверки электрических данных, экспортированных из подраздела 10.2. Пожалуйста, обратитесь к рисунку 4 для графического представления электрического сигнала, представляющего мембранную пермеабилизацию вследствие электропорации.- Загрузка данных в аналитическое программное обеспечение (MATLAB, Python и т.д.). Создайте график Текущее и Время для каждого пульсирующего состояния.
- Вручную определяют степень пермеабилизации клеточной мембраны (ΔIP/ΔIC). См. рисунок 4. Сгенерируйте карту пермеабилизации клеточной мембраны (ΔIP / ΔIC в сравнении с электрической энергией, рисунок 5) для всех тестируемых импульсных условий. Проверьте «оптимальное» пульсирующее состояние.
- эффективность электротрансфекции (eTE)
- После 24-часового инкубационного периода удалите электропорированные клетки из инкубатора.
- Выполните окрашивание живых клеток. Разбавить DRAQ5 1:1000 до конечной концентрации 5 мкМ в сосуде клеточной культуры. Аккуратно смешайте клетки/окрашивающий раствор и инкубируйте при 37 °C в течение 5-30 мин.
ПРИМЕЧАНИЕ: На этом шаге может быть реализовано другое пятно. Убедитесь, что флуоресцентные свойства не перекрываются с флуоресцентным маркером, указывающим на успешную электротрансфекцию (т. Е. GFP находится в зеленой длине волны, а DRAQ5 - в дальнем красном). - Включите эпифлуоресцентный микроскоп, лампу и фотоаппараты (см. Таблицу материалов).
- Извлеките клетки из инкубатора и сосредоточьте их на микроскопе.
- Захват фазово-контрастного изображения (яркого поля) выбранного поля.
- Захват эпифлуоресцентных изображений одного и того же поля с помощью фильтров FITC (GFP) и Far-Red (DRAQ5). Анализируйте наборы изображений вручную или с помощью алгоритма.
ПРИМЕЧАНИЕ: Обратитесь к рисунку 7 для репрезентативных изображений. - Подсчитайте общее количество GFP-положительных клеток на всех изображениях. Подсчитайте общее количество окрашенных клеток DRAQ5 во всех изображениях. Рассчитайте eTE (отношение GFP положительных клеток к окрашенным DRAQ5 клеткам).
Access restricted. Please log in or start a trial to view this content.
Результаты
На рисунке 4 показаны принципы работы, лежащие в основе определения пермеабилизации мембраны на уровне одной ячейки для одной амплитуды импульса. После начала эксперимента с электропорацией алгоритм обнаружения клеток определяет оптимальный порог для обнаружения кле...
Access restricted. Please log in or start a trial to view this content.
Обсуждение
Методология, представленная в этом протоколе, в первую очередь фокусируется на микропроизводстве микрофлюидного устройства, которое затем интегрируется в специализированную экспериментальную установку электропорации. Термин «рецепт», который часто используется при описании специ?...
Access restricted. Please log in or start a trial to view this content.
Раскрытие информации
Авторам нечего раскрывать.
Благодарности
Авторы хотели бы отметить финансовую поддержку Со стороны Национального научного фонда (NSF CBET 0967598, DBI IDBR 1353918) и Аспирантуры Министерства образования США в новых областях точной и персонализированной медицины (P200A150131) для финансирования аспиранта J.J.S. по стипендии.
Access restricted. Please log in or start a trial to view this content.
Материалы
| Name | Company | Catalog Number | Comments |
| 150-mm diameter petri dishes | VWR | 25384-326 | step 6.1.1 to secure wafer |
| 24-well tissue culture plates | VWR | 10062-896 | step 10.3.6 to plate electroporated cells |
| 33220A Waveform/Function generator | Agilent | step 9.2.3 electroporation pulse generator | |
| 4'' Si-wafers | University Wafer | subsection 2.1 for microfluidic channel fabrication | |
| 6-well tissue culture plates | VWR | 10062-892 | step 8.1.8 to plate cells |
| Acetone | Fisher Scientific | A18-4 | step 2.1.2 for cleaning and step 5.1 photoresist lift-off |
| Allegra X-22R Centrifuge | Beckman Coulter | steps 8.1.4 , 8.3.2. and 8.3.3. to spin down cells | |
| AutoCAD 2018 | Autodesk | subsection 1.1. to design transparency masks | |
| Buffered oxide etchant 10:1 | VWR | 901621-1L | subsection 3.1 for HF etching |
| CCD Monochrome microscope camera | Hamamatsu | Orca 285 C4742-96-12G04 | step 11.2.3. for imaging |
| CMOS camera- Sensicam QE 1.4MP | PCO | subsection 9.3 part of the experimental setup | |
| Conductive Epoxy | CircuitWorks | CW2400 | subsection 7.6. for wire attachement |
| Conical Centrifuge Tubes, 15 mL | Fisher Scientific | 14-959-70C | step 8.1.4. for cell centrifuging |
| Dektak 3ST Surface Profilometer | Veeco (Sloan/Dektak) | step 2.1.15 and 5.4 for surface profilometry | |
| Disposable biopsy punch, 0.75 mm | Robbins Instruments | RBP075 | step 6.2.3 for inlet access |
| Disposable biopsy punch, 3 mm | Robbins Instruments | RBP30P | step 6.2.3 for outlet access |
| DRAQ5 | abcam | ab108410 | step 11.2.2. for live cell staining |
| Dulbecco’s Modified Eagle’s Medium | ThermoFisher Scientific | 11885084 | step 8.1.2. part of media composition |
| E3631A Bipolar Triple DC power supply | Agilent | step 9.2.1.-9.2.2.part of the experimental setup | |
| Eclipse TE2000-U Inverted Microscope | Nikon | subsection 9.3. part of the experimental setup | |
| EVG620 UV Lithography System | EVG | step 2.1.9. and 2.2.7. for UV Exposure | |
| Fetal Bovine Serum | Neuromics | FBS001 | step 8.1.2. part of media composition |
| FS20 Ultrasonic Cleaner | Fisher Scientific | subsection 5.1. for photoresist lift-off | |
| Glass Media Bottle with Cap, 100mL | Fisher Scientific | FB800100 | step 8.2.1. for buffer storage |
| Glass Media Bottle with Cap, 500mL | Fisher Scientific | FB800500 | step 8.1.2.for media storage |
| HEK-293 cell line | ATCC | CRL-1573 | subsection 8.1 for cell culturing |
| HEPES buffer solution | Sigma Aldrich | 83264-100ML-F | step 8.2.1 part of electroporation buffer composition |
| Hexamethyldisilazane | Sigma Aldrich | 379212-25ML | step 2.2.3 adhesion promoter |
| HF2LI Lock-in Amplifier | Zurich Instruments | subsection 9.2 part of the experimental setup | |
| HF2TA Current amplifier | Zurich Instruments | subsection 9.2 part of the experimental setup | |
| Isopropyl Alcohol | Fisher Scientific | A459-1 | step 2.1.2 for cleaning, step 2.1.14 for rinsing wafer following SU-8 development, and step 6.3.1 for cleaning PDMS |
| IX81 fluorescence microscope | Olympus | step 11.2.3 for imaging | |
| L-Glutamine Solution | Sigma Aldrich | G7513-20ML | step 8.1.2. part of media composition |
| M16878/1BFA 22 gauge wire | AWC | B22-1 | subsection 7.5 for device fabrication |
| Magnesium chloride | Sigma Aldrich | 208337-100G | step 8.1.2 part of electroporation buffer composition |
| MF 319 Developer | Kayaku Advanced Materials | 10018042 | step 2.2.9. photoresist developer |
| Microposit S1818 photoresist | Kayaku Advanced Materials | 1136925 | step 2.2.4 positive photoresist for electrode patterning |
| Microscope slides, 75 x 25 mm | VWR | 16004-422 | step 2.2.1 electrode soda lime glass substrate |
| Model 2350 High voltage amplifier | TEGAM | 2350 | step 9.2.5. part of the experimental setup |
| National Instruments LabVIEW | National Instruments | data acquisition | |
| Needle, 30G x 1 in | BD Scientific | 305128 | step 10.1.1. part of the system priming |
| PA90 IC OPAMP Power circuit | Digi-key | 598-1330-ND | Part of the custom circuit |
| Penicillin-Streptomycin | Sigma Aldrich | P4458-20ML | step 8.1.2. part of media composition |
| Plasmid pMAX-GFP | Lonza | VCA-1003 | step 8.3.4. for intracellular delivery |
| Plastic tubing, 0.010'' x 0.030" | VWR | 89404-300 | step 10.1.2. for system priming |
| Platinum targets | Kurt J. Lesker | subsection 4.2. for physical vapor deposition | |
| Potassium chloride | Sigma Aldrich | P9333-500G | step 8.2.1. part of electroporation buffer composition |
| Pump 11 PicoPlus microfluidic syringe pump | Harvard Apparatus | MA1 70-2213 | step 10.1.4. for system priming |
| PVD75 Physical vapor deposition system | Kurt J. Lesker | subsection 4.1. for physical vapor deposition | |
| PWM32 Spinner System | Headway Research | steps 2.1.6 and 2.2.2. for substrate coating with photoresist | |
| PX-250 Plasma treatment system | March Instruments | subsection 7.2 for PDMS and glass substrate bonding | |
| SDG1025 Function/Waveform generator | Siglent | step 9.2.2. part of the experimental setup | |
| Sodium hydroxide | Sigma Aldrich | S8045-500G | step 8.2.1. part of electroporation buffer composition |
| SU-8 2010 negative photoresist | Kayaku Advanced Materials | Y111053 | step 2.1.7. for microfluidic channel patterning |
| SU-8 developer | Microchem | Y010200 | step 2.1.12. for photoresist developing |
| Sucrose | Sigma Aldrich | S7903-1KG | step 8.2.1. part of electroporation buffer composition |
| Sylgard 184 elastomer kit | Dow Corning | 3097358-1004 | step 6.2.1 10 : 1 mixture of PDMS polymer and hardening agent |
| Syringe, 1 ml | BD Scientific | 309628 | step 8.3.4. part of system priming |
| SZ61 Stereomicroscope System | Olympus | subsection 7.3. for channel and electrode alignment | |
| Tissue Culture Treated T25 Flasks | Falcon | 353108 | step 8.1.2 for cell culturing |
| Titanium targets | Kurt J. Lesker | subsection 4.2. for physical vapor deposition | |
| Transparency masks | CAD/ART Services | steps 2.1.9. and 2.2.7. for photolithography | |
| Trichloro(1H,1H,2H,2H-perfluorooctyl)silane | Sigma Aldrich | 448931-10G | step 6.1.2. for wafer silanization |
| Trypsin-EDTA solution | Sigma Aldrich | T4049-100ML | steps 8.1.3. and 8.3.1. for cell harvesting |
Ссылки
- Gao, Q. Q., et al. Therapeutic potential of CRISPR/Cas9 gene editing in engineered T-cell therapy. Cancer Medicine. 8 (9), 4254-4264 (2019).
- Aijaz, A., et al. Biomanufacturing for clinically advanced cell therapies. Nature Biomedical Engineering. 2 (6), 362-376 (2018).
- Milone, M. C., O'Doherty, U. Clinical use of lentiviral vectors. Leukemia. 32 (7), 1529-1541 (2018).
- Weaver, J. C., Chizmadzhev, Y. A. Theory of electroporation: A review. Bioelectrochemistry and Bioenergetics. 41 (2), 135-160 (1996).
- Kotnik, T., Rems, L., Tarek, M., Miklavcic, D. Membrane electroporation and electropermeabilization: mechanisms and models. Annual Review of Biophysics. 48, 63-91 (2019).
- Rosazza, C., Meglic, S. H., Zumbusch, A., Rols, M. P., Miklavcic, D. Gene electrotransfer: A mechanistic perspective. Current Gene Therapy. 16 (2), 98-129 (2016).
- Clauss, J., et al. Efficient non-viral T-cell engineering by sleeping beauty minicircles diminishing DNA toxicity and miRNAs silencing the endogenous T-cell receptors. Human Gene Therapy. 29 (5), 569-584 (2018).
- Sherba, J. J., et al. The effects of electroporation buffer composition on cell viability and electro-transfection efficiency. Scientific Reports. 10 (1), 3053(2020).
- Lu, H., Schmidt, M. A., Jensen, K. F. A microfluidic electroporation device for cell lysis. Lab on a Chip. 5 (1), 23-29 (2005).
- Kar, S., et al. Single-cell electroporation: current trends, applications and future prospects. Journal of Micromechanics and Microengineering. 28 (12), (2018).
- Shi, J. F., et al. A review on electroporation-based intracellular delivery. Molecules. 23 (11), (2018).
- Wang, S. N., Zhang, X. L., Wang, W. X., Lee, L. J. Semicontinuous flow electroporation chip for high-throughput transfection on mammalian cells. Analytical Chemistry. 81 (11), 4414-4421 (2009).
- Wei, W. J., et al. An implantable microelectrode array for simultaneous L-glutamate and electrophysiological recordings in vivo. Microsystems & Nanoengineering. 1, (2015).
- Maschietto, M., Dal Maschio, M., Girardi, S., Vassanelli, S. In situ electroporation of mammalian cells through SiO2 thin film capacitive microelectrodes. Scientific Reports. 11 (1), (2021).
- Wu, Q. R., et al. Organ-on-a-chip: recent breakthroughs and future prospects. Biomedical Engineering Online. 19 (1), (2020).
- Pandey, C. M., et al. Microfluidics Based Point-of-Care Diagnostics. Biotechnology Journal. 13 (1), (2018).
- Vigneshvar, S., Sudhakumari, C. C., Senthilkumaran, B., Prakash, H. Recent advances in biosensor technology for potential applications - An overview. Frontiers in Bioengineering and Biotechnology. 4, (2016).
- Nuxoll, E. BioMEMS in drug delivery. Advanced Drug Delivery Reviews. 65 (11-12), 1611-1625 (2013).
- Kang, S., Kim, K. H., Kim, Y. C. A novel electroporation system for efficient molecular delivery into Chlamydomonas reinhardtii with a 3-dimensional microelectrode. Scientific Reports. 5, (2015).
- Zheng, M. D., Shan, J. W., Lin, H., Shreiber, D. I., Zahn, J. D. Hydrodynamically controlled cell rotation in an electroporation microchip to circumferentially deliver molecules into single cells. Microfluidics and Nanofluidics. 20 (1), (2016).
- Santra, T. S., Kar, S., Chang, H. Y., Tseng, F. G. Nano-localized single-cell nano-electroporation. Lab on a Chip. 20 (22), 4194-4204 (2020).
- Lee, W. G., Demirci, U., Khademhosseini, A. Microscale electroporation: challenges and perspectives for clinical applications. Integrative Biology. 1 (3), 242-251 (2009).
- Santra, T. S., Chang, H. Y., Wang, P. C., Tseng, F. G. Impact of pulse duration on localized single-cell nano-electroporation. Analyst. 139 (23), 6249-6258 (2014).
- Geng, T., Lu, C. Microfluidic electroporation for cellular analysis and delivery. Lab on a Chip. 13 (19), 3803-3821 (2013).
- Hsi, P., et al. Acoustophoretic rapid media exchange and continuous-flow electrotransfection of primary human T cells for applications in automated cellular therapy manufacturing. Lab on a Chip. 19 (18), 2978-2992 (2019).
- Khine, M., Ionescu-Zanetti, C., Blatz, A., Wang, L. P., Lee, L. P. Single-cell electroporation arrays with real-time monitoring and feedback control. Lab on a Chip. 7 (4), 457-462 (2007).
- Ye, Y. F., et al. Single-cell electroporation and real-time electrical monitoring on a microfluidic chip. 2020 33rd Ieee International Conference on Micro Electro Mechanical Systems (Mems 2020). , 1040-1043 (2020).
- Huang, Y., Rubinsky, B. Microfabricated electroporation chip for single cell membrane permeabilization. Sensors and Actuators a-Physical. 89 (3), 242-249 (2001).
- Guo, X. L., Zhu, R. Controllable in-situ cell electroporation with cell positioning and impedance monitoring using micro electrode array. Scientific Reports. 6, (2016).
- Punjiya, M., Nejad, H. R., Mathews, J., Levin, M., Sonkusale, S. A flow through device for simultaneous dielectrophoretic cell trapping and AC electroporation. Scientific Reports. 9, (2019).
- Wang, H. Y., Lu, C. Microfluidic electroporation for delivery of small molecules and genes into cells using a common DC power supply. Biotechnology and Bioengineering. 100 (3), 579-586 (2008).
- Zheng, M. D., et al. Continuous-flow, electrically-triggered, single cell-level electroporation. Technology. 5 (1), 31-41 (2017).
- Batista Napotnik, T., Miklavcic, D. In vitro electroporation detection methods - An overview. Bioelectrochemistry. 120, 166-182 (2018).
- MICROPOSIT™ S1800® G2 Series Photoresists. KAYAKU. , Available from: https://kayakuam.com/wp-content/uploads/2019/09/S1800-G2.pdf (2021).
- SU-8 2000 Permanent Negative Epoxy Photoresist. KAYAKU. , Available from: https://kayakuam.com/wp-content/uploads/2020/08/KAM-SU-8-2000-2000.5-2015-Datasheet-8.13.20-final.pdf (2001).
- Substrate Preparation. MicroChemicals. , Available from: https://www.microchemicals.com/technical_information/subtrate_cleaning_adhesion_photoresist.pdf (2021).
- Lisinenkova, M., Hahn, L., Schulz, J. 4M 2006 - Second International Conference on Multi-Material Micro Manufacture. , Elsevier. 91-94 (2006).
- Beh, C. W., Zhou, W. Z., Wang, T. H. PDMS-glass bonding using grafted polymeric adhesive - alternative process flow for compatibility with patterned biological molecules. Lab on a Chip. 12 (20), 4120-4127 (2012).
- PA90 High Voltage Power Operational Amplifiers. APEX. , Available from: https://www.apexanalog.com/resources/products/pa90u.pdf (2021).
- Lissandrello, C. A., et al. High-throughput continuous-flow microfluidic electroporation of mRNA into primary human T cells for applications in cellular therapy manufacturing. Scientific Reports. 10 (1), 18045(2020).
Access restricted. Please log in or start a trial to view this content.
Перепечатки и разрешения
Запросить разрешение на использование текста или рисунков этого JoVE статьи
Запросить разрешениеСмотреть дополнительные статьи
This article has been published
Video Coming Soon
Авторские права © 2025 MyJoVE Corporation. Все права защищены