
Method Article
DNA 커튼은 상동 재조합 중 복잡한 분자 시스템을 조명합니다.
요약
DNA 커튼은 수백 또는 수천 개의 DNA 결합 단백질이 미세유체 시료 챔버 표면에 정렬된 DNA 분자와 상호 작용할 때 실시간으로 시각화할 수 있는 새로운 방법을 제시합니다.
초록
상동 재조합(HR)은 모든 유기체에서 이중 가닥 DNA 절단(DSB) 및 지연된 복제 분기점의 복구에 중요합니다. HR의 결함은 인간 세포의 게놈 무결성 손실 및 발암성 변형과 밀접한 관련이 있습니다. HR은 복잡한 단백질 집합의 조정된 작용을 포함하며, 그 중 많은 부분이 잘 이해되지 않은 상태로 남아 있습니다. 여기에 기술된 연구의 핵심은 "DNA 커튼"이라고 불리는 기술인데, 이 기술은 미세유체 시료 챔버의 표면에 정렬된 DNA 분자를 조립할 수 있게 하는 기술입니다. 그런 다음 전반사 형광 현미경(TIRFM)으로 시각화할 수 있습니다. DNA 커튼은 우리 실험실에서 개척되었으며 다른 방법론을 통해 쉽게 밝힐 수 없는 밀리초 시간 척도 및 나노미터 단위 분해능에서 시공간 정보에 직접 액세스할 수 있습니다. DNA 커튼의 주요 장점은 단일 분자 실험에서 통계적으로 관련된 데이터 수집을 단순화한다는 것입니다. 이 연구는 세포가 게놈 무결성을 조절하고 보존하는 방법에 대한 새로운 통찰력을 지속적으로 제공합니다.
서문
게놈 무결성의 유지는 모든 살아있는 세포의 적절한 기능에 매우 중요합니다1. 게놈 무결성의 결함은 다양한 유형의 암 및 노화 관련 퇴행성 질환을 포함한 심각한 건강 상태로 이어질 수 있습니다2. 상동 재조합(Homologous recombination, HR)은 주형 의존적 DNA 합성을 사용하여 DNA 이중 가닥 절단(DSB), 단일 가닥 DNA(ssDNA) 갭 및 가닥 간 DNA 가교결합을 복구합니다3. HR은 또한 중단되고 축소된 복제 포크 3,4의 복구를 위해 필요합니다. 또한, HR은 감수분열 5,6 동안 정확한 염색체 분리를 위해 필수적입니다.
HR은 복잡한 단백질 집합의 조정된 작용을 포함하며, 그 중 많은 부분이 잘 이해되지 않은 상태로 남아 있습니다1. 예를 들어 복제 단백질 A(RPA), Rad51 및 Rad54 등이 있습니다7. 원핵 세포와 진핵 세포 모두에서 HR 반응은 ssDNA 결합 단백질(원핵생물의 SSB 및 진핵생물의 RPA)에 의해 빠르게 코팅되는 ssDNA 중간체를 포함합니다8. 이 단백질은 nuclease로부터 ssDNA를 보호하고, 2차 구조를 제거하며, 다운스트림 인자의 동원을 촉진합니다 8,9. Rad51은 모든 살아있는 유기체에 존재하는 고도로 보존된 ATP 의존성 Rad51/RecA DNA 재조합효소 계열의 구성원입니다1. Rad51은 상동 dsDNA 공여체의 DNA 가닥 침입을 촉진합니다. Rad51의 중요성을 감안할 때, Rad51은 고도로 규제되고 있으며, 이러한 조절 과정의 결함은 일반적으로 게놈 무결성 손실 및 발암성 형질전환과 관련이 있습니다7. Rad54는 dsDNA 트랜스로케이저 및 염색질 리모델링기의 Swi2/Snf2 계열10,11에 속합니다. 이러한 단백질은 필수 Rad51 조절 인자로 작용합니다. 중요한 것은 Rad54가 가닥 침입의 dsDNA 산물에서 Rad51을 제거하며 염색질11에 Rad51의 오축적을 방지하는 데에도 필요하다는 것입니다. 효모세포와 박테리아 세포 모두에서 HR에 관여하는 단백질의 분자 활성은 HR에서 단백질의 기능을 밝혀 주었지만, 이들의 활동이 HR에 정확히 어떻게 기여하는지는 잘 알려져 있지 않다 12.
DNA 커튼은 그렇지 않으면 접근할 수 없는 분자 메커니즘 및 거대분자 역학에 직접 접근할 수 있는 고유한 플랫폼으로 부상했습니다13,14. DNA 커튼을 준비하기 위해 미세유체 챔버의 표면은 지질 이중층으로 코팅되고 DNA 분자는 비오틴-스트렙타비딘 결합을 통해 이중층에 연결됩니다. 이중층은 자연 세포막을 모방하여 표면을 불활성으로 만듭니다. 유체역학적 힘은 나노 가공 장벽을 따라 DNA를 정렬하여 전반사 형광 현미경(TIRFM)을 통해 단일 시야에서 수백 개의 분자를 시각화할 수 있습니다. 장벽은 전자빔 리소그래피에 의해 만들어지며, 장벽 설계의 변화로 DNA의 분포 및 테더링 기하학을 정밀하게 제어할 수 있습니다. 이러한 접근법은 ssDNA 또는 dsDNA 13,14,15,16,17,18에 쉽게 적용할 수 있습니다. 받침대는 또한 DNA의 양쪽 끝을 샘플 챔버 표면에 테더링하여 완충 흐름이 없는 상태에서 정상 상태 실험을 수행할 수 있도록 나노 제작(장벽과 함께)할 수 있습니다.
개별 단백질-핵산 복합체의 시간에 따른 변화는 실시간 비디오 검사를 통해 밝혀지며 시간이 지남에 따라 DNA에서 단백질의 위치 변화를 나타내는 카이모그래프를 사용하여 인쇄물로 표현됩니다. DNA 커튼 접근법의 중요한 측면은 개별 반응 성분의 거동을 실시간으로 관찰할 수 있기 때문에 분자 메커니즘에 대한 선험 적 모델이나 가정이 반드시 필요한 것은 아니라는 것입니다. 이를 통해 분자 거동을 직접 관찰할 수 있습니다. 본 명세서에서 이 프로토콜은 dsDNA 기질로 DNA 커튼을 준비하는 방법과 상동 재조합에서 중간 생성물을 연구하기 위한 응용에 대해 설명합니다.
프로토콜
1. 지질 스톡의 제조
- 클로로포름 10mL에 18:1(Δ9-Cis) PC 1g, 18:1 PEG2000 PE 100mg, 18:1 비오티닐 캡 PE 5mg을 용해합니다.
참고: DOPC와 DPPE는 모두 특성이 잘 규명되어 있으며 플로우 셀 표면에 대한 비특이적 흡착을 최소화할 수 있는 능력으로 인해 PEG 변형과 함께 선택되었습니다. 용존 지질 마스터 믹스는 -20°C에서 최대 12개월까지 분주하여 보관할 수 있습니다. - 클로로포름을 사용하여 유기 용매 유리 주사기를 세척한 다음 지질 마스터 믹스의 200μL(최종 원하는 부피의 10%)를 유리 바이알로 옮깁니다.
- 매우 부드러운 압축 질소 흐름을 사용하여 유리 바이알에서 클로로포름을 증발시킵니다. 클로로포름의 모든 흔적이 제거되도록 증발이 끝날 때까지 압력을 약간 높입니다.
알림: 증발 과정에서 바이알을 기울이고 회전시켜 지질이 벽에 균일한 흰색 필름 층으로 건조되도록 합니다. - 뚜껑을 열지 않은 바이알을 밤새 진공 상태에서 두십시오.
- 건조된 지질 필름에 2mL의 지질 완충액(10mM Tris-HCl[pH 8.0], 100mM NaCl)을 추가하고 바이알을 덮습니다. 실온(RT)에서 최소 1시간 동안 배양하고 용해될 때까지 와류를 일으킵니다.
- 용액을 5mL 폴리스티렌 둥근 바닥 튜브로 옮기고 진폭 = 50, 공정 시간 = 1.5분, 펄스 = 15초, 시간 차단 = 2분 매개변수와 함께 마이크로팁을 사용하여 얼음 위에서 초음파 처리합니다. 용액은 초음파 처리 후 명확해집니다 (총 에너지 = 1,500-2,000 J).
- 0.22μm PVDF 주사기 필터를 통해 용액을 여과하고 지질 스톡을 4°C에서 최대 1개월 동안 보관합니다.
2. dsDNA 기질의 제조
- λ-DNA 스톡(500μg/mL)을 65°C로 예열합니다.
- 1.6 pM의 λ-DNA, 100x 몰 초과량의 각 올리고뉴클레오티드(BioL: 5'Phos-AGG TCG CCG CCC-3Bio 및 DigR: 5'Phos-GGG CGG CGA CCT-3Dig_N) 및 60 μL의 10x T4 리가제 반응 완충액(총 부피 600 μL)을 포함하는 어닐링/ligation 혼합물을 준비합니다.
참고: BioL 및 DigR 올리고뉴클레오티드는 각각 비오틴화(Bio) 및 디곡시제닌(Dig)으로 변형됩니다. 비오틴화는 스트렙타비딘 결합을 통해 DNA 분자를 비오틴화된 지질에 부착할 수 있도록 합니다. Dig 변형은 받침대에 코팅된 anti-Dig 항체를 Dig 부분에 결합함으로써 이중 테더링을 가능하게 합니다. - 올리고뉴클레오티드를 65°C에서 10분 동안 반응물을 배양하여 λ-DNA cos-end에 어닐링한 다음 RT로 천천히 냉각시킵니다.
- 5 μL의 T4 DNA 리가아제를 추가하고 RT에서 밤새 배양합니다.
참고: 어닐링 및 결찰 단계는 열 순환기에서 수행할 수도 있습니다. - 300μL의 PEG-8000 용액(30% w/v PEG-8000 및 10mM MgCl2)을 첨가하여 λ-DNA를 침전시킵니다. 4 °C에서 최소 1 시간 동안 배양하십시오.
- 상온에서 5분 동안 14,000 x g 에서 원심분리기. 펠릿을 방해하지 않고 상층액을 조심스럽게 제거합니다. 선택적으로, 펠릿을 500μL의 냉각된 70% 에탄올로 1x 세척합니다.
- 펠릿을 40μL의 TE 완충액(10mM Tris-HCl, 1mM EDTA[pH 8.0])에 재현탁합니다.
3. 크롬 패턴의 나노 가공
- 탁상용 드릴 프레스에서 다이아몬드 코팅된 1.4mm 드릴 비트를 사용하여 1" x 3" 석영 현미경 슬라이드(~3cm 간격)의 중앙에 두 개의 구멍을 뚫습니다(드릴링 위치의 형상은 그림 1 참조).
- 천공된 슬라이드를 200mL의 피라냐 용액(75% 황산[97%] 및 25% 과산화수소)에 담가 청소합니다. 30분 동안 배양합니다.
주의 : 노출된 피부와의 모든 접촉을 피하기 위해 각별한 주의를 기울여야 합니다. 차가운 과산화수소를 황산에 매우 천천히 혼합하여 용액을 준비합니다. 피라냐 폐기물은 지정된 쓰레기통에 적절히 버리십시오. - 200mL의 이중 증류수에 담가 슬라이드를 헹굽니다. 2번 반복하여 남아 있는 피라냐를 제거합니다. 압축 질소의 부드러운 흐름으로 슬라이드를 건조시킵니다.
- 스핀 코터(4,000초 동안 45rpm, 램프 속도 300rpm/s)를 사용하여 다음과 같이 깨끗한 슬라이드를 포토레지스트와 전도성 폴리머로 코팅합니다.
- 3%(w/v) 폴리메틸메타크릴레이트(PMMA, 24.3K MW, 아니솔에 용해됨) 층으로 슬라이드를 코팅합니다.
- 1.5%(w/v) 폴리메틸메타크릴레이트(PMMA, 495K MW, 아니솔에 용해) 층으로 슬라이드를 코팅합니다.
- 코트 슬라이드는 전자빔 리소그래피를 위한 정전기 방지제 층으로 덮여 있습니다.
- 나노패턴 생성 시스템 소프트웨어가 장착된 주사 전자 현미경으로 전자빔 리소그래피를 사용하여 코팅된 슬라이드의 장벽과 받침대에 대한 사용자 정의 패턴을 작성합니다.
- 먼저 전자빔 리소그래피용 정전기 방지제를 이중 증류수로 헹궈 슬라이드를 현상합니다. 그런 다음 현상 용액(75% 메틸 이소부틸 케톤[MIBK] 및 25% 이소프로판올, -20°C에서 냉장 보관)이 들어 있는 50mL 튜브에 넣고 저전력으로 1분 동안 얼음 수조에서 초음파 처리합니다.
- 현상 용액을 이소프로판올로 헹구고 압축 질소의 부드러운 흐름으로 건조시킵니다.
- 전자빔 증발기를 사용하여 250 Å 두께의 크롬 층을 0.5 Å / s로 패턴 된 표면에 증착합니다.
- 분출 병의 아세톤을 사용하여 크롬과 남은 PMMA를 들어 올린 다음 아세톤 수조에서 10분 동안 슬라이드를 초음파 처리합니다. 분출 병에서 아세톤으로 다시 씻으십시오.
- 남아 있는 아세톤에서 침전물이 생기는 것을 방지하려면 이소프로필 알코올로 슬라이드를 헹구고 압축 질소의 부드러운 흐름으로 말리십시오.
4. flow cell의 조립
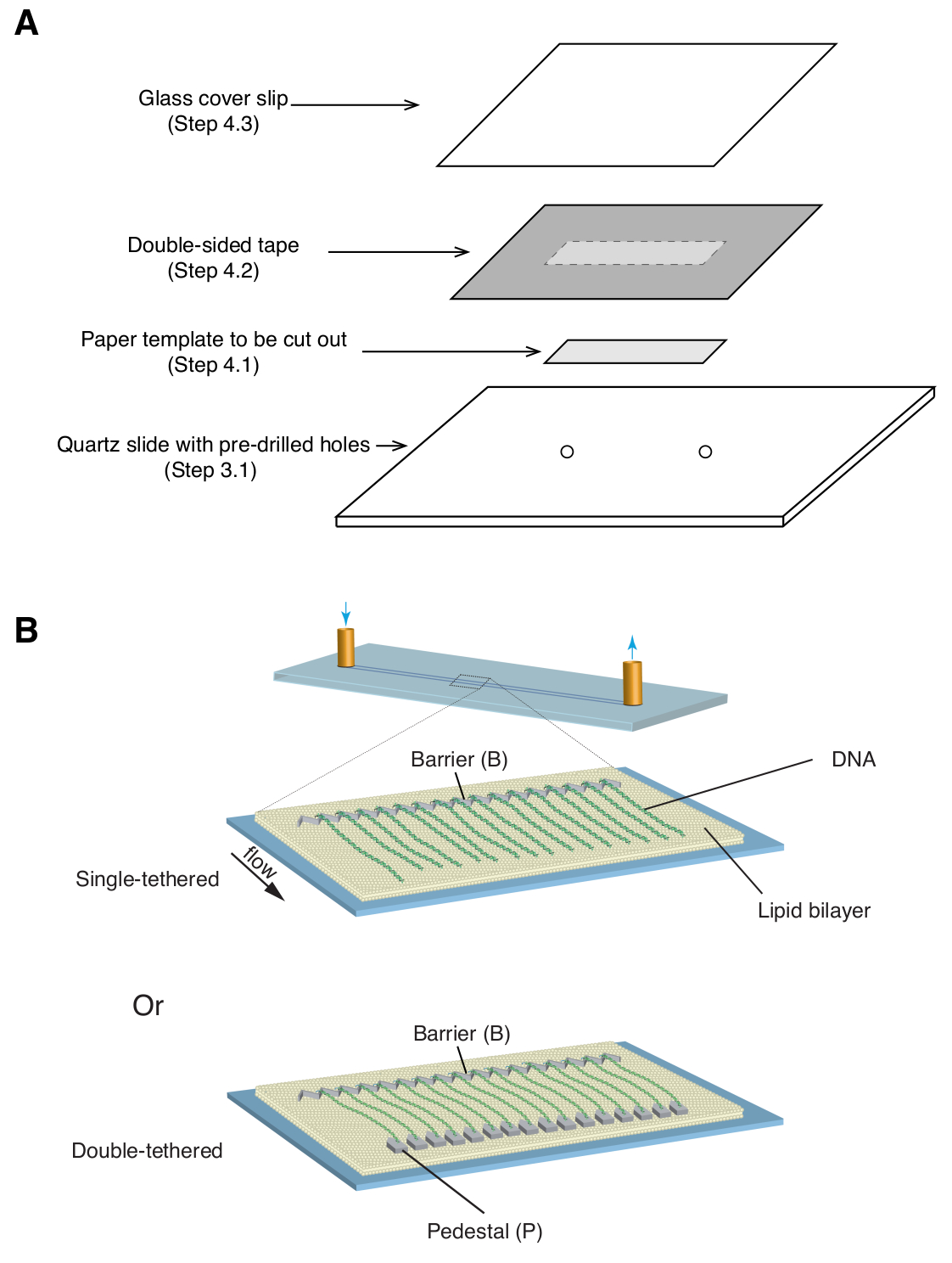
- 35mm x 5mm 직사각형 종이 템플릿(크롬 패턴과 미리 뚫린 입구/출구 구멍 포함)을 패턴이 있는 면이 위를 향하도록 석영 슬라이드 위에 놓습니다( 그림 1A).
- 종이 템플릿을 덮고 있는 석영 슬라이드 위에 양면 테이프(1mm 두께) 조각을 바른 다음 면도날을 사용하여 직사각형 종이 템플릿(그림 1A)을 잘라냅니다.
- 양면 테이프 위에 유리 커버슬립을 놓고 부드러운 압력을 가하여 커버슬립을 테이프에 밀봉합니다( 그림 1A). 양면 테이프로 밀봉된 커버슬립과 슬라이드 사이의 공간은 미세유체 챔버를 형성합니다.
알림: 커버 슬립과 슬라이드 사이의 밀봉이 실험 중 누출을 방지할 수 있을 만큼 충분히 고정되는 것이 중요합니다. - 유리 슬라이드 사이에 커버슬립 슬라이드 샌드위치를 놓고 네 면 모두에 바인더 클립을 부착하여 압력을 고르게 분산시킵니다. 어셈블리를 135°C에서 60분 동안 진공 오븐에 넣어 양면 테이프를 녹이고 챔버를 밀봉합니다.
- 오븐에서 꺼낸 다음 바인더 클립과 유리 슬라이드를 해제합니다.
- 접착된 어셈블리를 가열 블록(220°C)에 1분 동안 놓고 제거한 다음 미세유체 포트를 적절하게 밀봉하기 위해 부드러운 압력을 가하여 뜨거운 글루건으로 미리 뚫린 입구/출구 구멍에 미세유체 포트를 부착합니다. 계속하기 전에 접착제가 완전히 경화되도록 하십시오.
참고: 조립된 플로우 셀은 50mL 튜브에 넣고 진공 상태에서 보관할 수 있습니다.
5. dsDNA 커튼 조립
- 주입구/배출구 튜빙을 플로우 셀 미세유체 포트에 부착하고 10mL 주사기 2개를 사용하여 20mL의 이중 증류수로 챔버를 세척합니다.
- 이중 증류수가 채워진 10mL 주사기를 플로우 셀의 양쪽에 연결하여 주입구와 배출구 주사기 사이에 물을 밀거나 당기는 힘을 가합니다. 이 단계에서는 플로우 셀(flow cell)에서 모든 기포를 제거합니다.
- 3mL의 지질 완충액(10mM Tris-HCl[pH 8.0], 100mM NaCl)으로 챔버를 세척합니다.
참고: 플로우 셀에 대한 모든 연결부가 drop-to-drop인지 확인하여 플로우 셀에 기포가 유입되지 않도록 하십시오. - 40μL의 리포좀 용액과 1mL의 지질 완충액을 혼합합니다. 300μL의 지질 용액을 플로우 셀(flow cell)에 주입하고 5분 동안 배양합니다. 주입 및 배양을 2회 반복합니다.
참고: 주사기가 플로우 셀(flow cell)에 연결될 때마다 주입을 위해 주입구/배출구 쪽을 번갈아 가며 사용합니다. - 3mL의 지질 완충액으로 챔버를 세척하고 15분 동안 배양합니다.
참고: 이 배양 기간은 실험 일정에 따라 5분에서 2시간까지 다양할 수 있으며, 최종 커튼 조립에서는 눈에 띄는 차이가 없습니다. - 40μL의 1mg/mL 항 디곡시제닌 원액과 200μL의 지질 완충액을 혼합합니다. 용액 100μL를 주입하고 10분 동안 배양합니다. 3mL의 지질 완충액으로 세척하기 전에 주입 및 배양을 1회 반복합니다.
참고: 이 단계는 이중 테더링 dsDNA 커튼에만 필요합니다. 단일 테더링된 dsDNA 커튼의 경우 이 단계를 건너뜁니다. - 3mL의 BSA 완충액(40mM Tris-HCl[pH 8.0], 2mM MgCl2, 1mM DTT 및 0.2mg/mL BSA)으로 챔버를 세척합니다.
- 10μL의 1mg/mL 스트렙타비딘 원액과 990μL의 BSA 완충액을 혼합합니다. 각 주입 후 10분 동안 배양하면서 두 개의 개별 400μL 주입으로 주입합니다.
- 3mL의 BSA 완충액으로 챔버를 세척하여 과도한 스트렙타비딘을 제거합니다.
- 25ng의 dsDNA 기질 스톡을 1mL BSA 완충액에 희석합니다. DNA 용액을 한 번에 200μL씩 천천히 주입한 다음 각 주입 후 5분 동안 배양합니다. 필요한 DNA의 양은 특정 유형의 실험에 최적화될 수 있습니다.
- 플로우 셀을 현미경 스테이지에 장착하고 필요에 따라 초점을 조정합니다.
- 입력 및 출력 포트를 주사기 펌프와 고압 스위치 밸브로 구성된 시료 주입 시스템에 연결합니다.
- BSA 버퍼로 챔버를 0.8mL/분으로 5분 동안 연속적으로 세척하여 DNA를 받침대(12μm 테더 길이)에 이중 테더링합니다.
알림: 다른 이중 테더 길이에 맞게 최적화하기 위해 유속과 시간을 조정해야 할 수도 있습니다. 이 단계는 이중 테더링된 DNA 커튼에만 필요합니다. 단일 테더링된 dsDNA 커튼의 경우 이 단계를 건너뜁니다. - 이제 커튼이 실험할 준비가 되었습니다.
6. 플로우 셀(flow cell)의 재활용
- 면도날을 사용하여 미세유체 포트와 커버슬립을 조심스럽게 제거하기 전에 슬라이드를 에탄올에 최소 30분 동안 담그십시오. 커버슬립을 적절하게 폐기하고 미세유체 포트에 남아 있는 접착제를 청소하여 재사용할 수 있도록 합니다. 슬라이드의 접착제 잔여물을 문질러 청소합니다.
알림: 슬라이드 중앙의 패턴이 있는 부분이 긁히지 않도록 주의하세요. - 이중 증류수로 슬라이드를 철저히 헹구고 2% 알칼리성 액체 석영 세척 농축액에 밤새 담그고 저어줍니다.
- 이중 증류수로 슬라이드를 철저히 헹구고 80 °C에서 교반하면서 1 시간 동안 2 % 알칼리성 액체 석영 세척 농축 용액에 담근 다음 45 °C에서 15 분 동안 초음파 처리합니다.
- 이중 증류수로 슬라이드를 철저히 헹구고 저어주면서 30-60분 동안 1M NaOH에 담그십시오.
- 두 배 증류수를 가진 활주를 완전히 헹구고, 그 후에 이소프로필 알콜에서 담그고 45 °C에 15 분 동안 sonicate.
- 슬라이드를 에탄올로 철저히 헹구고 에탄올에 1시간 동안 담그고 저어줍니다.
- 슬라이드를 진공 오븐에 넣고 135°C에서 건조시킵니다. 이제 슬라이드를 플로우 셀로 재조립할 준비가 되었습니다.
결과
위에서 설명한 것은 DNA 복구 중간체에서 단백질-DNA 상호 작용을 연구하는 맥락에서 단일 및 이중 테더링된 dsDNA 커튼의 준비, 조립 및 이미징입니다. 그림 1A 는 flow cell의 모든 구성 요소를 조립된 순서대로 계층화한 것을 보여줍니다. 그림 1B 는 단일 또는 이중 테더링 DNA 커튼의 개략도를 보여줍니다. 지질 이중층(lipid bilayer)은 플로우 셀(flow cell)의 표면을 부동태화하는 데 사용됩니다. DNA 커튼은 한쪽 끝이 지질에 묶여 있고 크롬 장벽에 정렬되어 흐름 방향에 수직으로 배향된 DNA 분자의 병렬 배열로 구성됩니다. 이중 테더링 DNA 커튼의 경우, DNA의 다른 쪽 끝은 디곡시제닌(Digoxigenin)과 안티 디곡시제닌(anti-Digoxigenin) 상호작용을 통해 받침대에 테더링되어 DNA가 확장된 상태로 유지되는 동안 흐름이 없는 상태에서 이미징을 수행할 수 있습니다. dsDNA는 형광 염료 YoYo1로 염색하고 TIRF 현미경으로 시각화할 수 있습니다. 그림 2A 는 YoYo1 염색 이중 테더 DNA 커튼의 대표적인 광시야 이미지를 보여줍니다.
DNA 커튼 실험에서 수집된 타임랩스 이미지 시리즈는 일반적으로 ImageJ에서 관심 있는 각 DNA 분자에 대해 수직 축의 DNA 분자를 따른 위치와 수평 축의 시간을 표시하는 카이모그래프를 먼저 생성하여 분석됩니다. 도 2B는 단일 가닥 DNA 결합 단백질 RPA-mCherry(자홍색) 및 ATP가 있는 상태에서 효모 절제 기계 GFP-Sgs1(보이지 않음), Top3-Rmi1 및 Dna2에 의한 YoYo1 염색 단일 테더링 λ-DNA(녹색)의 절제를 보여주는 대표적인 카이모그래프입니다. YoYo1 신호는 dsDNA가 Sgs1-DNA2에 의해 자유단에서 절제됨에 따라 시간이 지남에 따라 손실됩니다. 동시에 mCherry 신호는 절제의 결과로 ssDNA가 생성되는 DNA 말단과 공동 국소화됩니다. 속도 및 진행도와 같은 절제의 생물물리학적으로 관련된 특성은 YoYo1 신호 손실 궤적의 기울기를 정량화하여 추출할 수 있습니다. 그림 2C,D는 Sgs1-Dna2에 의한 절제의 속도와 진행도의 분포를 각각 보여줍니다.

그림 1: 플로우 셀 어셈블리 및 DNA 커튼의 개략도. (A) 플로우 셀 어셈블리에 대한 단계적 그림. 석영 슬라이드 위에 양면 테이프를 붙이고, 종이 템플릿을 사용하여 중앙에서 직사각형 채널을 잘라낸 다음, 상단에 커버 슬립으로 밀봉하여 챔버를 형성합니다. (B) 완전히 조립된 DNA 커튼의 개략도. 중간 패널에는 단일 테더링 플로우 셀이 표시되고, 하단 패널에는 이중 테더링 플로우 셀이 표시됩니다. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
{kind=link}

그림 2: 단일분자 DNA 커튼 시각화 및 분석. (A) YoYo1(녹색)으로 염색된 이중 테더링 λ-DNA 커튼의 대표적인 광시야 이미지. (B) DNA2로 추적했을 때 미리 결합된 GFP-Sgs1(보이지 않음)이 있는 경우, RPA-mCherry(자홍색) 및 2mM ATP가 있는 상태에서 YoYo1 염색 dsDNA(녹색)의 절제를 보여주는 대표적인 카이모그래프. Sgs1-Dna2에 의한 dsDNA 절제의 속도 분포(C) 및 진행도 플롯(D)은 시간 경과에 따른 YoYo1 신호 손실의 속도와 범위를 정량화하여 얻었습니다. 패널 (B), (C) 및 (D)는 Xue et al.19에서 채택한 것입니다. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
{kind=link}
토론
DNA 커튼은 다양한 DNA 복구 과정을 연구하기 위한 다목적 플랫폼임이 입증되었습니다 13,14,15,16. 지속적인 완충액 흐름 또는 받침대에 대한 이중 테더링을 통해 실험 전반에 걸쳐 표면 근처에 확장된 상태로 남아 있는 DNA 분자는 단일 분자 수준에서 복구 중간체에서 단백질-DNA 상호 작용의 TIRFM 이미징을 가능하게 합니다. 이 방법은 수백 개의 DNA 분자가 표면에 특이적으로 부착되지 않고 나노 가공 장벽에 질서 정연하게 정렬되어 있기 때문에 실험 처리량과 예측 가능성을 개선하여 대규모 데이터 세트 처리에 더욱 도움이 됩니다.
생성된 타임 랩스 이미지 시리즈를 주의 깊게 분석하면 단백질-DNA 상호 작용(즉, 결합 수명, 위치, 화학량론, 해리 역학, 전좌 속도, 운동 단백질의 진행도, 단백질 및/또는 DNA 간의 공동 국소화 및 상호 작용)을 정량적으로 측정할 수 있습니다. DNA 분자를 표면에 직접 묶거나 상용 기기에서 공초점 이미징과 힘 조작을 결합하는 다른 단일 분자 이미징 기술과 비교할 때, 더 높은 처리량을 위한 DNA 커튼의 주요 절충안은 초기 설정에 필요한 기술을 확립하는 것입니다 13,14,15,16. 그러나 일단 확립되면 하나의 플로우 셀에서 수백 개의 분자에 대한 데이터를 동시에 수집할 수 있는 기능을 통해 데이터 수집에 소요되는 시간을 크게 절약할 수 있습니다.
다른 많은 단일 분자 이미징 방법과 마찬가지로 깨끗한 표면을 준비하는 능력은 DNA 커튼을 안정적으로 조립할 수 있는 사용자의 능력을 크게 좌우합니다. 이 경우 깨끗한 표면은 깨끗한 슬라이드로 시작해야 할 뿐만 아니라 유체 연결을 만드는 동안 챔버 내부에 미세한 기포가 유입되지 않도록 해야 합니다. 첫째, 슬라이드의 청결도와 관련하여 위의 섹션 6에 자세히 설명된 단계는 일상적인 청소 프로세스로 충분해야 합니다. 여과된 용액과 버퍼를 사용하는 것도 좋습니다. 둘째, 플로우 셀을 버퍼 주입 시스템에 부착하는 동안 drop-to-drop 연결을 만드는 것은 기포 유입을 방지하는 데 필수적입니다. 완충액을 만드는 데 사용되는 이중 증류수의 가스를 제거하는 것이 도움이 될 수 있습니다. 또한 기포가 없는지 확인하기 위해 주입 과정을 주의 깊게 관찰하는 것이 좋습니다. 미세한 기포가 중심 패턴 영역에 도달하기 전에 도입되면 반대쪽의 미세유체 포트에서 버퍼를 주입하여 기포를 다시 밀어냄으로써 해결할 수 있습니다.
형광 기반 실험에서 고려해야 할 또 다른 중요한 사항은 신호 대 잡음비입니다. 단일 형광 단백질(즉, GFP)을 관찰하는 열쇠는 깨끗한 표면에도 있습니다. 장기간 사용 후에는 일반적으로 단백질 응집체가 크롬 장벽과 받침대에 축적됩니다. 따라서 형광 단백질을 추가하기 전에 장벽에서 강한 형광 신호가 관찰되면 Piranha 용액에 10분 동안 담궈 슬라이드를 처리하는 것이 좋습니다. 크롬 기능이 침식되지 않도록 기간을 짧게 유지합니다. 피라냐 세척 후에는 정기적인 세척 절차를 거쳐야 합니다.
전자빔 리소그래피를 사용한 나노 제조는 장비 및 시설에 대한 요구 사항과 재료 과학에 대한 기존 전문 지식으로 인해 어려울 수 있습니다. 대안으로, DNA 커튼의 크롬 특징 제작을 위한 UV 리소그래피 기반 방법이 개발되었습니다20. 또한 이 프로토콜과 함께 12μm에서 람다 DNA를 이중 테더링하기 위한 기본 패턴이 제공됩니다(추가 정보 참조). 이 패턴의 설계의 여러 측면은 다양한 실험 요구에 맞게 조정될 수 있습니다. 여기에는 장벽과 받침대 사이의 거리, DNA 밀도를 제어하기 위한 장벽의 인접한 웰 사이의 간격, 이중 테더링 효율성을 최적화하고 이중 테더링 길이의 불확실성을 최소화하기 위한 받침대의 모양과 크기가 포함됩니다. 다른 실질적인 문제로는 단백질에 대한 형광 표지 전략 선택, 수동 데이터 분석의 편향 최소화, 보다 간소화된 데이터 처리를 위한 스크립트 개발 등이 있습니다.
여기에 설명된 프로토콜은 dsDNA 기질과 관련이 있지만 ssDNA를 기반으로 하는 DNA 커튼도 광범위하게 사용되었습니다 13,14,15,16. ssDNA 기질의 준비는 비오틴화 프라이머13을 사용한 M13 ssDNA 플라스미드의 롤링 서클 복제를 활용합니다. ssDNA 커튼을 조립하려면 2차 구조를 제거하고 ssDNA 기질을 확장하기 위해 요소 및 ssDNA 결합 단백질 RPA를 사용해야 합니다13. RNA 상호 작용 단백질의 연구를 위해 RNA 또는 DNA/RNA 하이브리드를 사용하도록 DNA 커튼을 확장하는 것도 가능할 수 있습니다. 현재 상태로, DNA 커튼 플랫폼은 섹션 5에 자세히 설명된 대로 조립 공정에서 자동화의 이점을 누릴 수 있으며, 대부분은 표준 버퍼 세트의 반복적인 주입입니다. 이는 여러 실험을 동시에 수행하는 데 효율성을 향상시킬 수 있습니다. 또한 잠재적으로 기계 학습 기반 분석 스크립트 제품군은 최소한의 바이어스로 데이터 처리 속도를 더욱 높일 수 있습니다.
공개
저자는 공개할 내용이 없습니다.
감사의 말
저자는 DNA 커튼 프로토콜을 개발, 최적화 및 논의하고 이 원고에 대해 논평해 준 Greene 연구소의 전현직 구성원에게 감사를 표합니다. 이 연구는 미국 국립과학재단(National Science Foundation, MCB1154511에서 E.C.G.까지)과 국립보건원(National Institutes of Health, R01CA217973에서 E.C.G.)의 보조금으로 지원되었습니다.
자료
| Name | Company | Catalog Number | Comments |
| nano pattern generation system software | JC Nabity Lithography Systems | N/A | NPGS |
| PFA Tubing, Natural 1/16" OD x .020" ID | IDEX | 1512L | |
| Poly(Methyl Methacrylate), Atactic (24.3K MW) | Polymer Source Inc. | P9790-MMA | |
| Quartz Microscope Slide | G. Finkenbeiner Inc. | N/A | 1" x 3" x 1 mm thick |
| Scanning Electron Microscope | FEI | N/A | Nova Nano SEM 450 |
| Scotch Double Sided Tape, 3/4 x 300 inches | 3M | N/A | |
| Sonicator | Misonix | S-4000 | For preparation of lipids |
| Sonicator | Branson | 1800 | For sonicating slides |
| Streptavidin from Streptomyces avidinii | Sigma-Aldrich | S4762 | Dissolved in 1X PBS to a final concentration of 1 mg/mL |
| Sulfuric Acid | Avantor - J.T.Baker | 9681-01 | |
| Syringe Pump | KD Scientific | 78-0200 | |
| Syringe, 500 µL, Model 750 RN SYR, Large Removable NDL, 22 ga, 2 in, point style 2 | Hamilton | 80830 | Reserve for use only with lipids |
| T4 DNA Ligase (400,000 units/ml) | NEB | M0202S |
참고문헌
- Kowalczykowski, S. C. An Overview of the Molecular Mechanisms of Recombinational DNA Repair. Cold Spring Harbor Perspectives in Biology. 7, 016410(2015).
- Ferlay, J., et al. Estimating the global cancer incidence and mortality in 2018: globocan sources and methods. International Journal of Cancer. 144, 1941-1953 (2018).
- Li, X., Heyer, W. D. Homologous recombination in DNA repair and DNA damage tolerance. Cell Research. 18, 99-113 (2008).
- Michel, B., Baoubakri, H., Baharoglu, Z., LeMasson, M., Lestini, R. Recombination proteins and rescue of arrested replication forks. DNA Repair. 6, 967-980 (2007).
- Paques, F., Haber, J. E. Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiology and Molecular Biology Reviews. 63, 349-404 (1999).
- Heyer, W. D., Ehmsen, K. T., Liu, J. Regulation of homologous recombination in eukaryotes. Annual Review of Genetics. 44, 113-139 (2010).
- Mason, J. M., et al. RAD54 family translocases counter genotoxic effects of RAD51 in human tumor cells. Nucleic Acids Research. 43, 3180-3196 (2015).
- Wold, M. S. Replication protein A: A heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annual Review of Biochemistry. 66, 61-92 (1997).
- Bianco, P. R. The tale of SSB. Progress in Biophysics and Molecular Biology. 127, 111-118 (2017).
- Hopfner, K. P., Gerhold, C. B., Lakomek, K., Wollmann, P. Swi2/Snf2 remodelers: hybrid views on hybrid molecular machines. Current Opinion in Structural Biology. 22, 225-233 (2012).
- Shah, P. P., et al. Swi2/Snf2-related translocases prevent accumulation of toxic Rad51 complexes during mitotic growth. Molecular Cell. 39, 862-872 (2010).
- Wright, W. D., Heyer, W. D. Rad54 functions as a heteroduplex DNA pump modulated by its DNA substrates and Rad51 during D loop formation. Molecular Cell. 53, 420-432 (2014).
- Ma, C. J., Steinfeld, J. B., Greene, E. C. Single-Stranded DNA Curtains for Studying Homologous Recombination. Methods in Enzymology. 582, 193-219 (2017).
- Qi, Z., et al. DNA sequence alignment by microhomology sampling during homologous recombination. Cell. 160, 856-869 (2015).
- Visnapuu, M. L., Greene, E. C. Single-molecule imaging of DNA curtains reveals intrinsic energy landscapes for nucleosome deposition. Nature Structural Molecular Biology. 16, 1056-1062 (2009).
- Kaniecki, K., De Tullio, L., Greene, E. C. A change of view: homologous recombination at single-molecule resolution. Nature Reviews Genetics. 19, 191-207 (2018).
- Redding, S., et al. Surveillance and Processing of Foreign DNA by the Escherichia coli CRISPR-Cas System. Cell. 163, 854-865 (2015).
- Lee, J. Y. Base triplet stepping by the Rad51/RecA family of recombinases. Science. 349, 977-981 (2015).
- Xue, C., et al. Regulatory control of Sgs1 and Dna2 during eukaryotic DNA end resection. Proceedings of the National Academy of Sciences USA. 116 (13), 6091-6100 (2019).
- Gallardo, I. F., Finkelstein, I. J. High-Throughput Universal DNA Curtain Arrays for Single-Molecule Fluorescence Imaging. Langmuir. 31 (37), 10310-10317 (2015).
재인쇄 및 허가
JoVE'article의 텍스트 или 그림을 다시 사용하시려면 허가 살펴보기
허가 살펴보기This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. 판권 소유