
Method Article
Integração de Wet and Processes bancada secos Otimiza alvejado Seqüenciamento Next-geração de baixa qualidade e biópsias de tumores de baixa quantidade
Neste Artigo
Resumo
An integrated system for targeted next-generation sequencing of oncology specimens is described. This cross-platform system is optimized for low-quality and low-quantity tumor biopsies, accommodates low DNA inputs, includes well-characterized multi-variant controls, and features a novel variant caller that is informed by quantitative pre-analytical quality control measures.
Resumo
Todos os procedimentos da próxima geração seqüenciamento (NGS) incluem ensaios realizados na bancada do laboratório ( "banco molhado") e dados de análises realizadas usando bioinformática condutas ( "banco seca"). Ambos os elementos são essenciais para produzir resultados exactos e fiáveis, que são particularmente críticos para laboratórios clínicos. tecnologias NGS direcionados têm encontrado cada vez mais favorável em aplicações de oncologia para ajudar objetivos de precisão medicina avanço, mas os métodos envolvem frequentemente desconectados e variável fluxos de trabalho de bancada secos e molhados e conjuntos de reagentes descoordenados. Neste relatório, nós descrevemos um método para a sequenciação desafiando amostras de cancro com um painel de 21 genes como um exemplo de um sistema global alvejado NGS. O sistema integra quantificação funcional DNA e qualificação, de tubo único multiplexado enriquecimento de PCR, e purificação biblioteca e normalização usando analiticamente verificados, reagentes de fonte única com um conjunto bioinformática independentes.Como resultado, a variante precisa chamadas a partir de baixa qualidade e baixa quantidade fixa em formalina (FFPE) e aspiração com agulha fina (FNA) biópsias de tumores pode ser alcançado embebido em parafina. O método pode rotineiramente avaliar variantes associadas a cancro a partir de uma entrada de 400 cópias de DNA amplific�eis, e é um projeto modular para acomodar novos conteúdos gene. Dois tipos diferentes de controles analiticamente definidos fornecer garantia de qualidade e precisão chamada ajuda salvaguarda com amostras clinicamente relevantes. A "tag" passo PCR flexível incorpora adaptadores específicos da plataforma e códigos de índice para permitir que o código de barras da amostra e compatibilidade com instrumentos NGS comum de bancada. Importantemente, o protocolo é simples e pode produzir bibliotecas de sequências 24-pronto em um único dia. Finalmente, a abordagem une processos banco secos e molhados, incorporando os resultados do controlo de qualidade da amostra pré-analítica directamente para a variante chamando algoritmos para melhorar a precisão da detecção de mutação e diferenciar falso-negativo e indetermchamadas inata. Este método NGS alvo utiliza avanços em wetware e software para atingir alta profundidade, sequenciamento multiplexados e análise de sensibilidade das amostras de câncer heterogêneos para aplicações de diagnóstico.
Introdução
medicina de precisão depende da individualização das opções diagnósticas e terapêuticas para os pacientes. A promessa de tratamentos personalizados é uma consequência directa de uma melhor compreensão das vias de doenças que podem informar a ligação de diagnóstico molecular e terapêutica alvo. Por exemplo, o uso de terapias molecularmente-alvo aumentou de 11% para 46% 2003-2013 1, e as drogas anti-câncer, como vemurafenib e crizotinib está aprovado pela FDA com testes de diagnóstico do companheiro. Com a sua capacidade de se recuperar com precisão alvos seqüência baixa abundância em toda conjuntos de amostras altamente multiplexados, sequenciamento de próxima geração (NGS) surgiu como um método de escolha para a avaliação de aberrações genéticas associadas ao câncer e identificar alvos moleculares para a medicina de precisão.
As biópsias de tumores sólidos mais comuns para teste molecular incluem fixado em formol e-embebidos em parafina (FFPE) e aspiração com agulha fina (FNA) SPECIMens. Estas amostras são repletas de baixa quantidade e / ou ácidos nucleicos que desafiam precisa NGS avaliações 2-5 de baixa qualidade. NGS comerciais atuais métodos de análise dessas amostras são baseados em uma colcha de retalhos de reagentes diferentes, protocolos e ferramentas informáticas que representam alvos em movimento de melhorias contínuas. Por exemplo, as mudanças na químicas de ensaio e / ou software ocorreu a cada 1-2 meses para os NGS kits direcionados mais utilizados 6. Esta instabilidade reflete uma falta de coerência na construção e verificação de um sistema unificado para NGS tipos de amostras difíceis, especialmente para testes de câncer, e coloca um fardo excessivo nos laboratórios para desenvolver protocolos coesas que são otimizados a partir da amostra-to-resultados. De fato, uma pesquisa recente de usuários NGS destacou as dificuldades destes "que mudam rapidamente" tecnologias, juntamente com os requisitos para estabelecido, o conteúdo medicamente acionável, a perícia bioinformática entrincheirado, um solidified e procedimento integrado que pode ser implementado rapidamente e simplificados fluxos de trabalho e protocolos simplificados que facilitam on-the-job training 7. Neste artigo, um sistema abrangente para NGS alvo é descrito que aborda estas lacunas.
A metodologia apresentada integra todas as etapas processuais de pré-analítica para a pós-analítica em bancos tanto secos e molhados para melhorar a precisão, sensibilidade e confiabilidade de quantificação alvo e detecção para NGS de loci gene do câncer clinicamente relevante. Esta abordagem começa com a quantificação do DNA "funcional" 4 de avaliar a qualidade do DNA, guia de entrada para a etapa de enriquecimento PCR, e proteção contra as chamadas falso-positivos que podem surgir a partir do interrogatório de muito baixas cópias do modelo. Um multiplex-único tubo PCR, em seguida, enriquece por 46 loci em 21 genes do cancro, utilizando apenas 400 cópias de DNA amplific�eis, seguido por incorporação de sequências específicas de plataforma para NGS using instrumentos de sequenciamento de desktop comum. Bibliotecas são purificados utilizando um procedimento grânulo magnético simples e quantificado com um romance, ensaio qPCR livre de calibração. Um conjunto bioinformática independentes, informados por amostra resultados DNA CQ a melhorar o desempenho chamada, fornece análise de sequência seguinte NGS. Apresentamos dados usando esta abordagem de sistemas para NGS direcionados para revelar mutações base-substituição, inserção / exclusão (Indels), e copiar o número de variantes (CNVs) em biópsias de baixa qualidade e tumor de baixo quantidade como FFPE e amostras Fna e prazo controlos.
Protocolo
Nota: Este protocolo descreve o processamento simultâneo de amostras usando um sistema MiSeq NGS, mas pode ser adaptado para o instrumento pessoais Genoma Máquina (PGM). Para a entrada de ADN mínima recomendada de 400 cópias molde amplificáveis, o ensaio é capaz de produzir, pelo menos, 3,000x cobertura mediana para cada uma das 96 amostras por NGS prazo, e a profundidade equivalente para a cobertura 24 amostras usando o PGM sobre um chip 318. O método também requer a utilização de um equipamento de PCR em tempo real.
1. DNA Quantificação Funcional e Controle de Qualidade (QC)
- reagentes descongelamento: 2x Mestre Mix, Primer Mix Probe, Inibição Mix Probe Primer, 6-carboxi-X-rodamina (ROX), diluente, e os quatro genômicas de normalização da curva de calibração DNA humano (DNA padrão (50 ng / mL), DNA Padrão (10 ng / jil), de ADN convencional (2 ng / ul), e ADN padrão (0,4 ng / mL)) (Tabela 1). Vortex todos os reagentes para 10 seg e centrifuga-se à velocidade máxima durante 10 segundos para recolher o conteúdo.Mantenha a mistura principal 2x no gelo.
- Prepara-se uma quantidade suficiente de mistura principal para o número total de amostras a serem testadas e incluem 10% mais volume para evitar a falta devido a pipetagem. Preparar a mistura de mestre num tubo de microcentrífuga utilizando os seguintes volumes por amostra: 5 ul 2x Master Mix, 0,5 ul de mistura de iniciadores de sonda, 0,5 ul de mistura de sonda Inibição Primer, 0,05 ul ROX e 2,95 ul de diluente. Vortex durante 10 seg e centrifuga-se à velocidade máxima durante 10 segundos para recolher o conteúdo.
- Adicionar 9 ul de mistura principal em poços de uma placa de 96 poços.
- Adicionar 1 ml de os padrões de ADN, em duplicado, para gerar uma curva de calibração. Misturar por pipetagem cima e para baixo 5 vezes.
- Assegurar que a amostra de ácidos nucleicos é bem misturada antes da utilização. Adicionar amostra de 1 ul a mistura principal e misture por pipetagem para cima e para baixo 5 vezes.
- Selar a placa, de vórtice durante 10 seg e centrifuga-se à velocidade máxima durante 10 segundos para recolher o conteúdo.
- Colocar a placa no sistema de PCR. Atribuir tanto FAM (quantificação funcional) e VIC (inibição funcional) detectores para cada amostra de acordo com as instruções do fabricante. Executar ciclos de PCR de 10 min a 95 ° C, e 40 ciclos (15 segundos a 95 ° C, 1 min a 60 ° C).
- Analisar os dados qPCR através da geração de uma trama de regressão linear para cada um dos padrões de ADN duplicado utilizando protocolos de software.
- Traça-se a log 10 do número de cópia para cada padrão de ADN no eixo X e o valor q FAM C correspondente no eixo dos y.
- Confirmar que os resultados da queda amostra de ácido nucleico dentro do intervalo dinâmico da curva de calibração padrões de ADN, e, em seguida, calcular a concentração de ADN desconhecida em "funcional" ou amplificável número de cópias por ul a partir da sua posição correspondente na curva padrão de referência. A Figura 1 mostra exemplos de curvas de calibração que passaram e falharam.
- Determinar se a amplificação ocorreu em cada reacção, verificando a presença do fragmento amplificado alvo não-humano no canal VIC.
Observação: Como controlo positivo, a mistura de sondas Inibição Primer contém iniciadores específicos para um alvo exógeno não-humano e a matriz correspondente. A inibição mistura de sonda iniciador é um componente da mistura principal, que é adicionada a cada reacção, incluindo o controlo sem modelo (NTC). Na ausência de um inibidor, o produto de PCR para o alvo não-humano deve sempre ser detectadas no canal VIC. Um "não detectado" C Q de uma amostra no canal VIC indica a presença de inibidores de PCR que podem beneficiar de limpeza subsequente da amostra antes do processamento ulterior.
2. Biblioteca Preparação: específicos do Gene (GS) PCR
- Prepare uma quantidade suficiente de mistura principal para o número total de amostras a serem testadas e incluem 10% mais volume para evitar a escassez devido a pipetaing. Preparar a mistura principal de PCR GS num tubo de microcentrífuga utilizando os seguintes volumes por amostra: 5 ul 2x Master Mix de amplificação (Tabela 1) e 1 uL do cancro Painel Pan-iniciador (Tabela 1). Misturar por pipetagem cima e para baixo, vortex por 10 segundos e centrifugar a velocidade máxima para 10 segundos para coletar conteúdo.
- Alíquota da GS 6 ul de PCR Master Mix para poços de uma placa de 96 poços. Adicionar 4 ul de cada amostra de ácido nucleico em poços individuais. Para outros poços, adicionar 4 mL do controle FFPE (Tabela 1), 4 uL de controlo com diversas variantes (Tabela 1), e 4 mL de água isenta de nuclease para um NTC processual. Para cada adição, misture pipetando para cima e para baixo 5 vezes.
- Selar a placa, de vórtice durante 10 seg e centrifuga-se à velocidade máxima durante 10 segundos para recolher o conteúdo.
- Colocar a placa no termociclador durante os ciclos de PCR seguintes: 5 min a 95 ° C,2 ciclos (15 seg a 95 ° C, 4 min a 60 ° C), 23 ciclos (15 seg a 95 ° C, 4 min a 72 ° C), e uma extensão final de 10 min a 72 ° C. Manter a 4 ° C.
Nota: Após conclusão do passo 2.4, a placa será referida como a placa de PCR GS.
3. Biblioteca Preparação: Tag PCR
- Reagentes descongelamento: 2x Mix Master Index (Tabela 1) e códigos de índice (Tabela 1). Vortex durante 10 seg e centrifuga-se à velocidade máxima durante 10 segundos para recolher o conteúdo.
Nota: Os códigos de indexação são pré-misturados para fornecer um conjunto único de índices de pares (códigos de barras) para cada amostra. - Em uma placa de 96 poços, adicionar 7,5 ml da Mix Master Índice de 2x e 5,5 ul de um código de índice para um bem especificado e misture por pipetagem para cima e para baixo 5 vezes.
- abra cuidadosamente a placa GS PCR e adicionar 2 produto mL GS PCR para a nova placa com a mistura principal. Misturar por pipetagem cima e para baixo 5 vezes. Para cada amostra, registre o ID da amostra e os correspondentes códigos de índice de pares. Selar a placa, de vórtice durante 10 seg e centrifuga-se à velocidade máxima durante 10 segundos para recolher o conteúdo.
- Colocar a placa no termociclador e a PCR durante 5 min a 95 ° C, 10 ciclos (30 segundos a 95 ° C, 30 seg a 55 ° C, 1 min a 72 ° C), e uma extensão final de 10 min a 72 ° C. Manter a 4 ° C.
Nota: Após conclusão do passo 3.4, a placa será referida como a placa de PCR Tag.
4. Biblioteca Purificação e seleção de tamanho
- Remover os Biblioteca puro Prep esferas magnéticas (Tabela 1) e tampão de eluição (Quadro 1) a partir de 2-8 ° C e deixar equilibrar à temperatura ambiente durante 30 min. Adicionar 9,6 ml de 100% de etanol ao tampão de lavagem (Tabela 1) recipiente, tampa e misture invertendo o frasco várias vezes.
- Vortex as esferas magnéticas para 10 seg e adicionar 11 ul em poços separados de uma 9placa de 6 poços.
- Abra a placa PCR Tag e adicione 10 ml de Tag produto de PCR até aos talões e pipeta de mistura 5 vezes. Incubar a mistura durante 4 minutos à temperatura ambiente.
- Colocar a placa de 96 poços no suporte magnético (Tabela 1) durante 4 min. Com a placa de 96 poços ainda no stand, remover e descartar o sobrenadante com uma pipeta.
- Remova a placa de 96 poços a partir do suporte magnético e adicionar 100 ul de tampão de lavagem contendo etanol a cada poço e misturar por pipetagem para cima e para baixo 5 vezes. Incubar durante 2 min.
- Colocar a placa de 96 poços no suporte magnético durante 2 min, em seguida, remover e descartar o sobrenadante com uma pipeta.
- Repetir o passo 4.5, para um total de 2 lavagens de etanol, a remoção da solução de lavagem o mais possível após a segunda lavagem.
- Com a placa de 96 poços no suporte magnético, secar os grânulos por 2 min à temperatura ambiente, em seguida, retire a placa da base.
- Ressuspender as contas pela adição de 20 ul eluição Buffer para cada poçol e pipeta de up-and-down 5 vezes.
- Incubar durante 2 min à temperatura ambiente.
- Colocar a placa de 96 poços para trás sobre o suporte magnético durante 4 min, e cuidadosamente remover e transferir 18 ul do sobrenadante límpido para um novo poço.
Nota: O procedimento pode ser interrompido com segurança neste passo e as amostras armazenadas a -15 a -30 ° C. Para reiniciar, descongelar as amostras congeladas no gelo antes de prosseguir.
5. Quantificação Biblioteca
- Thaw Biblioteca Quant (LQ) reagentes: 2x LQ Mestre Mix, LQ Primer / Probe Mix, LQ Padrão, LQ controlo positivo, LQ diluente e LQ ROX (Tabela 1). Vortex durante 10 seg e centrifuga-se à velocidade máxima durante 10 segundos para recolher o conteúdo.
- Usando produtos de biblioteca purificados, execute uma diluição em série de cada amostra individual na LQ diluente.
- Adicionar 2 mL (biblioteca do produto purificado) a 198 ul LQ diluente e misture para cima e para baixo com uma pipeta de 10 vezes.
- Adicionar 2 ul (diluição 1: 100) para uma98 ul LQ diluente e misture up-and-down com uma pipeta 10 vezes.
- Prepara-se uma quantidade suficiente de LQ mistura principal para o número total de amostras a serem testadas e incluem 10% mais volume para evitar a falta devido a pipetagem. Preparar a Mistura Principal LQ num tubo de microcentrífuga utilizando os seguintes volumes por amostra: 5 ul de 2X Master Mix LQ, 2 ul LQ mistura iniciador / sonda, 0,5 ul LQ padrão e 0,5 ul LQ ROX. Misturar por pipetagem cima e para baixo, vortex por 10 segundos e centrifugar a velocidade máxima para 10 segundos para coletar conteúdo.
- Adicionar 8 mL master mix LQ a um poço de uma placa de 96 poços óptico.
- Em poços separados, adicionar 2 mL de biblioteca diluída, 2 ul LQ Controle Positivo e 2 ul LQ diluente (NTC) e misture pipetando para cima e para baixo 5 vezes. Selar a placa com película adesiva óptica, de vórtice durante 10 segundos e centrifugar a 400 xg durante 10 seg para recolher conteúdos.
- Atribuir ambos FAM e VIC Detectors para cada amostra. Realizar a amplificação por PCR usando condições de ciclização de 5 min a 95 ° C, e 40 ciclos (15 segundos a 95 ° C, 1 min a 60 ° C).
- Determinar a concentração de cada amostra (nM) usando o método comparativo de C q. Calcula-se a diferença do conhecido LQ Padrão (C Q VIC) para a biblioteca desconhecido (C Q FAM). A concentração da amostra diluída (pM) é calculado usando a seguinte equação:
[Lib Conc] nM = 12,5 x 2 Δ Cq
Se usar um outro factor de diluição de 10.000, o cálculo da relação do factor de diluição para 10.000 alvo, e multiplicar este factor pelo resultado da equação em 5,7.
6. Biblioteca Normalização e Pooling Amostra
- Determinar a concentração mediana (nM) em todas as amostras (cada um contendo um único índice de pares) para ser reunidas.
- Determine o volum amostra individuale (l) para reunir-se multiplicando a média em todas as amostras por 5, em seguida, dividindo pela sua concentração individual (nM). Em volta do valor resultante para o número inteiro mais próximo. volumes redondas com valores de <2 ul a 2 ul e volumes> 15 ul e 15 ul.
- Adicionar o volume normalizado (ul) para cada amostra a um único tubo de microcentrífuga para criar o pool de amostra.
- Calcule a nova concentração para cada amostra usando valores inteiros arredondados e registrar os resultados.
- Para determinar a concentração da amostra piscina, calcular a soma de todas as concentrações individuais e gravar o valor resultante (nM).
- Dilui-se a amostra a piscina 1,25 nM usando Diluente A sequenciação (Tabela 1).
Nota: O procedimento pode ser interrompido com segurança neste passo e as amostras armazenadas a -15 a -30 ° C. Para reiniciar, descongelar as amostras congeladas no gelo antes de prosseguir.
7. Sequencing
- denature a piscina de exemplo na presença de Controlo phix V3 (quadro 1), incluindo as seguintes: 15 mL de 1,25 nM pool de amostras, 3 ul de 0,5 nM phix e 2 ul de NaOH 1 N. Vortex brevemente seguido por uma breve centrifugação e incubar durante 5 min à TA.
- Coloque a piscina amostra desnaturada no gelo.
- Adicionar 8 mL de biblioteca desnaturado a 992 ul de tampão HT1-Hib pré-arrefecido a um tubo de microcentrífuga. Vortex para misturar brevemente, seguida de uma breve centrifugação para recolher o conteúdo. Manter em gelo.
- Adicionar 600 ul da biblioteca desnaturado e diluiu-se para a posição # 17 do cartucho de reagente.
- Thaw Leia 1 Sequencing Primers (Tabela 1), índice de leitura de sequenciação Primers (Tabela 1), e leia 2 iniciadores de sequenciação (Tabela 1). Em tubos de microcentrífuga, diluir separadamente 4 ul iniciadores de sequenciação com 636 tampão HT1-Hib ul. Nota: tampão HT1-Hyb é fornecida with, o estojo de reagentes de sequenciação (Tabela 1).
- Misturar em vortex durante 10 seg e centrifuga-se à velocidade máxima durante 10 segundos para recolher o conteúdo. Adicionar 600 mL de diluído Leia 1 Sequencing Primers para posição # 18 do cartucho de reagente sequenciamento, a 600 ul de diluído índice lidas Primers para a posição # 19, e 600 mL de Primers Leia 2 Sequencing diluídos para a posição # 20.
- Carregar os reagentes no instrumento NGS (Tabela 1) e a sequência de acordo com as instruções do fabricante. Executar uma ponta emparelhado 2 x 150 ciclo de sequenciação prazo.
Análise 8. Dados
Nota: O software do aparelho NGS converte imagens de cluster para chamadas de base e índices de qualidade, e demultiplexes índices de pares para gerar FASTQ compactado pelo gzip indivíduo (* .fastq.gz) arquivos para cada amostra. Antes de analisar os arquivos desmultiplexados, o leitor deve baixar e instalar o software de bioinformática associada (Tabela1). O software pode ser instalado em um consumidor-grade do Windows PC e não requer hardware de computação especializada ou uma conexão com a internet para realizar a análise de dados.
- Dê um duplo clique no ícone no desktop de software.
- Entre para o sistema usando o nome de usuário e senha fornecidos no manual do software.
- Abra o painel do projeto, e clique em "New Project".
- Nome do projeto e fornecer uma descrição opcional projeto. Selecione o tipo de painel NGS alvejado e NGS tipo de instrumento. Clique em "Salvar e continuar".
- Faça o upload dos arquivos FASTQ comprimido para a frente e reverso lê. Não carregue as FASTQs "não atribuídos", que contêm lê que não conseguiu demultiplex. Clique em "Salvar e continuar".
- Introduzir o número de cópias de entrada funcionais utilizados para preparar cada biblioteca tal como determinado pelo ensaio de quantificação do ADN funcional (ver Passo 1 acima). adicionar manualmente valores ou valores de copiar e colar a partir de um spreadfolha na tabela de anotação. Clique em "Salvar e continuar".
- Reveja as bibliotecas anotados enviados para análise e clique em "Enviar análise" para iniciar a análise.
- Monitorizar o progresso da análise apresentada através do painel de projecto.
Nota: Uma indicação de status completa análise é apresentada quando os resultados estão prontos para análise. - Comente os resultados analisados para amostra métricas QC, incluindo a cobertura total por biblioteca, percentagem de leituras filtros de passagem, cobertura aprofundada amplicon e uniformidade. Comente a variante chama para cada biblioteca sequenciado com dbSNP, COSMIC, 1.000 genomas e outras fontes de anotação nível funcional e da população.
- Exportar os resultados brutos como tabelas de planilhas resumo, os arquivos * .bam e arquivos * .vcf para armazenamento a longo prazo ou a análise a jusante com ferramentas informáticas complementares.
Resultados
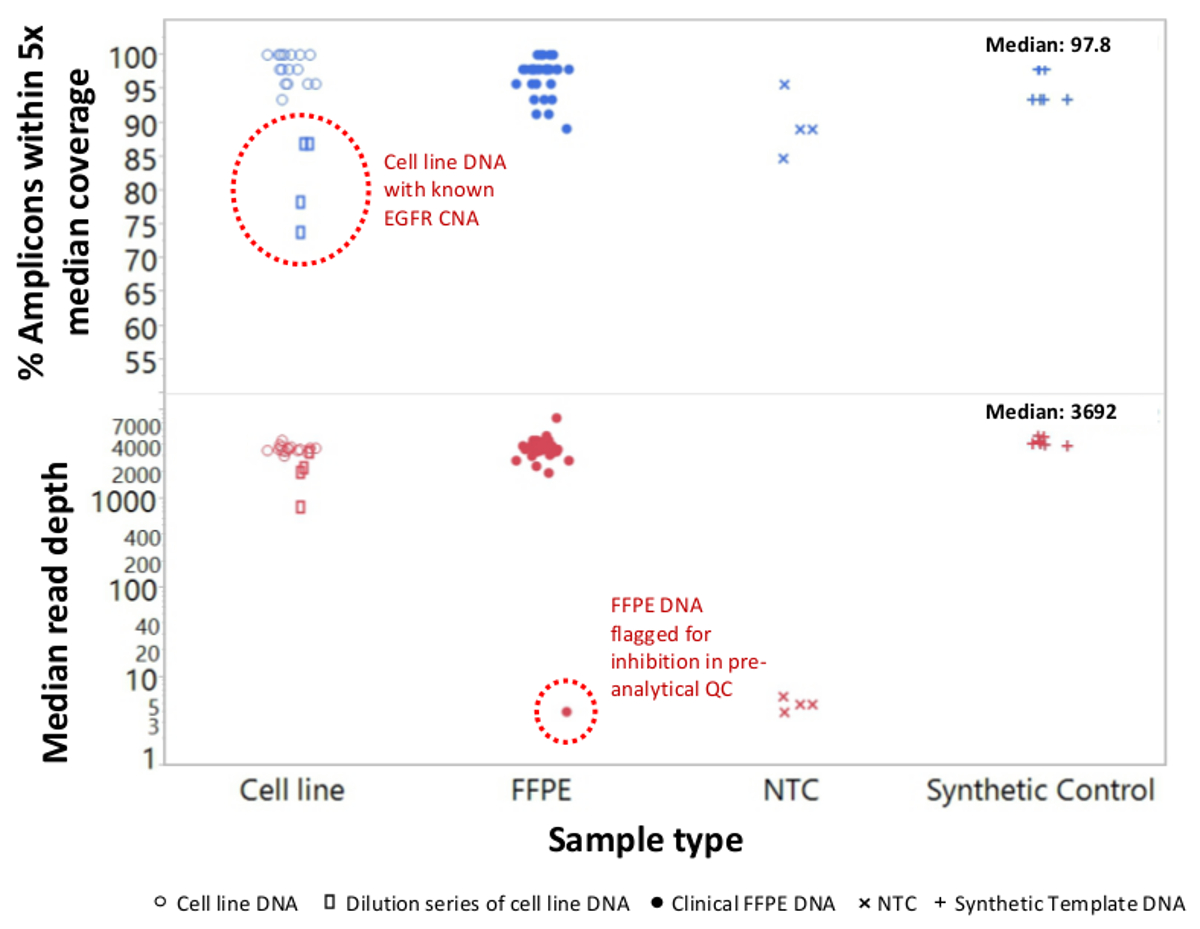
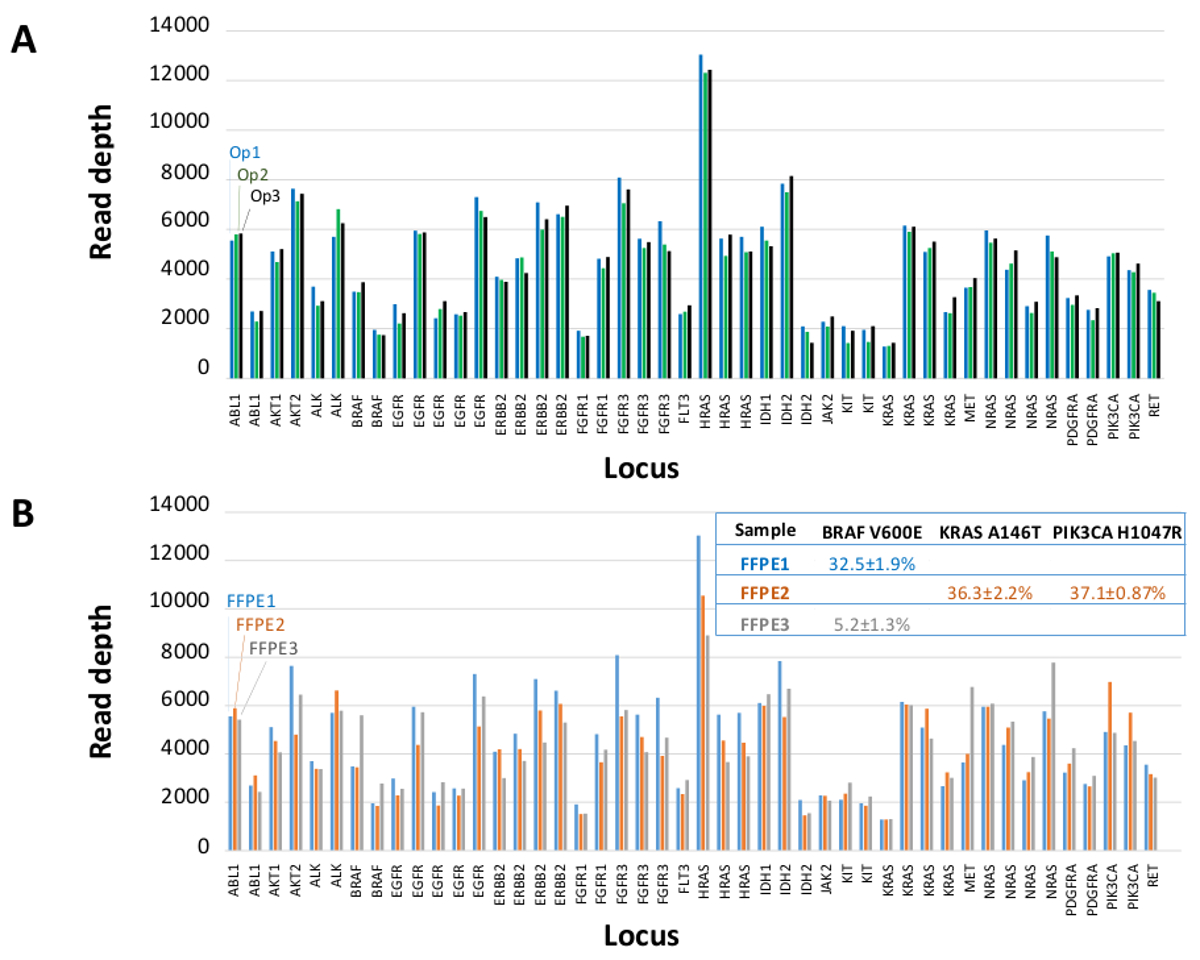
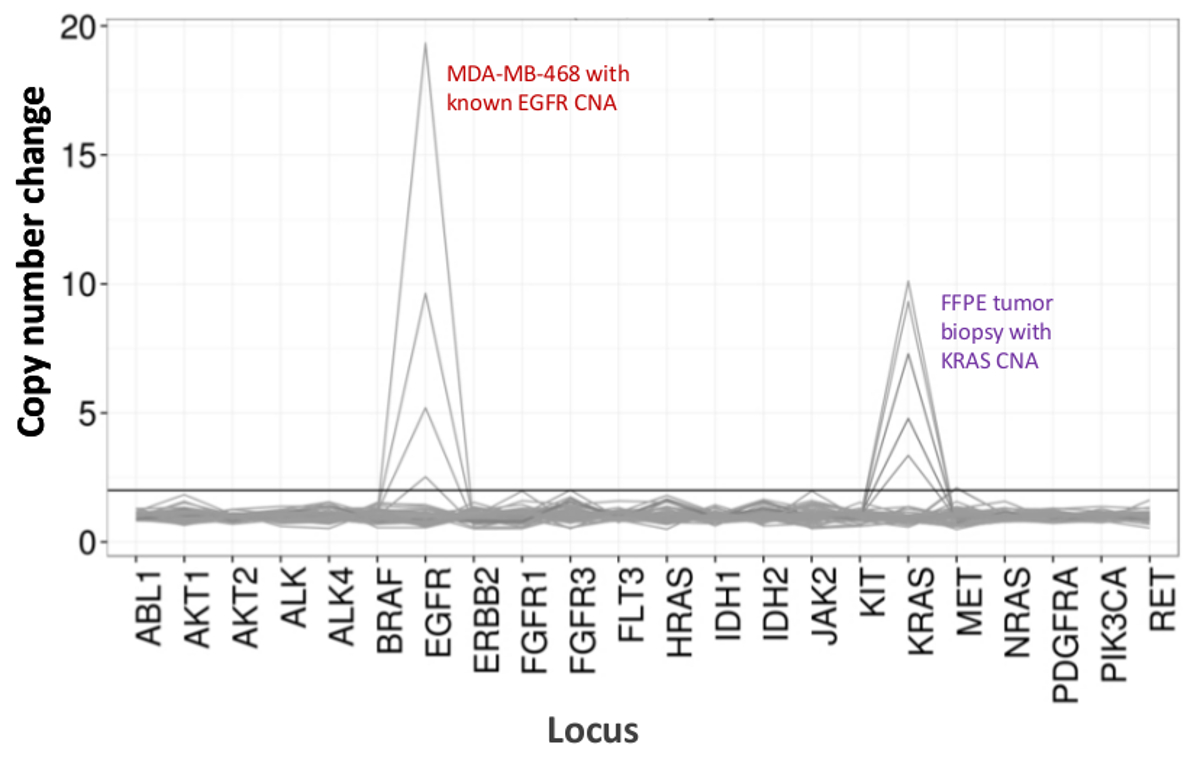
Um total de 90 amostras (74 original) que representam os controlos positivos e negativos, as linhas celulares caracterizadas anteriormente, e biópsias tumorais FFPE clínicos residuais foram avaliadas para o ADN amplificável, a entrada em multiplex enriquecimento de PCR, marcados com adaptadores de sequência, com código de barras, e analisados num único bancada NGS instrumento run (Figura 2) que produziu 19,1 M lê passando filtro. o pool de amostra equimolar resultou na sequenciação de profundidade elevada (3,692x leituras) e uma cobertura uniforme (97,8% de amplicões abrangidos dentro de cinco vezes a profundidade de leitura mediana). Outliers composto não-modelo controla, uma ADN-linha de células com uma grande amplificação do número de cópias, e um ADN que FFPE foi marcado para inibição da PCR através do ensaio de pré-QC analítica (Figura 3). Uniformidade de cobertura entre os 46 amplicons foi mantida utilizando três operadores diferentes (Figura 4A), e por amostras de DNA FFPE diferentes de baixa qualidade ( Figura 4B). Um controlo de ADN tumoral FFPE, formulada a partir de uma mistura de amostras clínicas residuais para atingir 5% V600E BRAF (quantificado por PCR digitais de gotas), foi relatado que têm a mutação BRAF alvo a abundância de 3,9, 5,3, e 6,5% por três operadores usando uma entrada de 400 cópias amplific�eis (e, portanto, apenas 20 cópias mutantes) (Figura 4B e "FFPE3" da tabela inserção). Além disso, uma mistura de 12 moldes de ADN sintéticos, cada um representando uma mutação de base de substituição conhecido "controlador", revelou as mutações esperados no intervalo pretendido de 9 - 17% de frequência de alelos significativo (Tabela 2). Diluição de-linha de células e as amostras de DNA FFPE com número de cópias amplificações demonstrado dependência da dose para as variantes em EGFR e KRAS, respectivamente (Figura 5). Importante, entrada de DNA FFPE poderia ser reduzida para tão pouco quanto 50 cópias amplificáveis ou 1,2 ng de DNA grandes quantidades, embora preservando a detecção de mutações conhecidas, sem falsos positivoschamadas (Figura 6). Entradas de ADN foram acomodados numa gama de 100 vezes de até, pelo menos, 50.000 cópias amplificáveis (Tabela 3). Neste e relacionados experimentos, variante chama em 22 FFPE e 20 espécimes FNA foram relatados de acordo com métodos independentes com cobertura mutação compartilhada (Tabela 4).
A sensibilidade e valor preditivo positivo para o ensaio foi determinada a partir de uma análise de 97 amostras, incluindo FFPE, FNA fresco congelado, e DNA de linha de célula, e um total de 195 resultados de sequenciação. Os resultados revelaram 365 verdadeiros chamadas positivas variantes, 4 chamadas falsos negativos, e uma chamada de falso positivo para uma sensibilidade de 98,9% (IC 95%: 97,1-99,7%) e um valor preditivo positivo (VPP) de 99,7% (IC 95% : 98,2-99,99%). As análises de indels foram realizadas para duas variantes comuns EGFR (p.E746_A750delELREA e p.V769_D770insASV) em 33 amostras-corre, demonstrando uma sensibilidade de 93,9% (IC 95%: 78,4-98,9%) e VPP de 100% (IC 95%: 86,3-100%) com variantes detectada numa gama de 2,4-84,8%.

Figura 1: Um Exemplo de Curves DNA Quantificação de calibração que aprovação e reprovação QC Criteria (A) A passagem da curva padrão.. (B) Uma curva padrão não. Neste caso, o problema foi causado por pipetagem duplicado do padrão de DNA de entrada mais baixo. Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}

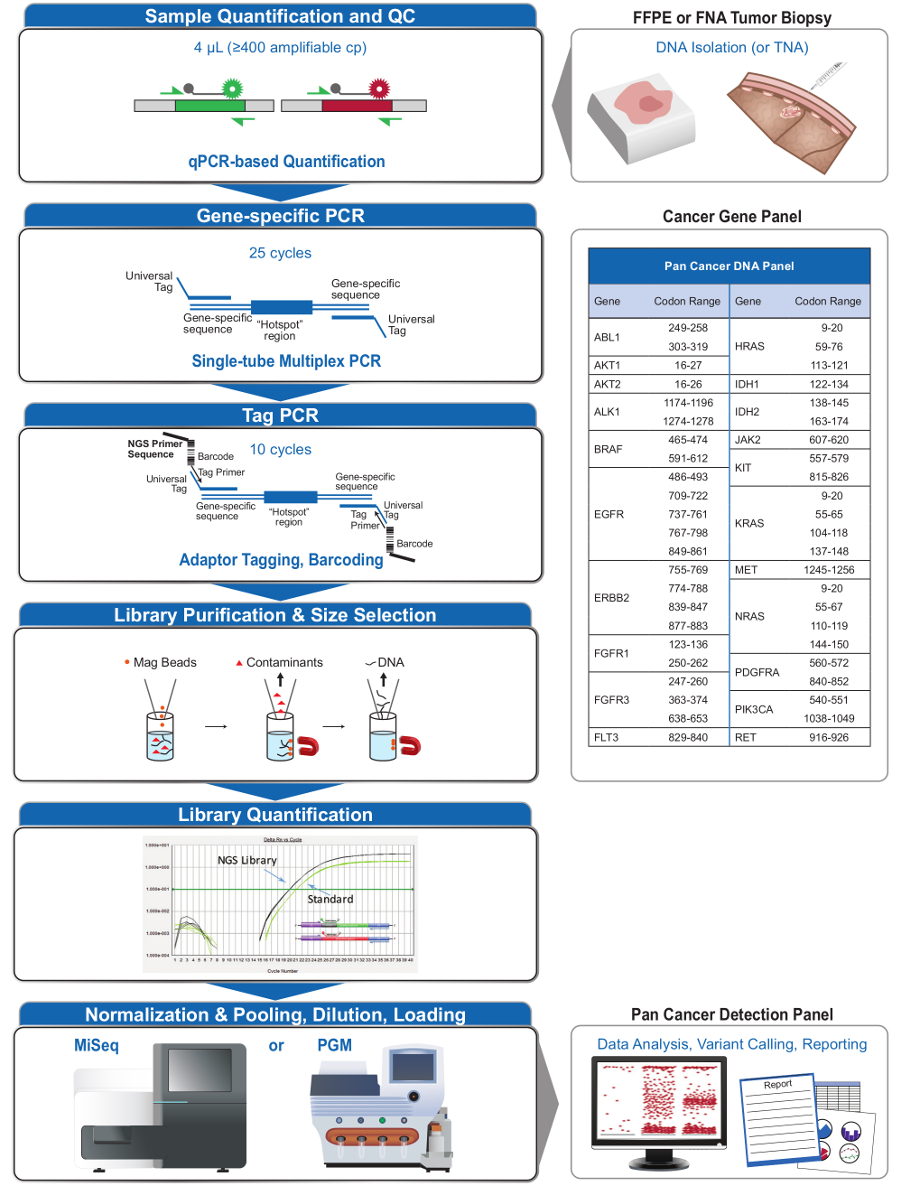
Figura 2: Vista geral de uma abrangente alvejado Sistema NGS for Applications Oncology que Integra Pré-analíticaOs fluxos de trabalho al, analítica e pós-analítica. Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 3: ler a cobertura e uniformidade para NGS-alvo de genes do cancro em DNA FFPE baixa qualidade em comparação com DNA-line celulares intactas e controla um total de 90 amostras que incluíam residual FFPE clínica (círculos fechados),-line celular (círculos abertos. ), e ADN sintético molde (mais símbolos) foram processados utilizando um único tubo, 21 para o gene de PCR multiplex de enriquecimento. Cada biblioteca fragmento amplificado foi marcado com sequências de adaptador para o instrumento NGS, códigos de barras com um código distinto de dupla indexação, purificado, quantificadas e normalizadas para uma concentração de 2,5 nM. A biblioteca de DNA foi sequenciado e analisado pelo software bioinformática de companhia. devi amostrações incluiu uma série de diluições de ADN de linha de células MDA-MB-468 tendo um número de amplificação cópia grande EGFR (superior, retângulos abertos dentro do círculo pontilhada) que distorceram a uniformidade de cobertura e uma amostra melanoma FFPE que não conseguiram gerar um número apreciável de lê devido a transição de inibidores da PCR da extração de DNA. O fracasso da amostra melanoma (parte inferior, círculo pontilhada) foi previsto pelo ensaio qPCR DNA QC pré-analítica. NTC, não-modelo de controle (x símbolos). Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 4: Amplicon-by-amplicon ler a cobertura, uniformidade e Detecção Variant no DNA tumoral FFPE Clínica Residual. (A) Ler cobertura em todos os loci enriquecido em uma amostra de DNA FFPE representante medidoatravés de três operadores diferentes. Operador 1, Op1 (barras azuis); Operador 2, Op2 (barras verdes); Operador 3, OP3 (barras pretas). Chamadas (B) uniformidade de cobertura e variantes avaliadas utilizando três amostras tumorais FFPE, incluindo uma mistura de controlo (FFPE3, bares cinza e texto), composta por um conhecido 5% BRAF c.1799T> Uma mutação. FFPE1 (barras azuis e texto), FFPE2 (barras de laranja e texto). Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 5:. Detecção dose-dependente do número de cópias variantes na linha celular e ADN da linha de células FFPE ADN MDA-MB-468 com um número de amplificação cópia EGFR bem caracterizada foi progressivamente diluída num fundo de uma célula de referência não mutado DNA -line para ilustrar a diminuição da mudança do número de cópias como umfunção da diluição. A percentagem de cada amostra de DNA de linha de célula é mostrada com uma linha distinta (0, 12,5, 25, 50, e 100%). Diluição de uma amostra de ovário tumor FFPE com uma amplificação KRAS conhecido revelaram um perfil similar usando a mesma série de titulação mas com repetições de DNA FFPE 100% para as duas linhas principais. Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 6: Detecção Precisa Mutação e quantificação de 50 cópias amplificável FFPE de ADN, ou 1,2 ng de DNA em massa o número de cópias amplificável de um cancro do cólon ADN FFPE foi determinada pelo ensaio de QC baseada-qPCR, e diluiu-se entre 400 e 25 cópias como um. entrada em multiplex enriquecimento PCR antes da sequenciação. O gasoduto bioinformática corretamente chamado tanto da Varian conhecidots até 50 cópias, ou o equivalente a ~ 10 modelos de mutantes. Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}
| Nome do material / materiais | companhia | Número de catálogo |
| 2x Master Mix Quantidex | Asuragen | 145345 |
| Quant Mix Probe Primer | Asuragen | 145336 |
| Inibição Mix Probe Primer | Asuragen | 145344 |
| ROX | Asuragen | 145346 |
| Diluente | Asuragen | 145339 |
| ADN padrão (50) | Asuragen | 145340 |
| ADN padrão (10) | Asuragen | 145341 |
| ADN padrão (2) | Asuragen | 145342 |
| ADN padrão (0,4) | Asuragen | 145343 |
| 2x Master Mix amplificação | Asuragen | 145348 |
| Painel Pan Cancer Primer | Asuragen | 145347 |
| Controle Pan Cancer FFPE | Asuragen | 145349 |
| Controle Pan Cancer Multi-Variant | Asuragen | 145350 |
| Biblioteca Beads Prep Pure | Asuragen | 145351 |
| Tampão de lavagem | Asuragen | 145352 |
| Tampão de eluição | Asuragen | 145353 |
| 2x Master Mix LQ | Asuragen | 145358 |
| LQ diluente | Asuragen | 145354 |
| LQ positivaAo controle | Asuragen | 145355 |
| LQ padrão | Asuragen | 145356 |
| LQ Primer Probe Mix / (ILMN) | Asuragen | 145357 |
| LQ ROX | Asuragen | 145359 |
| Códigos de índice (ILMN) - Conjunto A | Asuragen | 150004 |
| AIL001 - AIL048 (48) | ||
| Códigos de índice (ILMN) - Conjunto B | Asuragen | 150005 |
| AIL049 - AIL096 (48) | ||
| 2x Master Mix Índice | Asuragen | 145361 |
| Leia 1 Sequencing Primers | Asuragen | 150001 |
| Índice Leia Sequencing Primers | Asuragen | 150002 |
| Leia 2 Sequencing Primers | Asuragen | 150003 |
| diluente Sequencing | Asuragen | 145365 |
| Illumina MiSeq | Illumina | |
| MiSeq Kit de Reagentes V3 (600-ciclo) | Illumina | MS-102-3003 |
| MiSeq Reagente Nano Kit V2 (300-ciclo) | Illumina | MS-103-1001 |
| Phix Controle v3 | Illumina | FC-110-3001 |
| Stand-96 magnética (ou dispositivo equivalente) | Ambion | AM10027 |
| Quantidex Software Reporter | Asuragen |
Tabela 1:. Os reagentes e kits Após a primeira utilização do ROX, armazenar o frasco a 2-8 ° C. Não recongelar. Software pode ser baixado em www.asuragen.com.
| Geno | variante COSMIC | Aminoácido COSMIC | % Variant |
| ARN | c.182A> L | p.Q61R | 13,3 |
| ARN | c.35G> A | p.G12D | 15.2 |
| HRAS | c.182A> L | p.Q61R | 17,8 |
| HRAS | c.35G> A | p.G12D | 9.2 |
| KRAS | c.182A> L | p.Q61R | 13,5 |
| KRAS | c.35G> A | p.G12D | 19.1 |
| PIK3CA | c.1633G> A | p.E545K | 9.3 |
| PIK3CA | c.3140A> L | p.H1047R | 9.1 |
| KIT | c.2447A> T | p.D816V | 14,6 |
| EGFR | c.2369C> T | p.T790M | 11.3 |
| EGFR | c.2573T> L | p.L858R | 14,9 |
| BRAF | c.1799T> A | p.V600E | 17,3 |
Tabela 2: A Synthetic Controle Pooled é composto por 12 "Driver" Câncer Gene variantes que estão quantificados a 9-17% Abundância Uma mistura de 12 diferentes modelos sintéticos de cadeia dupla de rolamento 12 mutações distintas foi avaliada após o seqüenciamento.. Todas as variantes foram corretamente chamado sem falsos positivos.
>% Variant| amostra ID | Funcional cps | Gene | variante COSMIC | Aminoácido COSMIC | Profundidade de leitura médio | % Dentro de 5x mediana | |
| BCPAP | 400 | BRAF | c.1799T> A | p.V600E | 99,5 | 3289 | 96% |
| BCPAP | 10.000 | BRAF | c.1799T> A | p.V600E | 99,7 | 4040 | 98% |
| BCPAP | 25.000 | BRAF | c.1799T> A | p.V600E | 99,4 | 3687 | 96% |
| BCPAP | 50.000 | BRAF | c.1799T> A | p.V600E | 99,7 | 4611 | 93% |
Tabela 3: Cobertura e Variante de chamada são preservados em uma faixa> 100 vezes de entrada DNA. ADN amplificável a partir de uma linha celular foi BCPAP entrada para o enriquecimento de PCR em multiplex em 400 a 50.000 cópias e sequenciado. profundidade ler, uniformidade de cobertura, detecção de mutações, e precisão mutação foram preservados em toda a gama de entrada.

Tabela 4: Variante Calls em 22 FFPE e 20 biópsias de tumores FNA Concordo com os resultados de Independent Mutation Ensaios Um conjunto de 22 DNA tumoral FFPE com estado de mutação previamente determinado por ensaios ortogonais NGS alvo foi a entrada no 400 a 2.928 cópias amplific�eis para o enriquecimento de PCR. passo e sequenciados utilizando o painel Cancer Pan de 21 gene. Além disso, um grupo de 20 amostras de DNA de FNA caracterizada anteriormente utilizando um ensaio de mutação matriz do grânulo líquido 8 foi amplificado por PCR usando 156 a 36080 cópias de entrada amplificáveis e sequenciado. Todos os que se sobrepõem as chamadas entre o painel de Pan Cancer NGS eo REFERÊmétodos NCE estavam de acordo. Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}
Discussão
Tecnologias NGS redefiniram as expectativas para interrogar os perfis moleculares de biópsias de tumores em ambientes clínicos 9. Uma série de painéis NGS alvo foram desenvolvidos como tecnologias de pesquisa, testes de laboratório desenvolvido, e produtos comercialmente disponíveis e painéis personalizados para avaliar vários tipos de amostras clínicas 3,5,10-15. Relatórios de vários estudos têm demonstrado o valor da NGS como uma ferramenta clínica sensível e específico para a detecção de alterações genômicas 3,10-12,14. No entanto, estudos também têm demonstrado o risco de artefatos que podem causar resultados falso-positivos de biópsias de câncer desafiadoras, como FFPE espécimes 2-5,12,14. Além disso, publicações recentes têm destacado altas taxas de insucesso para NGS utilizando oncologia espécimes 16 e falso-positivos chamadas com painéis comerciais direcionados NGS que são agravadas pelo uso de DNA de baixa entrada de 12. Como resultado, alguns laboratories foram modificados ou adicionados freios e contrapesos adicionais para comercialmente disponíveis tecnologias NGS direcionados para melhorar o desempenho, muitas vezes para garantir a precisão ou confirmar os resultados da bioinformática análises 5,10,11.
O painel de 21-gene (Figura 2) foi desenvolvido como conteúdo direcionado para um sistema NGS abrangente para interrogar, mutações viáveis baseadas em evidências em tipos de amostras desafiadoras, como FFPE e biópsias de tumores FNA. O fluxo de trabalho tem uma série de benefícios: 1) consistência, oferecendo cobertura amplicon uniforme (Figuras 3 e 4, Tabela 3); 2) a facilidade de utilização, fornecendo pré-formulado, conjuntos de reagentes otimizados, e simplificando os requisitos de bioinformática; 3) a eficiência através da racionalização do fluxo de trabalho e reduzir o número de etapas de pipetagem, em comparação com outros métodos NGS comerciais; e 4) a precisão através da incorporação de um ensaio de DNA QC para avaliar o amplifinúmero de cópias de DNA capaz de assegurar a diversidade modelo aceitável e evitar flutuações estocásticos na detecção variante 4. A integração dos dados QC pré-analítica com a análise bioinformática permite a baixas entradas de DNA FFPE. Isto foi conseguido através da formação de um algoritmo de árvore de decisão utilizando diferentes cópias funcionais de entrada de DNA em toda 400 amostras FFPE com medidas independentes de verdade, e incorporar este algoritmo no software de bioinformática. Como resultado, a entrada recomendada de 400 cópias amplificáveis, tipicamente o equivalente a ~ 5-20 ng de DNA FFPE, compara-se favoravelmente com outros métodos de 10-12, incluindo o enriquecimento à base de hibridização, onde ~ 250 ng de DNA FFPE é recomendado 17,18 . Embora a técnica está descrita, para utilização numa plataforma MiSeq, ele pode ser modificado usando Tag iniciadores de PCR com os adaptadores específicos do aparelho para permitir a análise da sequência em outras plataformas NGS.
Vários passos são fundamentais para garantir o sucessodo processo. O ensaio de pré-QC analítica determina o número de cópias de ADN amplificável e relatórios inibição funcional. No entanto, se a menos de 400 cópias de DNA amplificáveis são usados no passo de enriquecimento de PCR, existe um risco aumentado de uma chamada falso-negativo a partir de amostras com mutações baixa abundância (Figura 6). Além disso, deve ser tomado cuidado durante a purificação biblioteca para evitar o excesso de secagem das esferas magnéticas durante os passos de lavagem ou de eluição. Além disso, a biblioteca de sucesso quantificação é altamente dependente da diluição exacta do ADN da biblioteca. Para o melhor resultado, a diferença do qPCR resultados para a biblioteca de samples (CQ FAM) em comparação com o LQ Padrão (CQ VIC) deve ser ≤3.3 Cq. Se a diferença é maior do que 3,3 Cq, re-diluição e teste da amostra é recomendado. Embora tenha sido observada uma excelente correlação entre este método qPCR competitiva e kits comerciais que oferecem quantificação absoluta utilizando uma curva padrão, Um deslocamento da entrada da biblioteca para o passo de amplificação clonal em relação a outros métodos podem ser necessários para alcançar densidade de sementeira óptima.
Alguns espécimes cancerosas são particularmente desafiador para sequenciar por causa de inibidores que persistem após o isolamento de DNA. Para identificar estas amostras antes da preparação da biblioteca, o ensaio de qPCR QC também detecta inibição da amplificação pela inclusão de um molde exógeno que serve como um controlo interno e uma sentinela para a inibição funcional. Um exemplo é apresentado na Figura 3 em que uma amostra de DNA de melanoma não conseguiu passar a inibição QC métrica antes da sequenciação e, em seguida, não conseguiu gerar uma biblioteca que pode ser sequenciado. A falha foi provavelmente uma consequência de contaminação de melanina, um inibidor de PCR conhecidos, realizado a partir da etapa de isolamento de DNA FFPE. As amostras que são identificados pelo ensaio de QC para estar em risco de insuficiência de amplificação pode ser recuperada através de um passo extra t limpezao remover potenciais inibidores.
O painel de 21 gene alvo se concentra em hotspots de genes baseados em evidências e fornece um sistema completo com reagentes otimizados e controles para DNA QC, NGS e bioinformática software que é informado pelo pré-analíticos resultados DNA de quantificação "funcionais". O método detecta com precisão mutações de base de substituição e indels a partir de ADN de baixo de entrada, e fornece um exemplo de um sistema de NGS com a opção de expandir o conteúdo do painel, para detectar variantes adicionais, tais como CNVs e ser adaptada para a sequenciação de ARN alvo.
Divulgações
JH, AH, RZ, BCH, e GJL estão empregados e têm a propriedade de ações em Asuragen, Inc. RZ, BCH, e GJL são co-inventores em um pedido de patente para melhorar variante chamando usando amplificável informações número de cópias determinado para cada amostra.
Agradecimentos
Agradecemos ao Dr. Annette Schlageter para revisão do manuscrito. Este trabalho foi apoiado em parte por CP120017 bolsa da Prevenção do Câncer e Instituto de Pesquisa do Texas (PI: GJL).
Materiais
| Name | Company | Catalog Number | Comments |
| 2x Quantidex Master Mix | Asuragen | 145345 | |
| Quant Primer Probe Mix | Asuragen | 145336 | |
| Inhibition Primer Probe Mix | Asuragen | 145344 | |
| ROX | Asuragen | 145346 | |
| Diluent | Asuragen | 145339 | |
| DNA Standard (50) | Asuragen | 145340 | |
| DNA Standard (10) | Asuragen | 145341 | |
| DNA Standard (2) | Asuragen | 145342 | |
| DNA Standard (0.4) | Asuragen | 145343 | |
| 2X Amplification Master Mix | Asuragen | 145348 | |
| Pan Cancer Primer Panel | Asuragen | 145347 | |
| Pan Cancer FFPE Control | Asuragen | 145349 | |
| Pan Cancer Multi-Variant Control | Asuragen | 145350 | |
| Library Pure Prep Beads | Asuragen | 145351 | |
| Wash Buffer | Asuragen | 145352 | |
| Elution Buffer | Asuragen | 145353 | |
| 2X LQ Master Mix | Asuragen | 145358 | |
| LQ Diluent | Asuragen | 145354 | |
| LQ Positive Control | Asuragen | 145355 | |
| LQ Standard | Asuragen | 145356 | |
| LQ Primer/Probe Mix (ILMN) | Asuragen | 145357 | |
| LQ ROX | Asuragen | 145359 | |
| Index Codes (ILMN) - Set A, AIL001 - AIL048 (48) | Asuragen | 150004 | |
| Index Codes (ILMN) - Set B, AIL049 - AIL096(48) | Asuragen | 150005 | |
| 2x Index Master Mix | Asuragen | 145361 | |
| Read 1 Sequencing Primers | Asuragen | 150001 | |
| Index Read Sequencing Primers | Asuragen | 150002 | |
| Read 2 Sequencing Primers | Asuragen | 150003 | |
| Sequencing Diluent | Asuragen | 145365 | |
| Illumina MiSeq | Illumina | ||
| MiSeq Reagent Kit v3 (600-cycle) | Illumina | MS-102-3003 | |
| MiSeq Reagent Nano Kit v2 (300-cycle) | Illumina | MS-103-1001 | |
| PhiX Control v3 | Illumina | FC-110-3001 | |
| Magnetic Stand-96 (Or equivalent device) | Ambion | AM10027 | |
| Quantidex Reporter Software | Asuragen |
Referências
- Chen, G., Mosier, S., Gocke, C. D., Lin, M. T., Eshleman, J. R. Cytosine deamination is a major cause of baseline noise in next-generation sequencing. Mol Diagn Ther. 18 (5), 587-593 (2014).
- Choudhary, A., et al. Evaluation of an integrated clinical workflow for targeted next-generation sequencing of low-quality tumor DNA using a 51-gene enrichment panel. BMC Med Genomics. 7, 62 (2014).
- Sah, S., et al. Functional DNA quantification guides accurate next-generation sequencing mutation detection in formalin-fixed, paraffin-embedded tumor biopsies. Genome Med. 5 (8), 77 (2013).
- Zhang, L., et al. Profiling cancer gene mutations in clinical formalin-fixed, paraffin-embedded colorectal tumor specimens using targeted next-generation sequencing. Oncologist. 19 (4), 336-343 (2014).
- Latham, G. J. Next-generation sequencing of formalin-fixed, paraffin-embedded tumor biopsies: navigating the perils of old and new technology to advance cancer diagnosis. Expert Rev Mol Diagn. 13 (8), 769-772 (2013).
- Crawford, J. M., et al. The business of genomic testing: a survey of early adopters. Genet Med. 16 (12), 954-961 (2014).
- Smith, D. L., et al. A multiplex technology platform for the rapid analysis of clinically actionable genetic alterations and validation for BRAF p.V600E detection in 1549 cytologic and histologic specimens. Arch Pathol Lab Med. 138 (3), 371-378 (2014).
- Thomas, F., Desmedt, C., Aftimos, P., Awada, A. Impact of tumor sequencing on the use of anticancer drugs. Curr Opin Oncol. 26 (3), 347-356 (2014).
- Singh, R. R., et al. Clinical validation of a next-generation sequencing screen for mutational hotspots in 46 cancer-related genes. J Mol Diagn. 15 (5), 607-622 (2013).
- Beadling, C., et al. Combining highly multiplexed PCR with semiconductor-based sequencing for rapid cancer genotyping. J Mol Diagn. 15 (2), 171-176 (2013).
- McCall, C. M., et al. False positives in multiplex PCR-based next-generation sequencing have unique signatures. J Mol Diagn. 16 (5), 541-549 (2014).
- Schleifman, E. B., et al. Next generation MUT-MAP, a high-sensitivity high-throughput microfluidics chip-based mutation analysis panel. PLoS One. 9 (3), e90761 (2014).
- Wong, S. Q., et al. Sequence artefacts in a prospective series of formalin-fixed tumours tested for mutations in hotspot regions by massively parallel sequencing. BMC Med Genomics. 7, 23 (2014).
- Narayan, A., et al. Ultrasensitive measurement of hotspot mutations in tumor DNA in blood using error-suppressed multiplexed deep sequencing. Cancer Res. 72 (14), 3492-3498 (2012).
- Hagemann, I. S., et al. Clinical next-generation sequencing in patients with non-small cell lung cancer. Cancer. 121 (4), 631-639 (2015).
- Won, H. H., Scott, S. N., Brannon, A. R., Shah, R. H., Berger, M. F. Detecting somatic genetic alterations in tumor specimens by exon capture and massively parallel sequencing. J Vis Exp. (80), e50710 (2013).
- Simen, B. B., et al. Validation of a next-generation-sequencing cancer panel for use in the clinical laboratory. Arch Pathol Lab Med. 139 (4), 508-517 (2015).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados